

Abstract
The widespread adoption of epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) for the first-line treatment of advanced EGFR-mutated non-small cell lung cancer (NSCLC) has resulted in acquired TKI resistance becoming a ubiquitous clinical problem. The identification of specific mechanisms of acquired resistance has allowed a better understanding of the biology and natural history of resistant disease, but is only now starting to impact treatment decisions. Strategies for managing acquired resistance in advanced NSCLC are complex and must be adapted to the individual characteristics of each patient’s cancer. While combination chemotherapy is the presumed standard of care for most patients, prospective trial data are lacking, highlighting the importance of offering patients participation in clinical trials in this setting. Emerging data from trials of third-generation mutant-specific EGFR kinase inhibitors suggests particular promise with this class of agents.
Keywords: NSCLC, targeted therapy, EGFR, acquired resistance
Introduction
Activating mutations in EGFR have come to define a distinct population of patients with NSCLC. Cancers that harbor these EGFR mutations have been demonstrated to possess profound sensitivity to EGFR TKIs,1–3 giving them a unique biology and natural history.4,5 Numerous studies have now demonstrated that patients with advanced NSCLC harboring specific EGFR activating mutations (exon 19 deletions or exon 21 L858R) should receive first line treatment with EGFR TKIs.6 These agents exhibit minimal toxicity and are broadly active with only 3–10% of patients exhibiting refractory disease with frank progression on TKI.6–8
The initial responses achieved with either standard first-generation EGFR kinase inhibitors (gefitinib, erlotinib) or recently approved alternative agents (icotinib, afatinib) are temporary and marred by the inevitable emergence of acquired treatment resistance.6,7,9,10 The management of acquired resistance has thus become the central challenge in the treatment of EGFR-mutant advanced NSCLC. Here we review current knowledge regarding the definition of acquired resistance, mechanisms of resistance and the optimal management thereof.
Defining resistance to EGFR kinase inhibitors
The development of acquired resistance to EGFR kinase inhibitors is both predictable and unavoidable. Importantly, acquired resistance is distinct in both mechanism and management from primary treatment resistance. The latter refers to a heterogeneous population of cancers lacking TKI sensitivity due to absence of an EGFR mutation, a distinct biology (e.g. presence of another oncogenic driver mutation), or due to baseline presence of a secondary mutation lending resistance (e.g. EGFR mutation plus EGFR T790M); primary resistance is outside of the scope of this review but has been reviewed recently elsewhere.11 In contrast, acquired resistance refers specifically to resistance that develops following initial EGFR TKI sensitivity. While a clinical definition of resistance was previously proposed which included non-genotyped patients with progressive disease after initial EGFR TKI response,12 the widespread adoption of EGFR genotyping has resulted in acquired resistance now loosely referring to EGFR-mutant lung cancers with progression on an EGFR kinase inhibitor after an initial period of response or stable disease.
Acquired resistance to EGFR kinase inhibitors is thought to be initiated by the emergence of clones possessing genomic alterations conferring survival advantage under the selective pressure of the TKI.13 The point at which resistant clones emerge relative to the initiation of TKI therapy remains controversial, particularly given technical challenges in detecting low prevalence resistance mutations prior to TKI therapy.14 No relationship has ever been demonstrated between detection of a pre-treatment resistance mutation in a minor population of cells and the ultimate acquired resistance mechanism. However, once resistant clones emerge they eventually grow to predominate and lead to clinically apparent disease progression (Figure 1).15,16
Figure 1.

Different clinical presentations of acquired resistance in EGFR mutant NSCLC can be due to different resistance mechanisms. TOP – isolated CNS progression can occur due to poor TKI penetration into the CNS secondary to the blood-brain barrier. MIDDLE – the emergence of a T790M clone can cause indolent progression on EGFR TKI, but re-growth of sensitive clones can occur upon TKI cessation. BOTTOM – the emergence of an alternative resistance mutation can produce rapidly growing clones and rapid progression.
One proposed criteria for defining resistance to EGFR kinase inhibitors has used radiographic progression as determined by RECIST criteria.12 The use of these criteria to define disease progression and thus acquired resistance has obvious utility in the conduct of clinical trials. However, caution must be applied in using these criteria to make treatment decisions in EGFR-mutant NSCLC with emerging acquired resistance. Patients with robust initial responses to EGFR TKIs may have minimal remaining disease, such that clinically insignificant changes on imaging may meet RECIST criteria for progression despite indolent growth and a lack of symptoms. Put differently, the determination of resistance based upon radiographic progression by does not necessarily imply clinically significant disease progression and treatment failure.17 The key clinical challenge is thus determining the point at which the degree of acquired resistance as manifested by radiographic progression has reached a threshold that warrants changing treatment. This important question is currently being investigated by the ASPIRATION trial, prospectively studying continued single-agent erlotinib beyond RECIST progression (NCT01310036).
Mechanisms of acquired resistance to EGFR kinase inhibitors
Several molecular mechanisms have been elucidated which are capable of triggering acquired resistance to EGFR TKIs. For the purposes of this review, we will broadly group these mechanisms into three categories based upon the degree to which each mechanism is potentially actionable or affects clinical management.
