

Abstract
Initially regarded as “epigenetic modifiers” acting predominantly through chromatin remodeling via histone acetylation, HDACIs, alternatively referred to as lysine deacetylase or simply deacetylase inhibitors, have since been recognized to exert multiple cytotoxic actions in cancer cells, often through acetylation of non-histone proteins. Some well-recognized mechanisms of HDACI lethality include, in addition to relaxation of DNA and de-repression of gene transcription, interference with chaperone protein function, free radical generation, induction of DNA damage, up-regulation of endogenous inhibitors of cell cycle progression, e.g., p21, and promotion of apoptosis. Intriguingly, this class of agents is relatively selective for transformed cells, at least in pre-clinical studies. In recent years, additional mechanisms of action of these agents have been uncovered. For example, HDACIs interfere with multiple DNA repair processes, as well as disrupt cell cycle checkpoints, critical to the maintenance of genomic integrity in the face of diverse genotoxic insults. Despite their pre-clinical potential, the clinical use of HDACIs remains restricted to certain subsets of T-cell lymphoma. Currently, it appears likely that the ultimate role of these agents will lie in rational combinations, only a few of which have been pursued in the clinic to date. This review focuses on relatively recently identified mechanisms of action of HDACIs, with particular emphasis on those that relate to the DNA damage response (DDR), and discuss synergistic strategies combining HDACIs with several novel targeted agents that disrupt the DDR or antagonize anti-apoptotic proteins that could have implications for the future use of HDACIs in patients with cancer.
Keywords: HDAC inhibitor, DNA damage response, DNA repair, cell cycle checkpoints, apoptosis, rational combinations
I. Introduction
In many respects, histone deacetylase inhibitors (HDACIs) represent prototypical “epigenetic” agents which act by modifying gene expression to restore the normal differentiation or death programs of transformed cells. In fact, the ability of HDACIs such as sodium butyrate to hyperacetylate histones and induce new gene expression in leukemia cells was discovered over thirty years ago (Reeves & Cserjesi, 1979). Over the ensuing decades, a large amount of data emerged concerning the mechanisms of action of these agents, culminating in the regulatory approval of two HDACIs, vorinostat (Zolinza®, Merck) and romidpesin (Istodax®, Celgene), for the treatment of patients with cutaneous and peripheral T-cell lymphoma (Watanabe, 2010). However, despite this large body of information, and intriguing evidence, both preclinical and clinical, suggesting a role for HDACIs in other malignancies (e.g., acute myeloid leukemia (AML), myelodysplastic syndromes (MDS) (Quintas-Cardama, Santos, & Garcia-Manero, 2011) and multiple myeloma (MM) (Richardson et al., 2013)), there is a general sense that HDACIs have not fully realized their potential as antineoplastic agents. One of the major barriers to this goal is continuing uncertainty about the mechanism of action by which these agents in fact trigger transformed cell death (Grant & Dai, 2012; Rosato & Grant, 2005). Complicating efforts to resolve this issue has been the emerging realization that HDACIs are truly pleiotropic agents which act through a wide variety of disparate and mutually interactive mechanisms. In this context, evidence that HDACIs modify gene expression, i.e., by altering chromatin structure, acetylating promoter regions, or disabling co-repressors, is undisputed. However, HDACIs target other nonhistone proteins which may, either directly or indirectly, influence cell fate, and it is highly likely that such actions intersect with those mediated by the canonical effects of HDACIs on gene expression (Grant & Dai, 2012; Rosato & Grant, 2005). Several excellent reviews of the mechanisms of action of this interesting group of agents in cancer cells have appeared in the literature (Bolden, Peart, & Johnstone, 2006; Dickinson, Johnstone, & Prince, 2010; Marks & Xu, 2009; Minucci & Pelicci, 2006; Schrump, 2009). The thrust of the present article is on recently identified mechanisms of HDACI lethality, particularly as they relate to the DNA damage response (DDR) network, paving the way for novel synergistic combination strategies with other targeted agents that exert complementary activities.
II. Mechanisms of HDACI lethality
DNA (chromatin) is wrapped around histone octamers to form nucleosomes, and these histones are reversibly modified in various ways in order to render DNA accessible to transcription factors, the best characterized of which is acetylation (Grant & Dai, 2012; Rosato & Grant, 2005). Acetylation of histones is reciprocally regulated by histone acetyltransferases (HATs) and HDACs (Grant & Dai, 2012; Rosato & Grant, 2005). Acetylation of positively charged N-terminal lysine residues in the histone tails interferes with their binding to negatively charged DNA, allowing a more open, or relaxed chromatin configuration that favors gene transcription (Rosato & Grant, 2005). Conversely, deacetylation of histones favors compaction of chromatin, which is generally associated with gene silencing (Rosato & Grant, 2005), although some genes can be down-regulated by histone acetylation, depending on the cellular context (Peart et al., 2005). HDACs, which are frequently dysregulated in cancer, represent the products of 18 genes, which can be subdivided into 4 classes (Grant & Dai, 2012). Classes I (HDACs 1, 2, 3 and 8), II (HDACs 4, 5, 6, 7, 9 and 10) and IV (HDAC 11) are zinc-dependent enzymes, whereas the class III enzymes (sirtuins) are zinc-independent but nicotinamide adenine dinucleotide (NAD)-dependent (Rosato & Grant, 2005). The class IIb HDAC, HDAC6 is distinguished by its ability to deacetylate tubulin, which has important mechanistic implications related to disruption of aggresome formation and down-regulation of chaperone proteins by HDAC6 inhibitors (Bali, Pranpat, Bradner, Balasis, Fiskus, Guo, Rocha, Kumaraswamy, Boyapalle, Atadja, Seto, & Bhalla, 2005b; Hideshima et al., 2005). Clinically relevant HDACIs represent different chemical classes, e.g., hydroxamic acids (vorinostat, dacinostat, panobinostat, belinostat), benzamides (entinostat, mocetinostat) and cyclic tetrapeptides (romidepsin) (Bolden et al., 2006; Dickinson et al., 2010). Some of these (the hydroxamic acids) act as pan-HDACIs, whereas others predominantly target the class I (entinostat, mocetinostat) or class IIb (e.g., ACY-1215) enzymes (Bolden et al., 2006; Grant & Dai, 2012).
Key determinants of HDACI lethality (Figure 1) include:
Figure 1.

Mechanisms of histone deacetylase inhibitor lethality.
down-regulation of anti-apoptotic proteins such as caspase inhibitors (e.g., X-linked inhibitor of apoptosis (XIAP), survivin and cellular FLICE-like inhibitory protein (c-FLIP)) (Aron et al., 2003; C. S. Mitsiades et al., 2004; Rosato et al., 2006; Rosato et al., 2007; Sanda et al., 2007), Bcl-w (Sanda et al., 2007) and myeloid cell leukemia-1 (Mcl-1, through reversal of microRNA silencing) (Sampath et al., 2012),
up-regulation of pro-apoptotic proteins such as Bim, Bmf and Noxa (through acetylation of p53) (S. Chen, Dai, Pei, & Grant, 2009b; Dai, Chen, Kramer et al., 2008; Dai, Chen, Wang, Pei, Kramer et al., 2011; Inoue, Riley, Gant, Dyer, & Cohen, 2007; Tan et al., 2006; Terui et al., 2003; Xargay-Torrent et al., 2011; Zhao et al., 2005),
activation of the death receptor pathway (Glick et al., 1999; Insinga et al., 2005; Nebbioso et al., 2005),
induction of Bid cleavage and activation (Lindemann et al., 2007; N. Mitsiades et al., 2003; Ruefli et al., 2001), thus linking the intrinsic and extrinsic pathways of apoptosis,
induction of the endogenous cyclin-dependent kinase (CDK) inhibitor p21 (H. Li & Wu, 2004; N. Mitsiades et al., 2003; Nawrocki et al., 2007; Richon, Sandhoff, Rifkind, & Marks, 2000; Sanda et al., 2007; H. Wang et al., 2012),
ROS generation (Sanda et al., 2007) and induction of DNA damage (Dai, Rahmani, Dent, & Grant, 2005; Dasmahapatra et al., 2010; Dasmahapatra et al., 2011; Gaymes et al., 2006; Hu et al., 2010; Petruccelli et al., 2011; Rosato, Almenara, & Grant, 2003; Rosato et al., 2008; Rosato et al., 2010; Ruefli et al., 2001; Ungerstedt et al., 2005; Xu, Ngo, Perez, Dokmanovic, & Marks, 2006),
disruption of chaperone protein (in particular, heat shock protein 90 (Hsp90)) function (via acetylation) (X. Yu et al., 2002), an effect that has been attributed to HDAC6 inhibition (Bali, Pranpat, Bradner, Balasis, Fiskus, Guo, Rocha, Kumaraswamy, Boyapalle, Atadja, Seto, & Bhalla, 2005b), leading to Hsp70-mediated proteasomal degradation of Hsp90 “client” oncoproteins (Nimmanapalli et al., 2003), and
inhibition of DNA repair through acetylation of Ku70 (Cohen et al., 2004; Rosato et al., 2008; Subramanian, Opipari, Bian, Castle, & Kwok, 2005), down-regulation of the DNA repair proteins Ku86, BRCA1, Chk1, RAD50, RAD51 and MRE11 (meiotic recombination 11) (Adimoolam et al., 2007; J. H. Lee, Choy, Ngo, Foster, & Marks, 2010; Rosato et al., 2008), interference with the S-phase checkpoint through loss of HDAC3 function (Bhaskara et al., 2008), disruption of both the homologous (Adimoolam et al., 2007; Kachhap et al., 2010) and non-homologous end-joining (NHEJ) (Miller et al., 2010) processes of DNA repair and interference with HDAC-mediated coordination of ATR (ATM and Rad3-related) checkpoint function, double strand break (DSB) processing and autophagy (T. Robert et al., 2011; Shubassi, Robert, Vanoli, Minucci, & Foiani, 2012).
The pleiotropic actions of HDACIs also include (Figure 1):
interference with the function of the co-repressor (e.g., Bcl-6 in diffuse large B-cell lymphoma (DLBCL) (Cerchietti et al., 2010)) and cofactor (e.g., NCOR1/SMRT (nuclear receptor corepressor 1/silencing mediator of retinoic acid and thyroid hormone receptor)) axis, critical for maintaining chromatin structure and genomic stability (Bhaskara et al., 2008; Bhaskara et al., 2010)),
promotion of proteotoxic and endoplasmic reticulum (ER) stress via disruption of aggresome formation via HDAC6 inhibition and acetylation of GRP78 (glucose regulated protein 78), a critical sensor of the ER stress response (Hideshima et al., 2005; Kawaguchi et al., 2003; Nawrocki et al., 2006; Nawrocki et al., 2007; Rao, Nalluri, Fiskus et al., 2010; Rao, Nalluri, Kolhe et al., 2010), as well as through inhibition of class I HDACs (Kahali, Sarcar, Prabhu, Seto, & Chinnaiyan, 2012),
disruption of cell cycle (especially mitotic spindle assembly) checkpoints (Ishii, Kurasawa, Wong, & Yu-Lee, 2008; Magnaghi-Jaulin, Eot-Houllier, Fulcrand, & Jaulin, 2007; Stevens, Beamish, Warrener, & Gabrielli, 2008b; Warrener et al., 2003), dysregulation of which is frequent in neoplastic cells (Kastan & Bartek, 2004),
c-Jun N-terminal kinase (JNK) activation (Dai et al., 2010; Dasmahapatra et al., 2010; Dasmahapatra et al., 2011; Vrana et al., 1999),
signal transducer and activator of transcription (STAT) 5 and 3 inhibition (Nguyen, Dai, Attkisson, Kramer, Jordan, Nguyen, Kolluri, Muschen, & Grant, 2011b; Rascle, Johnston, & Amati, 2003; Shao et al., 2010),
interference with proteasome function (Fotheringham et al., 2009; C. S. Mitsiades et al., 2004),
anti-angiogenic effects (Deroanne et al., 2002; Qian et al., 2004),
generation of the pro-apoptotic lipid second messenger ceramide (Maggio et al., 2004) and
induction of autophagy, possibly through acetylation of the autophagy signaling component Atg3 (Yi et al., 2012).
Autophagy induced by HDACIs can be cytoprotective (Carew et al., 2007; Gammoh et al., 2012), as can induction of p21 by these agents (Almenara, Rosato, & Grant, 2002), providing a basis for synergism with agents that block these phenomena (Almenara et al., 2002; Carew et al., 2007; Gammoh et al., 2012). The selective toxicity of HDACIs towards transformed cells may be explained in part by the ability of normal but not neoplastic cells to escape HDACI-induced oxidative injury by up-regulating thioredoxin (Ungerstedt et al., 2005), to repair HDACI-induced DNA damage (J. H. Lee et al., 2010) and by the activation of death receptor pathways in transformed cells (Insinga et al., 2005; Nebbioso et al., 2005).
III. The DDR signaling network, cell cycle checkpoints and HDACIs
The DDR represents a complex network of multiple signaling pathways involving cell cycle checkpoints, DNA repair, transcriptional programs, and apoptosis, through which cells maintain genomic integrity following various endogenous (metabolic) or environmental stresses (Dai & Grant, 2010). The cell cycle progresses in an orderly fashion and is monitored by safety mechanisms known as cell cycle checkpoints, which, on activation, function to halt cell division (A. N. Tse, Carvajal, & Schwartz, 2007). When DNA damage occurs, distinct, albeit overlapping and cooperating, checkpoint pathways are activated, which block S-phase entry (the G1/S-phase checkpoint), delay S-phase progression (the intra-S or S-phase checkpoint), or prevent mitotic entry (the G2/M-phase checkpoint). These events direct phase-specific DNA repair mechanisms through repair-specific gene transcription. If repair fails, checkpoints trigger apoptosis (Dai & Grant, 2010).
Components of the checkpoint mechanism include sensors, mediators, transducers and effectors, which work cooperatively in different phases of the cell cycle. Sensors recognize damage, and recruit the proximal transducers ATM (ataxia telangiectasia mutated) and ATR to lesions where they are initially activated. ATM and ATR, in turn, transduce signals to the distal transducers, checkpoint kinases 1 and 2 (Chk1 and Chk2). In general, ATM is activated by DSBs, resulting, for example, from ionizing radiation, whereas ATR is activated by DNA damage and DNA replication stress. The acetylation of ATM by the HAT Tip60 (Tat interactive protein 60) is a key step linking the detection of DNA damage and the activation of ATM kinase activity (Sun, Xu, Roy, & Price, 2007). Classically, ATM has been thought to activate Chk2, and ATR primarily Chk1. However, more recent data have challenged the classic model of ATM and ATR being activated by distinct lesions and triggering independent downstream pathways, and coordinated cross-talk definitely exists between the two pathways. These concepts have been reviewed (Bucher & Britten, 2008; Dai & Grant, 2010; A. N. Tse et al., 2007) and are summarized in Figure 2.
Figure 2.
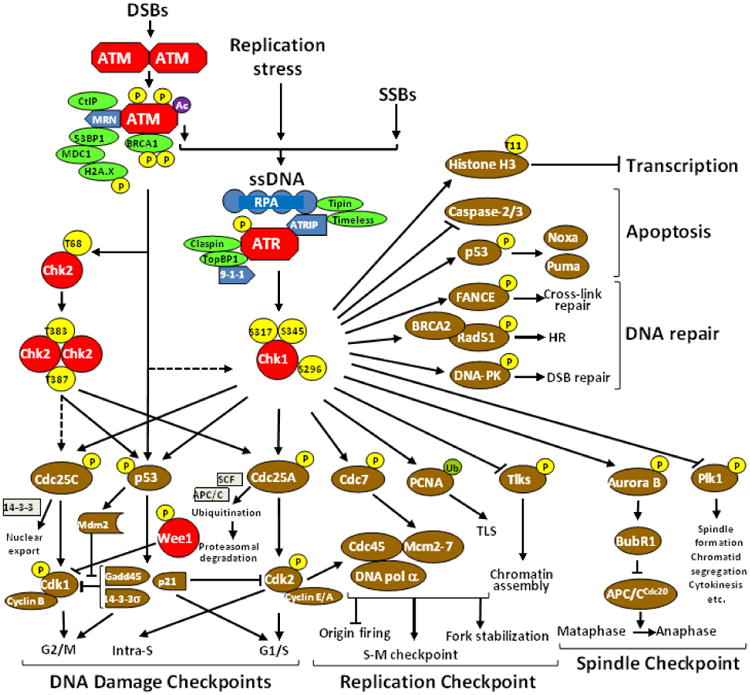
The DDR signaling network. DNA damage (e.g., DSBs, SSBs, and stalled replication forks) generates ssDNA that initiates ATR-mediated Chk1 activation. In this context, the ATR/ATRIP (ATR interacting protein) complex is recruited to ssDNA lesions via binding of ATRIP with RPA that recognizes and coats ssDNA. In conjunction with recruited/activated sensors and mediators, ATR phosphorylates Chk1 at two canonical sites (Ser345 and S317), directly leading to its activation without the homodimerization and intramolecular transautophosphorylation that is required for Chk2 activation. Activated Chk1 then phosphorylates diverse downstream effectors, which in turn are involved in cell cycle checkpoints (i.e., intra-S-phase, G2/M-phase, and G1/S-phase checkpoints), the DNA replication checkpoint, and the mitotic spindle checkpoint, as well as DNA repair, apoptosis, and transcription. Modified, with permission, from (Dai & Grant, 2010).
