

Abstract
Periventricular heterotopia (PH) is one of the most common malformations of cortical development (MCD). Nodules along the lateral ventricles of the brain, disruption of the ventricular lining, and a reduced brain size are hallmarks of this disorder. PH results in a disruption of the neuroependyma, inhibition of neural proliferation and differentiation, and altered neuronal migration. Human mutations in the genes encoding the actin-binding Filamin A (FLNA) and the vesicle trafficking Brefeldin A-associated guanine exchange factor 2 (BIG2 is encoded by the ARFGEF2 gene) proteins are implicated in PH formation. Recent studies have shown that the transition from proliferating neural progenitors to post-mitotic neurons relies on apical abscission along the neuroepithelium. This mechanism involves an actin dependent contraction of the apical portion of a neural progenitor along the ventricular lining to complete abscission. Actin also maintains stability of various cell adhesion molecules along the neuroependyma. Loss of cadherin directs disassembly of the primary cilium, which transduces sonic-hedgehog (Shh) signaling. Shh signaling is required for continued proliferation. In this context, apical abscission regulates neuronal progenitor exit and migration from the ventricular zone by detachment from the neuroependyma, relies on adhesion molecules that maintain the integrity of the neuroepithelial lining, and directs neural proliferation. Each of these processes is disrupted in PH, suggesting that genes causal for this MCD, may fundamentally mediate apical abscission in cortical development. Here we discuss several recent reports that demonstrate a coordinated role for actin and vesicle trafficking in modulating neural development along the neurepithelium, and potentially the neural stem cell to neuronal transition.
Keywords: abscission, brefeldin A inhibited guanine exchange factor 2, filamin, periventricular heterotopia
Introduction
Periventricular heterotopia (PH, Figure 1) is one of the most common malformations of cortical development (MCD) and causes seizures, dyslexia, and psychiatric disturbances.1 PH is characterized by bilateral ectopic neuronal nodules (arrows) found along the lateral ventricles.2 The nodules are caused by impaired migration from the ventricular zone (VZ) and disruption in the integrity of the neuroependyma.1 Mutations in the causative PH genes can also cause microcephaly (meaning small brain).3,4
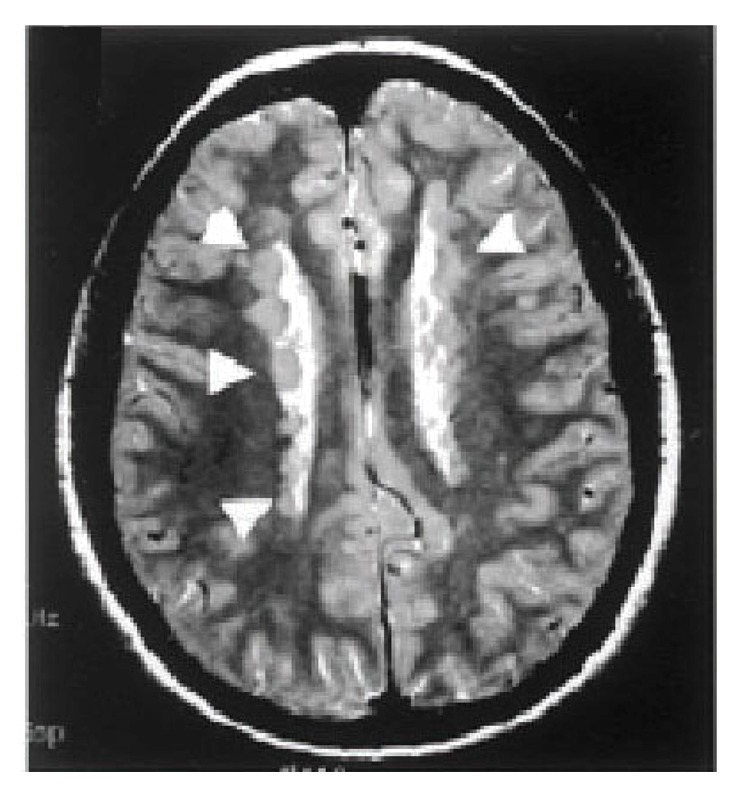
Figure 1. Periventricular Heterotopia. Coronal T2 MRI of the brain demonstrates bilateral heterotopic nodules (arrowheads) of neurons that are ectopically situated along the neuroepithelium of the lateral ventricles.
