

Abstract
This report describes the development of an enantioselective C–N bond-forming reaction to produce 1,2,3,4-tetrahydroisoquinoline-derived cyclic aminals catalyzed by chiral phosphate anions. Central to the success of this goal was the design of a library of 3,3′-triazolyl BINOL-derived phosphoric acids capable of forming attractive hydrogen-bonding interactions with the peptide-like substrate. We envision this work will offer an alternative to the conventional strategy of increasing catalyst steric bulk to improve enantioselectivity with BINOL-derived phosphoric acids.
In recent years, chiral anions have seen increasing application as stereocontrolling elements in catalytic asymmetric reactions.1 Theoretically, chiral counteranion catalysis has the potential to be a general strategy, as the only prerequisite for asymmetric induction is the presence of a cationic reaction intermediate capable of forming a tight ion pair with the chiral counteranion. Among the catalysts employed to this end, the conjugate bases of axially chiral phosphoric acids (PAs), initially reported by Akiyama and Terada,2 have enjoyed considerable success in both transition metal-catalyzed3 and metal-free reactions.4 Although controversy can exist as to whether the association between the catalyst and substrate is purely electrostatic in nature,1,5 this concept has provided an intriguing approach to reaction design.
Our group has an ongoing interest in applying this strategy to outstanding challenges in organic synthesis. To this end, we recognized that chiral counteranion catalysis might provide a novel approach to enantioselective cross-dehydrogenative coupling (CDC), a term coined by Li6a to describe the functionalization of C–H bonds alpha to heteroatoms in the presence of a hydrogen acceptor (Scheme 1A). To date, this methodology has proven amenable to a range of nucleophiles and oxidants,6 including several recent reports of enantioselective variants.7 Mechanistically, CDC reactions are proposed to proceed via initial substrate oxidation to a cationic, unsaturated intermediate (1) that undergoes subsequent inter- or intramolecular nucleophilic attack.8 Given our group’s recent success in developing enantioselective halogenation reactions by exploiting chiral phosphate anion phase-transfer catalysis with cationic halogenating reagents,9 we envisioned that a chiral phosphate (2, Scheme 1B) could undergo anion exchange with an appropriately chosen cationic oxidant, ensuring that the phosphate would be in close proximity to form a tight ion-pair (3) with the substrate upon oxidation. Preferential attack on one of the prochiral faces of the resulting ionic complex would provide an enantioenriched product (4) and release the chiral phosphate.
Scheme 1.

Enantioselective CDC Hypothesis
We elected to initiate our studies using oxopiperidinium salts as the cationic oxidants,10 as these reagents have demonstrated utility in a number of dehydrogenative coupling processes,11 although no asymmetric variants have been reported to date. Thus, beginning with amide-tethered tetrahydroisoquinoline substrate 5a and oxoammonium salt 7, we screened a number of conventional axially chiral PAs (Table 1). However, none of the catalysts initially tested afforded useful levels of enantioselectivity, despite providing promising conversions to the desired product 6a (Table 1, entries 1–5). We reasoned that although the putative substrate–phosphate complex 3 may be forming, the chiral environment created by these catalysts was inadequate to differentiate the prochiral faces of the substrate, leading us to consider alternative interactions beyond sterics that might influence enantioselectivity.
Table 1.
Selected Catalyst Optimization Data for Enantioselective CDC Amidationa
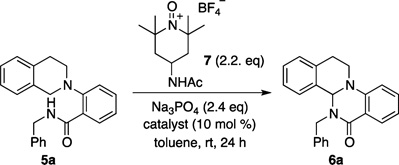 | |||
|---|---|---|---|
| entry | catalyst | conversion (%)b | ee (%)c |
| 1 | (S)-C8-TRIP | 86 | 8 |
| 2 | (S)-TCYP | 95 | 16 |
| 3 | (R)-STRIP | 64 | 30 |
| 4 | (R,R)-Ph-DAP | 69 | −10d |
| 5 | (S)-VAPOL PA | 97 | 31 |
| 6 | (S)-9b | 99 | −69 |
| 7 | (S)-9g | 92 | −78 |
| 8 | (S)-9h | 92 | −80 |
| 9 | (S)-9i | 91 | −77 |
| 10 | (S)-9l | 75 | −81 |
| 11e | (S)-9l | 91(83)f | −84 |
| 12 | none | 82 | nd |
See Supporting Information for complete optimization data.