The G1/S checkpoint is the first defense against genomic stress in cycling cells. In response to DNA damage, the G1 checkpoint prevents cells from entering the S-phase by inhibiting the initiation of DNA replication. At this checkpoint, Chk2 is activated by ATM to phosphorylate (and thereby inhibit) the cell division cycle (CDC)25A phosphatase, thus preventing activation of cyclin E(A)/CDK2 and temporarily halting the cell cycle. G1 arrest is sustained by ATM/Chk2-mediated phosphorylation of murine double minute homolog 2 (Mdm2) and p53, resulting in p53 stabilization and accumulation. p53 transcriptionally activates the endogenous CDK inhibitor p21, which in turn inhibits cyclin E(A)/CDK2 and preserves the association of the tumor suppressor Rb (retinoblastoma protein) with the transcription factor E2F (Bucher & Britten, 2008).
The intra-S or S-phase checkpoint is activated in response to structural DNA damage as well as stalled replication forks. Upon activation of this checkpoint, ATR/Chk1 and ATM/Chk2 phosphorylate CDC25A, resulting in enhanced proteolysis of the phosphatase and inhibition of its function through 14-3-3 σ binding. Cyclin E(A)/CDK2 is thereby inhibited, and progression through the S phase halted (A. N. Tse et al., 2007).
The G2/M checkpoint prevents mitotic entry of cells that have either incurred DNA damage during G2, or that have escaped the G1/S and intra-S checkpoints despite earlier genomic insults. The key downstream target of the G2/M checkpoint is the pro-mitotic cyclin B/CDK1 (cdc2) complex. During interphase, this complex is inactivated through phosphorylation by Myt1 and Wee1 (A. N. Tse et al., 2007). Chk1 may phosphorylate Wee1, mediating binding of Wee1 to 14-3-3 proteins, which in turn may stimulate the kinase activity of Wee1 against CDK1 (cdc2). Thus, both Chk1 and 14-3-3 proteins may act together as positive regulators of Wee1 (M. H. Lee & Yang, 2001). Activation of cyclin B/CDK1 (cdc2) requires dephosphorylation by the CDC25 phosphatases (A, B and C). Initiation of the G2/M checkpoint is mediated by ATR/Chk1, which phosphorylates (and thereby inhibits) CDC25A, -B and –C, whereas maintenance of this checkpoint requires p53 and its downstream effectors p21, 14-3-3 σ and growth arrest and DNA damage 45 (GADD45) (A. N. Tse et al., 2007).
Checkpoint dysfunction is common in human cancers and is considered a pathologic hallmark of neoplastic transformation (Kastan & Bartek, 2004). Conversely, agents used for cancer treatment, such as cytotoxic chemotherapy and ionizing radiation, also activate cell cycle checkpoints (A. N. Tse et al., 2007). Cancer cells are particularly dependent upon the S- and G2/M-phase checkpoints for repair of DNA damage due to pre-existing defects in G1/S checkpoint mechanisms, such as p53 and Rb mutations. Because the S-phase checkpoint facilitates slowing, rather than arrest, of the cell cycle, a cancer cell harboring DNA damage may progress through the S-phase checkpoint, only to halt at the G2/M checkpoint. The latter is therefore a key guardian of the cancer cell genome, and its abrogation should lead to enhanced tumor cell death, sparing normal cells, which maintain an intact G1/S-phase checkpoint. G2/M checkpoint abrogation prevents cancer cells from repairing DNA damage, forcing them into a premature and lethal mitosis (“mitotic catastrophe”) (Bucher & Britten, 2008). Hsp90 inhibitors down-regulate several key DDR proteins (e.g., ATR, Chk1 and Wee1) that are “clients” of the chaperone protein (Ha et al., 2011; Sugimoto et al., 2008; A. N. Tse, Sheikh, Alan, Chou, & Schwartz, 2009). Disruption of chaperone protein (Hsp90) function through acetylation of this protein, leading to proteasomal degradation of “client” oncoproteins (Nimmanapalli et al., 2003; X. Yu et al., 2002), is a widely recognized mechanism of action of HDACIs that has generally been attributed to HDAC6 inhibition (Bali, Pranpat, Bradner, Balasis, Fiskus, Guo, Rocha, Kumaraswamy, Boyapalle, Atadja, Seto, & Bhalla, 2005b), although similar effects have been observed with the class I selective HDACI entinostat (Nishioka et al., 2008). HDACIs have also been shown to down-regulate Chk1 and cause premature mitotic entry and apoptosis in lung cancer cells (Brazelle et al., 2010). Significantly, ATM forms a complex with HDAC1 and their mutual binding increases in response to ionizing radiation (G. D. Kim et al., 1999). HDAC4 has also been shown to be a component of the DDR (Basile, Mantovani, & Imbriano, 2006). HDAC4 shuttles from the cytoplasm into the nucleus following DNA damage independently of the activation of p53, colocalizes with p53 binding protein 1 (p53BP1) (Kao et al., 2003) and becomes associated with G2/M promoters through a p53-dependent mechanism (Basile et al., 2006), to epigenetically silence genes that control the G2/M transition. Silencing of HDAC4 via RNA interference abrogated DNA damage-induced G2 delay, and radiosensitized HeLa cells (Kao et al., 2003).
IV. Induction of DNA damage and inhibition of DNA repair by HDACIs
Although it has been reported that HDACIs can directly activate the DDR as a result of changes in chromatin structure without the requirement for DNA DSBs (Bakkenist & Kastan, 2003), ample evidence exists to support the notion that HDACIs directly induce DNA damage, resulting in DSBs (C. S. Chen et al., 2007; Dasmahapatra et al., 2010; Dasmahapatra et al., 2011; Gaymes et al., 2006; J. H. Lee et al., 2010; Miller et al., 2010; Namdar, Perez, Ngo, & Marks, 2010; Petruccelli et al., 2011; Rosato et al., 2008; Rosato et al., 2010). Furthermore, the chromatin remodeling and DNA damage signaling pathways are by no means mutually exclusive (Lukas, Lukas, & Bartek, 2011; Misteli & Soutoglou, 2009). Mechanistically, DNA damage and apoptosis induced by HDACIs is due, at least in part, to the generation of oxidative stress (Bhalla et al., 2009; Butler et al., 2002; Dai et al., 2005; Dasmahapatra et al., 2011; Petruccelli et al., 2011; Rahmani et al., 2005; Rosato et al., 2003; Rosato et al., 2006; Rosato et al., 2008; Rosato et al., 2010; Ruefli et al., 2001; Sanda et al., 2007; Ungerstedt et al., 2005; Xu et al., 2006). ROS generation by HDACIs has been attributed to down-regulation of antioxidant pathways (Butler et al., 2002; Ungerstedt et al., 2005). HDACIs have also been shown to down-regulate proteins that participate in base excision repair (BER) and nucleotide excision repair (NER), pathways for the repair of oxidative damage, and to trigger depolarization of the mitochondrial membrane (Rosato et al., 2008; Rosato et al., 2010), which can induce reactive oxygen species (ROS) generation. HDACI-induced nuclear factor kappa B (NFκB) activation(Dai, Rahmani, & Grant, 2003; Dai et al., 2005; Dai et al., 2010; Dai, Chen, Wang, Pei, Funk et al., 2011; Mayo et al., 2003; Rosato et al., 2010; Roychowdhury et al., 2004) could also, in part, explain the production of ROS by these agents, through the induction of pro-inflammatory cytokines (J. J. Kim, Lee, Park, & Yoo, 2010). Not surprisingly, HDACIs sensitize tumor cells to DNA-damaging agents and ionizing radiation (IR)(Camphausen et al., 2004; Miller et al., 2010; Munshi et al., 2005; Munshi et al., 2006). DNA damage resulting from HDACI treatment has also been attributed to profound changes in chromatin structure, exposing portions of DNA normally protected from damage to DNA-damaging agents, inhibition of DNA repair pathways and/or modulation of expression of DDR and cellular survival genes (C. Robert & Rassool, 2012). At pharmacologic concentrations, vorinostat slows down replication forks in cancer cells, activates dormant replication origins and induces replication-mediated DNA damage, likely through inhibition of HDAC3 (Conti et al., 2010).
DSBs represent the most serious form of DNA damage in mammalian cells (reviewed in (Khanna & Jackson, 2001)). Histone H1.2 plays an important role in transmitting apoptotic signals from the nucleus to the mitochondria following DNA DSBs (Konishi et al., 2003). As noted above, ATM is a key mediator in the activation of cell cycle checkpoints by DSBs. ATM is recruited to DSB sites by the MRN (MRE11/NBS1/RAD50) complex (Falck, Coates, & Jackson, 2005), followed by its phosphorylation and conversion from an inactive dimer to an active monomer (Bakkenist & Kastan, 2003)(Bakkenist & Kastan, 2003; Bakkenist & Kastan, 2003; Falck et al., 2005). Upon activation, ATM phosphorylates MRN and downstream effectors to activate the G1/S, intra-S and G2/M checkpoints (reviewed in (O'Driscoll & Jeggo, 2006)). Homologous recombination (HR) repairs replication-associated DSBs and is a generally error-free pathway of DNA repair that is active in late S-phase and in G2, and utilizes the undamaged sister chromatid as the template for repair (C. Robert & Rassool, 2012). Critical steps in HR include CtBP interacting protein (CtIP)-mediated resection of the DSB in a 5′-3′ fashion (Huertas & Jackson, 2009; Sartori et al., 2007), followed by the recruitment and assembly of RAD51 into a nucleoprotein filament, which involves RAD52, x-ray repair cross-complementing proteins (XRCC) 2 and 3 and BRCA2 (N. Liu et al., 1998; Petalcorin, Sandall, Wigley, & Boulton, 2006; Sonoda et al., 2007). DSB repair by the NHEJ pathway is initiated by the Ku70/Ku86 heterodimer, which binds to ends of DNA (Walker, Corpina, & Goldberg, 2001) and activates DNA protein kinase (PK) (Calsou et al., 1999; Gottlieb & Jackson, 1993; Singleton, Torres-Arzayus, Rottinghaus, Taccioli, & Jeggo, 1999). The key step in NHEJ is the physical juxtaposition of DNA ends (C. Robert & Rassool, 2012). There is also an alternative DNA end-joining repair pathway that has been implicated in certain cancers (Nussenzweig & Nussenzweig, 2007).
HDACIs inhibit multiple processes of DNA repair (C. Robert & Rassool, 2012). First, these agents transcriptionally down-regulate a large number of DNA DSB repair proteins, e.g., RAD50, MRE11, Ku70, Ku80, Ku86, DNA PK, BRCA1, RAD51, Chk1, and Bloom syndrome (BLM) (Kachhap et al., 2010; J. H. Lee et al., 2010; Munshi et al., 2005; Munshi et al., 2006; Rosato et al., 2008). HDACIs additionally impair the recruitment of these proteins to repair foci and decrease levels of the transcription factor E2F1, which promotes gene expression of RAD51, Chk1 and BRCA1 (Kachhap et al., 2010). Another major mechanism of regulation of a number of proteins important in DNA repair (e.g., p53, Ku70, flap structure-specific endonuclease 1 (FEN1), Werner syndrome (WRN), ATM, mediator of DNA damage checkpoint 1 (MDC1) and DNA PK) is through acetylation (Choudhary et al., 2009). HDACIs acetylate and thereby stabilize p53, leading to transcription of its target genes, including p21, triggering cell cycle arrest or apoptosis (J. Luo, Su, Chen, Shiloh, & Gu, 2000). The proteasomal degradation of p53 requires MDM2-HDAC1-mediated deacetylation (Ito et al., 2002). Ku70 acetylation by HDACIs reduces its binding to DNA and sensitizes prostate cancer cells to agents such as bleomycin, doxorubicin and etoposide, which induce DSBs (C. S. Chen et al., 2007). In addition, HDACIs disrupt the association of Ku70 with Bax by acetylating the former, releasing pro-apoptotic Bax and triggering apoptosis via the mitochondrial pathway (C. S. Chen et al., 2007). In colorectal cancer cells, acetylation of Ku70 by HDACIs disrupts the FLIP/Ku70 complex and triggers FLIP polyubiquitination and degradation by the proteasome, thus de-repressing the death receptor-mediated pathway (Kerr et al., 2012). Recently, HDAC1 and HDAC2 were shown to be rapidly recruited to sites of DSBs in osteosarcoma cells and to be responsible for deacetylation of the histone mark H3K56 (Miller et al., 2010). HDAC1- and 2-depleted cells were hypersensitive to DNA-damaging agents and showed sustained DNA-damage signaling and defective DSB repair, particularly by NHEJ (Miller et al., 2010). It was proposed that local condensation of chromatin as a consequence of histone deacetylation might be necessary for proper DSB repair through inhibition of transcription and prevention of Ku70 sliding off naked DNA (Miller et al., 2010). H3K56 acetylation has recently been shown to interfere with Ku localization at retrotransposons, leading to dissociation of condensin from retro-transposons, releasing condensin-mediated genomic associations during S-phase and upon DNA damage (A. Tanaka et al., 2012). ATR mediates the DDR of condensin-mediated genome organization (A. Tanaka et al., 2012). HDAC3 has also recently been shown to be essential for efficient DNA replication and repair, and for the maintenance of chromatin structure and genome stability (Bhaskara et al., 2008; Bhaskara et al., 2010). HDAC3 deacetylates histone marks H3K9, H3K14, H4K5 and H4K12 during late S-phase, and deletion of HDAC3 in cycling mouse embryonic fibroblasts (MEFs) led to a delay in cell-cycle progression, cell-cycle-dependent DNA damage, and apoptosis, which appeared to be associated with defective DNA DSB repair (Bhaskara et al., 2008; Bhaskara et al., 2010). HDAC9 and HDAC10 appear to participate in HR repair, although the precise mechanisms remain elusive (Kotian, Liyanarachchi, Zelent, & Parvin, 2011). The sirtuin SIRT6 participates in promoting DNA end resection, a crucial step in HR repair, the primary repair mechanism in S-phase (Branzei & Foiani, 2008), by deacetylating the DSB resection protein CtIP (Kaidi, Weinert, Choudhary, & Jackson, 2010). HDACIs promote acetylation of CtIP (Langerak, Mejia-Ramirez, Limbo, & Russell, 2011), triggering nuclear export and autophagic degradation (T. Robert et al., 2011). Adding further to the complexity of the mechanisms by which pharmacologic inhibition of HDACs might impact DNA repair processes in cancer cells is the fact that several HATs (e.g., HIV-1, TIP60, MOF) play critical roles in DNA repair (Sharma et al., 2010; Sun et al., 2009), acetylating histones and relaxing chromatin, thus improving accessibility of DNA repair proteins to DSB sites (Murr et al., 2006).
V. HDACIs and apoptotic pathways
Cancer cells invariably have abnormalities in one or more apoptotic pathways, determining a survival advantage for these cells over their normal counterparts (Hanahan & Weinberg, 2011). Furthermore, abnormalities in the apoptotic response also play a major role in the development of drug resistance by malignant cells (Davids & Letai, 2012). Most, if not all, anti-cancer drugs kill malignant cells through induction of apoptosis, and most tumor cells do retain their apoptotic machinery (Davids & Letai, 2012). Defects in apoptotic pathways thus represent an ideal target for therapeutic intervention.
At a molecular level, apoptosis is caused by the activation of caspases (Irvine, McMullin, & Ong, 2002; Schimmer, 2007; Testa & Riccioni, 2007). The extrinsic, or death receptor pathway is initiated by engagement of death receptors of the TNF (tumor necrosis factor) superfamily on the cell membrane by their respective ligands, such as Fas ligand (FasL), TNF or TRAIL (TNF-related apoptosis inducing ligand). c-FLIP is an endogenous inhibitor of this pathway. The intrinsic, or mitochondrial pathway of apoptosis is triggered by various cellular stresses, such as growth factor deprivation, DNA or microtubule damage, hypoxia, etc. and is the mechanism by which apoptosis is induced by cytotoxic chemotherapy. The seminal event in this pathway is mitochondrial outer membrane polarization (MOMP), which is controlled by the B-cell lymphoma 2 (Bcl-2) family of proteins and results from the formation of pores in the mitochondrial outer membrane (Davids & Letai, 2012). The inhibitors of apoptosis (IAPs) are cytoplasmic proteins that inhibit the action of caspases. The most important member of this family, XIAP, potently inhibits active caspases 9, 3 and 7, thus blocking apoptosis triggered by multiple caspase activation pathways (Datta et al., 2000).
The anti-apoptotic Bcl-2 family members are Bcl-2, Bcl-XL, Bcl-w, Mcl-1, Bfl-1/A1 and Bcl-B. The “multi-domain” pro-apoptotic members are Bax, Bak and Bok (Labi, Grespi, Baumgartner, & Villunger, 2008). The other pro-apoptotic members, the so-called “BH3-only” proteins, include Bim, Bid, Bad, Bik, Hrk, Bmf, Puma and Noxa (Davids & Letai, 2012). The multi-domain proteins Bax and Bak (apoptosis “effectors”) are absolutely essential for the pro-apoptotic function of the BH3-only proteins (Labi et al., 2008).