Importantly, pharmacokinetic failure of EGFR kinase inhibitors constitutes a separate mechanism of apparent resistance not well encompassed in this schema. This phenomenon has been described secondary to drug-drug interactions and smoking-related effects on EGFR TKI metabolism, where a patient with progression may be able to respond to increased EGFR kinase inhibitor dose.18,19 Isolated CNS progression due to poor drug penetration into the CSF represents another type of pharmacokinetic failure, where the blood-brain barrier limits drug penetration to sub-therapeutic doses allowing regrowth of EGFR-mutant disease (Figure 1).
Clinically actionable resistance mechanisms
The EGFR T790M mutation is the most common mechanism of acquired resistance, found in 49–63% of re-biopsies performed after resistance develops to EGFR TKIs.20–22 The T790M mutation alters the affinity of EGFR for ATP, dramatically reducing the ability of first- and second-generation TKIs to compete for binding.23,24 The presence of the T790M resistance mutation thus confers survival advantage to tumor cells when subjected to the selective pressure of EGFR kinase inhibitors. However, the growth kinetics of T790M-positive tumor cells are inferior to T790M-negative EGFR mutant tumor cells in the absence of EGFR TKI.15,16 This may explain, in part, the phenomenon of both tumor flare noted upon cessation of EGFR TKIs, as sensitive clones overgrow the resistant clones, as well as subsequent re-response of these sensitive clones to re-treatment with the same TKI (Figure 1).25,26
Clinically, T790M-mediated acquired resistance often exhibits a distinctive indolent pattern of progression,13,15,16 and in some series has been found to be associated with a favorable prognosis compared to T790M-negative resistance.15,16 In one of the largest re-biopsy series to date, presence of T790M was associated with a lower incidence of new metastatic sites, higher performance status and longer survival.15 Beyond its role as a prognostic marker, the T790M mutation also has an emerging role as a predictive biomarker given that early data on novel third-generation EGFR kinase inhibitors have suggested high response rates in T790M-positive lung cancers (Table 1).27,28
Table.
| Tier 1: Clinically actionable resistance mechanisms
| |||
|---|---|---|---|
| Mechanism | Prevalence | Potential Therapy | Efficacy data |
| EGFR T790M24 | 49–63%20,21,22 | CO-168628 | 66% RR in 9 T790M+ patients at highest dose level† |
| AZD929127 | 43% RR in 35 patients from all dose levels
|
||
| afatinib + cetuximab65 | 30% RR and median 4.7 month PFS in 96 patients at maximum tolerated dose
|
||
|
| |||
| Small Cell Transformation29 | 3–14%21,22 | platinum–etoposide21 | 60% RR in 5 patients |
| Tier 2: Clinical trials ongoing
| |||
|---|---|---|---|
| Mechanism | Prevalence | Potential Therapy | Ongoing Clinical Trials |
| MET amplification30,31 | 5–11%20,21,22 | cabozantinib + erlotinib LY2875358 +/− erlotinib INC280 + gefitinib |
Phase II (NCT01866410) Phase II (NCT01900652) Phase IB/II (NCT01610336) |
|
| |||
| HER2 amplification32 | 12–13%22,32 | High-dose intermittent afatinib dacomitinib intermittent dacomitinib |
Phase Ib (NCT01647711) Phase III versus placebo (NCT1000025), with a pre-planned subgroup analysis in EGFR-mutant cancers Phase II (NCT01858389) |
|
| |||
| PIK3CA mutation35 | 0–5%21,22 | BKM120 + gefitinib BKM120 + erlotinib |
Phase I (NCT01570296) Phase II (NCT01487265) |
|
| |||
| ERK amplification37 | N/a | selumetinib + gefitinib | PhaseIb/II (NCT02025114) |
|
| |||
| BRAF V600E33 | 1%33 | Combinations of BRAF and EGFR inhibitors are in development in colorectal cancer34 | |
|
| |||
| Tier 3: Pre-clinical (CRKL amplification, AXL overexpression, elevated HGF) 38–42 | |||
Response data presented only for T790M positive patients.
Small cell transformation is another discrete resistance mechanism found in a subset of cases of acquired resistance where neuroendocrine histological features are seen with the original EGFR mutation maintained.29 The clinical course of transformed disease has been difficult to study due to its rarity (3–14%), but anecdotally can be associated with aggressive behavior (Figure 1). One report found 3 of 5 patients with this type of transformed disease responded to standard platinum-etoposide chemotherapy.21
Potentially actionable resistance mechanisms
The second genomic mechanism discovered to mediate acquired resistance to EGFR kinase inhibitors was amplification of the MET gene and associated overexpression of the MET kinase.30,31 MET amplification bypasses reliance on the EGFR signaling pathway by alternatively activating the PI3K/AKT pathway via ErbB3 signaling. The prevalence of MET amplification in recent clinical series has ranged between 5 and 11%,20–22 lower than the 20% prevalence seen in smaller early reports.30,31 Several MET inhibitors have been developed and are now in clinical trials as both single agents and in combination with erlotinib (Table 1).