The most common form of PH is inherited in an X-linked dominant fashion from mutations in the filamin A (FLNA) gene.5-7 Hemizygous males are generally embryonic lethal with PH manifesting in females. FLNA encodes the cytoplasmic actin binding protein FLNA, which serves as a scaffold for over 30 proteins.8 A second form of autosomal recessive PH with microcephaly (ARPHM) has been associated with mutations in the ARFGEF2 gene. ARFGEF2 encodes brefeldin A inhibited guanine exchange factor-2 (BIG2).3 BIG2 is a protein kinase A anchoring protein (AKAP) which regulates Golgi-vesicle trafficking through its Sec7 domain. Recent work has identified cadherin receptor ligands in causing PH due to mutations in FAT4 and DCHS1.9
Some of the pathophysiological mechanisms giving rise to hallmarks of PH (diminished proliferation, impaired initial neural migration, and disruption of the neuropendymal lining causing heterotopia) have become apparent. Studies from this laboratory and others have demonstrated that a loss in cell-cell and cell-extracellular matrix (ECM) adhesion leading to loss of integrity along the ventricular lining causes heterotopia formation.10-12 The reduction in neuronal production seen with loss of FlnA is partially due to impaired degradation of cell cycle associated proteins, leading to a prolongation in the time to transit through the cell cycle and subsequent reduced proliferation.13 The migratory defects seen with loss of either FlnA or Big2 could be attributed to some disruption in actin dynamics.14 While these studies clarify individual cellular and molecular pathways for the specific PH phenotypes, they do not provide a unifying general mechanism for this disorder. Here we comment on recent experiments from this lab demonstrating both physical and functional interactions between FlnA and Big2. We suggest that coordinated interactions between actin and vesicle trafficking give rise to PH hallmarks and perhaps, serve primary roles in cell abscission along the neuroependyma.
Apical Abscission in Cortical Development
Normal development of the human cortex occurs through a series of well-described cellular events. During embryonic development, this process involves the proliferation, migration, and differentiation of neuronal progenitors located in the ventricular zone. Beginning along the lateral ventricles, neural ependymal progenitors undergo several rounds of cell division leading to the formation of the rapidly expanding ventricular zone. As this process continues, post-mitotic neurons attach and migrate along radial glial scaffolds out of the expanding ventricular zone, creating a new overlying layer of cells called the preplate. The laminar position of neurons is characteristic of their birthdate, such that younger neuroblasts migrate past their older counterparts to form the more superficial layers of the cortex. Finally, once neurons reach their cortical destinations, they differentiate ultimately forming the complex dendritic and axonal connections that are characteristic of fully mature cortical neurons.15
Recent studies have yielded new insights into how the transition from neural progenitor to post-mitotic neuron occurs along the neuroepithelium.16 A form of cell subdivision takes place along the neuroepithelium resulting in abscission of the apical membrane and detachment of the newly formed post-mitotic neuron from the ventricle (Fig. 2). Neuroepithelial progenitors contain a subapical actin cable that contracts to mediate apical abscission at the ventricular surface. N-cadherin is connected intracellularly to the contractile actin cable and maintains apical junctions and cell-cell adhesion, thereby inhibiting abscission. Maintenance of the neuroepithelium also preserves primary cilia function. Neuroepithelial cells contain a primary cilium which transduces sonic hedgehog (Shh) signaling required for continued progenitor proliferation. With loss of N-cadherin function and activation of actin, abscission is initiated. Apical abscission leads to a dismantling of the cilium, loss of Shh signaling, and decline in proliferation with progenitors exiting the cell cycle.17,18 Abscission also regulates cell polarity as apical proteins such as the Par-complex protein, atypical protein kinase C, remain with the abscissed portion along the ventricular neuroependyma and is absent in the post mitotic, migratory neuron.
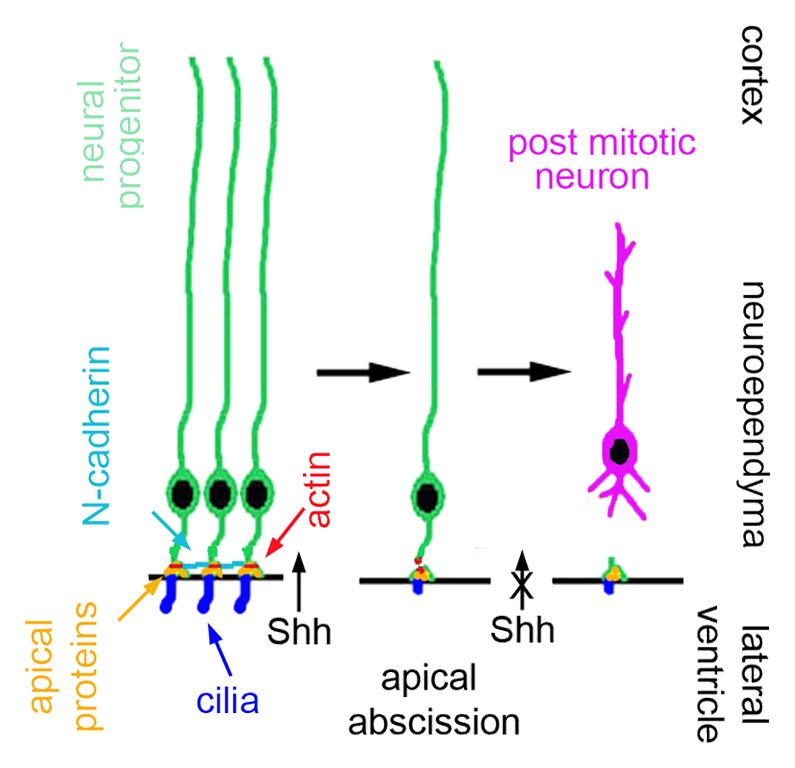
Figure 2. Schematic diagram of apical abscission. Cell adhesion molecules (N-cadherins) are connected to the contractile actin cable, maintain the integrity of the neuroepithlium, and inhibit apical abscission. With loss of N-cadherin expression, the actin cable contracts, and abscission follows. Apical proteins are separated from the main cell compartments and reside with the abscissed portion. Abscission also interrupts sonic hedgehog signaling (Shh) through the primary cilium, thereby inhibiting further proliferation. Following abscission, the newly generated post-mitotic neuron or progenitor transitions toward the cortical plate.