Determined by HPLC using 1,4-dinitrobenzene internal standard.
Determined by chiral HPLC.
Negative sign indicates that opposite enantiomer predominates.
p-Xylene was used as solvent with 5 mol % catalyst.
Value in parentheses reflects isolated yield.
In chiral PA/phosphate catalysis, asymmetric induction has generally been achieved by strategically installing bulky groups that serve to project the catalyst’s axial chirality (Figure 1A), coercing the prochiral substrate into a position within the chiral pocket that minimizes unfavorable steric interactions with the catalyst (Figure 1B).12 Thus, achieving high levels of enantioselectivity often hinges on the identification and synthesis of catalysts bearing increasingly large substituents. Although remarkable success has been realized using this strategy,13 the enzyme-inspired principle of preferentially stabilizing one diastereomeric transition state via attractive, non-covalent interactions14 has not been explored in the realm of PA catalysis, despite having seen great success with other classes of organic catalysts. In principle, judiciously chosen atoms or groups incorporated at the 3 and 3′ positions of the binaphthyl backbone would be appropriately positioned to form attractive interactions with a substrate at a site remote from the phosphate moiety, providing an alternative type of organizational element (Figure 1C). With respect to both modularity and function, the 1,2,3-triazole seems well suited to this purpose.15
Figure 1.

Axially chiral PA design principles.
The advent of click chemistry,16 in particular the copper-catalyzed azide/alkyne cycloaddition (CuAAC),17 has provided a tool for rapid generation of 1,2,3-triazole-containing compounds that have found widespread application. From the standpoint of modular catalyst design, the click chemistry approach is ideal in that the broad alkyne/azide scope allows for the facile preparation of numerous sterically and electronically diverse derivatives from a common intermediate. In addition to synthetic accessibility, the 1,2,3-triazole displays interesting hydrogen-bonding properties, as exemplified by its ability to serve as an effective anion binder18 and peptide bond surrogate.19 With respect to this latter feature, we hypothesized that, in addition to the ionic phosphate–substrate association, the amide bond-containing substrate 5 might undergo attractive hydrogen-bonding interaction with the triazole, resulting in additional rigidity within the chiral pocket created by the catalyst and stabilization of one diastereomeric transition state.
To this end, a library of 3,3′-triazolyl-BINOL-derived PAs was prepared using an operationally simple two-pot procedure (Scheme 2, 9a–l). In this manner, all derivatives were synthesized from a single intermediate (bis-alkyne 8), allowing rapid access to catalysts bearing a range of N-1 aryl and alkyl substituents. It was subsequently determined that lipophilic groups at the 6 and 6′ positions were necessary in order to improve the solubility of 9 in common organic solvents.
Scheme 2.

Synthesis of Catalyst Librarya
aSee Supporting Information for detailed experimental procedures and reaction conditions.
We were pleased to find that when these novel catalysts were tested in our model reaction, a significant increase in enantioselectivity was observed while maintaining excellent levels of conversion (Table 1, entries 6–11). Notably, the major enantiomer produced using the triazolyl PA catalysts was opposite to that produced using the purely steric PA catalysts in all cases (vide infra). Furthermore, we were surprised to discover that the reaction reached comparable conversion even in the absence of the PA (Table 1, entry 12), suggesting a remarkable acceleration of the enantiodetermining step in the presence of the catalyst. A series of experiments revealed that N-acyl oxoammonium salt 7, tribasic sodium phosphate, and p-xylene provided the optimal balance of yield and enantioselectivity (see Supporting Information for details).