Individual BH3-only proteins differ markedly in their ability to bind to different anti-apoptotic Bcl-2 relatives. For example, Bad and Bmf bind strongly with Bcl-2, Bcl-XL, and Bcl-w, and Noxa binds only with high affinity to Mcl-1 and Bfl-1/A1. Thus, BH3-only proteins with different selective binding patterns (e.g., Bad plus Noxa) synergize in apoptosis induction, an important finding from co-transfection assays of great translational relevance (Kuroda & Taniwaki, 2009).
A link between the extrinsic and intrinsic pathways is provided by the cleavage of the proapoptotic Bcl-2 family “BH3-only” protein Bid by caspase 8 to produce a truncated molecule, tBid, that is able to directly activate the multi-domain apoptosis effectors Bax and Bak. Mitochondrial translocation of Bax (normally a cytosolic protein) and oligomerization of Bax and Bak lead to MOMP (Ott, Norberg, Zhivotovsky, & Orrenius, 2009).
HDACIs interact at multiple levels with both the intrinsic (mitochondrial) and extrinsic (death receptor-mediated) pathways of apoptosis, up-regulating pro-apoptotic and down-regulating anti-apoptotic proteins (Grant & Dai, 2012). The transcriptional signature of vorinostat in MM reveals down-regulation of caspase inhibitors (C. S. Mitsiades et al., 2004). HDACIs have been shown to down-regulate Bcl-xL, Mcl-1 and XIAP in both solid tumor (Gillespie, Borrow, Zhang, & Hersey, 2006) and malignant hematopoietic cells (Jona et al., 2011; Rosato et al., 2006; Rosato et al., 2007). Down-regulation of survivin and XIAP by HDACIs has been demonstrated in human glioma cells to be mediated through inhibition of cdc2 (CDK1) (E. H. Kim et al., 2005). Reversal of epigenetic silencing of critical microRNAs by HDACIs, leading to activation of the latter, may underlie the ability of HDACIs to down-regulate Mcl-1 (Saito et al., 2009; Sampath et al., 2012). In chronic lymphocytic leukemia (CLL) cells, romidepsin activates the extrinsic pathway of apoptosis via down-regulation of c-FLIP (Aron et al., 2003). E2F1-dependent up-regulation of the pro-apoptotic BH3-only protein Bim via induction of ASK1 (apoptosis signal-regulating kinase 1) is a major mediator of apoptosis in response to HDACIs (Tan et al., 2006; Zhao et al., 2005). Bim up-regulation by HDACIs plays a critical role in sensitizing human leukemia and myeloma cells to the BH3-mimetic ABT-737 (S. Chen, Dai, Pei, & Grant et al., 2009b). Reversal of epigenetic silencing of Bim by HDACIs may overcome resistance to glucocorticoids in pediatric acute lymphoblastic leukemia (ALL) (Bachmann et al., 2010) and to chemotherapy in BL (Richter-Larrea et al., 2010). Bim up-regulation by HDACIs is also implicated in synergistic interactions between this class of agents and proteasome inhibitors (PIs) (Dai, Chen, Kramer et al., 2008; Dai, Chen, Wang, Pei, Kramer et al., 2011). Recently, MAPK (mitogen activated protein kinase) pathway activation was identified as a key mechanism of resistance to romidepsin in cutaneous T-cell lymphoma (CTCL) cells, and the addition of MEK (MAPK kinase) inhibitors synergistically induced apoptosis in these resistant cells via restoration of Bim (Chakraborty et al., 2013). Besides Bim, a key role for induction of other pro-apoptotic BH3-only proteins such as Bmf and Noxa in HDACI-induced apoptosis and synergism with ABT-737 has been proposed (Inoue et al., 2007; Wiegmans et al., 2011). Up-regulation of Noxa by HDACIs may be due to acetylation of p53 (Terui et al., 2003). In mantle cell lymphoma (MCL) cells, vorinostat has been shown to hyperacetylate the promoter regions of Bim, Bmf and Noxa, thereby activating their transcription, and to synergize with ABT-263 (navitoclax) (C. Tse et al., 2008), the orally available analog of ABT-737 (Xargay-Torrent et al., 2011). HDACIs induce expression of TRAIL in human AML cells and activate the death receptor pathway of apoptosis (Insinga et al., 2005; Nebbioso et al., 2005). Synergism between HDACIs and TRAIL has been reported in preclinical studies in a number of hematologic (Al-Yacoub et al., 2012; Guo et al., 2004; Inoue et al., 2004) and solid organ malignancies (Bangert et al., 2012; Kauh et al., 2010; E. H. Kim et al., 2005), in some cases involving down-regulation of c-FLIP (Al-Yacoub et al., 2012; Bangert et al., 2012; Kauh et al., 2010). Finally, Bid cleavage and activation appears to be an important mechanism of HDACI lethality in malignant cells (Gillenwater, Zhong, & Lotan, 2007; Lindemann et al., 2007; Ruefli et al., 2001). In summary, HDACIs promote apoptosis induction via both the mitochondrial (intrinsic) and death receptor (extrinsic) pathways, down-regulating key anti-apoptotic proteins such as Mcl-1, Bcl-xL, XIAP and c-FLIP, up-regulating critical pro-apoptotic proteins, e.g., Bim, Bmf and Noxa, and facilitating cross-talk between the two pathways through cleavage and activation of Bid.
VI. Rational combinations involving HDACIs and other novel targeted agents involving disruption of the DDR
Given the ability of HDACIs to induce DNA damage and the many ways in which these agents impair DNA repair mechanisms, it is not surprising that these drugs potentiate the effects of DNA-damaging chemotherapy (C. S. Chen et al., 2007; Rosato et al., 2008), in some cases in a sequence-dependent fashion (Marchion et al., 2004). Particularly in the case of topoisomerase II inhibitors, synergism may require pre-treatment with HDACIs (Marchion et al., 2004; Shiozawa et al., 2009). Some of these observations (Sanchez-Gonzalez et al., 2006; Shiozawa et al., 2009) have led to clinical trials combining HDACIs with genotoxic agents in AML and high-risk MDS (Garcia-Manero et al., 2012), and encouraging results from such single-institution studies have served as the basis of large, cooperative group-led phase III studies that are currently ongoing (e.g., NCT01802333). While such approaches undoubtedly hold promise and continue to be explored, the remainder of this review will focus on the potential for mechanism-based combinations of HDACIs involving novel, targeted molecules that disrupt the DDR or promote apoptosis via antagonism of anti-apoptotic protein function.
The frequency of DNA repair defects and cell cycle checkpoint dysfunction in cancer cells makes targeting the DDR a particularly attractive anti-cancer strategy, often in concert with agents capable of inducing DNA damage (Bouwman & Jonkers, 2012). For example, certain genetically defined subsets of AML, e.g., those harboring fms-like tyrosine kinase-internal tandem duplication (FLT3-ITD) mutations (approximately 25% of cases of AML) are characterized by defective NHEJ DNA repair pathways (Fan, Li, Small, & Rassool, 2010; L. Li et al., 2011). Similarly, certain leukemia-associated fusion proteins, such as promyelocytic leukemia zinc finger-retinoic acid receptor alpha (PLZF-RARα), promyelocytic leukemia-retinoic acid receptor alpha (PML-RARα) and runt related transcription factor 1-eight twenty one (RUNX1-ETO, AML1-ETO) confer a DNA repair-deficient phenotype, and expression of these fusion proteins increases sensitivity of AML cells to vorinostat-induced DNA damage and apoptosis (Petruccelli et al., 2013). Thus far, most efforts to develop G2/M checkpoint abrogators such as Chk1 inhibitors therapeutically have focused on combination strategies involving genotoxic agents, but clinical efficacy has been modest (Karp et al., 2012). Since HDACIs down-regulate Chk1 and Wee1, induce DNA damage and inhibit DNA repair pathways by multiple mechanisms (see above discussion), there is a strong theoretical rationale for combining these agents with G2/M checkpoint abrogators. Indeed, the novel Chk1 inhibitor MK-8776 synergizes with vorinostat in inducing apoptosis of AML cell lines and primary AML blasts, particularly the putative leukemia-initiating cell (LIC) population (CD34+CD38−CD123+) while relatively sparing normal cord blood CD34+ cells, accompanied by interference with the intra-S checkpoint, disruption of DNA replication, down-regulation of proteins involved in DNA replication (e.g., chromatin licensing and DNA replication factor 1 (CDT1)) and repair (e.g., CtIP and BRCA1) and sharp increases in DNA damage (Dai et al., 2013). The MK-8776/vorinostat combination was effective against AML cells with intact or deficient p53 function, as well as those bearing the FLT3-ITD mutation (Dai et al., 2013). Similar results were obtained using the Wee1 inhibitor MK-1775 in combination with vorinostat and validated in a mouse xenograft model (Zhang, Y, et al. Proc Am Assoc Can Res (Abstr), 55: 4556, 2014). The MK-1775/vorinostat combination was particularly active against FLT3-ITD AML, and demonstrated activity regardless of p53 mutational status (Zhang, Y, et al. Proc Am Assoc Can Res (Abstr), 55: 4556, 2014). Indeed, genomic instability, with increased ROS production, DNA DSBs and defective repair may be a hallmark of myeloid malignancies (Sallmyr, Fan, & Rassool, 2008).
Disruption by HDACIs of mitotic spindle checkpoints in neoplastic cells (Qiu et al., 2000) and their down-regulation of oncogenic Hsp90 client proteins (Bali, Pranpat, Bradner, Balasis, Fiskus, Guo, Rocha, Kumaraswamy, Boyapalle, Atadja, Seto, & Bhalla, 2005a) such as Raf, Akt, FLT3 and Bcr-Abl (breakpoint cluster region-Abelson) underlie the ability of these agents to potentiate the actions of small-molecule inhibitors of aurora (Dai, Chen, Venditti et al., 2008; Fiskus et al., 2008; Nguyen, Dai, Attkisson, Kramer, Jordan, Nguyen, Kolluri, Muschen, & Grant, 2011a), FLT3 (Bali et al., 2004) and Bcr-Abl (Fiskus, Pranpat, Balasis et al., 2006; Fiskus, Pranpat, Bali et al., 2006; Nimmanapalli, Fuino, Stobaugh, Richon, & Bhalla, 2003; C. Yu, Rahmani, Almenara et al., 2003) kinases. Additionally, HDACIs induce “mitotic slippage” (Stevens, Beamish, Warrener, & Gabrielli, 2008a) and synergize with Janus-associated kinase (JAK)2 inhibitors (Novotny-Diermayr et al., 2012; Y. Wang et al., 2009). The aurora kinases, serine-threonine protein kinases that are overexpressed in a variety of cancer types, regulate various processes including chromosome alignment, segregation, centrosomal maturation, mitotic spindle formation, and cytokinesis during mitosis (Kelly et al., 2011). Their fundamental role in cell cycle regulation and aberrant expression in a broad range of malignancies has prompted the development of a number of small-molecule inhibitors (Kelly et al., 2011). HDACIs have been shown to enhance the lethality of dual aurora/Bcr-Abl tyrosine kinase inhibitors (TKIs) such as MK-0457 (VX-680) (Dai, Chen, Venditti et al., 2008; Fiskus et al., 2008) and KW-2449 (Nguyen, Dai, Attkisson, Kramer, Jordan, Nguyen, Kolluri, Muschen, & Grant, 2011a) against Bcr-Abl+ cells, both sensitive and resistant to imatinib, including those bearing the T315 “gatekeeper” mutation. The latter two multi-targeted TKIs are no longer in clinical development. However, synergistic anti-leukemic interactions between AT9283, a small-molecule inhibitor of aurora, Bcr-Abl, FLT3 and Janus kinases (Dawson et al., 2010; Santo et al., 2011; R. Tanaka et al., 2010) and entinostat have been observed in Bcr-Abl+ (CML and Philadelphia chromosome-positive ALL) cells, including those bearing T315I, as well as in AML cells, both FLT3-mutated and –wild type (Nguyen and Grant, unpublished observations).
Polo-like kinase 1 (PLK-1) is a conserved serine-threonine kinase involved in mitotic progression through interactions with cyclin B, the CDC25C phosphatase, and Wee1 (Archambault & Glover, 2009). It has been implicated in progression into M-phase, mitotic spindle formation, cytokinesis, and chromosome segregation (Lapenna & Giordano, 2009). PLK-1 plays an important role in the DDR (Smits et al., 2000), including recovery from the G2/M DNA damage checkpoint (Takaki, Trenz, Costanzo, & Petronczki, 2008). Moreover, interactions between PLK-1 and multiple checkpoint proteins, including Chk1/2, p53, claspin, and FoxM1 (forkhead box protein M1), have been described (X. S. Liu, Song, & Liu, 2010). Co-treatment of imatinib-sensitive and –resistant Bcr-Abl+ cells with the PLK-1 inhibitor BI2536 and vorinostat synergistically induced cell death with minimal toxicity towards normal cells (Dasmahapatra, Patel, Nguyen, Attkisson, & Grant, 2013). The combination triggered pronounced mitochondrial dysfunction, inhibition of phospho-BCR/ABL, caspase activation, PARP (poly ADP ribose polymerase) cleavage, ROS generation and DNA damage, and was dramatically effective in a xenograft model of Bcr-Abl+ leukemia (Dasmahapatra et al., 2013). Although Bcr-Abl signals downstream to PLK-1 (Gleixner et al., 2010), enhanced Bcr-Abl pathway inhibition was not felt to be the sole mechanism for lethality of the BI2536/vorinostat regimen; rather, potentiation of DNA damage and disabling of the DDR appeared to predominate (Dasmahapatra et al., 2013). Potentiation by vorinostat of BI2536-mediated inhibition of PLK-1 phosphorylation and enhanced ROS generation were noted, and abrogation of G0/G1 arrest with striking accumulation of cells in G2/M phase was observed (Dasmahapatra et al., 2013). The strategy of combined PLK and HDAC inhibition also affords considerable promise in DLBCL and in AML, in which PLK-1 is highly expressed (L. Liu, Zhang, & Zou, 2007; Renner et al., 2009), at least in preclinical studies (Dasmahapatra and Grant, unpublished observations). Significantly, the novel PLK inhibitor volasertib was recently granted “breakthrough” status (Three more drugs judged “breakthroughs”.2013) by the US FDA (Food and Drug Administration) based on encouraging efficacy in AML (Maertens et al., 2012). Synergism between PLK inhibitors and HDACIs has also been demonstrated in vitro against prostate cancer cells (Wissing et al., 2013); however, there are no ongoing clinical trials investigating this combination. Based on striking synergism observed in preclinical studies (Dasmahapatra and Grant, unpublished observations), phase I trials combining volasertib with HDACIs are planned in patients with relapsed/refractory NHL and myeloid malignancies.
The combination of PIs with HDACIs remains an attractive one for several reasons, and has been extensively studied. A central theme underlying synergism between these classes of agents is the activation of the NF-κB survival pathway by HDACIs, and its inhibition by PIs, which prevent degradation of IκB (I kappa B), the negative regulator of NF-κB. The NF-κB pathway mediates a cytoprotective response strongly implicated in cancer development and progression (reviewed in (Karin, 2006)) that is activated by HDACIs and limits their lethality (Dai et al., 2005; Dai, Chen, Wang, Pei, Funk et al., 2011; Rosato et al., 2010). Deacetylation of the RelA subunit of NF-κB by HDAC3 promotes its association with IκBα, the physiologic inhibitor of NF-κB, thus acting as an intra-nuclear molecular switch that controls the duration of the NF-κB transcriptional response (L. Chen, Fischle, Verdin, & Greene, 2001). The activation of NF-κB by HDACIs has served as the basis of many synergistic strategies combining these agents with those that inhibit NF-κB by various mechanisms, such as the CDK inhibitor flavopiridol (alvocidib) (Gao, Dai, Rahmani, Dent, & Grant, 2004; Rosato et al., 2007), the IKK (I kappa B kinase) inhibitor parthenolide (Dai et al., 2010), the SIRT1 (sirtuin 1) agonist resveratrol (Yaseen et al., 2012) and PIs (Dai, Chen, Kramer et al., 2008; Dai, Chen, Wang, Pei, Kramer et al., 2011; Dasmahapatra et al., 2010; Dasmahapatra et al., 2011; Pei, Dai, & Grant, 2004; C. Yu, Rahmani, Conrad et al., 2003). A number of these have been explored in clinical trials (Badros et al., 2009; B. Holkova et al., 2013; B. Holkova et al., 2011; B. Holkova et al., 2012; Kmieciak et al., 2013). Attention has also focused on the formation of aggresomes in response to proteasome inhibition, and the inhibition of aggresome function by HDACIs, at least by those exhibiting tubulin acetylase activity (Catley et al., 2006; Hideshima et al., 2005; Nawrocki et al., 2006), leading to enhanced proteotoxic stress and apoptosis; a related HDACI effect is the disruption of chaperone protein function, further adding to the cellular burden of mis-folded proteins and leading to ER stress and the unfolded protein response (reviewed in (Hideshima, Richardson, & Anderson, 2011)). However, PIs lead to ROS generation (Ling, Liebes, Zou, & Perez-Soler, 2003; Perez-Galan et al., 2006; C. Yu, Rahmani, Dent, & Grant, 2004) in multiple tumor types and consequently to DNA damage, and may also inhibit DNA repair (Vaziri et al., 2005). Specifically, bortezomib has been demonstrated to sensitize cells to apoptosis induced by DNA-damaging chemotherapy in a p53- and sequence-dependent manner involving down-regulation of 14-3-3 σ and survivin, both of which play critical roles in regulating G2/M progression and apoptosis (Vaziri et al., 2005). With respect to clinical development, the PI/HDACI combination strategy has advanced the furthest (phase III) in MM (Richardson, Schlossman et al., 2013), and multiple phase I and II trials are investigating this combination in a variety of hematologic malignancies and solid tumors (NCT00798720, NCT01276717, NCT00901147, NCT00667082). Promising results with the combination of panobinostat, bortezomib and dexamethasone in heavily pretreated patients with MM participating in the PANORAMA (Panobinostat or Placebo With Bortezomib and Dexamethasone in Patients With Relapsed Multiple Myeloma) trials, including those with bortezomib-refractory disease (Richardson, Schlossman et al., 2013), raise the possibility that panobinostat may gain regulatory approval for use in this patient population.
