

Abstract
This work describes a non-enzymatic, isothermal genotyping method based on the kinetic differences exhibited in the dehybridization of perfectly matched (PM) and single-base mismatched (MM) DNA duplexes in an alkaline solution. Multifunctional encoded hydrogel particles incorporating allele-specific oligonucleotide (ASO) probes in two distinct regions were fabricated using microfluidic-based stop-flow lithography. Each particle contained two distinct ASO probe sequences differing at a single base position, and thus each particle was capable of simultaneously probing two distinct target alleles. Fluorescently labeled target alleles were annealed to both probe regions of a particle, and the rate of duplex dehybridization was observed using fluorescence microscopy. Duplex dehybridization was achieved through an alkaline stimulus using either a pH step function or a temporal pH gradient. When a single target probe sequence was used, the rate of mismatch duplex dehybridization could be discriminated from the rate of perfect match duplex dehybridization. In a more demanding application in which two distinct probe sequences were used, we found that the rate profiles provided a means to discriminate probe dehybridizations from both of the two mismatched duplexes as well as to distinguish at high certainty the dehybridization of the two perfectly matched duplexes. These results demonstrate an ability of alkaline dehybridization to correctly discriminate the rank hierarchy of thermodynamic stability among four sets of perfect match and single-base mismatch duplexes. We further demonstrate that these rate profiles are strongly temperature dependent and illustrate how the sensitivity can be compensated beneficially via the use of an actuating gradient pH field.
Introduction
A single-nucleotide polymorphism (SNP) is a type of genetic variation in which two or more alternative bases may occur at a single position in the genome.[1] SNPs are the most commonly encountered DNA sequence variation,[2] and have been widely associated with genetic disorders.[3] The development of low-cost and high-throughput SNP detection methods is therefore important to progress in numerous fields impacting healthcare and biomedical research.[4] Many currently used SNP detection strategies such as primer extension, oligonucleotide ligation, and endonuclease cleavage are based on enzyme activity,[5] and generally require reagents which can be expensive and more often chemically sensitive. The development of non-enzymatic SNP discrimination is therefore an active area of research.[6]
Most non-enzymatic SNP-discrimination methods use temperature as the driving force for discrimination.[1] In these methods, target DNA is captured by multiple allele-specific oligonucleotide (ASO) probes to form either perfectly matched (PM) or single-base mismatched (MM) duplexes.[7] These duplexes are then distinguished based on their differing thermodynamic stability at some specific temperature.[1] One of the most widely used techniques, the Affymetrix Genechip, requires careful optimization of the assay and reaction conditions so that a single temperature may be used to discriminate many SNPs on an array.[8] Newer methods such as Dynamic Allele-Specific Hybridization (DASH)[6a, 9]avoid this constraint by monitoring the dehybridization process dynamically throughout the application of a temporal[6a, 9-10] or spatial[11] temperature gradient. This enables the application of the optimal discrimination conditions for several different SNPs in a single experiment, with these conditions occurring at different points in the gradient. The kinetic melting curve obtained from DASH provides rich and quantitative information on the dehybridization of the target DNA.[12] This technique, however, requires precise temporal or spatial control over temperature. Given recent advances in isothermal DNA amplification[13] and the analysis of unamplified genomic samples,[14] there remains a value in exploring the advantages of non-enzymatic SNP discrimination techniques which do not use temperature as a driving force.
Several alternative discrimination methods have been exploited towards this end. Ng et al. used a denaturing reagent (50% aldehyde and 100 mmol NaCl) to discriminate SNPs,[15] while Sosnowski et al. found that MM duplexes could be selectively dehybridized by attaching the DNA duplexes to an electrode and applying a controlled negative bias.[16] However, the mechanism of this latter discrimination is not clear. The authors evaluated three possible factors: electric field, Joule heating (or temperature) and the change in local pH resulting from the applied field. They found that electric field (300 Vm-1) and temperature (< 37°C) generated in this particular experiment were insufficient for driving DNA dehybridization, and disregarded the possibility of alkaline discrimination because the pH range over which thermodynamic discrimination may be performed is quite narrow (ΔpH < 0.3). We have previously shown, however, that SNPs can be discriminated by the difference in kinetics exhibited in the dehybridization of PM and MM DNA duplexes in an alkaline solution.[17]
In the current work we show a significant improvement in that alkaline dehybridization protocol, one that may be combined with the use of an encoded hydrogel particle array to perform SNP discrimination in a versatile, non-enzymatic and isothermal technique. Encoded hydrogel particles were fabricated in a microfluidic system using stop-flow lithography (SFL) such that each particle included two spatially separated ASO probe regions.[18] Each particle contained two distinct ASO probe sequences differing at a single base position, and thus each particle was capable of simultaneously probing two distinct target alleles. Fluorescently tagged target DNA oligomers were annealed to these probes in either a direct labeling or sandwich capture scheme, and multiple labeled assay particles were placed in a microfluidic device for multiplexed genotyping. A dehybridization stimulus was applied via the introduction of an alkaline solution, with this stimulus structured to produce either a pH step function or a temporal pH gradient. Fluorescence microscopy was used to observe the change in fluorescent signal as the stimulus was applied, and the kinetics of the duplex dehybridization were compared to achieve discrimination between duplex thermodynamic stabilities.