Using this set of conditions, the scope of this process was explored. Benzylic groups bearing both electron-rich and electron-poor groups at the para position were tolerated on the nucleophilic nitrogen atom (Table 2, 6a–d), resulting in both good yields and enantioselectivities. Furthermore, ortho-methoxy-substituted benzylic substrate 6e afforded the product in excellent enantiomeric excess (ee). We next turned our attention to substrates bearing N-aryl groups. Although only modest selectivities were observed using N-phenyl substrate 6f and its 3-methyl-substituted analogue 6g, switching to ortho substitution (6h) resulted in a dramatic increase in enantioselectivity, although reactivity was sluggish. A range of ortho functional groups were tolerated, including naphthyl (6i), chloro (6j), and, perhaps surprisingly, hydroxyl (6k). Moreover, substrates with bulky alkyl substituents underwent the transformation with good enantioselectivity (6l, m). Additionally, the tetrahydroisoquinoline N-aryl ring could be substituted with both electron-donating and -withdrawing groups, while still maintaining excellent levels of enantioselectivity (6n–p).
Table 2.
Substrate Scope for Enantioselective CDC
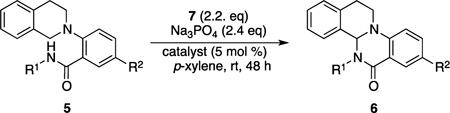 | |||||
|---|---|---|---|---|---|
| entry | R1 | R2 | catalyst | yielda | eeb |
| 1 | Bn (6a) | H | 9l | 83 | 84 |
| 2 | 4-Me-Bn (6b) | H | 9gc | 81 | 87 |
| 3 | 4-OMe-Bn (6c) | H | 9g | 89 | 86 |
| 4 | 4-NO2Bn (6d) | H | 9g | 90 | 88 |
| 5 | 2-OMe-Bn (6e) | H | 9h | 81 | 94 |
| 6 | Ph (6f) | H | 9l | 86 | 63 |
| 7 | 3-Me-Ph (6g) | H | 9l | 91 | 60 |
| 8 | 2-Me-Ph (6h) | H | 9l | 65 | 91 |
| 9 | 1-naphthyl (6i) | H | 9l | 69 | 82 |
| 10 | 2-Cl-Ph (6j) | H | 9l | 42 | 87 |
| 11 | 2-OH-Ph (6k) | H | 9b | 38 | 80 |
| 12 | tBu (6l) | H | 9l | 82 | 73 |
| 13 | c-C6H11 (6m) | H | 9g | 93 | 80 |
| 14 | 2-Me-Ph (6n) | Me | 9l | 49 | 90 |
| 15 | 2-Me-Ph (6o) | Br | 9l | 55 | 93 |
| 16 | 2-Me-Ph (6p) | CF3 | 9l | 51 | 90 |
| 17 | H | 9h | 88 | (7:1 d.r.)d | |
| 18 | H | 9h | 91 | (1:3 d.r.)d | |
Isolated yield. All reactions conducted on 0.1 mmol scale with respect to substrate.
Determined by chiral HPLC.
Catalyst 9l gave the product with 64% ee.
Diastereomeric ratio (d.r.) determined by analysis of crude 1H NMR spectrum.
Finally, both antipodes of the amino acid valine were incorporated into the substrate scaffold as their corresponding methyl esters and subjected to the standard reaction conditions (Table 2, entries 17 and 18). In the absence of catalyst, a 1:1 mixture of diastereomers was produced. However, when 9h was employed, l-valine-derived substrate 5q produced the product 6q as a 7:1 mixture of diastereomers, while d-valine-derived substrate 5r afforded a 3:1 mixture in favor of the opposite diastereomer 6r, demonstrating the catalyst’s ability to control the absolute configuration of the newly formed stereocenter in the presence of an existing one.