Although recognized for some time, synergism between HDACIs and BH3-mimetics provides a promising avenue of investigation that remains unexplored therapeutically. HDACIs up-regulate Bim, which is largely sequestered by Bcl-2 and Bcl-xL, and can be released from such binding by the bona fide BH3-mimetic ABT-737 (Oltersdorf et al., 2005), triggering Bax/Bak activation and MOMP, leading to apoptosis (S. Chen, Dai, Pei, & Grant et al., 2009b). Besides Bim, HDACIs release pro-apoptotic Bax from its binding with Ku70, and induce Bmf and Noxa. HDACIs also down-regulate Mcl-1 (Gillespie et al., 2006; Rosato et al., 2006; Rosato et al., 2007; Saito et al., 2009; Sampath et al., 2012), the primary determinant of resistance to ABT-737 (Konopleva et al., 2006; van Delft et al., 2006). Synergistic induction of apoptosis and autophagy has been observed in leukemia cell lines and primary AML cells exposed to the combination of HDACIs and the BH3-mimetic obatoclax (Wei et al., 2010). Both ABT-737 and obatoclax have also been shown to synergistically enhance entinostat-induced apoptosis in Hodgkin's lymphoma cell lines (Jona et al., 2011). In MCL cell lines and primary cells, vorinostat activated transcription (through promoter hyperacetylation) of Bim, Bmf and Noxa, and induced apoptosis with navitoclax in synergistic fashion (Xargay-Torrent et al., 2011). However, the development of both obatoclax and navitoclax has been discontinued due to the occurrence of infusional central nervous system toxicity and dose-dependent thrombocytopenia, respectively, despite promising efficacy of navitoclax in CLL (Roberts et al., 2012). ABT-199 (GDC-0199) is an orally available Bcl-2-selective BH3-mimetic developed by reverse engineering of navitoclax that exhibits potent anti-tumor activity while sparing platelets (due to lack of Bcl-xL inhibition) (Souers et al., 2013). Although it has been suggested that Bak must be released from both Mcl-1 and Bcl-xL, but not Bcl-2, in order to induce apoptosis (Willis et al., 2005), this drug has been highly effective in a large number of tumor types preclinically (Pan et al., 2014; Touzeau et al., 2014; Vaillant et al., 2013; Vandenberg & Cory, 2013; Vogler, Dinsdale, Dyer, & Cohen, 2013) and in patients with CLL or small lymphocytic lymphoma (BCL-2 inhibitor yields high response in CLL and SLL.2014). Information is not currently available on the results of combining this agent with HDACIs. Similarly, published information is lacking on combination studies of HDACIs and XIAP antagonists (Smacmimetics), another rational combination based on the ability of HDACIs to down-regulate survivin and XIAP.
A potentially interesting combination is that of HDACIs with MLN4924 (Soucy et al., 2009), a first-in-class inhibitor of protein neddylation, a critical pathway of protein degradation located upstream of the proteasome (reviewed in (Rabut & Peter, 2008)). Targeting protein neddylation allows more selective interference with protein turnover than is possible with proteasome inhibition, potentially translating to a better therapeutic index for this drug compared with PIs (Nawrocki, Griffin, Kelly, & Carew, 2012; M. Wang et al., 2011). MLN4924 exhibits impressive activity in preclinical studies in DLBCL (Milhollen et al., 2010), AML (R. T. Swords et al., 2010a) and ovarian cancer (Jazaeri et al., 2013; Nawrocki et al., 2013), and has demonstrated single-agent activity in patients with AML (R. T. Swords et al., 2010b). Like PIs, MLN4924 potently inhibits the NF-κB pathway (Milhollen et al., 2010; R. T. Swords et al., 2010b), and like HDACIs, leads to ROS generation (R. T. Swords et al., 2010a). MLN4924 interferes with the turnover of the cullin-RING ligase substrate CDT1, a critical DNA replication licensing factor, thus inducing DNA re-replication, an irreversible cellular insult that results in apoptosis (Milhollen et al., 2011). This phenomenon appears to be most marked in S-phase (Lin, Milhollen, Smith, Narayanan, & Dutta, 2010). Besides down-regulating checkpoint proteins activated in response to DNA damage, HDACIs inhibit both HR repair and NHEJ (Koprinarova, Botev, & Russev, 2011). Finally, MLN4924 triggers a cytoprotective autophagic response (Z. Luo et al., 2012; Z. Luo, Pan, Jeong, Liu, & Jia, 2012) that may be counteracted by HDACIs (T. Robert et al., 2011; Shubassi et al., 2012).
While each of the combinatorial approaches involving HDACIs and other novel, targeted agents discussed above is supported by strong theoretical rationale and, in most cases, clear demonstration of synergism at the preclinical level, not all are equally likely to be developed clinically. For example, the multi-targeted TKIs MK0457 (VX-680) and KW-2449 are no longer in development, and the future development of AT9283 is unclear. Similarly, a number of Chk1/Chk2 inhibitors have been discontinued or deprioritized by the respective manufacturers (e.g., AZD7762, PF-00477736, XL844, MK-8776), although there are others in early phases of testing (e.g., LY2603618, LY2606368, GDC-0425, reviewed in (Thompson & Eastman, 2013)). However, the recent licensing by Merck of its Wee1 inhibitor MK-1775 to Astra Zeneca has raises the possibility of a clinical trial involving this agent (now AZD-1775) in combination with an HDACI. The combination of PIs with HDACIs has been extensively tested, both preclinically and clinically, and it is conceivable that the combination of panobinostat, bortezomib and dexamethasone will be granted approval by the FDA for use in patients with relapsed/refractory MM. On theoretical grounds, the combination of MLN4924 and an HDACI appears particularly appealing, and AML might be the most logical choice for clinical investigation of this combination given the promising single agent activity of MLN4924 in this disease (R. T. Swords et al., 2010a; R. T. Swords et al., 2010b). Finally, the FDA's granting of “breakthrough” status to volasertib for AML (Three more drugs judged “breakthroughs”.2013) and the evidence of clinical activity of ABT-199 (GDC-0199) in patients with resistant lymphoid malignancies (BCL-2 inhibitor yields high response in CLL and SLL.2014) raise the possibility of regulatory approval for these agents, increasing the chances that combination trials with HDACIs will be pursued, given the compelling preclinical data with these classes of agents (S. Chen, Dai, Pei, & Grant, 2009a; Dasmahapatra et al., 2013; Wei et al., 2010; Wissing et al., 2013).
VII. Conclusions
It has become abundantly clear in recent years that HDACIs are unlikely to make a substantial impact in the clinic when used alone, and that the future of this very promising class of agents lies in rational combination therapy. A very large number of rational, HDACI-based combinations is possible, and new ones continue to emerge as the pleiotropic mechanisms of action of these drugs are progressively elucidated. In the therapeutic arena, the HDACI-based combinations that have advanced the farthest include those with cytotoxic chemotherapy (Garcia-Manero et al., 2012; Gojo et al., 2013), hypomethylating agents (Fandy et al., 2009; Juergens et al., 2011; Stathis et al., 2011)and PIs (Badros et al., 2009; Richardson, Schlossman et al., 2013).
However, although many trials have been completed (e.g., NCT00667082, NCT01105377, NCT00895934) or are ongoing (e.g., NCT01802333, NCT01305499, NCT00948064), none of these combinations has met with regulatory approval yet or been widely adopted in clinical practice. It follows from the above discussion that many other exciting avenues for rational combination exist, e.g., with agents that interfere with cell cycle checkpoints, mitosis, proteasome activity, protein neddylation and the function of anti-apoptotic proteins. Hopefully, the theoretical potential of these combinations and encouraging preclinical data will successfully be translated into novel therapies for patients with cancer in the near future.
Acknowledgments
Role of the funding source: This work was supported in part by the following awards (SG): R01 CA167708-01A1 and R01 CA100866-09 from the National Institutes of Health, and an award from the Leukemia and Lymphoma Society.
The authors acknowledge Michael S. Batalo, M.D. for providing Figure 1, and Kevin T. Hogan, Ph.D. for helping develop the table of contents and list of abbreviations for this article.
Abbreviations
- ALL
acute lymphoblastic leukemia
- AML
acute myeloid leukemia
- APAF-1
apoptotic protease activating factor 1
- ASK1
apoptosis signal-regulating kinase 1
- ATM
ataxia telangiectasia mutated
- ATR
ATM and Rad3-related
- ATRIP
ATR interacting protein
- Bcl-2
B-cell lymphoma-2
- Bcl-xL
Bcl-extra long
- Bcr-Abl
breakpoint cluster region-Abelson
- Bad
Bcl-2 antagonist of cell death
- BER
base excision repair
- Bim
Bcl-2 interacting mediator of cell death
- Bid
Bcl-2 interacting domain death agonist
- Bik
Bcl-2 interacting killer
- BLM
Bloom syndrome
- Bmf
Bcl-2 modifying factor
- CDC25
cell division cycle 25
- CDK
cyclin-dependent kinase
- CDT1
chromatin licensing and DNA replication factor 1
- c-FLIP
cellular FLICE-like inhibitory protein
- Chk
checkpoint kinase
- CLL
chronic lymphocytic leukemia
- CTCL
cutaneous T-cell lymphoma
- CtIP
CtBP interacting protein
- DNA
deoxyribonucleic acid
- DDR
DNA damage response
- DIABLO
direct IAP-binding protein with low pI
- DISC
death-inducing signaling complex
- DLBCL
diffuse large B-cell lymphoma
- DSB
double strand break
- ER
endoplasmic reticulum
- ETO
eight twenty one
- Fadd
Fas-associated protein with death domain
- FasL
Fas ligand
- FDA
Food and Drug Administration
- FEN1
flap structure-specific endonuclease 1
- FLT3
fms-like tyrosine kinase 3
- FoxM1
forkhead box protein M1
- GRP78
glucose regulated protein 78
- HAT
histone acetyltransferase
- HDAC
histone deacetylase
- HDACI
histone deacetylase inhibitor
- HR
homologous recombination
- Hrk
harakiri
- Hsp
heat shock protein
- IAP
inhibitor of apoptosis
- IκB
I kappa B
- IKK
I kappa B kinase
- ITD
internal tandem duplication
- JAK
Janus-associated kinase
- JNK
c-Jun N-terminal kinase
- LIC
leukemia-initiating cell
- MAPK
mitogen activated protein kinase
- MCL
mantle cell lymphoma
- Mcl-1
myeloid cell leukemia-1
- MDC1
mediator of DNA damage checkpoint 1
- Mdm2
murine double minute homolog 2
- MDS
myelodysplastic syndromes
- MEF
mouse embryonic fibroblast
- MEK
mitogen activated protein kinase kinase
- MM
multiple myeloma
- MOMP
mitochondrial outer membrane polarization
- MRE11
meiotic recombination 11
- MRN
MRE11/NBS1/RAD50
- NAD
nicotinamide adenine dinucleotide
- NCOR1
nuclear receptor corepressor 1
- NER
nucleotide excision repair
- NF-κB
nuclear factor kappa B
- NHEJ
nonhomologous end-joining
- PANORAMA
Panobinostat or Placebo With Bortezomib and Dexamethasone in Patients With Relapsed Multiple Myeloma
- PARP
poly ADP ribose polymerase
- p53BP1
p53 binding protein 1
- Ph
Philadelphia chromosome
- PI
proteasome inhibitor
- PK
protein kinase
- PLK-1
polo-like kinase 1
- PLZF
promyelocytic leukemia zinc finger
- PML
promyelocytic leukemia
- Puma
p53-upregulated modulator of apoptosis
- RARα
retinoic acid receptor alpha
- Rb
retinoblastoma protein
- RNA
ribonucleic acid
- ROS
reactive oxygen species
- RUNX1
runt related transcription factor 1
- SIRT1
sirtuin 1
- SMAC
second mitochondria-derived activator of caspases
- SMRT
silencing mediator of retinoic acid and thyroid hormone receptor
- STAT
signal transducer and activator of transcription
- Tip60
Tat interactive protein 60
- TKI
tyrosine kinase inhibitor
- TNF
tumor necrosis factor
- TRAIL
TNF-related apoptosis inducing ligand
- WRN
Werner syndrome
- XIAP
X-linked inhibitor of apoptosis
- XRCC
x-ray repair cross-complementing proteins
Footnotes
Conflict of interest statement: The authors report no conflicts of interest relevant to this article.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adimoolam S, Sirisawad M, Chen J, Thiemann P, Ford JM, Buggy JJ. HDAC inhibitor PCI-24781 decreases RAD51 expression and inhibits homologous recombination. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(49):19482–19487. doi: 10.1073/pnas.0707828104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almenara J, Rosato R, Grant S. Synergistic induction of mitochondrial damage and apoptosis in human leukemia cells by flavopiridol and the histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) Leukemia. 2002;16(7):1331–1343. doi: 10.1038/sj.leu.2402535. [DOI] [PubMed] [Google Scholar]
- Al-Yacoub N, Fecker LF, Mobs M, Plotz M, Braun FK, Sterry W, et al. Apoptosis induction by SAHA in cutaneous T-cell lymphoma cells is related to downregulation of c-FLIP and enhanced TRAIL signaling. The Journal of investigative dermatology. 2012;132(9):2263–2274. doi: 10.1038/jid.2012.125. [DOI] [PubMed] [Google Scholar]
- Archambault V, Glover DM. Polo-like kinases: Conservation and divergence in their functions and regulation. Nature reviews Molecular cell biology. 2009;10(4):265–275. doi: 10.1038/nrm2653. [DOI] [PubMed] [Google Scholar]
- Aron JL, Parthun MR, Marcucci G, Kitada S, Mone AP, Davis ME, et al. Depsipeptide (FR901228) induces histone acetylation and inhibition of histone deacetylase in chronic lymphocytic leukemia cells concurrent with activation of caspase 8-mediated apoptosis and down-regulation of c-FLIP protein. Blood. 2003;102(2):652–658. doi: 10.1182/blood-2002-12-3794. [DOI] [PubMed] [Google Scholar]
- Bachmann PS, Piazza RG, Janes ME, Wong NC, Davies C, Mogavero A, et al. Epigenetic silencing of BIM in glucocorticoid poor-responsive pediatric acute lymphoblastic leukemia, and its reversal by histone deacetylase inhibition. Blood. 2010;116(16):3013–3022. doi: 10.1182/blood-2010-05-284968. [DOI] [PubMed] [Google Scholar]
- Badros A, Burger AM, Philip S, Niesvizky R, Kolla SS, Goloubeva O, et al. Phase I study of vorinostat in combination with bortezomib for relapsed and refractory multiple myeloma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009;15(16):5250–5257. doi: 10.1158/1078-0432.CCR-08-2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421(6922):499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- Bali P, George P, Cohen P, Tao J, Guo F, Sigua C, et al. Superior activity of the combination of histone deacetylase inhibitor LAQ824 and the FLT-3 kinase inhibitor PKC412 against human acute myelogenous leukemia cells with mutant FLT-3. Clinical cancer research : an official journal of the American Association for Cancer Research. 2004;10(15):4991–4997. doi: 10.1158/1078-0432.CCR-04-0210. [DOI] [PubMed] [Google Scholar]
- Bali P, Pranpat M, Bradner J, Balasis M, Fiskus W, Guo F, et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: A novel basis for antileukemia activity of histone deacetylase inhibitors. The Journal of biological chemistry. 2005a;280(29):26729–26734. doi: 10.1074/jbc.C500186200. [DOI] [PubMed] [Google Scholar]
- Bali P, Pranpat M, Bradner J, Balasis M, Fiskus W, Guo F, et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: A novel basis for antileukemia activity of histone deacetylase inhibitors. The Journal of biological chemistry. 2005b;280(29):26729–26734. doi: 10.1074/jbc.C500186200. [DOI] [PubMed] [Google Scholar]
- Bangert A, Cristofanon S, Eckhardt I, Abhari BA, Kolodziej S, Hacker S, et al. Histone deacetylase inhibitors sensitize glioblastoma cells to TRAIL-induced apoptosis by c-myc-mediated downregulation of cFLIP. Oncogene. 2012;31(44):4677–4688. doi: 10.1038/onc.2011.614. [DOI] [PubMed] [Google Scholar]
- Basile V, Mantovani R, Imbriano C. DNA damage promotes histone deacetylase 4 nuclear localization and repression of G2/M promoters, via p53 C-terminal lysines. The Journal of biological chemistry. 2006;281(4):2347–2357. doi: 10.1074/jbc.M507712200. [DOI] [PubMed] [Google Scholar]
- BCL-2 inhibitor yields high response in CLL and SLL. Cancer discovery. 2014;4(2) doi: 10.1158/2159-8290.CD-NB2013-178. OF5-8290.CD-NB2013-178. Epub 2013 Dec 19. [DOI] [PubMed] [Google Scholar]