Periventricular Heterotopia: A Genetic Disorder of Endocytosis
FLNA is a 280 kD actin-binding phosphoprotein represented by an N-terminal actin-binding domain, followed by Immunoglobulin (IG) like repeat domains, that contain the receptor binding region at the C-terminus. FLNA also contains a PKA-dependent phosphorylation site at serine 2152 that has been shown to regulate its redistribution to the cell membrane.19,20 FLNA homodimers regulate the actin cytoskeleton through interactions derived from its multiple receptor binding regions, thereby directing cell stability, protrusion, and motility.21,22 Filamin also promotes actin branching, tethers large actin filaments and holds them in a perpendicular arrangement.23,24 The actin filaments represent a characteristic cortical actin structure at the leading edge of migrating cells. FLNA has also been shown to interact with numerous other proteins, suggesting many potential mechanistic roles that could influence cortical development. These interactors demonstrate great functional diversity including (1) regulation of cortical actin networks through molecules including β-integrin and the Rho family of small GTPases, (2) interaction with transmembrane receptors and signaling molecules such as the dopamine and G protein coupled calcium sensing receptors, and (3) serving as signaling scaffolds with diverse intracellular cell signaling kinases, phosphatases and adaptor molecules such as SHIP-2 or SEK1.25-28 In a general sense, filamins serve to stabilize various receptors and cell signaling partners near the cell membrane and also promote co-localization of interacting proteins and molecules to facilitate potential activation of signaling cascades.
BIG2 is one of the three large Sec7 proteins isolated in mammalian cells (GBF1, BIG1/2) and distinguished by their sensitivity to the fungal metabolite Brefeldin A (BFA).29,30 The Sec7 domain of BIG2 gives it the ability to bind and activate ADP-Ribosylation Factor (ARF) proteins by accelerating the replacement of ARF-bound GDP to GTP.31 ARF proteins belong to the Ras-superfamily of GTPases and are primarily involved in intracellular vesicle trafficking through the trans-Golgi network, recycling endosome, and plasma membrane.32,33 BIG2 therefore regulates intracellular vesicle trafficking by activating ARF proteins (specifically ARFs 1 and 3). Additionally, the N-terminal domain of BIG2 binds Exo70, a member of the exocyst complex involved in vesicle exocytosis.34 Several AKAP (A-kinase anchoring protein) binding sites were identified within the Exo70 domain. These residues interact with PKA (Protein Kinase A) subunits to regulate BIG2 activation of the ARFs.35 Finally, at its C-terminal, BIG2 interacts with the β subunit of GABA receptors.36 Overall, these data might suggest that BIG2 can coordinate vesicle trafficking (through ARF and Exo70 proteins) with cAMP-dependent signaling (through the A-kinase anchoring domain).
Recent studies from this laboratory have begun to reconcile how these two seemingly dissimilar proteins can give rise to PH.14,37 Either acute or chronic loss of either Big2 or FlnA leads to impairments in neural migration during development of the cortex. Migratory neural cells show defects in filopodia extension and attachment onto extracellular matrix coated surfaces. Both proteins physically bind and interact within neural progenitors, and loss of protein expression of either FlnA or Big2 leads to compensatory upregulation of the other. As with many proteins that FlnA binds, Big2 localization is dependent on phosphorylation of the actin binding protein which redirects Big2 from the Golgi to the cell membrane. Relocalization to the membrane allows Big2 to activate Arf1. Arfs have been shown to reside at the cell surface with ARF1 and ARF3 mediating endocytosis.38,39 This reciprocal regulation likely reflects a negative feedback, where loss of Big2 promotes phospho-FlnA expression to enhance redistribution of Big2 to the membrane. Conversely, loss of FlnA enhances Big2 expression to allow for Big2 delivery to the membrane. These studies begin to suggest an integral relationship between FlnA dependent actin dynamics and Big2 dependent regulation of vesicle formation and trafficking.