Two other highly targetable oncogenes, HER2 and BRAF, have also been identified to mediate acquired resistance in a small subset of cases.32,33 Amplification of HER2 has previously been postulated as a mechanism of acquired resistance, and was recently identified by fluorescence in-situ hybridization (FISH) in 3 patients in a re-biopsy series of 24 patients.32 Mutations in BRAF have been demonstrated to confer acquired resistance in pre-clinical models and have also been identified in a small number of patients (2 of 195 patients) in a recent re-biopsy study.33 While these resistance mechanisms may be too rare for dedicated clinical trials, there are trials of the pan-HER kinase inhibitors afatinib and dacomitinib ongoing for acquired resistance (Table 1), and synergy between EGFR and BRAF kinase inhibitors is an area of active investigation in colorectal cancer.34 PIK3CA mutations have similarly been demonstrated to confer gefitinib resistance in vitro and were identified in a small number of patients (2 of 37 patients) in one re-biopsy series;21 this has not been identified in subsequent re-biopsy studies and the role of these mutations in acquired resistance remains controversial.20,22,35 Hoping to identify some synergy, early phase clinical trial of PI3K inhibitors combined with erlotinib have moved forward in patients with acquired resistance.36 Reactivation of ERK signaling through ERK amplification has also been identified in pre-clinical studies as resistance mechanism to third-generation EGFR kinase inhibitors targeting T790M and a trial combining selumetinib and gefitinib is underway.37
Additional resistance mechanisms with targeted agents in preclinical development
Several potential mechanisms for acquired resistance have been identified in primarily preclinical studies and small re-biopsy series including CRKL amplification, AXL kinase overexpression and increased levels of hepatocyte growth factor.38–41 These pre-clinical targets, although promising, are beyond the scope of this article and have recently been reviewed elsewhere.42
Managing acquired resistance to EGFR kinase inhibitors
Much has been learned regarding the management of acquired resistance since the early studies of TKIs in patients with EGFR-mutant lung cancer.43,44 Before EGFR kinase inhibitors, the only systemic therapies for treatment of advanced NSCLC were cytotoxic chemotherapies, drugs with modest efficacy and significant toxicity, often necessitating discontinuing treatment at the first sign of progression. In contrast, EGFR TKIs are given daily by mouth and have remarkable efficacy for EGFR-mutant lung cancer with modest toxicity. These differences have altered the basic balance between continued treatment versus treatment cessation in patients with acquired resistance.
The first question posited upon apparent disease progression must be whether a patient’s progression is clinically significant enough to warrant initiating a new line of therapy (Figure 2). Indolent, asymptomatic progression of existing metastatic disease, without the involvement of new organ systems, is seen often when acquired resistance initially develops.15 Continuing treatment with a TKI beyond progression in this context has been suggested to delay the need for chemotherapy.45 What’s more, stopping treatment prematurely may carry a risk of disease flare, particularly if immediate treatment with systemic chemotherapy is not planned. Disease flare causing hospitalization or death was noted in 23% of patients stopping EGFR TKIs for subsequent clinical trial enrolment in one series.25
Figure 2.
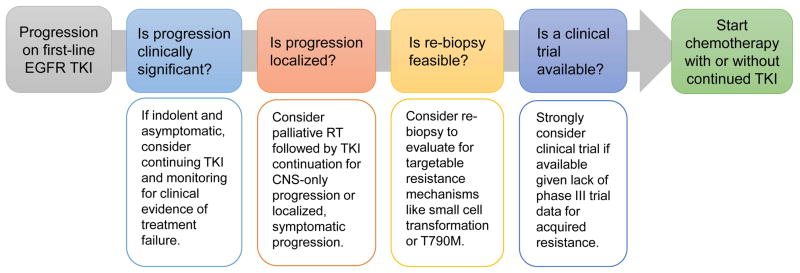
Approach to the management of EGFR mutant NSCLC with progression on first-line EGFR TKI. Here we propose a step-wise approach that considers progression characteristics and clinical trial availability before initiating second-line chemotherapy. RT: Radiation therapy
The next important issue to consider in patients exhibiting progression is whether their disease progression is localized, and potentially controlled with palliative local therapy (Figure 2). Isolated CNS progression on TKI therapy may, in some cases, be due to limited drug penetration into the CNS due to the blood-brain barrier. Palliative radiation followed by re-treatment with TKI has the potential to regain control of the disease; in one series of 51 patients, an additional median PFS of 6.2 months was reported.46 If systemic control and prevention of flare is a concern, it is also possible to continue EGFR TKI during whole brain radiation (WBRT), which was demonstrated to be safe in a phase II study of erlotinib plus WBRT.47 The RTOG 0320 examined the combination of erlotinib, whole brain and sterotactic radiotherapy in NSCLC with limited brain metastases. This study demonstrated increased incidence of grade 3–5 toxicity among those patients receiving erlotinib and stereotactic radiotherapy and underscores the risk of combining these strategies.48 Alternative strategies using pulsed high-dose EGFR kinase inhibitors to overcome the blood-brain barrier are currently investigational with variable rates of clinical benefit described in several series.49,50
Acquired treatment resistance may also manifest as limited oligometastatic disease progression outside of the CNS. Locally ablative therapies such as palliative radiation can certainly be beneficial in the context of symptomatic oligometastatic disease. However, whether the treatment of oligometastatic disease with local ablation changes the natural history of acquired resistance or improves long term outcomes remains unclear. Uncontrolled studies have described favorable outcomes in selected patients with acquired resistance receiving aggressive surgery or radiation to focal sites of disease,46,51 but evidence from properly controlled randomized studies will be needed before broad adoption of this strategy.