Results and Discussion
Hydrogel particle synthesis via SFL
Multifunctional hydrogel particles incorporating ASO probes in distinct regions were fabricated using stop-flow lithography (SFL, Figure 1a).[19] Three monomer streams were flowed side-by-side in a microchannel in the laminar flow regime in order to minimize mixing between the individual streams. The central stream contained 60% polyethylene glycol diacrylate (PEGDA) loaded with an acrylate-modified dye. Each side stream contained 20% PEGDA, 40% polyethylene glycol(PEG), and a selected acrylate-modified 21-base DNA probe. Exposing the monomer streams to a burst of UV light through a photomask produced 2D extruded particles in which the cross-section and encoding of the particles (Figure 1b and 1c) were determined by the photomask. The DNA probe regions on the particle were visualized after hybridizing fluorescently labeled target DNA oligomers (Figure 1d). Several sets of particles were mixed and gently injected into a microfluidic channel with a PDMS dam (Figure 1e; the fabrication protocol for the two-layered PDMS channel is detailed in the Experimental Section) in order to form a monolayer particle array for easy visualization (Figure 1f). The particles were then ejected out of the channel using a steep, pulsed increase of the fluid flow rate.
Figure 1.
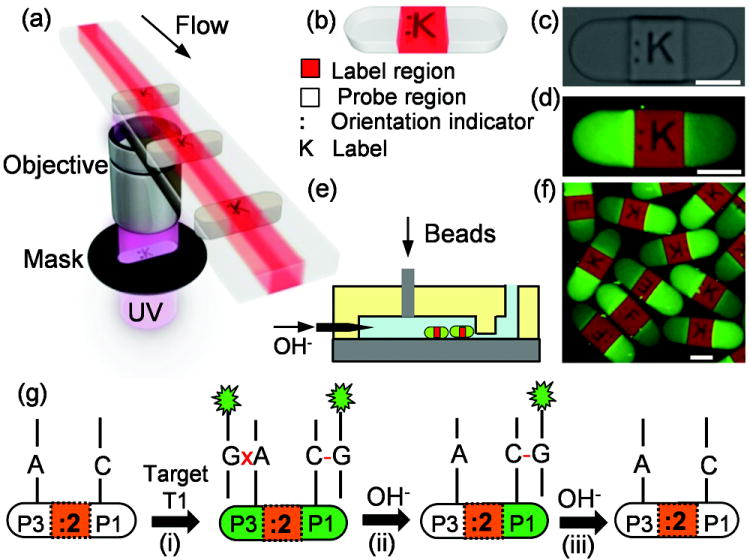
a) Stop-flow lithography (SFL) is illustrated, in which we photocure co-flowing PEG-based monomer streams to produce multifunctional encoded hydrogel particles (b, c) which contain DNA probes on either side and a fluorescent dye in the center. Probe regions are visualized by fluorescence microscopy (d) after hybridization with fluorescently labeled (green) target DNA. Particles are immobilized by a dam in a microfluidic channel (e) to form a multiplexed particle assay (f). g) a scheme illustrating a genotyping method using these particles in an alkaline dehybridization protocol. Scale bars are 100 μm.
Using a hydrogel network as a solid support for co-polymerized DNA probes provides an ideal environment for quantitative DNA analysis with fast kinetics, low fluorescence background, and high target capacity.[20]The PEGDA/PEG mixture forms a semi-interpenetrating network with tunable porosity, enabling control over mass transfer through the hydrogel-water interface.[20b] Fabrication in the laminar flow regime allows immobilization of allele-specific probes in distinct regions for easy comparison, and SFL enables high-throughput fabrication with little variation between particles within a single batch. In this work, numbers and letters were used as a graphical encoding scheme for straightforward identification of particles, but more complex encoding schemes such as barcodes may be used for automated identification.[18]
SNP discrimination via pH step function alkaline dehybridization
A schematic for genotyping using alkaline dehybridization and encoded particles is illustrated in Figure 1g. When particles incorporating probes P3 and P1 (labeled “:2”) are mixed with a fluorescently labeled homozygote (T1), both sides of the particle hybridize and fluoresce if a stringency condition is not applied (Figure 1g(i)). The specific sequences of P3, P1, and T1 are given in Table 1. The “colon” side of the :2 particle (i.e., P3) forms MM duplexes with a mismatch site in the center of the duplex while the “label” side (i.e., P1) forms PM duplexes. Increasing the pH of the surrounding medium results in dehybridization of the target T1 from both sides, but the rate at which T1 dehybridizes is expected to be faster for the MM case.[17] Within an appropriate time scale and pH range, the difference in dehybridization kinetics provides an easily measured temporal difference in the fluorescence intensity between the two sides (Figure 1g(ii)) which evolves until both sides dehybridize completely (Figure 1g(iii)). Dehybridization of the other homozygote (T3) provides a fluorescence difference with opposite sign, while a heterozygous sample (i.e., a mixture of hybridized T1 and T3) is expected to produce little or no difference between the two sides. The present work demonstrates that the temporal differences in fluorescence intensity between the two ends can be used to effectively discriminate genotypes.