Changes in Arf-dependent endocytosis have the potential to disrupt several cell developmental processes. With respect to migration, Big2 and FlnA interactions regulate the stability and turnover of integrin/paxillin at the cell membrane, thereby directly impacting neural migration. Endocytosis also mediates the stability and turnover of cell-cell and cell-ECM adhesion molecules such as cadherins, and mediate neuroependymal integrity. Finally, changes in the expression and stability of receptors such as cadherins at the membrane would directly affect the processing, signaling and degradation of downstream effectors such as catenins, as these molecules are trafficked either back to the membrane or into lysosomal compartments. In this manner, a more generalized defect in vesicle trafficking may account for the multiple PH phenotypes.
Apical Abscission and PH: Development and Disease through a Common Pathway?
The gross anatomical features seen with PH suggest that it may reflect a disorder along the neuroependymal lining. Although a cell motility problem is in part implicated in heterotopia formation,14,37 the neuronal nodules along the lateral ventricles are primarily composed of later-born neurons.11 Impairments in cell autonomous motility would not be expected to favor different neuronal birth dates. We also observe a denudation of the neuroependymal lining adjacent to the heterotopia, suggesting that loss of the neuroepithelium could lead to disruption of the radial glial fibers and heterotopias formation. Another hallmark of PH is a reduction in brain size, consistent with a proliferative defect. Neural progenitor proliferation occurs within the ventricular zone of the cortex and abnormalities in the integrity could also disrupt neural stem cell proliferation. Thus, a defect in neural stem cell development along the neuroependyma would collectively account for all the neurological phenotypes seen in this disorder.
Given that apical abscission takes place along the neuroependyma, genes implicated in PH might be expected to regulate this fundamental process. Both FlnA and Big2 are highly expressed along the apical portion of the neuroepithelium during cortical development, suggesting a primary function along the neuroependyma.40 Loss of FlnA or Big2 also leads to diminished expression of various cell adhesion-associated proteins along the neuroepitheium such as cadherins and paxillin.11,41 Disruption of cell-cell and cell-ECM interactions through cadherins or integrins would alter abscission through cadherin-actin interactions and we find that loss of FlnA leads to diminished subapical actin cable expression (Fig. 3). Additionally, FlnA forms a functional complex with meckelin, a protein that directs centriole migration to the apical membrane and formation of the primary cilium. Disruption of this interaction impairs ciliogenesis and basal body positioning.42 Finally, apical abscission mirrors abscission during the final steps of cell cytokinesis, a process by which the cytoplasm of a single cell is divided into two daughter cells.43 FlnA mediates this process by stabilizing the ring canal assembly, the structure through which intracellular material passes.44,45 Our current work also suggests that FlnA-Big2 interact to regulate endocytosis, and endosomal recycling has emerged as one of the key components required for the successful completion of cytokinesis.43,46 Membrane recycling through vesicle trafficking is required for advancement of the cleavage furrow and separation of a single cell into daughter progeny. A similar process is likely invoked with apical abscission.
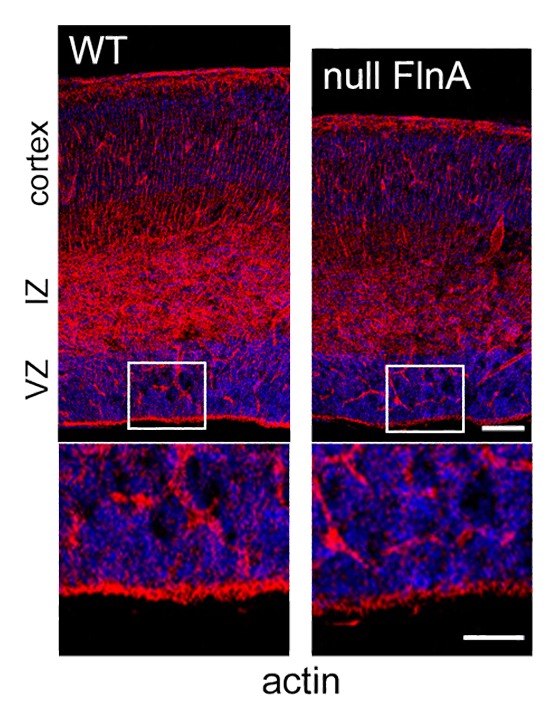
Figure 3. FlnA mediates subapical actin cable expression. Immunofluorescent photomicrographs of murine cortex shows that actin staining with phalloidin (rhodamine) is reduced along the apical portion of the neuroepithelium of null FlnA compared with wild type mice. Higher magnification is shown below. VZ = ventricular zone, IZ = intermediate zone. Scale bar in upper panel = 100 microns, lower panel 50 microns.