- Bhalla S, Balasubramanian S, David K, Sirisawad M, Buggy J, Mauro L, et al. PCI-24781 induces caspase and reactive oxygen species-dependent apoptosis through NFkappaB mechanisms and is synergistic with bortezomib in lymphoma cells. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009;15(10):3354–3365. doi: 10.1158/1078-0432.CCR-08-2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaskara S, Chyla BJ, Amann JM, Knutson SK, Cortez D, Sun ZW, et al. Deletion of histone deacetylase 3 reveals critical roles in S phase progression and DNA damage control. Molecular cell. 2008;30(1):61–72. doi: 10.1016/j.molcel.2008.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaskara S, Knutson SK, Jiang G, Chandrasekharan MB, Wilson AJ, Zheng S, et al. Hdac3 is essential for the maintenance of chromatin structure and genome stability. Cancer cell. 2010;18(5):436–447. doi: 10.1016/j.ccr.2010.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nature reviews Drug discovery. 2006;5(9):769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- Bouwman P, Jonkers J. The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nature reviews Cancer. 2012;12(9):587–598. doi: 10.1038/nrc3342. [DOI] [PubMed] [Google Scholar]
- Branzei D, Foiani M. Regulation of DNA repair throughout the cell cycle. Nature reviews Molecular cell biology. 2008;9(4):297–308. doi: 10.1038/nrm2351. [DOI] [PubMed] [Google Scholar]
- Brazelle W, Kreahling JM, Gemmer J, Ma Y, Cress WD, Haura E, et al. Histone deacetylase inhibitors downregulate checkpoint kinase 1 expression to induce cell death in non-small cell lung cancer cells. PloS one. 2010;5(12):e14335. doi: 10.1371/journal.pone.0014335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucher N, Britten CD. G2 checkpoint abrogation and checkpoint kinase-1 targeting in the treatment of cancer. British journal of cancer. 2008;98(3):523–528. doi: 10.1038/sj.bjc.6604208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler LM, Zhou X, Xu WS, Scher HI, Rifkind RA, Marks PA, et al. The histone deacetylase inhibitor SAHA arrests cancer cell growth, up-regulates thioredoxin-binding protein-2, and down-regulates thioredoxin. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(18):11700–11705. doi: 10.1073/pnas.182372299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calsou P, Frit P, Humbert O, Muller C, Chen DJ, Salles B. The DNA-dependent protein kinase catalytic activity regulates DNA end processing by means of ku entry into DNA. The Journal of biological chemistry. 1999;274(12):7848–7856. doi: 10.1074/jbc.274.12.7848. [DOI] [PubMed] [Google Scholar]
- Camphausen K, Burgan W, Cerra M, Oswald KA, Trepel JB, Lee MJ, et al. Enhanced radiation-induced cell killing and prolongation of gammaH2AX foci expression by the histone deacetylase inhibitor MS-275. Cancer research. 2004;64(1):316–321. doi: 10.1158/0008-5472.can-03-2630. [DOI] [PubMed] [Google Scholar]
- Carew JS, Nawrocki ST, Kahue CN, Zhang H, Yang C, Chung L, et al. Targeting autophagy augments the anticancer activity of the histone deacetylase inhibitor SAHA to overcome bcr-abl-mediated drug resistance. Blood. 2007;110(1):313–322. doi: 10.1182/blood-2006-10-050260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catley L, Weisberg E, Kiziltepe T, Tai YT, Hideshima T, Neri P, et al. Aggresome induction by proteasome inhibitor bortezomib and alpha-tubulin hyperacetylation by tubulin deacetylase (TDAC) inhibitor LBH589 are synergistic in myeloma cells. Blood. 2006;108(10):3441–3449. doi: 10.1182/blood-2006-04-016055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerchietti LC, Hatzi K, Caldas-Lopes E, Yang SN, Figueroa ME, Morin RD, et al. BCL6 repression of EP300 in human diffuse large B cell lymphoma cells provides a basis for rational combinatorial therapy. The Journal of clinical investigation. 2010 doi: 10.1172/JCI42869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty AR, Robey RW, Luchenko VL, Zhan Z, Piekarz RL, Gillet JP, et al. MAPK pathway activation leads to bim loss and histone deacetylase inhibitor resistance: Rationale to combine romidepsin with an MEK inhibitor. Blood. 2013;121(20):4115–4125. doi: 10.1182/blood-2012-08-449140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CS, Wang YC, Yang HC, Huang PH, Kulp SK, Yang CC, et al. Histone deacetylase inhibitors sensitize prostate cancer cells to agents that produce DNA double-strand breaks by targeting Ku70 acetylation. Cancer research. 2007;67(11):5318–5327. doi: 10.1158/0008-5472.CAN-06-3996. [DOI] [PubMed] [Google Scholar]
- Chen L, Fischle W, Verdin E, Greene WC. Science. 5535. Vol. 293. New York, N.Y.: 2001. Duration of nuclear NF-kappaB action regulated by reversible acetylation; pp. 1653–1657. [DOI] [PubMed] [Google Scholar]
- Chen S, Dai Y, Pei XY, Grant S. Bim upregulation by histone deacetylase inhibitors mediates interactions with the bcl-2 antagonist ABT-737: Evidence for distinct roles for bcl-2, bcl-xL, and mcl-1. Molecular and cellular biology. 2009a;29(23):6149–6169. doi: 10.1128/MCB.01481-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Dai Y, Pei XY, Grant S. Bim upregulation by histone deacetylase inhibitors mediates interactions with the bcl-2 antagonist ABT-737: Evidence for distinct roles for bcl-2, bcl-xL, and mcl-1. Molecular and cellular biology. 2009b;29(23):6149–6169. doi: 10.1128/MCB.01481-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, et al. Science. 5942. Vol. 325. New York, N.Y.: 2009. Lysine acetylation targets protein complexes and co-regulates major cellular functions; pp. 834–840. [DOI] [PubMed] [Google Scholar]
- Cohen HY, Lavu S, Bitterman KJ, Hekking B, Imahiyerobo TA, Miller C, et al. Acetylation of the C terminus of Ku70 by CBP and PCAF controls bax-mediated apoptosis. Molecular cell. 2004;13(5):627–638. doi: 10.1016/s1097-2765(04)00094-2. [DOI] [PubMed] [Google Scholar]
- Conti C, Leo E, Eichler GS, Sordet O, Martin MM, Fan A, et al. Inhibition of histone deacetylase in cancer cells slows down replication forks, activates dormant origins, and induces DNA damage. Cancer research. 2010;70(11):4470–4480. doi: 10.1158/0008-5472.CAN-09-3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Y, Chen S, Kmieciak M, Zhou L, Lin H, Pei XY, et al. The novel Chk1 inhibitor MK-8776 sensitizes human leukemia cells to HDAC inhibitors by targeting the intra-S checkpoint and DNA replication and repair. Molecular cancer therapeutics. 2013;12(6):878–889. doi: 10.1158/1535-7163.MCT-12-0902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Y, Chen S, Kramer LB, Funk VL, Dent P, Grant S. Interactions between bortezomib and romidepsin and belinostat in chronic lymphocytic leukemia cells. Clinical cancer research : an official journal of the American Association for Cancer Research. 2008;14(2):549–558. doi: 10.1158/1078-0432.CCR-07-1934. [DOI] [PubMed] [Google Scholar]
- Dai Y, Chen S, Venditti CA, Pei XY, Nguyen TK, Dent P, et al. Vorinostat synergistically potentiates MK-0457 lethality in chronic myelogenous leukemia cells sensitive and resistant to imatinib mesylate. Blood. 2008;112(3):793–804. doi: 10.1182/blood-2007-10-116376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Y, Chen S, Wang L, Pei XY, Funk VL, Kramer LB, et al. Disruption of IkappaB kinase (IKK)-mediated RelA serine 536 phosphorylation sensitizes human multiple myeloma cells to histone deacetylase (HDAC) inhibitors. The Journal of biological chemistry. 2011;286(39):34036–34050. doi: 10.1074/jbc.M111.284216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Y, Chen S, Wang L, Pei XY, Kramer LB, Dent P, et al. Bortezomib interacts synergistically with belinostat in human acute myeloid leukaemia and acute lymphoblastic leukaemia cells in association with perturbations in NF-kappaB and bim. British journal of haematology. 2011;153(2):222–235. doi: 10.1111/j.1365-2141.2011.08591.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Y, Grant S. New insights into checkpoint kinase 1 in the DNA damage response signaling network. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010;16(2):376–383. doi: 10.1158/1078-0432.CCR-09-1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Y, Guzman ML, Chen S, Wang L, Yeung SK, Pei XY, et al. The NF (nuclear factor)-kappaB inhibitor parthenolide interacts with histone deacetylase inhibitors to induce MKK7/JNK1-dependent apoptosis in human acute myeloid leukaemia cells. British journal of haematology. 2010;151(1):70–83. doi: 10.1111/j.1365-2141.2010.08319.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Y, Rahmani M, Dent P, Grant S. Blockade of histone deacetylase inhibitor-induced RelA/p65 acetylation and NF-kappaB activation potentiates apoptosis in leukemia cells through a process mediated by oxidative damage, XIAP downregulation, and c-jun N-terminal kinase 1 activation. Molecular and cellular biology. 2005;25(13):5429–5444. doi: 10.1128/MCB.25.13.5429-5444.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Y, Rahmani M, Grant S. Cell cycle. 5. Vol. 2. Georgetown, Tex.: 2003. An intact NF-kappaB pathway is required for histone deacetylase inhibitor-induced G1 arrest and maturation in U937 human myeloid leukemia cells; pp. 467–472. [PubMed] [Google Scholar]
- Dasmahapatra G, Lembersky D, Kramer L, Fisher RI, Friedberg J, Dent P, et al. The pan-HDAC inhibitor vorinostat potentiates the activity of the proteasome inhibitor carfilzomib in human DLBCL cells in vitro and in vivo. Blood. 2010;115(22):4478–4487. doi: 10.1182/blood-2009-12-257261. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Dasmahapatra G, Lembersky D, Son MP, Attkisson E, Dent P, Fisher RI, et al. Carfilzomib interacts synergistically with histone deacetylase inhibitors in mantle cell lymphoma cells in vitro and in vivo. Molecular cancer therapeutics. 2011;10(9):1686–1697. doi: 10.1158/1535-7163.MCT-10-1108. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Dasmahapatra G, Patel H, Nguyen T, Attkisson E, Grant S. PLK1 inhibitors synergistically potentiate HDAC inhibitor lethality in imatinib mesylate-sensitive or -resistant BCR/ABL+ leukemia cells in vitro and in vivo. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19(2):404–414. doi: 10.1158/1078-0432.CCR-12-2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta R, Oki E, Endo K, Biedermann V, Ren J, Kufe D. XIAP regulates DNA damage-induced apoptosis downstream of caspase-9 cleavage. The Journal of biological chemistry. 2000;275(41):31733–31738. doi: 10.1074/jbc.M910231199. [DOI] [PubMed] [Google Scholar]
- Davids MS, Letai A. Targeting the B-cell lymphoma/leukemia 2 family in cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2012;30(25):3127–3135. doi: 10.1200/JCO.2011.37.0981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson MA, Curry JE, Barber K, Beer PA, Graham B, Lyons JF, et al. AT9283, a potent inhibitor of the aurora kinases and Jak2, has therapeutic potential in myeloproliferative disorders. British journal of haematology. 2010;150(1):46–57. doi: 10.1111/j.1365-2141.2010.08175.x. [DOI] [PubMed] [Google Scholar]
- Deroanne CF, Bonjean K, Servotte S, Devy L, Colige A, Clausse N, et al. Histone deacetylases inhibitors as anti-angiogenic agents altering vascular endothelial growth factor signaling. Oncogene. 2002;21(3):427–436. doi: 10.1038/sj.onc.1205108. [DOI] [PubMed] [Google Scholar]
- Dickinson M, Johnstone RW, Prince HM. Histone deacetylase inhibitors: Potential targets responsible for their anti-cancer effect. Investigational new drugs. 2010;28(Suppl 1):S3–20. doi: 10.1007/s10637-010-9596-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falck J, Coates J, Jackson SP. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature. 2005;434(7033):605–611. doi: 10.1038/nature03442. [DOI] [PubMed] [Google Scholar]
- Fan J, Li L, Small D, Rassool F. Cells expressing FLT3/ITD mutations exhibit elevated repair errors generated through alternative NHEJ pathways: Implications for genomic instability and therapy. Blood. 2010;116(24):5298–5305. doi: 10.1182/blood-2010-03-272591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fandy TE, Herman JG, Kerns P, Jiemjit A, Sugar EA, Choi SH, et al. Early epigenetic changes and DNA damage do not predict clinical response in an overlapping schedule of 5-azacytidine and entinostat in patients with myeloid malignancies. Blood. 2009;114(13):2764–2773. doi: 10.1182/blood-2009-02-203547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiskus W, Pranpat M, Balasis M, Bali P, Estrella V, Kumaraswamy S, et al. Cotreatment with vorinostat (suberoylanilide hydroxamic acid) enhances activity of dasatinib (BMS-354825) against imatinib mesylate-sensitive or imatinib mesylate-resistant chronic myelogenous leukemia cells. Clinical cancer research : an official journal of the American Association for Cancer Research. 2006;12(19):5869–5878. doi: 10.1158/1078-0432.CCR-06-0980. [DOI] [PubMed] [Google Scholar]
- Fiskus W, Pranpat M, Bali P, Balasis M, Kumaraswamy S, Boyapalle S, et al. Combined effects of novel tyrosine kinase inhibitor AMN107 and histone deacetylase inhibitor LBH589 against bcr-abl-expressing human leukemia cells. Blood. 2006;108(2):645–652. doi: 10.1182/blood-2005-11-4639. [DOI] [PubMed] [Google Scholar]
- Fiskus W, Wang Y, Joshi R, Rao R, Yang Y, Chen J, et al. Cotreatment with vorinostat enhances activity of MK-0457 (VX-680) against acute and chronic myelogenous leukemia cells. Clinical cancer research : an official journal of the American Association for Cancer Research. 2008;14(19):6106–6115. doi: 10.1158/1078-0432.CCR-08-0721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fotheringham S, Epping MT, Stimson L, Khan O, Wood V, Pezzella F, et al. Genome-wide loss-of-function screen reveals an important role for the proteasome in HDAC inhibitor-induced apoptosis. Cancer cell. 2009;15(1):57–66. doi: 10.1016/j.ccr.2008.12.001. [DOI] [PubMed] [Google Scholar]
- Gammoh N, Lam D, Puente C, Ganley I, Marks PA, Jiang X. Role of autophagy in histone deacetylase inhibitor-induced apoptotic and nonapoptotic cell death. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(17):6561–6565. doi: 10.1073/pnas.1204429109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao N, Dai Y, Rahmani M, Dent P, Grant S. Contribution of disruption of the nuclear factor-kappaB pathway to induction of apoptosis in human leukemia cells by histone deacetylase inhibitors and flavopiridol. Molecular pharmacology. 2004;66(4):956–963. doi: 10.1124/mol.104.002014. [DOI] [PubMed] [Google Scholar]
- Garcia-Manero G, Tambaro FP, Bekele NB, Yang H, Ravandi F, Jabbour E, et al. Phase II trial of vorinostat with idarubicin and cytarabine for patients with newly diagnosed acute myelogenous leukemia or myelodysplastic syndrome. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2012;30(18):2204–2210. doi: 10.1200/JCO.2011.38.3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaymes TJ, Padua RA, Pla M, Orr S, Omidvar N, Chomienne C, et al. Histone deacetylase inhibitors (HDI) cause DNA damage in leukemia cells: A mechanism for leukemia-specific HDI-dependent apoptosis? Molecular cancer research : MCR. 2006;4(8):563–573. doi: 10.1158/1541-7786.MCR-06-0111. [DOI] [PubMed] [Google Scholar]
- Gillenwater AM, Zhong M, Lotan R. Histone deacetylase inhibitor suberoylanilide hydroxamic acid induces apoptosis through both mitochondrial and fas (Cd95) signaling in head and neck squamous carcinoma cells. Molecular cancer therapeutics. 2007;6(11):2967–2975. doi: 10.1158/1535-7163.MCT-04-0344. [DOI] [PubMed] [Google Scholar]
- Gillespie S, Borrow J, Zhang XD, Hersey P. Bim plays a crucial role in synergistic induction of apoptosis by the histone deacetylase inhibitor SBHA and TRAIL in melanoma cells. Apoptosis : An International Journal on Programmed Cell Death. 2006;11(12):2251–2265. doi: 10.1007/s10495-006-0283-6. [DOI] [PubMed] [Google Scholar]
- Gleixner KV, Ferenc V, Peter B, Gruze A, Meyer RA, Hadzijusufovic E, et al. Polo-like kinase 1 (Plk1) as a novel drug target in chronic myeloid leukemia: Overriding imatinib resistance with the Plk1 inhibitor BI 2536. Cancer research. 2010;70(4):1513–1523. doi: 10.1158/0008-5472.CAN-09-2181. [DOI] [PubMed] [Google Scholar]