Table I.
Sequences of DNA probes and targets
| DNA | Sequence[a] |
|---|---|
| P1 | 5’-/Acryd/CCT GGG AAA GTC CCC TCA ACA /-3’ |
| T1 | 3’-/GGA CCC TTT CAG GGG AGT TGT/Org488/5’ |
| P3 | 5’-/Acryd/CCT GGG AAA GTA CCC TCA ACA / -3’ |
| T3 | 3’-/GGA CCC TTT CAT GGG AGT TGT/Org488/5’ |
| P4 | 5’-/Acryd/CCT AGG AAA GTC CCC TCA ACA /-3’ |
| T4 | 3’-/GGA TCC TTT CAG GGG AGT TGT/Org488/5’ |
Nucleobases different from P1 or T1 are underlined
To test this concept, three sets of P3/P1 particles labeled :2, :3 and :4 were fabricated and hybridized with synthetic DNA targets T1, T3, and a 50/50 mixture of T1 and T3, respectively. These hybridized particles were then mixed together and injected into a microfluidic channel for assay. Phosphate buffer (pH 11.20) was injected into the channel at room temperature (22°C), and the fluorescence intensity of the DNA duplexes during alkaline dehybridization was monitored using time-lapse fluorescence microscopy. Normalization and background correction were carried out by calculating a time-dependent fluorescence retention ratio, (I(t)-Imin)/(Imax-Imin), with maximum and minimum intensities calculated separately for each probe region. Representative time series of the fluorescence retention ratio for each particle set are shown in Figure S1 in the Supporting Information. We found that within a 10 minute dehybridization process, a significant intensity contrast was produced between the two sides of both particle sets :2 and :3 (i.e., Figure 2a and Figure 2b), while particle set :4 showed a much weaker intensity contrast (Figure 2c). To quantify the time-dependent intensity contrast for a given particle, we calculated the difference in the fluorescence retention ratio (Δ, Equation 1) between the “colon” and the “label” sides at all times and compared the different particle sets. The Δ curves for each of the three particles sets are distinctly different (Figure 2d). The negative peak in the Δ curve of particle set :2 reflects that the DNA duplexes at the “colon” side dehybridize faster than the “label” side, while the positive peak in particle set :3 indicates faster dehybridization on the “label” side.
Figure 2.
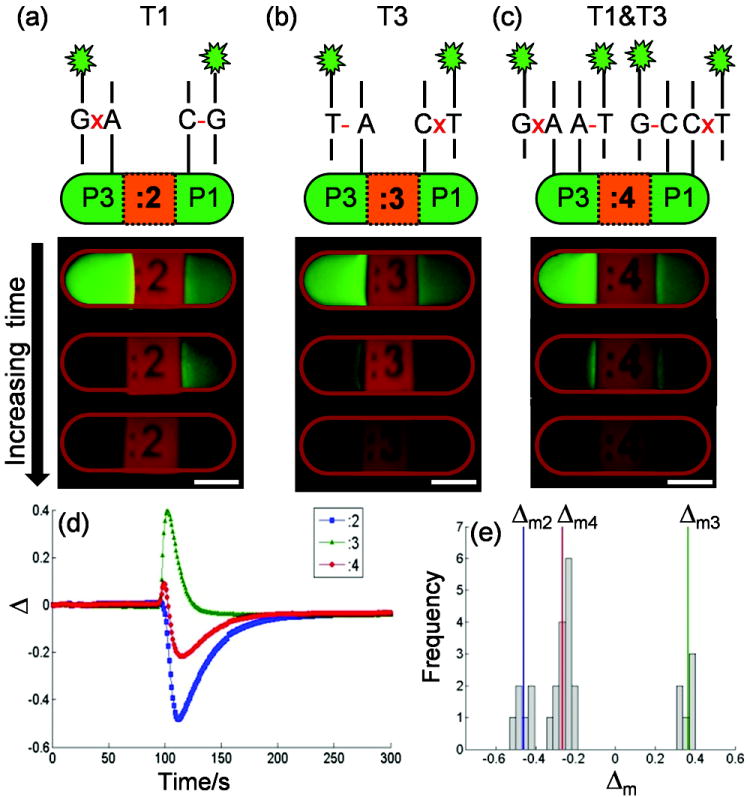
Three batches of particles containing allele-specific probes P1 and P3 (5’-CCTGGGAAAGT(C/A)CCCTCAACA-3’, alternatives shown in parentheses) form perfectly matched (PM) or mismatched (MM) duplexes on either end of a particle when they hybridize, respectively, with synthetic DNA targets T1 (a), T3 (b), and a 50/50 mixture of T1 and T3 (c), simulating three possible genotypes. A time-dependent difference in fluorescence retention ratio, Δ, across each particle is generated when subjected to an alkaline condition (pH 11.20 phosphate buffer). Δ curves for each hybridization scheme are shown in (d) and the peak values Δm are found for a mix of particles and plotted in a histogram (e). Δm fall into natural groups for each hybridization scheme. Scale bars are 100 μm.