Having established the utility of catalysts 9 in this CDC amidation reaction, we wished to gain insight into the role that the triazole substituents may be playing in asymmetric induction. Thus, pyrazolyl (10a) and imidazolyl (10b) PAs were prepared (Scheme 3), which were expected to impose steric environments similar to that of 9l, but with inherently different electronic properties. When these isosteric analoges were tested in the CDC reaction with substrate 5a, similar levels of conversion were observed, but with significantly reduced enantioselectivity compared to 9l, albeit with the same sense of enantioinduction. The observation that 9a–l (Table 1, entries 1–5 and Supporting Information), 10a, and 10b all provide the product enantiomer opposite that given by conventional PAs may be rationalized by either (1) attractive interactions between the heterocyclic substituents and substrate that override the selectivity based purely on sterics or (2) a fundamentally different steric environment created by these catalysts (Table 1, entries 1–5).20,21 However, the diminished enantioselectivites afforded by 10a and 10b vs 9l suggest that electronic rather than steric properties of the triazolyl substituents in catalysts 9 are primarily responsible for the high enantioselectivities reported herein.22
Scheme 3.

Enantioselective CDC Using Heterocyclic 1,2,3-Triazole Isosteres
In conclusion, we have used click chemistry to synthesize a library of axially chiral PAs based on BINOL bearing 1,2,3-triazoles at the 3 and 3′ positions. Using these molecules as catalysts, we were able to achieve enantioselectivities in a novel C–N bond-forming reaction that were unattainable using conventional PAs. These results suggest that, in addition to being an easily modified catalyst substituent, the triazole may serve an organizational function other than simply creating a sterically hindered environment around the catalyst active site. Future work will be aimed at elucidating the specific interactions responsible for this divergent selectivity.
Supplementary Material
ACKNOWLEDGMENTS
We thank the University of California, Berkeley, Amgen and the NIHGMS (RO1 GM104534) for financial support. J.P.H. gratefully acknowledges the support from the German National Academy of Sciences, Leopoldina, through a postdoctoral fellowship.
Footnotes
ASSOCIATED CONTENT
Supporting Information
Experimental details and characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- 1.For recent reviews on chiral anions in asymmetric catalysis, see: Phipps RJ, Hamilton GL, Toste FD. Nature Chem. 2012;4:603. doi: 10.1038/nchem.1405. Mahlau M, List B. Angew Chem., Int. Ed. 2013;52:518. doi: 10.1002/anie.201205343. Brak K, Jacobsen EN. Angew Chem., Int. Ed. 2013;52:534. doi: 10.1002/anie.201205449.
- 2.For recent reviews on axially chiral PA catalysts, see: Terada M. Curr. Org. Chem. 2011;15:2227. Terada M. Synthesis. 2010;12:1929. Akiyama T. Chem. Rev. 2007;107:5744. doi: 10.1021/cr068374j.