It remains to be seen what aspects of apical abscission are disrupted following FlnA or Big2 inhibition. Prior to abscission, Shh signaling is maintained through the primary cilium.16 Maintenance of N-cadherin expression and actin cables stabilize the neuroepithelium and inhibit cell abscission. Downregulation of cadherins through transcription factors such as FoxP2/4, promotes apical abscission, and presumptive loss in Shh signaling with subsequent dismantling of the primary cilium. Loss of Shh signaling leads to withdrawal from the cell cycle and a decline in proliferation. FlnA loss would be expected to impair Shh signaling and lead to diminished proliferation, if it truly affects primary cilium function. Loss of cadherin stability and expression through disruption of PH-associated proteins would also likely increase the number of cells undergoing abscission, inhibit Shh signaling, and promote withdrawal from the cell cycle. With respect to neuronal migration into the cortical plate, disruption of FlnA or Big2 dependent endocytosis could lead to failed abscission or a delay in the time required for separation of the main cell body from the abscissed portion at the apical surface. A delay in abscission would impede exit of post-mitotic neurons from the ventricular zone, leading to heterotopia formation.
Open Questions
The basic mechanisms underlying the transition from neural progenitor to neuron along the neuroepithlial lining may extend beyond normal corticogenesis and MCD. More broadly, apical abscission regulates an epithelial to mesenchymal type transformation (EMT) of various organ systems. As with the neuroependyma, epithelial cells in general maintain an apical to basal polarity, lack motility and form organized sheets. Conversely, mesenchymal cells lack this polarity and are loosely organized allowing such cells to dislodge and migrate to distant sites- a process similar to post-mitotic neurons which dislodge from the neuroepithelium and migrate to the cortical plate. Mesenchymal cells therefore possess greater malignant and metastatic potential. Genes associated with PH are implicated in these same processes. Beyond the brain, FlnA directs the development of various progenitors along the lining of various organ systems (i.e., gut, blood vessels, lung, etc). FlnA is also highly expressed and secreted in certain cancers.47 FlnA loss has been shown to inhibit the progression of cancers, consistent with the notion that it might inhibit EMT.48 Similarly, BIG2 levels are increased in certain cancer cells and its effector ARF1 has been shown to regulate growth and migration of cancer cells.49 It remains to be seen whether genes associated with PH coordinate actin dynamics and vesicle formation in apical abscission and how these processes might mediate other disease pathogenesis, including cancer.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported in part by the National Institutes of Health [NS063997–01 to VLS].
References
- 1.Sheen VL. Periventricular Heterotopia: Shuttling of Proteins through Vesicles and Actin in Cortical Development and Disease. Scientifica (Cairo) 2012;2012:480129. doi: 10.6064/2012/480129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lu J, Sheen V. Periventricular heterotopia. Epilepsy Behav. 2005;7:143–9. doi: 10.1016/j.yebeh.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 3.Sheen VL, Ganesh VS, Topcu M, Sebire G, Bodell A, Hill RS, Grant PE, Shugart YY, Imitola J, Khoury SJ, et al. Mutations in ARFGEF2 implicate vesicle trafficking in neural progenitor proliferation and migration in the human cerebral cortex. Nat Genet. 2004;36:69–76. doi: 10.1038/ng1276. [DOI] [PubMed] [Google Scholar]
- 4.Sheen VL, Topçu M, Berkovic S, Yalnizoglu D, Blatt I, Bodell A, Hill RS, Ganesh VS, Cherry TJ, Shugart YY, et al. Autosomal recessive form of periventricular heterotopia. Neurology. 2003;60:1108–12. doi: 10.1212/01.WNL.0000055898.00349.02. [DOI] [PubMed] [Google Scholar]