Equation 1: Difference in retention ratio
In contrast to the single-peak behavior observed in each of the homozygous curves, the heterozygous curve shows a more complex double-peak behavior. The Δ curve for particle set :4 shows an initial positive peak followed by a negative peak, both with lower magnitude than the homozygous single-peak curves. This double-peak behavior appears to combine the behaviors observed in the two homozygous experiments, suggesting the presence of both PM and MM duplexes on each side (Figure 2c). It is interesting to note that the retention ratio observed on either side of the :4 particle set may be approximated by a linear combination of the observed retention ratios in the corresponding probe regions of the homozygous experiments(see more detail in Figure S2, Supporting Information). Further quantitative studies are necessary to better quantify this approximately additive behavior.
To simplify comparisons between the data sets, we calculated the peak value Δm of each curve (i.e., Δmax or Δmin depending on the absolute magnitude); for the “double-peak” heterozygous experiment we use the higher-magnitude negative peak. These Δm values for each particle in the array fall into three distinct groups corresponding to the three particle sets, as shown by the histogram in Figure 2e. The use of Δm compensates to some extent for various types of system heterogeneity and can be used autonomously as a single metric to make genotyping determinations.
We performed similar experiments (using pH 11.20 dehybridization buffer) at 10°C and 37°C to evaluate the method’s temperature sensitivity (Figure S1), and found the major difference to be in the time required to complete an experiment. The time required for the fluorescence intensity to fall to 5% of the initial value is approximately 60 minutes at 10°C, 7 minutes at 22°C, and 2 minutes at 37°C. Our calculated Δm, however, was only modestly insensitive to changes in temperature over this range and remains a viable genotyping criterion. We verified statistical significance of this result using Welch’s two-sample t-test, which showed the means of each Δm distribution to be significantly different from the other two in that experiment at 95% confidence.
The sensitivity of the dehybridization rate to temperature presents some difficulties. At higher temperatures the temporal resolution of the experiment may be insufficient for resolving the intensity contrast, while at lower temperatures the time required for experiments is greatly increased. This suggests that a multiplexed assay would require a detailed optimization for the different time scales involved. However, there exist modified procedures that could make such optimization unnecessary. These are discussed below.
SNP discrimination via pH gradient alkaline dehybridization
We further tested the dehybridization protocol by applying a temporal pH gradient to the particle assay so that an optimal stringency condition can be achieved for each SNP at any specific measurement temperature. To generate a temporal pH gradient, we injected a 0.02 M NaOH solution at an increasing rate, mixing completely with a constant flow of water before reaching the particle assay (NaOH injection profile and calculated [OH-] and pH values are provided in Figure S3 in the Supporting Information). Applying the same gradient at 10°C, 22°C and 37°C for the three sets of P3/P1 particles showed similar discrimination to the constant pH buffer (Figure 3a), but with better discrimination at the higher temperature (Figure 3b). The same protocols were applied to DNA duplexes with the SNP site located three bases from the 5’ end of the probe strand, rather than at the center (as above). We fabricated three sets of particles incorporating probes P4 and P1 (Table 1), labeled :E, :F, and :K (Figure 1f), and hybridized separately with synthetic targets simulating three genotypes (Table 1, and key for Figure 3d). Both a constant pH 11.20 buffer (Figure 3c) and a pH gradient (Figure 3d) were applied to mixed sets of these particles, and Δm was calculated for each particle, as previously described. As before, these values fall into clearly separated groups. In each of the twelve DNA/temperature/protocol combinations shown in Figure 3, the differences in the mean values of the Δm distributions within an experiment are statistically significant at the 95% confidence level. This result is most significant given that it is well known that SNPs located near the end of a DNA sequence are especially difficult to discriminate using thermal methods.[6a] The clear discrimination shown here demonstrates the high sensitivity of this kinetic technique even in this challenging context.
Figure 3.
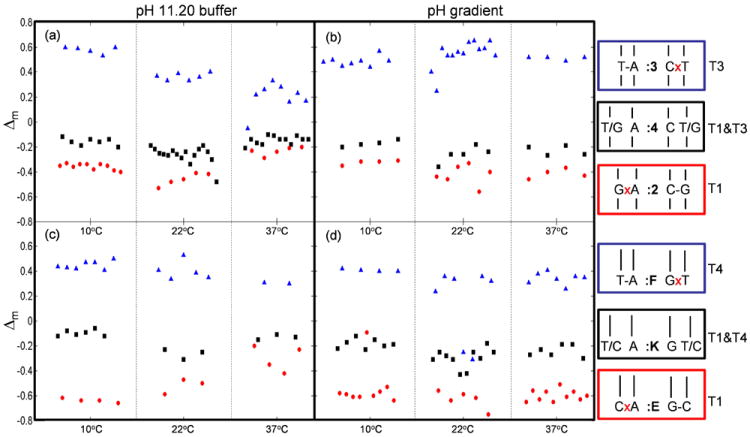
Genotyping is studied at different temperatures (10°C, 22°C, and 37°C) using a pH 11.20 buffer (a and c) or a pH gradient to drive dehybridization (b and d). (a) and (b) are for DNA with the variant base located in the center, while (c) and (d) show results when the variant base located at three base away from the end.