- Glick RD, Swendeman SL, Coffey DC, Rifkind RA, Marks PA, Richon VM, et al. Hybrid polar histone deacetylase inhibitor induces apoptosis and CD95/CD95 ligand expression in human neuroblastoma. Cancer research. 1999;59(17):4392–4399. [PubMed] [Google Scholar]
- Gojo I, Tan M, Fang HB, Sadowska M, Lapidus R, Baer MR, et al. Translational phase I trial of vorinostat (suberoylanilide hydroxamic acid) combined with cytarabine and etoposide in patients with relapsed, refractory, or high-risk acute myeloid leukemia. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19(7):1838–1851. doi: 10.1158/1078-0432.CCR-12-3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb TM, Jackson SP. The DNA-dependent protein kinase: Requirement for DNA ends and association with ku antigen. Cell. 1993;72(1):131–142. doi: 10.1016/0092-8674(93)90057-w. [DOI] [PubMed] [Google Scholar]
- Grant S, Dai Y. Histone deacetylase inhibitors and rational combination therapies. Advances in Cancer Research. 2012;116:199–237. doi: 10.1016/B978-0-12-394387-3.00006-9. [DOI] [PubMed] [Google Scholar]
- Guo F, Sigua C, Tao J, Bali P, George P, Li Y, et al. Cotreatment with histone deacetylase inhibitor LAQ824 enhances apo-2L/tumor necrosis factor-related apoptosis inducing ligand-induced death inducing signaling complex activity and apoptosis of human acute leukemia cells. Cancer research. 2004;64(7):2580–2589. doi: 10.1158/0008-5472.can-03-2629. [DOI] [PubMed] [Google Scholar]
- Ha K, Fiskus W, Rao R, Balusu R, Venkannagari S, Nalabothula NR, et al. Hsp90 inhibitor-mediated disruption of chaperone association of ATR with hsp90 sensitizes cancer cells to DNA damage. Molecular cancer therapeutics. 2011;10(7):1194–1206. doi: 10.1158/1535-7163.MCT-11-0094. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hideshima T, Bradner JE, Wong J, Chauhan D, Richardson P, Schreiber SL, et al. Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(24):8567–8572. doi: 10.1073/pnas.0503221102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hideshima T, Richardson PG, Anderson KC. Mechanism of action of proteasome inhibitors and deacetylase inhibitors and the biological basis of synergy in multiple myeloma. Molecular cancer therapeutics. 2011;10(11):2034–2042. doi: 10.1158/1535-7163.MCT-11-0433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holkova B, Supko JG, Ames MM, Reid JM, Shapiro GI, Perkins EB, et al. A phase I trial of vorinostat and alvocidib in patients with relapsed, refractory, or poor prognosis acute leukemia, or refractory anemia with excess blasts-2. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19(7):1873–1883. doi: 10.1158/1078-0432.CCR-12-2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holkova B, Perkins EB, Sokol L, Richards KL, Parekh S, Elstrom R, et al. A phase II trial of bortezomib and vorinostat in mantle cell lymphoma and diffuse large B-cell lymphoma. ASH Annual Meeting Abstracts. 2011;118(21):779. [Google Scholar]
- Holkova B, Shea TC, Bose P, Tombes MB, Shrader E, Wan W, et al. Phase I study of bortezomib and romidepsin in patients with chronic lymphocytic Leukemia/Small lymphocytic lymphoma, indolent B-cell lymphoma, or peripheral T-cell lymphoma. ASH Annual Meeting Abstracts. 2012;120(21):1794. [Google Scholar]
- Hu Y, Lu W, Chen G, Zhang H, Jia Y, Wei Y, et al. Overcoming resistance to histone deacetylase inhibitors in human leukemia with the redox modulating compound beta-phenylethyl isothiocyanate. Blood. 2010;116(15):2732–2741. doi: 10.1182/blood-2009-11-256354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huertas P, Jackson SP. Human CtIP mediates cell cycle control of DNA end resection and double strand break repair. The Journal of biological chemistry. 2009;284(14):9558–9565. doi: 10.1074/jbc.M808906200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue S, MacFarlane M, Harper N, Wheat LM, Dyer MJ, Cohen GM. Histone deacetylase inhibitors potentiate TNF-related apoptosis-inducing ligand (TRAIL)-induced apoptosis in lymphoid malignancies. Cell death and differentiation. 2004;11(Suppl 2):S193–206. doi: 10.1038/sj.cdd.4401535. [DOI] [PubMed] [Google Scholar]
- Inoue S, Riley J, Gant TW, Dyer MJ, Cohen GM. Apoptosis induced by histone deacetylase inhibitors in leukemic cells is mediated by bim and noxa. Leukemia. 2007;21(8):1773–1782. doi: 10.1038/sj.leu.2404760. [DOI] [PubMed] [Google Scholar]
- Insinga A, Monestiroli S, Ronzoni S, Gelmetti V, Marchesi F, Viale A, et al. Inhibitors of histone deacetylases induce tumor-selective apoptosis through activation of the death receptor pathway. Nature medicine. 2005;11(1):71–76. doi: 10.1038/nm1160. [DOI] [PubMed] [Google Scholar]
- Irvine AE, McMullin MF, Ong YL. Hematology. 1. Vol. 7. Amsterdam, Netherlands: 2002. Bcl-2 family members as prognostic indicators in AML; pp. 21–31. [DOI] [PubMed] [Google Scholar]
- Ishii S, Kurasawa Y, Wong J, Yu-Lee LY. Histone deacetylase 3 localizes to the mitotic spindle and is required for kinetochore-microtubule attachment. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(11):4179–4184. doi: 10.1073/pnas.0710140105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito A, Kawaguchi Y, Lai CH, Kovacs JJ, Higashimoto Y, Appella E, et al. MDM2-HDAC1-mediated deacetylation of p53 is required for its degradation. The EMBO journal. 2002;21(22):6236–6245. doi: 10.1093/emboj/cdf616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jazaeri AA, Shibata E, Park J, Bryant JL, Conaway MR, Modesitt SC, et al. Overcoming platinum resistance in preclinical models of ovarian cancer using the neddylation inhibitor MLN4924. Molecular cancer therapeutics. 2013;12(10):1958–1967. doi: 10.1158/1535-7163.MCT-12-1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jona A, Khaskhely N, Buglio D, Shafer JA, Derenzini E, Bollard CM, et al. The histone deacetylase inhibitor entinostat (SNDX-275) induces apoptosis in Hodgkin lymphoma cells and synergizes with bcl-2 family inhibitors. Experimental hematology. 2011;39(10):1007–1017.e1. doi: 10.1016/j.exphem.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juergens RA, Wrangle J, Vendetti FP, Murphy SC, Zhao M, Coleman B, et al. Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer. Cancer discovery. 2011;1(7):598–607. doi: 10.1158/2159-8290.CD-11-0214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kachhap SK, Rosmus N, Collis SJ, Kortenhorst MS, Wissing MD, Hedayati M, et al. Downregulation of homologous recombination DNA repair genes by HDAC inhibition in prostate cancer is mediated through the E2F1 transcription factor. PloS one. 2010;5(6):e11208. doi: 10.1371/journal.pone.0011208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahali S, Sarcar B, Prabhu A, Seto E, Chinnaiyan P. Class I histone deacetylases localize to the endoplasmic reticulum and modulate the unfolded protein response. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2012;26(6):2437–2445. doi: 10.1096/fj.11-193706. Electronic version. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaidi A, Weinert BT, Choudhary C, Jackson SP. Science. 5997. Vol. 329. New York, N.Y.: 2010. Human SIRT6 promotes DNA end resection through CtIP deacetylation; pp. 1348–1353. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Kao GD, McKenna WG, Guenther MG, Muschel RJ, Lazar MA, Yen TJ. Histone deacetylase 4 interacts with 53BP1 to mediate the DNA damage response. The Journal of cell biology. 2003;160(7):1017–1027. doi: 10.1083/jcb.200209065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441(7092):431–436. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- Karp JE, Thomas BM, Greer JM, Sorge C, Gore SD, Pratz KW, et al. Phase I and pharmacologic trial of cytosine arabinoside with the selective checkpoint 1 inhibitor sch 900776 in refractory acute leukemias. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18(24):6723–6731. doi: 10.1158/1078-0432.CCR-12-2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432(7015):316–323. doi: 10.1038/nature03097. [DOI] [PubMed] [Google Scholar]
- Kauh J, Fan S, Xia M, Yue P, Yang L, Khuri FR, et al. c-FLIP degradation mediates sensitization of pancreatic cancer cells to TRAIL-induced apoptosis by the histone deacetylase inhibitor LBH589. PloS one. 2010;5(4):e10376. doi: 10.1371/journal.pone.0010376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell. 2003;115(6):727–738. doi: 10.1016/s0092-8674(03)00939-5. [DOI] [PubMed] [Google Scholar]
- Kelly KR, Ecsedy J, Mahalingam D, Nawrocki ST, Padmanabhan S, Giles FJ, et al. Targeting aurora kinases in cancer treatment. Current Drug Targets. 2011;12(14):2067–2078. doi: 10.2174/138945011798829410. [DOI] [PubMed] [Google Scholar]
- Kerr E, Holohan C, McLaughlin KM, Majkut J, Dolan S, Redmond K, et al. Identification of an acetylation-dependant Ku70/FLIP complex that regulates FLIP expression and HDAC inhibitor-induced apoptosis. Cell death and differentiation. 2012;19(8):1317–1327. doi: 10.1038/cdd.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna KK, Jackson SP. DNA double-strand breaks: Signaling, repair and the cancer connection. Nature genetics. 2001;27(3):247–254. doi: 10.1038/85798. [DOI] [PubMed] [Google Scholar]
- Kim EH, Kim HS, Kim SU, Noh EJ, Lee JS, Choi KS. Sodium butyrate sensitizes human glioma cells to TRAIL-mediated apoptosis through inhibition of Cdc2 and the subsequent downregulation of survivin and XIAP. Oncogene. 2005;24(46):6877–6889. doi: 10.1038/sj.onc.1208851. [DOI] [PubMed] [Google Scholar]
- Kim GD, Choi YH, Dimtchev A, Jeong SJ, Dritschilo A, Jung M. Sensing of ionizing radiation-induced DNA damage by ATM through interaction with histone deacetylase. The Journal of biological chemistry. 1999;274(44):31127–31130. doi: 10.1074/jbc.274.44.31127. [DOI] [PubMed] [Google Scholar]
- Kim JJ, Lee SB, Park JK, Yoo YD. TNF-alpha-induced ROS production triggering apoptosis is directly linked to Romo1 and bcl-X(L) Cell death and differentiation. 2010;17(9):1420–1434. doi: 10.1038/cdd.2010.19. [DOI] [PubMed] [Google Scholar]
- Kmieciak M, Bose P, Barr PM, Tombes MB, Shrader E, Cebula E, et al. Phase I trial of carfilzomib in combination with vorinostat (SAHA) in patients with Relapsed/Refractory B-cell lymphomas. Blood. 2013;122(21):4375–4375. doi: 10.3109/10428194.2015.1075019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi A, Shimizu S, Hirota J, Takao T, Fan Y, Matsuoka Y, et al. Involvement of histone H1.2 in apoptosis induced by DNA double-strand breaks. Cell. 2003;114(6):673–688. doi: 10.1016/s0092-8674(03)00719-0. [DOI] [PubMed] [Google Scholar]
- Konopleva M, Contractor R, Tsao T, Samudio I, Ruvolo PP, Kitada S, et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer cell. 2006;10(5):375–388. doi: 10.1016/j.ccr.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Koprinarova M, Botev P, Russev G. Histone deacetylase inhibitor sodium butyrate enhances cellular radiosensitivity by inhibiting both DNA nonhomologous end joining and homologous recombination. DNA repair. 2011;10(9):970–977. doi: 10.1016/j.dnarep.2011.07.003. [DOI] [PubMed] [Google Scholar]
- Kotian S, Liyanarachchi S, Zelent A, Parvin JD. Histone deacetylases 9 and 10 are required for homologous recombination. The Journal of biological chemistry. 2011;286(10):7722–7726. doi: 10.1074/jbc.C110.194233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda J, Taniwaki M. Involvement of BH3-only proteins in hematologic malignancies. Critical reviews in oncology/hematology. 2009;71(2):89–101. doi: 10.1016/j.critrevonc.2008.10.004. [DOI] [PubMed] [Google Scholar]
- Labi V, Grespi F, Baumgartner F, Villunger A. Targeting the bcl-2-regulated apoptosis pathway by BH3 mimetics: A breakthrough in anticancer therapy? Cell death and differentiation. 2008;15(6):977–987. doi: 10.1038/cdd.2008.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langerak P, Mejia-Ramirez E, Limbo O, Russell P. Release of ku and MRN from DNA ends by Mre11 nuclease activity and Ctp1 is required for homologous recombination repair of double-strand breaks. PLoS genetics. 2011;7(9):e1002271. doi: 10.1371/journal.pgen.1002271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapenna S, Giordano A. Cell cycle kinases as therapeutic targets for cancer. Nature reviews Drug discovery. 2009;8(7):547–566. doi: 10.1038/nrd2907. [DOI] [PubMed] [Google Scholar]
- Lee JH, Choy ML, Ngo L, Foster SS, Marks PA. Histone deacetylase inhibitor induces DNA damage, which normal but not transformed cells can repair. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(33):14639–14644. doi: 10.1073/pnas.1008522107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MH, Yang HY. Negative regulators of cyclin-dependent kinases and their roles in cancers. Cellular and molecular life sciences : CMLS. 2001;58(12-13):1907–1922. doi: 10.1007/PL00000826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Wu X. Histone deacetylase inhibitor, trichostatin A, activates p21WAF1/CIP1 expression through downregulation of c-myc and release of the repression of c-myc from the promoter in human cervical cancer cells. Biochemical and biophysical research communications. 2004;324(2):860–867. doi: 10.1016/j.bbrc.2004.09.130. [DOI] [PubMed] [Google Scholar]
- Li L, Zhang L, Fan J, Greenberg K, Desiderio S, Rassool FV, et al. Defective nonhomologous end joining blocks B-cell development in FLT3/ITD mice. Blood. 2011;117(11):3131–3139. doi: 10.1182/blood-2010-05-286070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JJ, Milhollen MA, Smith PG, Narayanan U, Dutta A. NEDD8-targeting drug MLN4924 elicits DNA rereplication by stabilizing Cdt1 in S phase, triggering checkpoint activation, apoptosis, and senescence in cancer cells. Cancer research. 2010;70(24):10310–10320. doi: 10.1158/0008-5472.CAN-10-2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindemann RK, Newbold A, Whitecross KF, Cluse LA, Frew AJ, Ellis L, et al. Analysis of the apoptotic and therapeutic activities of histone deacetylase inhibitors by using a mouse model of B cell lymphoma. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(19):8071–8076. doi: 10.1073/pnas.0702294104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling YH, Liebes L, Zou Y, Perez-Soler R. Reactive oxygen species generation and mitochondrial dysfunction in the apoptotic response to bortezomib, a novel proteasome inhibitor, in human H460 non-small cell lung cancer cells. The Journal of biological chemistry. 2003;278(36):33714–33723. doi: 10.1074/jbc.M302559200. [DOI] [PubMed] [Google Scholar]
- Liu L, Zhang M, Zou P. Expression of PLK1 and survivin in diffuse large B-cell lymphoma. Leukemia & lymphoma. 2007;48(11):2179–2183. doi: 10.1080/10428190701615918. [DOI] [PubMed] [Google Scholar]
- Liu N, Lamerdin JE, Tebbs RS, Schild D, Tucker JD, Shen MR, et al. XRCC2 and XRCC3, new human Rad51-family members, promote chromosome stability and protect against DNA cross-links and other damages. Molecular cell. 1998;1(6):783–793. doi: 10.1016/s1097-2765(00)80078-7. [DOI] [PubMed] [Google Scholar]
- Liu XS, Song B, Liu X. The substrates of Plk1, beyond the functions in mitosis. Protein & cell. 2010;1(11):999–1010. doi: 10.1007/s13238-010-0131-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukas J, Lukas C, Bartek J. More than just a focus: The chromatin response to DNA damage and its role in genome integrity maintenance. Nature cell biology. 2011;13(10):1161–1169. doi: 10.1038/ncb2344. [DOI] [PubMed] [Google Scholar]
- Luo J, Su F, Chen D, Shiloh A, Gu W. Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature. 2000;408(6810):377–381. doi: 10.1038/35042612. [DOI] [PubMed] [Google Scholar]
- Luo Z, Pan Y, Jeong LS, Liu J, Jia L. Inactivation of the cullin (CUL)-RING E3 ligase by the NEDD8-activating enzyme inhibitor MLN4924 triggers protective autophagy in cancer cells. Autophagy. 2012;8(11):1677–1679. doi: 10.4161/auto.21484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Z, Yu G, Lee HW, Li L, Wang L, Yang D, et al. The Nedd8-activating enzyme inhibitor MLN4924 induces autophagy and apoptosis to suppress liver cancer cell growth. Cancer research. 2012;72(13):3360–3371. doi: 10.1158/0008-5472.CAN-12-0388. [DOI] [PubMed] [Google Scholar]