- 5.Fox JW, Lamperti ED, Ekşioğlu YZ, Hong SE, Feng Y, Graham DA, Scheffer IE, Dobyns WB, Hirsch BA, Radtke RA, et al. Mutations in filamin 1 prevent migration of cerebral cortical neurons in human periventricular heterotopia. Neuron. 1998;21:1315–25. doi: 10.1016/S0896-6273(00)80651-0. [DOI] [PubMed] [Google Scholar]
- 6.Sheen VL, Dixon PH, Fox JW, Hong SE, Kinton L, Sisodiya SM, Duncan JS, Dubeau F, Scheffer IE, Schachter SC, et al. Mutations in the X-linked filamin 1 gene cause periventricular nodular heterotopia in males as well as in females. Hum Mol Genet. 2001;10:1775–83. doi: 10.1093/hmg/10.17.1775. [DOI] [PubMed] [Google Scholar]
- 7.Sheen VL, Jansen A, Chen MH, Parrini E, Morgan T, Ravenscroft R, Ganesh V, Underwood T, Wiley J, Leventer R, et al. Filamin A mutations cause periventricular heterotopia with Ehlers-Danlos syndrome. Neurology. 2005;64:254–62. doi: 10.1212/01.WNL.0000149512.79621.DF. [DOI] [PubMed] [Google Scholar]
- 8.Feng S, Reséndiz JC, Lu X, Kroll MH. Filamin A binding to the cytoplasmic tail of glycoprotein Ibalpha regulates von Willebrand factor-induced platelet activation. Blood. 2003;102:2122–9. doi: 10.1182/blood-2002-12-3805. [DOI] [PubMed] [Google Scholar]
- 9.Cappello S, Gray MJ, Badouel C, Lange S, Einsiedler M, Srour M, Chitayat D, Hamdan FF, Jenkins ZA, Morgan T, et al. Mutations in genes encoding the cadherin receptor-ligand pair DCHS1 and FAT4 disrupt cerebral cortical development. Nat Genet. 2013;45:1300–8. doi: 10.1038/ng.2765. [DOI] [PubMed] [Google Scholar]
- 10.Carabalona A, Beguin S, Pallesi-Pocachard E, Buhler E, Pellegrino C, Arnaud K, Hubert P, Oualha M, Siffroi JP, Khantane S, et al. A glial origin for periventricular nodular heterotopia caused by impaired expression of Filamin-A. Hum Mol Genet. 2012;21:1004–17. doi: 10.1093/hmg/ddr531. [DOI] [PubMed] [Google Scholar]
- 11.Ferland RJ, Batiz LF, Neal J, Lian G, Bundock E, Lu J, Hsiao YC, Diamond R, Mei D, Banham AH, et al. Disruption of neural progenitors along the ventricular and subventricular zones in periventricular heterotopia. Hum Mol Genet. 2009;18:497–516. doi: 10.1093/hmg/ddn377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sarkisian MR, Bartley CM, Chi H, Nakamura F, Hashimoto-Torii K, Torii M, Flavell RA, Rakic P. MEKK4 signaling regulates filamin expression and neuronal migration. Neuron. 2006;52:789–801. doi: 10.1016/j.neuron.2006.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lian G, Lu J, Hu J, Zhang J, Cross SH, Ferland RJ, Sheen VL. Filamin a regulates neural progenitor proliferation and cortical size through Wee1-dependent Cdk1 phosphorylation. J Neurosci. 2012;32:7672–84. doi: 10.1523/JNEUROSCI.0894-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang J, Neal J, Lian G, Shi B, Ferland RJ, Sheen V. Brefeldin A-inhibited guanine exchange factor 2 regulates filamin A phosphorylation and neuronal migration. J Neurosci. 2012;32:12619–29. doi: 10.1523/JNEUROSCI.1063-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Götz M, Huttner WB. The cell biology of neurogenesis. Nat Rev Mol Cell Biol. 2005;6:777–88. doi: 10.1038/nrm1739. [DOI] [PubMed] [Google Scholar]
- 16.Das RM, Storey KG. Apical abscission alters cell polarity and dismantles the primary cilium during neurogenesis. Science. 2014;343:200–4. doi: 10.1126/science.1247521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rousso DL, Pearson CA, Gaber ZB, Miquelajauregui A, Li S, Portera-Cailliau C, Morrisey EE, Novitch BG. Foxp-mediated suppression of N-cadherin regulates neuroepithelial character and progenitor maintenance in the CNS. Neuron. 2012;74:314–30. doi: 10.1016/j.neuron.2012.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Louvi A, Grove EA. Cilia in the CNS: the quiet organelle claims center stage. Neuron. 2011;69:1046–60. doi: 10.1016/j.neuron.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vadlamudi RK, Li F, Adam L, Nguyen D, Ohta Y, Stossel TP, Kumar R. Filamin is essential in actin cytoskeletal assembly mediated by p21-activated kinase 1. Nat Cell Biol. 2002;4:681–90. doi: 10.1038/ncb838. [DOI] [PubMed] [Google Scholar]
- 20.Jay D, García EJ, de la Luz Ibarra M. In situ determination of a PKA phosphorylation site in the C-terminal region of filamin. Mol Cell Biochem. 2004;260:49–53. doi: 10.1023/B:MCBI.0000026052.76418.55. [DOI] [PubMed] [Google Scholar]