The ability of alkaline dehybridization to discriminate the rank hierarchy of thermodynamic stability among PM and MM DNA duplexes was analyzed by examining the time-dependent fluorescence retention ratios from the homozygous beads, i.e. :2, :3, :E, and :F. Plotting the signals for the room-temperature pH-gradient dehybridization experiments (Figure 4) shows a clear dehybridization order for each SNP location. In the case where the SNP is located in the center of the strand (Figure 4a), we can clearly see that the P1xT3 MM duplex dehybridizes first, followed by the P3xT1 MM duplex, the P3/T3 PM duplex, and the P1/T1 PM duplex in succession. DNA melting simulations carried out with RNAStructure[21] and UNAFold[22] for each of these duplexes show that this order corresponds to the rank ordering of the duplex stabilities as measured by the hybridization Gibbs free energy: P1xT3 is the least stable duplex with the smallest free energy difference, P3xT1 the next least stable, P3/T3 next, and P1/T1 the most stable with the largest free energy difference. It is worth noting that dynamic alkaline dehybridization easily distinguishes the two PM duplexes, a difficult procedure to accomplish via thermal discrimination methods. Our experiment in the case where the SNP is located at the end of the strand (Figure 4b) also produces a clear dehybridization sequence, with the P4xT1 MM duplex dehybridizing first, followed by the P1xT4 MM duplex, the P4/T4 PM duplex, and the P1/T1 PM duplex in succession. This again corresponds to the rank order of calculated thermodynamic stabilities by Gibbs free energy, and once again alkaline dehybridization shows a clear discrimination of both PM and MM duplexes. It is significant to note that the difference in DNA melting temperature between P1/T1 and P4/T4 as calculated by UNAfold is only 0.3°C, but we may still clearly see the separation between these duplexes in the plotted retention ratios.
Figure 4.
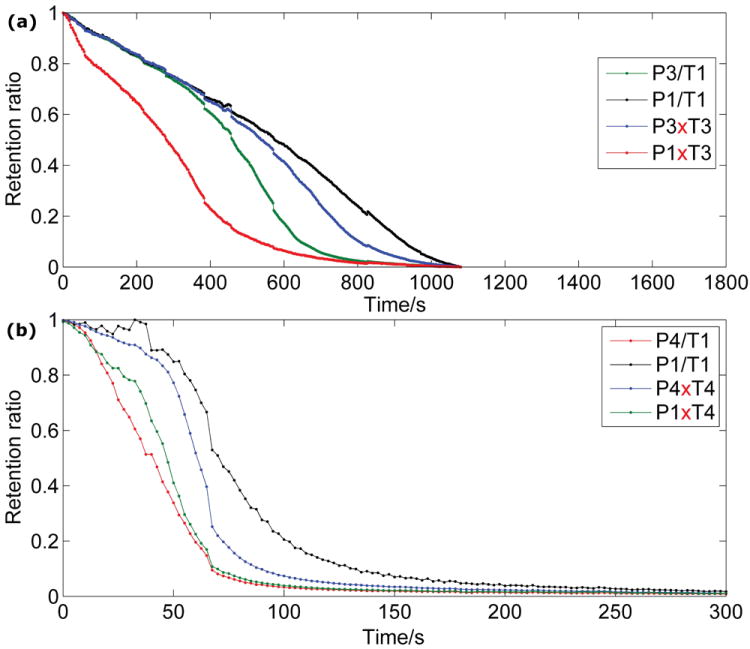
The time-dependent fluorescence retention ratios for the various perfect match and single-base mismatch DNA duplexes that were studied are plotted as a function of time during ph-gradient alkaline dehybridization. a) The four PM and MM duplex combinations formed from probe oligomers P1 and P3 and target oligomers T1 and T3. b) The four PM and MM duplex combinations formed from probe oligomer P1 and P4 and target oligomers T1 and T4.
SNP discrimination of model clinically relevant mutations
We further tested this genotyping approach using a “sandwich” tagging method (Figure 5a) on three clinically relevant mutations associated with thrombotic disorders, MTHFR (C→T), Factor II (G→A) and Factor V (G→A).[23] We fabricated three sets of particles, labeled :M, :FII, and :FV, each containing two 21-base ASO probes for the corresponding SNP (Table S1, Supporting Information). Probes for the wild-type sequence are always found on the “label” side, with the mutant sequence at the “colon” side. We prepared three mixtures of synthetic non-fluorescent target DNA oligomers (~70 base long, see Experimental Section) that simulate three combinations of homozygous or heterozygous samples. The target mixtures were hybridized with particle assays and rinsed (Figure 5a(i)) before adding a mixture of three gene-specific, fluorescently labeled, secondary probes (30-base) (Figure 5a(ii)). By carrying out an alkaline gradient experiment (Figure 5a(iii)), we were able to straightforwardly determine the genotypes from the particle fluorescence at peak contrasts and Δm values (Figure 5b). These results correspond correctly to the target composition.