- 3.(a) Hamilton GL, Kang EJ, Mba M, Toste FD. Science. 2007;317:496. doi: 10.1126/science.1145229. [DOI] [PubMed] [Google Scholar]; (b) Mukherjee S, List B. J. Am. Chem. Soc. 2007;129:11336. doi: 10.1021/ja074678r. [DOI] [PubMed] [Google Scholar]; (c) Rueping M, Antonchick A, Brinkmann C. Angew. Chem., Int. Ed. 2007;46:6903. doi: 10.1002/anie.200702439. [DOI] [PubMed] [Google Scholar]; (d) Liao S, List B. J. Am. Chem. Soc. 2010;49:628. doi: 10.1002/anie.200905332. [DOI] [PubMed] [Google Scholar]; (e) Jiang G, Halder R, Fang Y, List B. Angew. Chem., Int. Ed. 2011;50:9752. doi: 10.1002/anie.201103843. [DOI] [PubMed] [Google Scholar]; (f) Rauniyar V, Wang Z, Burks H, Toste FD. J. Am. Chem. Soc. 2011;133:8486. doi: 10.1021/ja202959n. [DOI] [PubMed] [Google Scholar]; (g) Chai Z, Rainey T. J. Am. Chem. Soc. 2012;134:3615. doi: 10.1021/ja2102407. [DOI] [PubMed] [Google Scholar]; (h) Zbieg J, Yamaguchi E, McInturff E, Krische M. Science. 2012;336:324. doi: 10.1126/science.1219274. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Ohmatsu K, Ito M, Kunieda T, Ooi T. Nature Chem. 2012;4:473. doi: 10.1038/nchem.1311. [DOI] [PubMed] [Google Scholar]
- 4.For seminal work, see: Mayer S, List B. Angew. Chem., Int. Ed. 2006;45:4193. doi: 10.1002/anie.200600512. For selected recent examples, see: Wang X, List B. Angew. Chem., Int. Ed. 2008;47:1119. doi: 10.1002/anie.200704185. Hamilton GL, Kanai T, Toste FD. J. Am. Chem. Soc. 2008;130:14984. doi: 10.1021/ja806431d. Rueping M, Uria U, Lin M-Y, Atodiresei I. J. Am. Chem. Soc. 2011;133:3732. doi: 10.1021/ja110213t.
- 5.For a discussion of H-bonding/ion-pair continuum, see: Fleischmann M, Drettwan D, Sugiono E, Rueping M, Gschwind R. Angew. Chem., Int. Ed. 2011;50:6364. doi: 10.1002/anie.201101385.
- 6.(a) Li C-J. Acc. Chem. Res. 2009;42:335. doi: 10.1021/ar800164n. [DOI] [PubMed] [Google Scholar]; (b) Scheuerrmann CJ. Chem. Asian J. 2010;5:436. doi: 10.1002/asia.200900487. [DOI] [PubMed] [Google Scholar]; (c) Yeung CS, Dong VM. Chem. Rev. 2011;111:1215. doi: 10.1021/cr100280d. [DOI] [PubMed] [Google Scholar]
- 7.(a) Li Z, Li C-J. Org. Lett. 2004;6:4997. doi: 10.1021/ol047814v. [DOI] [PubMed] [Google Scholar]; (b) Guo C, Song J, Gong L-Z. Angew. Chem., Int. Ed. 2010;49:5558. doi: 10.1002/anie.201002108. [DOI] [PubMed] [Google Scholar]; (c) Zhang G, Zhang Y, Wang R. Angew. Chem., Int. Ed. 2011;50:10429. doi: 10.1002/anie.201105123. [DOI] [PubMed] [Google Scholar]; (d) Zhang J, Tiwari B, Xing C, Chen X, Chi Y. Angew. Chem., Int. Ed. 2012;51:3649. doi: 10.1002/anie.201109054. [DOI] [PubMed] [Google Scholar]; (e) Zhang G, Ma Y, Wang S, Zhang Y, Wang R. J. Am. Chem. Soc. 2012;134:12334. doi: 10.1021/ja303333k. [DOI] [PubMed] [Google Scholar]; (f) Zhang G, Ma Y, Wang S, Kong W, Wang R. Chem. Sci. 2013;4:2645. [Google Scholar]; (g) Cui Y, Villafane L, Clausen D, Floreancig P. Tetrahedron. 2013;69:7618. doi: 10.1016/j.tet.2013.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boess E, Schmitz C, Klussmann M. J. Am. Chem. Soc. 2012;134:5317. doi: 10.1021/ja211697s. [DOI] [PubMed] [Google Scholar]