- 21.Gorlin JB, Yamin R, Egan S, Stewart M, Stossel TP, Kwiatkowski DJ, Hartwig JH. Human endothelial actin-binding protein (ABP-280, nonmuscle filamin): a molecular leaf spring. J Cell Biol. 1990;111:1089–105. doi: 10.1083/jcb.111.3.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cunningham CC, Gorlin JB, Kwiatkowski DJ, Hartwig JH, Janmey PA, Byers HR, Stossel TP. Actin-binding protein requirement for cortical stability and efficient locomotion. Science. 1992;255:325–7. doi: 10.1126/science.1549777. [DOI] [PubMed] [Google Scholar]
- 23.Hartwig JH, Shevlin P. The architecture of actin filaments and the ultrastructural location of actin-binding protein in the periphery of lung macrophages. J Cell Biol. 1986;103:1007–20. doi: 10.1083/jcb.103.3.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hartwig JH, Tyler J, Stossel TP. Actin-binding protein promotes the bipolar and perpendicular branching of actin filaments. J Cell Biol. 1980;87:841–8. doi: 10.1083/jcb.87.3.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feng Y, Walsh CA. The many faces of filamin: a versatile molecular scaffold for cell motility and signalling. Nat Cell Biol. 2004;6:1034–8. doi: 10.1038/ncb1104-1034. [DOI] [PubMed] [Google Scholar]
- 26.Dyson JM, O’Malley CJ, Becanovic J, Munday AD, Berndt MC, Coghill ID, Nandurkar HH, Ooms LM, Mitchell CA. The SH2-containing inositol polyphosphate 5-phosphatase, SHIP-2, binds filamin and regulates submembraneous actin. J Cell Biol. 2001;155:1065–79. doi: 10.1083/jcb.200104005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marti A, Luo Z, Cunningham C, Ohta Y, Hartwig J, Stossel TP, Kyriakis JM, Avruch J. Actin-binding protein-280 binds the stress-activated protein kinase (SAPK) activator SEK-1 and is required for tumor necrosis factor-alpha activation of SAPK in melanoma cells. J Biol Chem. 1997;272:2620–8. doi: 10.1074/jbc.272.5.2620. [DOI] [PubMed] [Google Scholar]
- 28.Woo MS, Ohta Y, Rabinovitz I, Stossel TP, Blenis J. Ribosomal S6 kinase (RSK) regulates phosphorylation of filamin A on an important regulatory site. Mol Cell Biol. 2004;24:3025–35. doi: 10.1128/MCB.24.7.3025-3035.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Achstetter T, Franzusoff A, Field C, Schekman R. SEC7 encodes an unusual, high molecular weight protein required for membrane traffic from the yeast Golgi apparatus. J Biol Chem. 1988;263:11711–7. [PubMed] [Google Scholar]
- 30.Sata M, Donaldson JG, Moss J, Vaughan M. Brefeldin A-inhibited guanine nucleotide-exchange activity of Sec7 domain from yeast Sec7 with yeast and mammalian ADP ribosylation factors. Proc Natl Acad Sci U S A. 1998;95:4204–8. doi: 10.1073/pnas.95.8.4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jones HD, Moss J, Vaughan M. BIG1 and BIG2, brefeldin A-inhibited guanine nucleotide-exchange factors for ADP-ribosylation factors. Methods Enzymol. 2005;404:174–84. doi: 10.1016/S0076-6879(05)04017-6. [DOI] [PubMed] [Google Scholar]
- 32.Shinotsuka C, Waguri S, Wakasugi M, Uchiyama Y, Nakayama K. Dominant-negative mutant of BIG2, an ARF-guanine nucleotide exchange factor, specifically affects membrane trafficking from the trans-Golgi network through inhibiting membrane association of AP-1 and GGA coat proteins. Biochem Biophys Res Commun. 2002;294:254–60. doi: 10.1016/S0006-291X(02)00456-4. [DOI] [PubMed] [Google Scholar]
- 33.Shinotsuka C, Yoshida Y, Kawamoto K, Takatsu H, Nakayama K. Overexpression of an ADP-ribosylation factor-guanine nucleotide exchange factor, BIG2, uncouples brefeldin A-induced adaptor protein-1 coat dissociation and membrane tubulation. J Biol Chem. 2002;277:9468–73. doi: 10.1074/jbc.M112427200. [DOI] [PubMed] [Google Scholar]