Figure 5.
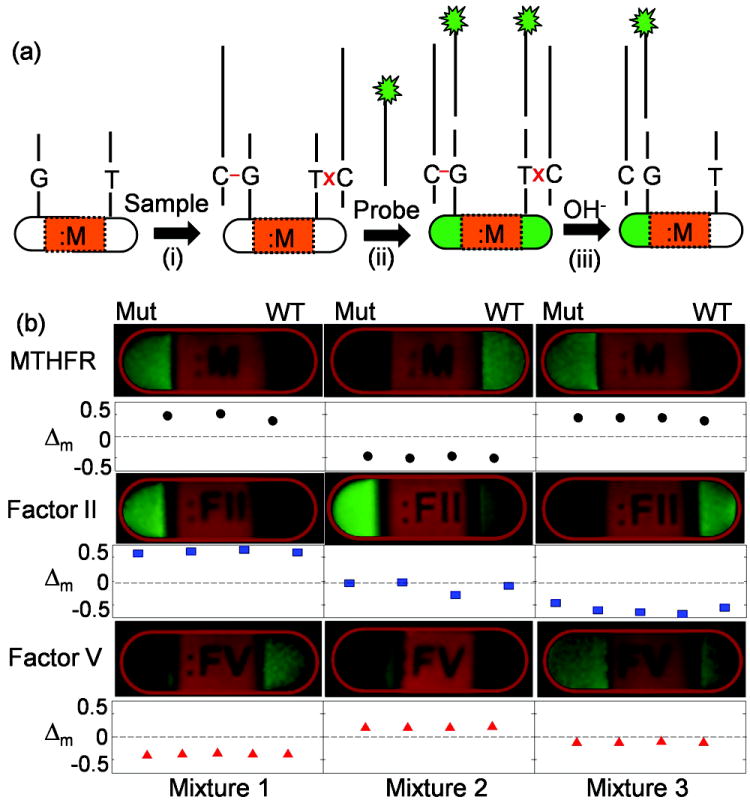
a) Synthetic DNA targets of genes associated with thrombotic disorders (MTHFR (C→T), Factor II (G→A), and Factor V (G→A)) and fluorescently labeled gene-specific secondary probes are sequentially hybridized to three types of particles containing allele-specific probes. b) The target mixture 1 contains MTHFR mutant, Factor II mutant, and Factor V wild-type. Target mixture 2 contains MTHFR wild-type, 50/50 mixture of Factor II wild-type and mutant, and Factor V mutant. Target mixture for test 3 contains MTHFR mutant, Factor II wild-type, and 50/50 mixture of Factor V wild-type and mutant.
Conclusion
In summary, we demonstrate alkaline dehybridization as an effective alternative to temperature-based discrimination of SNPs. Genotyping is carried out via a microfluidic alkaline stimulus over a multiplexed particle array composed of multifunctional encoded hydrogel particles. The kinetic difference of PM and MM duplex dehybridization under alkaline conditions is effectively measured by the difference in fluorescence retention ratio, and the peak value of the difference in fluorescence retention ratio can be used as a simple metric for genotyping. We show the utility of a pH gradient to stimulate duplex dehybridization over a range of temperatures in order to discriminate target DNA sequences with varying SNP insertion points. When two distinct probe sequences were used, not only could the rates of the two mismatch duplex dehybridizations be discriminated from the rates of the two perfect match duplex dehybridizations, but the rates of the two different perfect match dehybridizations could also be discriminated from one another. The ability of alkaline dehybridization to correctly discriminate the rank hierarchy of thermodynamic stability among four sets of perfect match and single-base mismatch duplexes demonstrates the power of a gradient pH field to compensate for temperature in genotyping research.
Experimental Section
Microfluidic device fabrication
Microchannels for SFL and genotyping were fabricated by molding polydimethylsiloxane (PDMS, Sylgard 184, Dow Corning) off of photoresist masters. Single-layer masters for SFL channels were produced by spin-coating SU-8 50 (Microchem) at 2000 rpm for 30s, with a 5 min soft bake at 120°C, a 40 s 300 W UV exposure through a photomask, and a 5 min post-bake at 120°C. Two-layer masters incorporating a 40 μm high dam for genotyping were similarly fabricated in two steps as above (spin-coat at 3500 rpm for 30 s for step), with an alignment step preceding the second exposure. PDMS elastomer and curing agent were mixed at a 10:1 ratio by weight and poured over masters, then baked at least 3 hours at 70°C and peeled up to form the top surface of the channel. The bottom surface of the microchannel was fabricated by spin-coating PDMS at 1500 rpm onto a glass coverslip (Gold Seal) and baking for 6 hours at 70°C. The two surfaces were bonded by exposure to UV/Ozone for 5 minutes.