One question to consider in a patient with progressive disease on EGFR TKI is whether re-biopsy may be valuable (Figure 2). Several large studies have shown re-biopsy to be feasible to characterize the molecular mechanisms of resistance.20–22 Re-biopsy can provide prognostic information given the favorable prognosis seen in two series with T790M-mediated acquired resistance.15,16 Further, re-biopsy is particularly valuable when considering a clinical trial – if pathology shows small cell transformation, immediate chemotherapy makes more sense, while detection of specific resistance mutations outlined previously could steer a patient towards a trial of a specific targeted therapy (Table).27,28 The importance of enrolling patients onto clinical trials for acquired resistance cannot be over emphasized, given that this is a setting where no positive phase III trial has ever been performed and the standard of care remains loosely defined.
For patients with systemic progression and no clinical trial available, cytotoxic chemotherapy is an appropriate second-line therapy after failure of EGFR TKI given the demonstrated efficacy of platinum-based chemotherapy in NSCLC. However, published data is lacking on the activity of cytotoxic chemotherapy after TKI failure. Two retrospective studies described response rates of 15% and 18% to chemotherapy using a variety of regimens.52,53 But a recently presented Japanese study reported a more favorable response rate of 40% (95%CI: 22–58%) to carboplatin, paclitaxel and bevacizumab in 30 patients with acquired resistance.54 More data on this topic is needed. Many clinicians add cytotoxic chemotherapy to continued EGFR TKI for patients with acquired resistance based upon preclinical work describing synergy, believed to occur through controlling the portion of cells that remain TKI sensitive.13 Indeed the only published prospective trial of chemotherapy for acquired resistance, to our knowledge, is a single-arm phase II trial of pemetrexed plus continued TKI with a RR of 26% and median PFS of 7 months.55 A retrospective study of 78 patients receiving chemotherapy for acquired resistance found an improved response rate for those continuing TKI with chemotherapy (41% vs. 18%) but there was no difference in PFS.53 The potential synergy of continuing EGFR inhibition when chemotherapy is started for acquired resistance is the subject of the ongoing IMPRESS trial of cisplatin-pemetrexed with or without gefitinib (NCT01544179).
Targeted therapies for acquired resistance to EGFR kinase inhibitors
Targeted therapies for EGFR TKI resistance have been an area of active investigation for nearly a decade; the earliest studies, many of which were negative, have been reviewed elsewhere recently.56 Here we will focus on therapies that currently are commercially available or under active investigation (Table).
Second-generation irreversible EGFR kinase inhibitors have been investigated as therapies for acquired resistance based on preclinical data suggesting increased activity against models with EGFR T790M.57 Afatinib is the most studied drug in this class, its use in acquired resistance having been evaluated in the phase II/III LUX-Lung 1 trial. This study was negative for its primary endpoint of overall survival (10.8 vs. 12 months, p=0.74),58 but demonstrated a statistically significant improvement in PFS (3.3 vs. 1.1 months, p>0.0001) and response rate (7% versus 0.5%, p=0.007).58 Because afatinib inhibits wildtype EGFR more potently than T790M (Figure 3),57 it has been hypothesized that EGFR-related toxicity impairs the clinical delivery of this drug at levels sufficient to block T790M-mediated signalling. For this reason, a study of intermittent high-dose afatinib is underway. Dacomitinib is another irreversible pan-HER inhibitor with promising activity in early studies in resistant disease.59 Of note, both afatinib and dacomitinib have activity against the HER2 kinase and therefore could be active against resistance mediated by HER2 amplification. The NCIC Br.26 trial randomized 720 patients that had progressed on standard therapy to either dacomitinib or placebo, including a pre-planned EGFR-mutant subgroup with acquired TKI resistance; the trial has reportedly failed to meet its primary survival endpoint though the outcome in the EGFR-mutant subgroup remains to be reported.60
Figure 3.

Relative potency of different EGFR kinase inhibitors against different EGFR genotypes in vitro. The Y-axis represents the relative IC50 normalized to the IC50 against EGFR sensitizing mutations (L858R or exon 19 deletion).27,57,62 Second-generation EGFR TKIs like afatinib are more potent against T790M than gefitinib, but dosing in the clinic is limited by wildtype inhibition (and toxicity) at a relatively lower dose. Third-generation EGFR TKIs like CO-1686 and AZD9291 selectively inhibit EGFR T790M well below the dose at which wildtype EGFR is inhibited and have the potential to yield reduced toxicity as a result.
Third-generation EGFR kinase inhibitors comprise a particularly promising class of investigational drugs for acquired resistance. These agents are structurally distinct from first and second generation inhibitors and were designed specifically to inhibit EGFR T790M while sparing wild-type EGFR (Figure 3).61,62 Several agents of this class have entered phase I clinical trials, the most studied being CO-1686 and AZD9291, both of which have demonstrated impressive responses in TKI-resistant disease. The CO-1686 study has reported on 9 patients with T790M from the highest dosing cohort (900mg BID), and 6 had a partial response.28 The AZD9291 study reported on 34 patients across all dosing cohorts (20–80 mg daily) and 15 had a partial response, including 9 of 18 positive for T790M and only 1 of 5 negative for T790M.27 No significant rash or diarrhea has been seen, consistent with a lack of inhibition of wildtype EGFR. Further studies of the relative effectiveness of these agents among both T790M positive and negative patients will serve to elucidate the potential role of T790M as a predictive marker for this new class of agents.