- 34.Xu KF, Shen X, Li H, Pacheco-Rodriguez G, Moss J, Vaughan M. Interaction of BIG2, a brefeldin A-inhibited guanine nucleotide-exchange protein, with exocyst protein Exo70. Proc Natl Acad Sci U S A. 2005;102:2784–9. doi: 10.1073/pnas.0409871102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuroda F, Moss J, Vaughan M. Regulation of brefeldin A-inhibited guanine nucleotide-exchange protein 1 (BIG1) and BIG2 activity via PKA and protein phosphatase 1gamma. Proc Natl Acad Sci U S A. 2007;104:3201–6. doi: 10.1073/pnas.0611696104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Charych EI, Yu W, Miralles CP, Serwanski DR, Li X, Rubio M, De Blas AL. The brefeldin A-inhibited GDP/GTP exchange factor 2, a protein involved in vesicular trafficking, interacts with the beta subunits of the GABA receptors. J Neurochem. 2004;90:173–89. doi: 10.1111/j.1471-4159.2004.02481.x. [DOI] [PubMed] [Google Scholar]
- 37.Zhang J, Neal J, Lian G, Hu J, Lu J, Sheen V. Filamin A regulates neuronal migration through brefeldin A-inhibited guanine exchange factor 2-dependent Arf1 activation. J Neurosci. 2013;33:15735–46. doi: 10.1523/JNEUROSCI.1939-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dong C, Zhang X, Zhou F, Dou H, Duvernay MT, Zhang P, Wu G. ADP-ribosylation factors modulate the cell surface transport of G protein-coupled receptors. J Pharmacol Exp Ther. 2010;333:174–83. doi: 10.1124/jpet.109.161489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kondo Y, Hanai A, Nakai W, Katoh Y, Nakayama K, Shin HW. ARF1 and ARF3 are required for the integrity of recycling endosomes and the recycling pathway. Cell Struct Funct. 2012;37:141–54. doi: 10.1247/csf.12015. [DOI] [PubMed] [Google Scholar]
- 40.Lu J, Tiao G, Folkerth R, Hecht J, Walsh C, Sheen V. Overlapping expression of ARFGEF2 and Filamin A in the neuroependymal lining of the lateral ventricles: insights into the cause of periventricular heterotopia. J Comp Neurol. 2006;494:476–84. doi: 10.1002/cne.20806. [DOI] [PubMed] [Google Scholar]
- 41.Feng Y, Chen MH, Moskowitz IP, Mendonza AM, Vidali L, Nakamura F, Kwiatkowski DJ, Walsh CA. Filamin A (FLNA) is required for cell-cell contact in vascular development and cardiac morphogenesis. Proc Natl Acad Sci U S A. 2006;103:19836–41. doi: 10.1073/pnas.0609628104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Adams M, Simms RJ, Abdelhamed Z, Dawe HR, Szymanska K, Logan CV, Wheway G, Pitt E, Gull K, Knowles MA, et al. A meckelin-filamin A interaction mediates ciliogenesis. Hum Mol Genet. 2012;21:1272–86. doi: 10.1093/hmg/ddr557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schiel JA, Childs C, Prekeris R. Endocytic transport and cytokinesis: from regulation of the cytoskeleton to midbody inheritance. Trends Cell Biol. 2013;23:319–27. doi: 10.1016/j.tcb.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sokol NS, Cooley L. Drosophila filamin encoded by the cheerio locus is a component of ovarian ring canals. Curr Biol. 1999;9:1221–30. doi: 10.1016/S0960-9822(99)80502-8. [DOI] [PubMed] [Google Scholar]
- 45.Li MG, Serr M, Edwards K, Ludmann S, Yamamoto D, Tilney LG, Field CM, Hays TS. Filamin is required for ring canal assembly and actin organization during Drosophila oogenesis. J Cell Biol. 1999;146:1061–74. doi: 10.1083/jcb.146.5.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Feng B, Schwarz H, Jesuthasan S. Furrow-specific endocytosis during cytokinesis of zebrafish blastomeres. Exp Cell Res. 2002;279:14–20. doi: 10.1006/excr.2002.5579. [DOI] [PubMed] [Google Scholar]
- 47.Alper O, Stetler-Stevenson WG, Harris LN, Leitner WW, Ozdemirli M, Hartmann D, Raffeld M, Abu-Asab M, Byers S, Zhuang Z, et al. Novel anti-filamin-A antibody detects a secreted variant of filamin-A in plasma from patients with breast carcinoma and high-grade astrocytoma. Cancer Sci. 2009;100:1748–56. doi: 10.1111/j.1349-7006.2009.01244.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Savoy RM, Ghosh PM. The dual role of filamin A in cancer: can’t live with (too much of) it, can’t live without it. Endocr Relat Cancer. 2013;20:R341–56. doi: 10.1530/ERC-13-0364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Boulay PL, Cotton M, Melançon P, Claing A. ADP-ribosylation factor 1 controls the activation of the phosphatidylinositol 3-kinase pathway to regulate epidermal growth factor-dependent growth and migration of breast cancer cells. J Biol Chem. 2008;283:36425–34. doi: 10.1074/jbc.M803603200. [DOI] [PMC free article] [PubMed] [Google Scholar]