Hydrogel particle formation
Multifunctional hydrogel particles were fabricated by SFL[18, 19b] using three-inlet Y-junction microchannels of polydimethylsiloxane (PDMS) in the laminar flow regime. Applied pressures were 1 psi in the central stream and 3 psi on either side. The central “label” stream consisted of a 60 vol% aqueous solution of poly(ethylene glycol) diacrylate (PEGDA, Sigma Aldrich, Mn = 700) with 5 vol% photoinitiator (Darocur 1173, Ciba) and 0.0005 wt% methacryloxyethyl thiocarbamoyl rhodamine B (PolyFluor 570, Polysciences) as the fluorescent dye. The “probe” streams on either side consisted of an aqueous solution containing 20 vol% PEGDA, 40% poly(ethylene glycol) (Polysciences, Mn=200), 5 vol% photoinitiator and 30 vol% of acrylate-modified single-strand DNA probe (IDT DNA) at 0.15 mmol in TE buffer. Solid particles were produced by photopolymerization using UV light from a Hg lamp and a photomask incorporating the particle label at the microscope field stop through a 20x objective lens. Fabrication was carried out with a flow time of 1 s, a pause after flow of 1 s, and an exposure time of 0.3 s. Particles were collected in a reservoir containing a mixture of 2/3 PEGDA and 1/3 TE buffer by volume and pipetted into microcentrifuge tubes. The particles were separated from solution by centrifugation at 5000 rpm for 1 min. They were then rinsed three times by 0.05% TWEEN20 in PBS buffer (PBST), and stored in PBST at room temperature until use.
Dehybridization experiment
Standard assay particles were hybridized with Oregon Green-488 or Alexa488-tagged target DNA (100 μL, 1 μmol) for at least 2 hr at room temperature. Sandwich assay particles were hybridized with a mixture of target DNAs (300 μL, the total concentration of each type of target DNA is 0.33 μmol) at 37°C overnight, rinsed with copious PBST at least 3x, and then hybridized with fluorescently tagged secondary DNA probes (100 μL, 1 μmol) for 2 hr. Particle samples were then mixed and injected gently into a microchannel (900 μm wide, 80 μm high) containing a PDMS dam (40 μm high) to form a monolayer assay. In single-buffer experiments, alkaline dehybridization was carried out by flowing over the particles a pH 11.20 phosphate buffer at a rate of 10 μLmin-1. In a pH gradient experiment, pH 7 deionized water was injected at a rate of 20 μLmin-1 and allowed to mix completely with a time-varying stream of 0.02 M NaOH using a programmable syringe pump (PHD 22/2000, Harvard Apparatus, Holliston, MA, USA). The injection rate of NaOH and calculated [OH-] and pH is given in Figure S2 (Supporting Information). Dehybridization was monitored by a Zeiss Axiovert 200M fluorescence microscope (Carl Zeiss, Thornwood, NY) with a home-build temperature control stage at 5X magnification and frame rates of 0.3-0.5 fps (Cascade: 512B imaging system, Photometrics). The resulting images were analyzed using custom software in Matlab to locate the particles and determine the intensity in probe regions.
Supplementary Material
Acknowledgments
We thank Mayandi Sivaguru, Institute for Genomic Biology, UIUC, for his help in imaging. This work was supported by the National Science Foundation (grant CHE0704153 and DMR-0652424), and based upon work supported by the U.S. Department of Energy, Division of Materials Sciences under Award No. DE-FG02-07ER46471, through the Frederick Seitz Materials Research Laboratory at the University of Illinois at Urbana-Champaign. A. J. DeConinck is partially supported by the Army Research Office under the National Defense Science and Engineering Graduate Fellowship. P.S. Doyle acknowledges partial support from grant R21EB008814 from the NIH.