- 9.(a) Rauniyar V, Lackner AD, Hamilton GL, Toste FD. Science. 2011;334:1681. doi: 10.1126/science.1213918. [DOI] [PubMed] [Google Scholar]; (b) Phipps R, Hiramatsu K, Toste FD. J. Am. Chem. Soc. 2012;134:8376. doi: 10.1021/ja303959p. [DOI] [PubMed] [Google Scholar]; (c) Wang Y-M, Wu J, Hoong C, Rauniyar V, Toste FD. J. Am. Chem. Soc. 2012;134:12928. doi: 10.1021/ja305795x. [DOI] [PubMed] [Google Scholar]; (d) Phipps R, Toste FD. J. Am. Chem. Soc. 2013;135:1268. doi: 10.1021/ja311798q. [DOI] [PubMed] [Google Scholar]; (e) Shunatona HP, Fruh N, Wang Y-M, Rauniyar V, Toste FD. Angew. Chem., Int. Ed. 2013;52:7724. doi: 10.1002/anie.201302002. [DOI] [PubMed] [Google Scholar]; (f) Wu J, Wang Y-M, Drljevic A, Rauniyar V, Phipps RJ, Toste FD. Proc. Natl. Acad. Sci. 2013;110:13729. doi: 10.1073/pnas.1304346110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bobbit J, Brückner C, Merbouh N. Org. React. 2010;74:103. [Google Scholar]
- 11.For a recent review of oxoammonium salts used in dehydrogenative coupling reactions, see: Mancheño O, Stopka T. Synthesis. 2013;45:1602. For recent examples, see: Richter H, Mancheño O. Eur. J. Org. Chem. 2010:4460. Richter H, Mancheño O. Org. Lett. 2011;13:6066. doi: 10.1021/ol202552y. Wertz S, Kodama S, Studer A. Angew. Chem., Int. Ed. 2011;50:11511. doi: 10.1002/anie.201104735.
- 12.Simón L, Goodman J. J. Org. Chem. 2011;76:1775. doi: 10.1021/jo102410r. [DOI] [PubMed] [Google Scholar]
- 13.(a) Uraguchi D, Sorimachi K, Terada M. J. Am. Chem. Soc. 2004;126:11804. doi: 10.1021/ja046185h. [DOI] [PubMed] [Google Scholar]; (b) Cheng X, Vellalath S, Goddard R, List B. J. Am. Chem. Soc. 2008;130:15786. doi: 10.1021/ja8071034. [DOI] [PubMed] [Google Scholar]; (c) Bachu P, Akiyama T. Chem. Commun. 2010;46:4112. doi: 10.1039/c000862a. [DOI] [PubMed] [Google Scholar]; (d) Shapiro ND, Rauniyar V, Hamilton GL, Wu J, Toste FD. Nature. 2011;470:245. doi: 10.1038/nature09723. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Čorić I, List B. Nature. 2012;483:315. doi: 10.1038/nature10932. [DOI] [PubMed] [Google Scholar]
- 14.(a) Doyle AG, Jacobsen EN. Chem. Rev. 2007;107:5713. doi: 10.1021/cr068373r. [DOI] [PubMed] [Google Scholar]; (b) Knowles RR, Lin S, Jacobsen EN. J. Am. Chem. Soc. 2010;132:5030. doi: 10.1021/ja101256v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Knowles RR, Jacobsen EN. Proc. Natl. Acad. Sci. U.S.A. 2010;107:20678. doi: 10.1073/pnas.1006402107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.For examples of incorporating 1,2,3-triazoles into the BINOL scaffold, see: Beckendorf S, Mancheño OG. Synthesis. 2012;44:2162. Recsei C, McErlean SP. Tetrahedron. 2012;68:464.
- 16.Kolb HC, Finn MG, Sharpless KB. Angew. Chem., Int. Ed. 2001;40:2004. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 17.For seminal examples, see: Tornøe C, Christensen C, Meldal M. J. Org. Chem. 2002;67:3057. doi: 10.1021/jo011148j. Rostovtsev V, Green L, Fokin V, Sharpless KB. Angew. Chem., Int. Ed. 2002;41:2596. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. For a recent review, see: Hein JE, Fokin VV. Chem. Soc. Rev. 2010;39:1302. doi: 10.1039/b904091a.