- Maertens J, Lubbert M, Fiedler W, Fouillard L, Haaland A, Brandwein JM, et al. Phase I/II study of volasertib (BI 6727), an intravenous polo-like kinase (plk) inhibitor, in patients with acute myeloid leukemia (AML): Results from the randomized phase II part for volasertib in combination with low-dose cytarabine (LDAC) versus LDAC monotherapy in patients with previously untreated AML ineligible for intensive treatment. ASH Annual Meeting Abstracts. 2012;120(21):411. [Google Scholar]
- Maggio SC, Rosato RR, Kramer LB, Dai Y, Rahmani M, Paik DS, et al. The histone deacetylase inhibitor MS-275 interacts synergistically with fludarabine to induce apoptosis in human leukemia cells. Cancer research. 2004;64(7):2590–2600. doi: 10.1158/0008-5472.can-03-2631. [DOI] [PubMed] [Google Scholar]
- Magnaghi-Jaulin L, Eot-Houllier G, Fulcrand G, Jaulin C. Histone deacetylase inhibitors induce premature sister chromatid separation and override the mitotic spindle assembly checkpoint. Cancer research. 2007;67(13):6360–6367. doi: 10.1158/0008-5472.CAN-06-3012. [DOI] [PubMed] [Google Scholar]
- Marchion DC, Bicaku E, Daud AI, Richon V, Sullivan DM, Munster PN. Sequence-specific potentiation of topoisomerase II inhibitors by the histone deacetylase inhibitor suberoylanilide hydroxamic acid. Journal of cellular biochemistry. 2004;92(2):223–237. doi: 10.1002/jcb.20045. [DOI] [PubMed] [Google Scholar]
- Marks PA, Xu WS. Histone deacetylase inhibitors: Potential in cancer therapy. Journal of cellular biochemistry. 2009;107(4):600–608. doi: 10.1002/jcb.22185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayo MW, Denlinger CE, Broad RM, Yeung F, Reilly ET, Shi Y, et al. Ineffectiveness of histone deacetylase inhibitors to induce apoptosis involves the transcriptional activation of NF-kappa B through the akt pathway. The Journal of biological chemistry. 2003;278(21):18980–18989. doi: 10.1074/jbc.M211695200. [DOI] [PubMed] [Google Scholar]
- Milhollen MA, Narayanan U, Soucy TA, Veiby PO, Smith PG, Amidon B. Inhibition of NEDD8-activating enzyme induces rereplication and apoptosis in human tumor cells consistent with deregulating CDT1 turnover. Cancer research. 2011;71(8):3042–3051. doi: 10.1158/0008-5472.CAN-10-2122. [DOI] [PubMed] [Google Scholar]
- Milhollen MA, Traore T, Adams-Duffy J, Thomas MP, Berger AJ, Dang L, et al. MLN4924, a NEDD8-activating enzyme inhibitor, is active in diffuse large B-cell lymphoma models: Rationale for treatment of NF-{kappa}B-dependent lymphoma. Blood. 2010;116(9):1515–1523. doi: 10.1182/blood-2010-03-272567. [DOI] [PubMed] [Google Scholar]
- Miller KM, Tjeertes JV, Coates J, Legube G, Polo SE, Britton S, et al. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nature structural & molecular biology. 2010;17(9):1144–1151. doi: 10.1038/nsmb.1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nature reviews Cancer. 2006;6(1):38–51. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- Misteli T, Soutoglou E. The emerging role of nuclear architecture in DNA repair and genome maintenance. Nature reviews Molecular cell biology. 2009;10(4):243–254. doi: 10.1038/nrm2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsiades CS, Mitsiades NS, McMullan CJ, Poulaki V, Shringarpure R, Hideshima T, et al. Transcriptional signature of histone deacetylase inhibition in multiple myeloma: Biological and clinical implications. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(2):540–545. doi: 10.1073/pnas.2536759100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsiades N, Mitsiades CS, Richardson PG, McMullan C, Poulaki V, Fanourakis G, et al. Molecular sequelae of histone deacetylase inhibition in human malignant B cells. Blood. 2003;101(10):4055–4062. doi: 10.1182/blood-2002-11-3514. [DOI] [PubMed] [Google Scholar]
- Munshi A, Kurland JF, Nishikawa T, Tanaka T, Hobbs ML, Tucker SL, et al. Histone deacetylase inhibitors radiosensitize human melanoma cells by suppressing DNA repair activity. Clinical cancer research : an official journal of the American Association for Cancer Research. 2005;11(13):4912–4922. doi: 10.1158/1078-0432.CCR-04-2088. [DOI] [PubMed] [Google Scholar]
- Munshi A, Tanaka T, Hobbs ML, Tucker SL, Richon VM, Meyn RE. Vorinostat, a histone deacetylase inhibitor, enhances the response of human tumor cells to ionizing radiation through prolongation of gamma-H2AX foci. Molecular cancer therapeutics. 2006;5(8):1967–1974. doi: 10.1158/1535-7163.MCT-06-0022. [DOI] [PubMed] [Google Scholar]
- Murr R, Loizou JI, Yang YG, Cuenin C, Li H, Wang ZQ, et al. Histone acetylation by trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nature cell biology. 2006;8(1):91–99. doi: 10.1038/ncb1343. [DOI] [PubMed] [Google Scholar]
- Namdar M, Perez G, Ngo L, Marks PA. Selective inhibition of histone deacetylase 6 (HDAC6) induces DNA damage and sensitizes transformed cells to anticancer agents. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(46):20003–20008. doi: 10.1073/pnas.1013754107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawrocki ST, Carew JS, Douglas L, Cleveland JL, Humphreys R, Houghton JA. Histone deacetylase inhibitors enhance lexatumumab-induced apoptosis via a p21Cip1-dependent decrease in survivin levels. Cancer research. 2007;67(14):6987–6994. doi: 10.1158/0008-5472.CAN-07-0812. [DOI] [PubMed] [Google Scholar]
- Nawrocki ST, Carew JS, Pino MS, Highshaw RA, Andtbacka RH, Dunner K, Jr, et al. Aggresome disruption: A novel strategy to enhance bortezomib-induced apoptosis in pancreatic cancer cells. Cancer research. 2006;66(7):3773–3781. doi: 10.1158/0008-5472.CAN-05-2961. [DOI] [PubMed] [Google Scholar]
- Nawrocki ST, Griffin P, Kelly KR, Carew JS. MLN4924: A novel first-in-class inhibitor of NEDD8-activating enzyme for cancer therapy. Expert opinion on investigational drugs. 2012;21(10):1563–1573. doi: 10.1517/13543784.2012.707192. [DOI] [PubMed] [Google Scholar]
- Nawrocki ST, Kelly KR, Smith PG, Espitia CM, Possemato A, Beausoleil SA, et al. Disrupting protein NEDDylation with MLN4924 is a novel strategy to target cisplatin resistance in ovarian cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19(13):3577–3590. doi: 10.1158/1078-0432.CCR-12-3212. [DOI] [PubMed] [Google Scholar]
- Nebbioso A, Clarke N, Voltz E, Germain E, Ambrosino C, Bontempo P, et al. Tumor-selective action of HDAC inhibitors involves TRAIL induction in acute myeloid leukemia cells. Nature medicine. 2005;11(1):77–84. doi: 10.1038/nm1161. [DOI] [PubMed] [Google Scholar]
- Nguyen T, Dai Y, Attkisson E, Kramer L, Jordan N, Nguyen N, et al. HDAC inhibitors potentiate the activity of the BCR/ABL kinase inhibitor KW-2449 in imatinib-sensitive or -resistant BCR/ABL+ leukemia cells in vitro and in vivo. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011a;17(10):3219–3232. doi: 10.1158/1078-0432.CCR-11-0234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T, Dai Y, Attkisson E, Kramer L, Jordan N, Nguyen N, et al. HDAC inhibitors potentiate the activity of the BCR/ABL kinase inhibitor KW-2449 in imatinib-sensitive or -resistant BCR/ABL+ leukemia cells in vitro and in vivo. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011b;17(10):3219–3232. doi: 10.1158/1078-0432.CCR-11-0234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmanapalli R, Fuino L, Bali P, Gasparetto M, Glozak M, Tao J, et al. Histone deacetylase inhibitor LAQ824 both lowers expression and promotes proteasomal degradation of bcr-abl and induces apoptosis of imatinib mesylate-sensitive or -refractory chronic myelogenous leukemia-blast crisis cells. Cancer research. 2003;63(16):5126–5135. [PubMed] [Google Scholar]
- Nimmanapalli R, Fuino L, Stobaugh C, Richon V, Bhalla K. Cotreatment with the histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) enhances imatinib-induced apoptosis of bcr-abl-positive human acute leukemia cells. Blood. 2003;101(8):3236–3239. doi: 10.1182/blood-2002-08-2675. [DOI] [PubMed] [Google Scholar]
- Nishioka C, Ikezoe T, Yang J, Takeuchi S, Koeffler HP, Yokoyama A. MS-275, a novel histone deacetylase inhibitor with selectivity against HDAC1, induces degradation of FLT3 via inhibition of chaperone function of heat shock protein 90 in AML cells. Leukemia research. 2008;32(9):1382–1392. doi: 10.1016/j.leukres.2008.02.018. [DOI] [PubMed] [Google Scholar]
- Novotny-Diermayr V, Hart S, Goh KC, Cheong A, Ong LC, Hentze H, et al. The oral HDAC inhibitor pracinostat (SB939) is efficacious and synergistic with the JAK2 inhibitor pacritinib (SB1518) in preclinical models of AML. Blood cancer journal. 2012;2(5):e69. doi: 10.1038/bcj.2012.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussenzweig A, Nussenzweig MC. A backup DNA repair pathway moves to the forefront. Cell. 2007;131(2):223–225. doi: 10.1016/j.cell.2007.10.005. [DOI] [PubMed] [Google Scholar]
- O'Driscoll M, Jeggo PA. The role of double-strand break repair - insights from human genetics. Nature reviews Genetics. 2006;7(1):45–54. doi: 10.1038/nrg1746. [DOI] [PubMed] [Google Scholar]
- Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, et al. An inhibitor of bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435(7042):677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- Ott M, Norberg E, Zhivotovsky B, Orrenius S. Mitochondrial targeting of tBid/Bax: A role for the TOM complex? Cell death and differentiation. 2009;16(8):1075–1082. doi: 10.1038/cdd.2009.61. [DOI] [PubMed] [Google Scholar]
- Pan R, Hogdal LJ, Benito JM, Bucci D, Han L, Borthakur G, et al. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer discovery. 2014 doi: 10.1158/2159-8290.CD-13-0609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peart MJ, Smyth GK, van Laar RK, Bowtell DD, Richon VM, Marks PA, et al. Identification and functional significance of genes regulated by structurally different histone deacetylase inhibitors. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(10):3697–3702. doi: 10.1073/pnas.0500369102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei XY, Dai Y, Grant S. Synergistic induction of oxidative injury and apoptosis in human multiple myeloma cells by the proteasome inhibitor bortezomib and histone deacetylase inhibitors. Clinical cancer research : an official journal of the American Association for Cancer Research. 2004;10(11):3839–3852. doi: 10.1158/1078-0432.CCR-03-0561. [DOI] [PubMed] [Google Scholar]
- Perez-Galan P, Roue G, Villamor N, Montserrat E, Campo E, Colomer D. The proteasome inhibitor bortezomib induces apoptosis in mantle-cell lymphoma through generation of ROS and noxa activation independent of p53 status. Blood. 2006;107(1):257–264. doi: 10.1182/blood-2005-05-2091. [DOI] [PubMed] [Google Scholar]
- Petalcorin MI, Sandall J, Wigley DB, Boulton SJ. CeBRC-2 stimulates D-loop formation by RAD-51 and promotes DNA single-strand annealing. Journal of Molecular Biology. 2006;361(2):231–242. doi: 10.1016/j.jmb.2006.06.020. [DOI] [PubMed] [Google Scholar]
- Petruccelli LA, Dupere-Richer D, Pettersson F, Retrouvey H, Skoulikas S, Miller WH., Jr Vorinostat induces reactive oxygen species and DNA damage in acute myeloid leukemia cells. PloS one. 2011;6(6):e20987. doi: 10.1371/journal.pone.0020987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petruccelli LA, Pettersson F, Del Rincon SV, Guilbert C, Licht JD, Miller WH., Jr Expression of leukemia-associated fusion proteins increases sensitivity to histone deacetylase inhibitor-induced DNA damage and apoptosis. Molecular cancer therapeutics. 2013;12(8):1591–1604. doi: 10.1158/1535-7163.MCT-12-1039. [DOI] [PubMed] [Google Scholar]
- Qian DZ, Wang X, Kachhap SK, Kato Y, Wei Y, Zhang L, et al. The histone deacetylase inhibitor NVP-LAQ824 inhibits angiogenesis and has a greater antitumor effect in combination with the vascular endothelial growth factor receptor tyrosine kinase inhibitor PTK787/ZK222584. Cancer research. 2004;64(18):6626–6634. doi: 10.1158/0008-5472.CAN-04-0540. [DOI] [PubMed] [Google Scholar]
- Qiu L, Burgess A, Fairlie DP, Leonard H, Parsons PG, Gabrielli BG. Histone deacetylase inhibitors trigger a G2 checkpoint in normal cells that is defective in tumor cells. Molecular biology of the cell. 2000;11(6):2069–2083. doi: 10.1091/mbc.11.6.2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintas-Cardama A, Santos FP, Garcia-Manero G. Histone deacetylase inhibitors for the treatment of myelodysplastic syndrome and acute myeloid leukemia. Leukemia. 2011;25(2):226–235. doi: 10.1038/leu.2010.276. [DOI] [PubMed] [Google Scholar]
- Rabut G, Peter M. Function and regulation of protein neddylation. ‘protein modifications: Beyond the usual suspects’ review series. EMBO reports. 2008;9(10):969–976. doi: 10.1038/embor.2008.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahmani M, Reese E, Dai Y, Bauer C, Payne SG, Dent P, et al. Coadministration of histone deacetylase inhibitors and perifosine synergistically induces apoptosis in human leukemia cells through akt and ERK1/2 inactivation and the generation of ceramide and reactive oxygen species. Cancer research. 2005;65(6):2422–2432. doi: 10.1158/0008-5472.CAN-04-2440. [DOI] [PubMed] [Google Scholar]
- Rao R, Nalluri S, Fiskus W, Savoie A, Buckley KM, Ha K, et al. Role of CAAT/enhancer binding protein homologous protein in panobinostat-mediated potentiation of bortezomib-induced lethal endoplasmic reticulum stress in mantle cell lymphoma cells. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010;16(19):4742–4754. doi: 10.1158/1078-0432.CCR-10-0529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao R, Nalluri S, Kolhe R, Yang Y, Fiskus W, Chen J, et al. Treatment with panobinostat induces glucose-regulated protein 78 acetylation and endoplasmic reticulum stress in breast cancer cells. Molecular cancer therapeutics. 2010;9(4):942–952. doi: 10.1158/1535-7163.MCT-09-0988. Electronic version. [DOI] [PubMed] [Google Scholar]
- Rascle A, Johnston JA, Amati B. Deacetylase activity is required for recruitment of the basal transcription machinery and transactivation by STAT5. Molecular and cellular biology. 2003;23(12):4162–4173. doi: 10.1128/MCB.23.12.4162-4173.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves R, Cserjesi P. Sodium butyrate induces new gene expression in friend erythroleukemic cells. The Journal of biological chemistry. 1979;254(10):4283–4290. [PubMed] [Google Scholar]
- Renner AG, Dos Santos C, Recher C, Bailly C, Créancier L, Kruczynski A, et al. Polo-like kinase 1 is overexpressed in acute myeloid leukemia and its inhibition preferentially targets the proliferation of leukemic cells. Blood. 2009;114(3):659–662. doi: 10.1182/blood-2008-12-195867. [DOI] [PubMed] [Google Scholar]
- Richardson PG, Mitsiades CS, Laubach JP, Hajek R, Spicka I, Dimopoulos MA, et al. Preclinical data and early clinical experience supporting the use of histone deacetylase inhibitors in multiple myeloma. Leukemia research. 2013;37(7):829–837. doi: 10.1016/j.leukres.2013.03.006. [DOI] [PubMed] [Google Scholar]
- Richardson PG, Schlossman RL, Alsina M, Weber DM, Coutre SE, Gasparetto C, et al. PANORAMA 2: Panobinostat in combination with bortezomib and dexamethasone in patients with relapsed and bortezomib-refractory myeloma. Blood. 2013;122(14):2331–2337. doi: 10.1182/blood-2013-01-481325. [DOI] [PubMed] [Google Scholar]
- Richon VM, Sandhoff TW, Rifkind RA, Marks PA. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(18):10014–10019. doi: 10.1073/pnas.180316197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter-Larrea JA, Robles EF, Fresquet V, Beltran E, Rullan AJ, Agirre X, et al. Reversion of epigenetically mediated BIM silencing overcomes chemoresistance in burkitt lymphoma. Blood. 2010;116(14):2531–2542. doi: 10.1182/blood-2010-02-268003. [DOI] [PubMed] [Google Scholar]
- Robert C, Rassool FV. HDAC inhibitors: Roles of DNA damage and repair. Advances in Cancer Research. 2012;116:87–129. doi: 10.1016/B978-0-12-394387-3.00003-3. [DOI] [PubMed] [Google Scholar]