References
- 1.Syvaneen A-C. Nat Rev Genet. 2001;2:930. doi: 10.1038/35103535. [DOI] [PubMed] [Google Scholar]
- 2.Altshuler D, Brooks L, Chakravarti A, Collins F, Daly M, Donnelly P. Nature. 2005;437:1299. [Google Scholar]
- 3.Ragoussis J. Annu Rev Genomics Hum Genet. 2009;10:117. doi: 10.1146/annurev-genom-082908-150116. [DOI] [PubMed] [Google Scholar]
- 4.a) Perkel J. Nat Meth. 2008;5:447. [Google Scholar]; b) Holland CA, Kiechle FL. Curr Opin Microbiol. 2005;8:504. doi: 10.1016/j.mib.2005.08.001. [DOI] [PubMed] [Google Scholar]; c) Twyman RM. Curr Top Med Chem. 2004;4:1421. doi: 10.2174/1568026043387656. [DOI] [PubMed] [Google Scholar]; d) Dobson MG, Galvin P, Barton DE. Expert Rev Mol Diagn. 2007;7:359. doi: 10.1586/14737159.7.4.359. [DOI] [PubMed] [Google Scholar]
- 5.Kim S, Misra A. Annu Rev Biomed Eng. 2007;9:289. doi: 10.1146/annurev.bioeng.9.060906.152037. [DOI] [PubMed] [Google Scholar]
- 6.a) Howell WM, Job M, Gyllensten U, Brookes AJ. Nat Biotechnol. 1999;17:87. doi: 10.1038/5270. [DOI] [PubMed] [Google Scholar]; b) Ogasawara S, Fujimoto K. Angew Chem Int Ed. 2006;45:4512. doi: 10.1002/anie.200600790. [DOI] [PubMed] [Google Scholar]; c) Bowler FrankR, DiazMochon JuanJ, Swift MichaelD, Bradley M. Angew Chem Int Ed. 2010;49:1809. doi: 10.1002/anie.200905699. [DOI] [PubMed] [Google Scholar]; d) Taton TA, Mirkin CA, Letsinger RL. Science. 2000;289:1757. doi: 10.1126/science.289.5485.1757. [DOI] [PubMed] [Google Scholar]
- 7.Kwok P-Y. Annu Rev Genomics Hum Genet. 2001;2:235. doi: 10.1146/annurev.genom.2.1.235. [DOI] [PubMed] [Google Scholar]
- 8.a) Wang DG, Fan J-B, Siao C-J, Berno A, Young P, Sapolsky R, Ghandour G, Perkins N, Winchester E, Spencer J, Kruglyak L, Stein L, Hsie L, Topaloglou T, Hubbell E, Robinson E, Mittmann M, Morris MS, Shen N, Kilburn D, Rioux J, Nusbaum C, Rozen S, Hudson TJ, Lipshutz R, Chee M, Lander ES. Science. 1998;280:1077. doi: 10.1126/science.280.5366.1077. [DOI] [PubMed] [Google Scholar]; b) Vainrub A, Pettitt BM. Biopolymers. 2004;73:614. doi: 10.1002/bip.20008. [DOI] [PubMed] [Google Scholar]
- 9.Jobs M, Howell WM, Stroemqvist L, Mayr T, Brookes AJ. Genome Res. 2003;13:916. doi: 10.1101/gr.801103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meuzelaar LS, Hopkins K, Liebana E, Brookes AJ. J Mol Diagn. 2007;9:30. doi: 10.2353/jmoldx.2007.060057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crews N, Wittwer CT, Montgomery J, Pryor R, Gale B. Anal Chem. 2009;81:2053. doi: 10.1021/ac801495w. [DOI] [PubMed] [Google Scholar]
- 12.Prince JA, Feuk L, Howell WM, Jobs M, Emahazion T, Blennow K, Brookes AJ. Genome Res. 2001;11:152. doi: 10.1101/gr.150201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gill P, Ghaemi A. Nucleosides, Nucleotides Nucleic Acids. 2008;27:224. doi: 10.1080/15257770701845204. [DOI] [PubMed] [Google Scholar]
- 14.Bao YP, Huber M, Wei T-F, Marla SS, Storhoff JJ, Müller UR. Nucl Acids Res. 2005;33:e15. doi: 10.1093/nar/gni017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ng JK-K, Feng H, Liu W-T. Anal Chim Acta. 2007;582:295. doi: 10.1016/j.aca.2006.09.016. [DOI] [PubMed] [Google Scholar]
- 16.Sosnowski RG, Tu E, Butler WF, O’Connell JP, Heller MJ. Proc Natl Acad Sci U S A. 1997;94:1119. doi: 10.1073/pnas.94.4.1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang H, Mitrovski SM, Nuzzo RG. Anal Chem. 2007;79:9014. doi: 10.1021/ac701660x. [DOI] [PubMed] [Google Scholar]
- 18.Pregibon DC, Toner M, Doyle PS. Science. 2007;315:1393. doi: 10.1126/science.1134929. [DOI] [PubMed] [Google Scholar]
- 19.a) Dendukuri D, Pregibon DC, Collins J, Hatton TA, Doyle PS. Nat Mater. 2006;5:365. doi: 10.1038/nmat1617. [DOI] [PubMed] [Google Scholar]; b) Dendukuri D, Gu SS, Pregibon DC, Hatton TA, Doyle PS. Lab Chip. 2007;7:818. doi: 10.1039/b703457a. [DOI] [PubMed] [Google Scholar]
- 20.a) Nolan JP, Sklar LA. Trends Biotechnol. 2002;20:9. doi: 10.1016/s0167-7799(01)01844-3. [DOI] [PubMed] [Google Scholar]; b) Pregibon DC, Doyle PS. Anal Chem. 2009;81:4873. doi: 10.1021/ac9005292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reuter J, Mathews D. BMC Bioinformatics. 2010;11:129. doi: 10.1186/1471-2105-11-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Markham NR, Zuker M. Methods Mol Biol. 2008;453:3. doi: 10.1007/978-1-60327-429-6_1. [DOI] [PubMed] [Google Scholar]
- 23.Lefferts JA, Jannetto P, Tsongalis GJ. Exp Mol Pathol. 2009;87:105. doi: 10.1016/j.yexmp.2009.06.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.