Another approach used to maximize inhibition of EGFR signalling in acquired resistance has been the combination of an EGFR kinase inhibitor with an EGFR-targeted antibody, a synergy that was initially discovered through studies of genetically-engineered mouse models.63 Though a phase II trial combining erlotinib and cetuximab revealed minimal activity,64 preliminary results of a phase Ib trial of afatinib and cetuximab in 60 patients with acquired resistance revealed a 30% response rate and 4.7 month median PFS,65 with grade 3 rash and diarrhea seen in 18% and 7% of patients, respectively (Table). Responses were seen irrespective of T790M status. Preclinical work shows that the combination of an EGFR antibody and a covalent kinase inhibitor markedly reduces both EGFR signalling as well as total levels of EGFR protein.63 Additional clinical trials with this regimen are in development.
Several other combinations are under investigation for acquired resistance, most commonly adding a new targeted agent to a reversible EGFR TKI (Table). Several studies are evaluating MET inhibitors (either antibodies or kinase inhibitors) added to an EGFR TKI, though none at this time are limited to MET-mediated resistance. The HSP90 inhibitor AUY922 is being studied in acquired resistance with response rates around 18% (12/66 patients) with AUY922 alone and 16% (4/25 patients) when combined with erlotinib.66,67 A phase II study is ongoing randomizing patients with TKI resistant disease to AUY922 versus single-agent chemotherapy (NCT01646125). The combination of an EGFR TKI with the PI3K inhibitor BKM120 is also under investigation with toxicities including hyperglycemia, rash and diarrhea.68 In general these combination studies have included all patients with acquired resistance, a molecularly heterogeneous population, rather than focusing on a biomarker-enriched subset of EGFR-mutant lung cancers more likely to respond.
Lastly, the use of PD-1 and PD-L1 inhibitors for acquired resistance must be considered given the promising early data in advanced NSCLC.69 Recent pre-clinical data has suggested that EGFR mutation-positive lung cancer may preferentially utilize PD-1/PD-L1 mediated mechanisms to evade immune surveillance.70 To investigate whether there might be synergy in EGFR-mutant lung cancers, several phase 1 studies are currently or will soon be combining EGFR TKI with PD-1 or PDL-1 antibodies (NCT01454102, NCT02039674 and NCT02013219).
Questions ahead
The coming year is expected to bring greater availability of third-generation EGFR TKIs as a clinical trial options for patients with acquired resistance. This exciting therapeutic development raises an important drug development question: when planning a phase III trial for patients with acquired resistance, what standard therapy should be given to the control arm? A single phase III trial has been performed for acquired resistance, the third/fourth-line LUX-Lung1 trial, and its primary endpoint was negative.58 Some might consider treating patients progressing on first-line TKI as “second-line NSCLC” and support a minimalist standard therapy such as single-agent pemetrexed. Others might consider these patients platinum naïve and support an aggressive standard therapy containing cisplatin or bevacizumab. Still others, believing in erlotinib continuation post-progression, might support erlotinib with chemotherapy as standard. This lack of a clear standard is certain to add complexity to drug development in this space.
A second important question raised by the emergence of EGFR T790M inhibitors is how to incorporate re-biopsy and T790M testing into the standard management of acquired resistance. While technically feasible at academic centers,20–22 re-biopsy for molecular analysis creates technical and resource allocation challenges, particularly at community centers. Exciting technologies are emerging that may allow noninvasive genotyping of cell-free plasma DNA, and have been proposed as a potential replacement for either initial EGFR genotyping or subsequent identification of the T790M resistance mutation.71,72 Such noninvasive T790M genotyping has the potential to be implemented more broadly than current strategies requiring biopsy tissue.
Acknowledgments
The research of the authors of this manuscript has been supported in part by by the National Cancer Institute of the National Institutes of Health (R01CA114465 and P50CA090578) and by the Conquer Cancer Foundation of the American Society of Clinical Oncology.
References
- 1.Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350(21):2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 2.Paez JG, Jänne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304(5676):1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 3.Pao W, Miller V, Zakowski M, et al. EGF receptor gene mutations are common in lung cancers from "never smokers" and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101(36):13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shigematsu H, Lin L, Takahashi T, et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst. 2005;97(5):339–346. doi: 10.1093/jnci/dji055. [DOI] [PubMed] [Google Scholar]
- 5.Johnson ML, Sima CS, Chaft J, et al. Association of KRAS and EGFR mutations with survival in patients with advanced lung adenocarcinomas. Cancer. 2013;119(2):356–362. doi: 10.1002/cncr.27730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hirsch FR, Jänne PA, Eberhardt WE, et al. Epidermal growth factor receptor inhibition in lung cancer: status 2012. J Thorac Oncol. 2013;8(3):373–384. doi: 10.1097/JTO.0b013e31827ed0ff. [DOI] [PubMed] [Google Scholar]