- 18.(a) Hua Y, Flood A. Chem. Soc. Rev. 2010;39:1262. doi: 10.1039/b818033b. [DOI] [PubMed] [Google Scholar]; (b) Juríček M, Kouwer PH, Rowan A. Chem. Commun. 2011;47:8740. doi: 10.1039/c1cc10685f. [DOI] [PubMed] [Google Scholar]; (c) Kumar A, Pandey P. Org. Lett. 2008;10:165. doi: 10.1021/ol702457w. [DOI] [PubMed] [Google Scholar]; (d) Leigh D, Robertson C, Slawin A, Thompson P. J. Am. Chem. Soc. 2013;135:9939. doi: 10.1021/ja404504m. [DOI] [PubMed] [Google Scholar]; (e) Beckendorf S, Asmus S, Mück-Lichtenfeld C, Mancheño OG. Chem.—Eur. J. 2013;19:1581. doi: 10.1002/chem.201203360. [DOI] [PubMed] [Google Scholar]
- 19.(a) Valverde I, Mindt T. Chimia. 2013;67:262. doi: 10.2533/chimia.2013.262. [DOI] [PubMed] [Google Scholar]; (b) Thirumurugan P, Matosiuk D, Jozwiak K. Chem. Rev. 2013;113:4905. doi: 10.1021/cr200409f. [DOI] [PubMed] [Google Scholar]; (c) Kolb HC, Sharpless KB. Drug Discov. Today. 2003;8:1128. doi: 10.1016/s1359-6446(03)02933-7. [DOI] [PubMed] [Google Scholar]; (d) Brik A, Alexandratos J, Lin Y-C, Elder J, Olson AJ, Wlodawer A, Goodsell DS, Wong ChemBioChem. 2005;6:1167. doi: 10.1002/cbic.200500101. [DOI] [PubMed] [Google Scholar]
- 20.For reviews on selectivity reversals in asymmetric catalysis, see: Escorihuela J, Burguete MI, Luis SV. Chem. Soc. Rev. 2013;42:5595. doi: 10.1039/c3cs60068h. Bartók M. Chem. Rev. 2010;110:1663. doi: 10.1021/cr9002352. For examples involving chiral PAs, see: Li N, Chen X-H, Song J, Luo S-W, Fan W, Gong L-Z. J. Am. Chem. Soc. 2009;131:15301. doi: 10.1021/ja905320q. Jiang J, Xu H-D, Xi J-B, Ren B-Y, Lv F-P, Guo X, Jiang L-Q, Zhang Z-Y, Hu W-H. J. Am. Chem. Soc. 2011;133:8428. doi: 10.1021/ja201589k.
-
21.In order to ascertain the generality of the selectivity switch observed in Table 1, the following previosly reported (ref 23) PA-catalyzed cyclic aminal-forming reaction was performed with (S)-TRIP and (S)-9h. The observed selectivity reversal may be explained by a fundamentally different steric environment created by 9l, or attractive interactions between the substrate and catalyst that override the selectivity based purely on sterics. The absolute stereochemistry of 11 was assigned on the basis of comparison of HPLC data with those reported in the literature (ref 24).
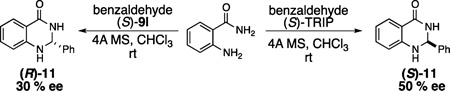
- 22.Given the anion-binding affinity of the C5–H bond of 1,4-disubstituted 1,2,3-triazoles (ref 18), one possibility may be that the triazole substituents interact with the anionic phosphate moiety, affecting the active conformation of the catalyst in a way that is not possible with 10a and 10b, which lack this feature.
- 23. Rueping M, Antonchick AP, Sugiono E, Grenader K. Angew. Chem., Int. Ed. 2009;48:908. doi: 10.1002/anie.200804770. and ref 13b.
- 24.Honjo T, Phipps RJ, Rauniyar V, Toste FD. Angew. Chem., Int. Ed. 2012;51:9684. doi: 10.1002/anie.201205383. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.