- 7.Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362(25):2380–2388. doi: 10.1056/NEJMoa0909530. [DOI] [PubMed] [Google Scholar]
- 8.Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13(3):239–246. doi: 10.1016/S1470-2045(11)70393-X. [DOI] [PubMed] [Google Scholar]
- 9.Sequist LV, Yang JC, Yamamoto N, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol. 2013;31(27):3327–3334. doi: 10.1200/JCO.2012.44.2806. [DOI] [PubMed] [Google Scholar]
- 10.Shi Y, Zhang L, Liu X, et al. Icotinib versus gefitinib in previously treated advanced non-small-cell lung cancer (ICOGEN): a randomised, double-blind phase 3 non-inferiority trial. Lancet Oncol. 2013;14(10):953–961. doi: 10.1016/S1470-2045(13)70355-3. [DOI] [PubMed] [Google Scholar]
- 11.Ohashi K, Maruvka YE, Michor F, Pao W. Epidermal growth factor receptor tyrosine kinase inhibitor-resistant disease. J Clin Oncol. 2013;31(8):1070–1080. doi: 10.1200/JCO.2012.43.3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jackman D, Pao W, Riely GJ, et al. Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. J Clin Oncol. 2010;28(2):357–360. doi: 10.1200/JCO.2009.24.7049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chmielecki J, Foo J, Oxnard GR, et al. Optimization of dosing for EGFR-mutant non-small cell lung cancer with evolutionary cancer modeling. Sci Transl Med. 2011;3(90):90ra59. doi: 10.1126/scitranslmed.3002356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ye X, Zhu ZZ, Zhong L, et al. High T790M detection rate in TKI-naive NSCLC with EGFR sensitive mutation: truth or artifact? J Thorac Oncol. 2013;8(9):1118–1120. doi: 10.1097/JTO.0b013e31829f691f. [DOI] [PubMed] [Google Scholar]
- 15.Oxnard GR, Arcila ME, Sima CS, et al. Acquired resistance to EGFR tyrosine kinase inhibitors in EGFR-mutant lung cancer: distinct natural history of patients with tumors harboring the T790M mutation. Clin Cancer Res. 2011;17(6):1616–1622. doi: 10.1158/1078-0432.CCR-10-2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hata A, Katakami N, Yoshioka H, et al. Rebiopsy of non-small cell lung cancer patients with acquired resistance to epidermal growth factor receptor-tyrosine kinase inhibitor: Comparison between T790M mutation-positive and mutation-negative populations. Cancer. 2013;119(24):4325–4332. doi: 10.1002/cncr.28364. [DOI] [PubMed] [Google Scholar]
- 17.Oxnard GR, Morris MJ, Hodi FS, et al. When progressive disease does not mean treatment failure: reconsidering the criteria for progression. J Natl Cancer Inst. 2012;104(20):1534–1541. doi: 10.1093/jnci/djs353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mir O, Blanchet B, Goldwasser F. Drug-induced effects on erlotinib metabolism. N Engl J Med. 2011;365(4):379–380. doi: 10.1056/NEJMc1105083. [DOI] [PubMed] [Google Scholar]
- 19.Hamilton M, Wolf JL, Rusk J, et al. Effects of smoking on the pharmacokinetics of erlotinib. Clin Cancer Res. 2006;12(7 Pt 1):2166–2171. doi: 10.1158/1078-0432.CCR-05-2235. [DOI] [PubMed] [Google Scholar]
- 20.Arcila ME, Oxnard GR, Nafa K, et al. Rebiopsy of lung cancer patients with acquired resistance to EGFR inhibitors and enhanced detection of the T790M mutation using a locked nucleic acid-based assay. Clin Cancer Res. 2011;17(5):1169–1180. doi: 10.1158/1078-0432.CCR-10-2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sequist LV, Waltman BA, Dias-Santagata D, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3(75):75ra26. doi: 10.1126/scitranslmed.3002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu HA, Arcila ME, Rekhtman N, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res. 2013;19(8):2240–2247. doi: 10.1158/1078-0432.CCR-12-2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kobayashi S, Boggon TJ, Dayaram T, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352(8):786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 24.Pao W, Miller VA, Politi KA, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2(3):e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chaft JE, Oxnard GR, Sima CS, Kris MG, Miller VA, Riely GJ. Disease flare after tyrosine kinase inhibitor discontinuation in patients with EGFR-mutant lung cancer and acquired resistance to erlotinib or gefitinib: implications for clinical trial design. Clin Cancer Res. 2011;17(19):6298–6303. doi: 10.1158/1078-0432.CCR-11-1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Watanabe S, Tanaka J, Ota T, et al. Clinical responses to EGFR-tyrosine kinase inhibitor retreatment in non-small cell lung cancer patients who benefited from prior effective gefitinib therapy: a retrospective analysis. BMC Cancer. 2011;11:1. doi: 10.1186/1471-2407-11-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ranson M, Pao W, Kim D, et al. AZD9291: an irreversible, potent and selective tyrosine kinase inhibitor of activating EGFR and resistance T790M mutations in advanced NSCLC. World Conference on Lung Cancer; 2013; Sydney, Australia. [Google Scholar]
- 28.Soria J, Sequist LV, Gadgeel S, et al. First-in-human evaluation of CO-1686, an irreversible highly selective TKI of mutations of EGFR (activating and T790M). World Conference on Lung Cancer; 2013; Sydney, Australia. [Google Scholar]
- 29.Zakowski MF, Ladanyi M, Kris MG Group MS-KCCLCO. EGFR mutations in small-cell lung cancers in patients who have never smoked. N Engl J Med. 2006;355(2):213–215. doi: 10.1056/NEJMc053610. [DOI] [PubMed] [Google Scholar]
- 30.Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316(5827):1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 31.Bean J, Brennan C, Shih JY, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A. 2007;104(52):20932–20937. doi: 10.1073/pnas.0710370104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takezawa K, Pirazzoli V, Arcila ME, et al. HER2 amplification: a potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFRT790M mutation. Cancer Discov. 2012;2(10):922–933. doi: 10.1158/2159-8290.CD-12-0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ohashi K, Sequist LV, Arcila ME, et al. Lung cancers with acquired resistance to EGFR inhibitors occasionally harbor BRAF gene mutations but lack mutations in KRAS, NRAS, or MEK1. Proc Natl Acad Sci U S A. 2012;109(31):E2127–2133. doi: 10.1073/pnas.1203530109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Corcoran RB, Ebi H, Turke AB, et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012;2(3):227–235. doi: 10.1158/2159-8290.CD-11-0341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Engelman JA, Mukohara T, Zejnullahu K, et al. Allelic dilution obscures detection of a biologically significant resistance mutation in EGFR-amplified lung cancer. J Clin Invest. 2006;116(10):2695–2706. doi: 10.1172/JCI28656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jänne P, Cohen R, Laird A, et al. Phase I Safety and Pharmacokinetic Study of the PI3K/mTOR Inhibitor SAR245409 (XL765) in Combination with Erlotinib in Patients with Advanced Solid Tumors. J Thorac Oncol. 2014 doi: 10.1097/JTO.0000000000000088. In Press. [DOI] [PubMed] [Google Scholar]
- 37.Ercan D, Xu C, Yanagita M, et al. Reactivation of ERK signaling causes resistance to EGFR kinase inhibitors. Cancer Discov. 2012;2(10):934–947. doi: 10.1158/2159-8290.CD-12-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cheung HW, Du J, Boehm JS, et al. Amplification of CRKL induces transformation and epidermal growth factor receptor inhibitor resistance in human non-small cell lung cancers. Cancer Discov. 2011;1(7):608–625. doi: 10.1158/2159-8290.CD-11-0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang Z, Lee JC, Lin L, et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet. 2012;44(8):852–860. doi: 10.1038/ng.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yano S, Wang W, Li Q, et al. Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor-activating mutations. Cancer Res. 2008;68(22):9479–9487. doi: 10.1158/0008-5472.CAN-08-1643. [DOI] [PubMed] [Google Scholar]
- 41.Turke AB, Zejnullahu K, Wu YL, et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell. 2010;17(1):77–88. doi: 10.1016/j.ccr.2009.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chong CR, Jänne PA. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat Med. 2013;19(11):1389–1400. doi: 10.1038/nm.3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sequist LV, Martins RG, Spigel D, et al. First-line gefitinib in patients with advanced non-small-cell lung cancer harboring somatic EGFR mutations. J Clin Oncol. 2008;26(15):2442–2449. doi: 10.1200/JCO.2007.14.8494. [DOI] [PubMed] [Google Scholar]
- 44.Rosell R, Moran T, Queralt C, et al. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med. 2009;361(10):958–967. doi: 10.1056/NEJMoa0904554. [DOI] [PubMed] [Google Scholar]
- 45.Oxnard GR, Lo P, Jackman DM, et al. Delay of chemotherapy through use of post-progression erlotinib in patients with EGFR-mutant lung cancer. American Society of Clinical Oncology Annual Meeting; 2012; Chicago, United States. [Google Scholar]
- 46.Weickhardt AJ, Scheier B, Burke JM, et al. Local ablative therapy of oligoprogressive disease prolongs disease control by tyrosine kinase inhibitors in oncogene-addicted non-small-cell lung cancer. J Thorac Oncol. 2012;7(12):1807–1814. doi: 10.1097/JTO.0b013e3182745948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Welsh JW, Komaki R, Amini A, et al. Phase II trial of erlotinib plus concurrent whole-brain radiation therapy for patients with brain metastases from non-small-cell lung cancer. J Clin Oncol. 2013;31(7):895–902. doi: 10.1200/JCO.2011.40.1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sperduto PW, Wang M, Robins HI, et al. A phase 3 trial of whole brain radiation therapy and stereotactic radiosurgery alone versus WBRT and SRS with temozolomide or erlotinib for non-small cell lung cancer and 1 to 3 brain metastases: Radiation Therapy Oncology Group 0320. Int J Radiat Oncol Biol Phys. 2013;85(5):1312–1318. doi: 10.1016/j.ijrobp.2012.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grommes C, Oxnard GR, Kris MG, et al. "Pulsatile" high-dose weekly erlotinib for CNS metastases from EGFR mutant non-small cell lung cancer. Neuro Oncol. 2011;13(12):1364–1369. doi: 10.1093/neuonc/nor121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jackman DM, Mach SL, Heng JC, et al. Pulsed dosing of erlotinib for CNS progression in EGFR-mutant NSCLC. American Society of Clinical Oncology Annual Meeting; 2013; Chicago, United States. [Google Scholar]
- 51.Yu HA, Sima CS, Huang J, et al. Local therapy with continued EGFR tyrosine kinase inhibitor therapy as a treatment strategy in EGFR-mutant advanced lung cancers that have developed acquired resistance to EGFR tyrosine kinase inhibitors. J Thorac Oncol. 2013;8(3):346–351. doi: 10.1097/JTO.0b013e31827e1f83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu JY, Shih JY, Yang CH, et al. Second-line treatments after first-line gefitinib therapy in advanced nonsmall cell lung cancer. Int J Cancer. 2010;126(1):247–255. doi: 10.1002/ijc.24657. [DOI] [PubMed] [Google Scholar]
- 53.Goldberg SB, Oxnard GR, Digumarthy S, et al. Chemotherapy with Erlotinib or chemotherapy alone in advanced non-small cell lung cancer with acquired resistance to EGFR tyrosine kinase inhibitors. Oncologist. 2013;18(11):1214–1220. doi: 10.1634/theoncologist.2013-0168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Namba Y, Hattori T, Satouchi M, et al. A phase II study of bevacizumab in combination with carboplatin and paclitaxel in patients with non-squamous NSCLC harboring mutations of EGFR gene after failing first-line EGFR-TKIs. World Conference on Lung Cancer; 2013; Sydney, Australia. [Google Scholar]
- 55.Yoshimura N, Okishio K, Mitsuoka S, et al. Prospective assessment of continuation of erlotinib or gefitinib in patients with acquired resistance to erlotinib or gefitinib followed by the addition of pemetrexed. J Thorac Oncol. 2013;8(1):96–101. doi: 10.1097/JTO.0b013e3182762bfb. [DOI] [PubMed] [Google Scholar]
- 56.Oxnard GR, Arcila ME, Chmielecki J, Ladanyi M, Miller VA, Pao W. New strategies in overcoming acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in lung cancer. Clin Cancer Res. 2011;17(17):5530–5537. doi: 10.1158/1078-0432.CCR-10-2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li D, Ambrogio L, Shimamura T, et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene. 2008;27(34):4702–4711. doi: 10.1038/onc.2008.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Miller VA, Hirsh V, Cadranel J, et al. Afatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-Lung 1): a phase 2b/3 randomised trial. Lancet Oncol. 2012;13(5):528–538. doi: 10.1016/S1470-2045(12)70087-6. [DOI] [PubMed] [Google Scholar]
- 59.Engelman JA, Zejnullahu K, Gale CM, et al. PF00299804, an irreversible pan-ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res. 2007;67(24):11924–11932. doi: 10.1158/0008-5472.CAN-07-1885. [DOI] [PubMed] [Google Scholar]
- 60.Pfizer announces two top-line results from two phase III trials of dacomitinib in patients with refractory advanced non-small cell lung cancer. Pfizer. 2014 Jan 27; Press Release. [Google Scholar]
- 61.Zhou W, Ercan D, Chen L, et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature. 2009;462(7276):1070–1074. doi: 10.1038/nature08622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Walter AO, Sjin RT, Haringsma HJ, et al. Discovery of a Mutant-Selective Covalent Inhibitor of EGFR that Overcomes T790M-Mediated Resistance in NSCLC. Cancer Discov. 2013;3(12):1404–1415. doi: 10.1158/2159-8290.CD-13-0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Regales L, Gong Y, Shen R, et al. Dual targeting of EGFR can overcome a major drug resistance mutation in mouse models of EGFR mutant lung cancer. J Clin Invest. 2009;119(10):3000–3010. doi: 10.1172/JCI38746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Janjigian YY, Azzoli CG, Krug LM, et al. Phase I/II trial of cetuximab and erlotinib in patients with lung adenocarcinoma and acquired resistance to erlotinib. Clin Cancer Res. 2011;17(8):2521–2527. doi: 10.1158/1078-0432.CCR-10-2662. [DOI] [PubMed] [Google Scholar]
- 65.Janjigian YY, Smit EF, Horn L, et al. Activity of afatinib/cetuximab in patients (pts) with EGFR mutant non-small cell lung cancer (NSCLC) and acquired resistance (AR) to EGFR inhibitors. European Society for Medical Oncology Congress; 2012; Vienna, Austria. [Google Scholar]
- 66.Yu HA, Johnson ML, Urman A, et al. A phase II study of HSP90 inhibitor and erlotinib in patients with EGFR-mutant lung cancer and acquired resistance to EGFR tyrosine kinase inhibitors. World Conference on Lung Cancer; 2013; Sydney, Australia. [Google Scholar]
- 67.Barlesi F, Besse B, Chu Q, et al. Phase II activity of the HSP90 inhibitor AUY922 in patients with EGFR-mutant advanced non-small cell lung cancer (NSCLC). World Conference on Lung Cancer; 2013; Sydney, Australia. [Google Scholar]
- 68.Grana B, Burris H, Ahnert J, et al. Oral PI3 kinase inhibitor BKM120 monotherapy in patients (pts) with advanced solid tumors: An update on safety and efficacy. ASCO Annual Meeting; 2011; Chicago, United States. [Google Scholar]
- 69.Zielinski C, Knapp S, Mascaux C, Hirsch F. Rationale for targeting the immune system through checkpoint molecule blockade in the treatment of non-small-cell lung cancer. Ann Oncol. 2013;24(5):1170–1179. doi: 10.1093/annonc/mds647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Akbay EA, Koyama S, Carretero J, et al. Activation of the PD-1 Pathway Contributes to Immune Escape in EGFR-Driven Lung Tumors. Cancer Discov. 2013;3(12):1355–1363. doi: 10.1158/2159-8290.CD-13-0310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kuang Y, Rogers A, Yeap BY, et al. Noninvasive detection of EGFR T790M in gefitinib or erlotinib resistant non-small cell lung cancer. Clin Cancer Res. 2009;15(8):2630–2636. doi: 10.1158/1078-0432.CCR-08-2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Oxnard GR, Paweletz CP, Kuang Y, et al. Noninvasive detection of response and resistance in EGFR-mutant lung cancer using quantitative next-generation genotyping of cell-free plasma DNA. Clinical Cancer Research. 2014 doi: 10.1158/1078-0432.CCR-13-2482. Epublished ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]