- Robert T, Vanoli F, Chiolo I, Shubassi G, Bernstein KA, Rothstein R, et al. HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature. 2011;471(7336):74–79. doi: 10.1038/nature09803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AW, Seymour JF, Brown JR, Wierda WG, Kipps TJ, Khaw SL, et al. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: Results of a phase I study of navitoclax in patients with relapsed or refractory disease. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2012;30(5):488–496. doi: 10.1200/JCO.2011.34.7898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosato RR, Almenara JA, Grant S. The histone deacetylase inhibitor MS-275 promotes differentiation or apoptosis in human leukemia cells through a process regulated by generation of reactive oxygen species and induction of p21CIP1/WAF1 1. Cancer research. 2003;63(13):3637–3645. [PubMed] [Google Scholar]
- Rosato RR, Almenara JA, Kolla SS, Maggio SC, Coe S, Gimenez MS, et al. Mechanism and functional role of XIAP and mcl-1 down-regulation in flavopiridol/vorinostat antileukemic interactions. Molecular cancer therapeutics. 2007;6(2):692–702. doi: 10.1158/1535-7163.MCT-06-0562. [DOI] [PubMed] [Google Scholar]
- Rosato RR, Almenara JA, Maggio SC, Coe S, Atadja P, Dent P, et al. Role of histone deacetylase inhibitor-induced reactive oxygen species and DNA damage in LAQ-824/fludarabine antileukemic interactions. Molecular cancer therapeutics. 2008;7(10):3285–3297. doi: 10.1158/1535-7163.MCT-08-0385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosato RR, Grant S. Histone deacetylase inhibitors: Insights into mechanisms of lethality. Expert opinion on therapeutic targets. 2005;9(4):809–824. doi: 10.1517/14728222.9.4.809. [DOI] [PubMed] [Google Scholar]
- Rosato RR, Kolla SS, Hock SK, Almenara JA, Patel A, Amin S, et al. Histone deacetylase inhibitors activate NF-kappaB in human leukemia cells through an ATM/NEMO-related pathway. The Journal of biological chemistry. 2010;285(13):10064–10077. doi: 10.1074/jbc.M109.095208. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Rosato RR, Maggio SC, Almenara JA, Payne SG, Atadja P, Spiegel S, et al. The histone deacetylase inhibitor LAQ824 induces human leukemia cell death through a process involving XIAP down-regulation, oxidative injury, and the acid sphingomyelinase-dependent generation of ceramide. Molecular pharmacology. 2006;69(1):216–225. doi: 10.1124/mol.105.017145. [DOI] [PubMed] [Google Scholar]
- Roychowdhury S, Baiocchi RA, Vourganti S, Bhatt D, Blaser BW, Freud AG, et al. Selective efficacy of depsipeptide in a xenograft model of epstein-barr virus-positive lymphoproliferative disorder. Journal of the National Cancer Institute. 2004;96(19):1447–1457. doi: 10.1093/jnci/djh271. [DOI] [PubMed] [Google Scholar]
- Ruefli AA, Ausserlechner MJ, Bernhard D, Sutton VR, Tainton KM, Kofler R, et al. The histone deacetylase inhibitor and chemotherapeutic agent suberoylanilide hydroxamic acid (SAHA) induces a cell-death pathway characterized by cleavage of bid and production of reactive oxygen species. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(19):10833–10838. doi: 10.1073/pnas.191208598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito Y, Suzuki H, Tsugawa H, Nakagawa I, Matsuzaki J, Kanai Y, et al. Chromatin remodeling at alu repeats by epigenetic treatment activates silenced microRNA-512-5p with downregulation of mcl-1 in human gastric cancer cells. Oncogene. 2009;28(30):2738–2744. doi: 10.1038/onc.2009.140. [DOI] [PubMed] [Google Scholar]
- Sallmyr A, Fan J, Rassool FV. Genomic instability in myeloid malignancies: Increased reactive oxygen species (ROS), DNA double strand breaks (DSBs) and error-prone repair. Cancer letters. 2008;270(1):1–9. doi: 10.1016/j.canlet.2008.03.036. [DOI] [PubMed] [Google Scholar]
- Sampath D, Liu C, Vasan K, Sulda M, Puduvalli VK, Wierda WG, et al. Histone deacetylases mediate the silencing of miR-15a, miR-16, and miR-29b in chronic lymphocytic leukemia. Blood. 2012;119(5):1162–1172. doi: 10.1182/blood-2011-05-351510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Gonzalez B, Yang H, Bueso-Ramos C, Hoshino K, Quintas-Cardama A, Richon VM, et al. Antileukemia activity of the combination of an anthracycline with a histone deacetylase inhibitor. Blood. 2006;108(4):1174–1182. doi: 10.1182/blood-2005-09-008086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanda T, Okamoto T, Uchida Y, Nakagawa H, Iida S, Kayukawa S, et al. Proteome analyses of the growth inhibitory effects of NCH-51, a novel histone deacetylase inhibitor, on lymphoid malignant cells. Leukemia. 2007;21(11):2344–2353. doi: 10.1038/sj.leu.2404902. [DOI] [PubMed] [Google Scholar]
- Santo L, Hideshima T, Cirstea D, Bandi M, Nelson EA, Gorgun G, et al. Antimyeloma activity of a multitargeted kinase inhibitor, AT9283, via potent aurora kinase and STAT3 inhibition either alone or in combination with lenalidomide. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011;17(10):3259–3271. doi: 10.1158/1078-0432.CCR-10-3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, et al. Human CtIP promotes DNA end resection. Nature. 2007;450(7169):509–514. doi: 10.1038/nature06337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schimmer AD. Novel therapies targeting the apoptosis pathway for the treatment of acute myeloid leukemia. Current treatment options in oncology. 2007;8(4):277–286. doi: 10.1007/s11864-007-0037-x. [DOI] [PubMed] [Google Scholar]
- Schrump DS. Cytotoxicity mediated by histone deacetylase inhibitors in cancer cells: Mechanisms and potential clinical implications. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009;15(12):3947–3957. doi: 10.1158/1078-0432.CCR-08-2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao W, Growney JD, Feng Y, O'Connor G, Pu M, Zhu W, et al. Activity of deacetylase inhibitor panobinostat (LBH589) in cutaneous T-cell lymphoma models: Defining molecular mechanisms of resistance. International journal of cancer Journal international du cancer. 2010;127(9):2199–2208. doi: 10.1002/ijc.25218. [DOI] [PubMed] [Google Scholar]
- Sharma GG, So S, Gupta A, Kumar R, Cayrou C, Avvakumov N, et al. MOF and histone H4 acetylation at lysine 16 are critical for DNA damage response and double-strand break repair. Molecular and cellular biology. 2010;30(14):3582–3595. doi: 10.1128/MCB.01476-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiozawa K, Nakanishi T, Tan M, Fang H, Wang W, Edelman MJ, et al. Preclinical studies of vorinostat (suberoylanilide hydroxamic acid) combined with cytosine arabinoside and etoposide for treatment of acute leukemias. Clinical Cancer Research. 2009;15(5):1698–1707. doi: 10.1158/1078-0432.CCR-08-1587. [DOI] [PubMed] [Google Scholar]
- Shubassi G, Robert T, Vanoli F, Minucci S, Foiani M. Acetylation: A novel link between double-strand break repair and autophagy. Cancer research. 2012;72(6):1332–1335. doi: 10.1158/0008-5472.CAN-11-3172. [DOI] [PubMed] [Google Scholar]
- Singleton BK, Torres-Arzayus MI, Rottinghaus ST, Taccioli GE, Jeggo PA. The C terminus of Ku80 activates the DNA-dependent protein kinase catalytic subunit. Molecular and cellular biology. 1999;19(5):3267–3277. doi: 10.1128/mcb.19.5.3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits VA, Klompmaker R, Arnaud L, Rijksen G, Nigg EA, Medema RH. Polo-like kinase-1 is a target of the DNA damage checkpoint. Nature cell biology. 2000;2(9):672–676. doi: 10.1038/35023629. [DOI] [PubMed] [Google Scholar]
- Sonoda E, Zhao GY, Kohzaki M, Dhar PK, Kikuchi K, Redon C, et al. Collaborative roles of gammaH2AX and the Rad51 paralog Xrcc3 in homologous recombinational repair. DNA repair. 2007;6(3):280–292. doi: 10.1016/j.dnarep.2006.10.025. [DOI] [PubMed] [Google Scholar]
- Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458(7239):732–736. doi: 10.1038/nature07884. [DOI] [PubMed] [Google Scholar]
- Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nature medicine. 2013;19(2):202–208. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- Stathis A, Hotte SJ, Chen EX, Hirte HW, Oza AM, Moretto P, et al. Phase I study of decitabine in combination with vorinostat in patients with advanced solid tumors and non-hodgkin's lymphomas. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011;17(6):1582–1590. doi: 10.1158/1078-0432.CCR-10-1893. [DOI] [PubMed] [Google Scholar]
- Stevens FE, Beamish H, Warrener R, Gabrielli B. Histone deacetylase inhibitors induce mitotic slippage. Oncogene. 2008a;27(10):1345–1354. doi: 10.1038/sj.onc.1210779. [DOI] [PubMed] [Google Scholar]
- Stevens FE, Beamish H, Warrener R, Gabrielli B. Histone deacetylase inhibitors induce mitotic slippage. Oncogene. 2008b;27(10):1345–1354. doi: 10.1038/sj.onc.1210779. [DOI] [PubMed] [Google Scholar]
- Subramanian C, Opipari AW, Jr, Bian X, Castle VP, Kwok RP. Ku70 acetylation mediates neuroblastoma cell death induced by histone deacetylase inhibitors. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(13):4842–4847. doi: 10.1073/pnas.0408351102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto K, Sasaki M, Isobe Y, Tsutsui M, Suto H, Ando J, et al. Hsp90-inhibitor geldanamycin abrogates G2 arrest in p53-negative leukemia cell lines through the depletion of Chk1. Oncogene. 2008;27(22):3091–3101. doi: 10.1038/sj.onc.1210978. [DOI] [PubMed] [Google Scholar]
- Sun Y, Jiang X, Xu Y, Ayrapetov MK, Moreau LA, Whetstine JR, et al. Histone H3 methylation links DNA damage detection to activation of the tumour suppressor Tip60. Nature cell biology. 2009;11(11):1376–1382. doi: 10.1038/ncb1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Xu Y, Roy K, Price BD. DNA damage-induced acetylation of lysine 3016 of ATM activates ATM kinase activity. Molecular and cellular biology. 2007;27(24):8502–8509. doi: 10.1128/MCB.01382-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swords RT, Kelly KR, Smith PG, Garnsey JJ, Mahalingam D, Medina E, et al. Inhibition of NEDD8-activating enzyme: A novel approach for the treatment of acute myeloid leukemia. Blood. 2010a;115(18):3796–3800. doi: 10.1182/blood-2009-11-254862. [DOI] [PubMed] [Google Scholar]
- Swords RT, Erba HP, DeAngelo DJ, Smith PG, Pickard MD, Dezube BJ, et al. The novel, investigational NEDD8-activating enzyme inhibitor MLN4924 in adult patients with acute myeloid leukemia (AML) or high-grade myelodysplastic syndromes (MDS): A phase 1 study. ASH Annual Meeting Abstracts. 2010b;116(21):658. [Google Scholar]
- Takaki T, Trenz K, Costanzo V, Petronczki M. Polo-like kinase 1 reaches beyond mitosis--cytokinesis, DNA damage response, and development. Current opinion in cell biology. 2008;20(6):650–660. doi: 10.1016/j.ceb.2008.10.005. [DOI] [PubMed] [Google Scholar]
- Tan J, Zhuang L, Jiang X, Yang KK, Karuturi KM, Yu Q. Apoptosis signal-regulating kinase 1 is a direct target of E2F1 and contributes to histone deacetylase inhibitor-induced apoptosis through positive feedback regulation of E2F1 apoptotic activity. The Journal of biological chemistry. 2006;281(15):10508–10515. doi: 10.1074/jbc.M512719200. [DOI] [PubMed] [Google Scholar]
- Tanaka A, Tanizawa H, Sriswasdi S, Iwasaki O, Chatterjee AG, Speicher DW, et al. Epigenetic regulation of condensin-mediated genome organization during the cell cycle and upon DNA damage through histone H3 lysine 56 acetylation. Molecular cell. 2012;48(4):532–546. doi: 10.1016/j.molcel.2012.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka R, Squires MS, Kimura S, Yokota A, Nagao R, Yamauchi T, et al. Activity of the multitargeted kinase inhibitor, AT9283, in imatinib-resistant BCR-ABL-positive leukemic cells. Blood. 2010;116(12):2089–2095. doi: 10.1182/blood-2009-03-211466. [DOI] [PubMed] [Google Scholar]
- Terui T, Murakami K, Takimoto R, Takahashi M, Takada K, Murakami T, et al. Induction of PIG3 and NOXA through acetylation of p53 at 320 and 373 lysine residues as a mechanism for apoptotic cell death by histone deacetylase inhibitors. Cancer research. 2003;63(24):8948–8954. [PubMed] [Google Scholar]
- Testa U, Riccioni R. Deregulation of apoptosis in acute myeloid leukemia. Haematologica. 2007;92(1):81–94. doi: 10.3324/haematol.10279. [DOI] [PubMed] [Google Scholar]
- Thompson R, Eastman A. The cancer therapeutic potential of Chk1 inhibitors: How mechanistic studies impact on clinical trial design. British journal of clinical pharmacology. 2013;76(3):358–369. doi: 10.1111/bcp.12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Three more drugs judged ‘breakthroughs’. Cancer discovery. 2013;3(11) OF2-8290.CDNB2013-142. Epub 2013 Oct 3. [Google Scholar]
- Touzeau C, Dousset C, Le Gouill S, Sampath D, Leverson JD, Souers AJ, et al. The bcl-2 specific BH3 mimetic ABT-199: A promising targeted therapy for t(11;14) multiple myeloma. Leukemia. 2014;28(1):210–212. doi: 10.1038/leu.2013.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse AN, Carvajal R, Schwartz GK. Targeting checkpoint kinase 1 in cancer therapeutics. Clinical cancer research : an official journal of the American Association for Cancer Research. 2007;13(7):1955–1960. doi: 10.1158/1078-0432.CCR-06-2793. [DOI] [PubMed] [Google Scholar]
- Tse AN, Sheikh TN, Alan H, Chou TC, Schwartz GK. 90-kDa heat shock protein inhibition abrogates the topoisomerase I poison-induced G2/M checkpoint in p53-null tumor cells by depleting Chk1 and Wee1. Molecular pharmacology. 2009;75(1):124–133. doi: 10.1124/mol.108.050807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, et al. ABT-263: A potent and orally bioavailable bcl-2 family inhibitor. Cancer research. 2008;68(9):3421–3428. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- Ungerstedt JS, Sowa Y, Xu WS, Shao Y, Dokmanovic M, Perez G, et al. Role of thioredoxin in the response of normal and transformed cells to histone deacetylase inhibitors. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(3):673–678. doi: 10.1073/pnas.0408732102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaillant F, Merino D, Lee L, Breslin K, Pal B, Ritchie ME, et al. Targeting BCL-2 with the BH3 mimetic ABT-199 in estrogen receptor-positive breast cancer. Cancer cell. 2013;24(1):120–129. doi: 10.1016/j.ccr.2013.06.002. [DOI] [PubMed] [Google Scholar]
- van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE, et al. The BH3 mimetic ABT-737 targets selective bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if mcl-1 is neutralized. Cancer cell. 2006;10(5):389–399. doi: 10.1016/j.ccr.2006.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberg CJ, Cory S. ABT-199, a new bcl-2-specific BH3 mimetic, has in vivo efficacy against aggressive myc-driven mouse lymphomas without provoking thrombocytopenia. Blood. 2013;121(12):2285–2288. doi: 10.1182/blood-2013-01-475855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaziri SA, Hill J, Chikamori K, Grabowski DR, Takigawa N, Chawla-Sarkar M, et al. Sensitization of DNA damage-induced apoptosis by the proteasome inhibitor PS-341 is p53 dependent and involves target proteins 14-3-3sigma and survivin. Molecular cancer therapeutics. 2005;4(12):1880–1890. doi: 10.1158/1535-7163.MCT-05-0222. [DOI] [PubMed] [Google Scholar]
- Vogler M, Dinsdale D, Dyer MJ, Cohen GM. ABT-199 selectively inhibits BCL2 but not BCL2L1 and efficiently induces apoptosis of chronic lymphocytic leukaemic cells but not platelets. British journal of haematology. 2013;163(1):139–142. doi: 10.1111/bjh.12457. [DOI] [PubMed] [Google Scholar]
- Vrana JA, Decker RH, Johnson CR, Wang Z, Jarvis WD, Richon VM, et al. Induction of apoptosis in U937 human leukemia cells by suberoylanilide hydroxamic acid (SAHA) proceeds through pathways that are regulated by bcl-2/Bcl-XL, cjun, and p21CIP1, but independent of p53. Oncogene. 1999;18(50):7016–7025. doi: 10.1038/sj.onc.1203176. [DOI] [PubMed] [Google Scholar]
- Walker JR, Corpina RA, Goldberg J. Structure of the ku heterodimer bound to DNA and its implications for double-strand break repair. Nature. 2001;412(6847):607–614. doi: 10.1038/35088000. [DOI] [PubMed] [Google Scholar]
- Wang H, Zhou W, Zheng Z, Zhang P, Tu B, He Q, et al. The HDAC inhibitor depsipeptide transactivates the p53/p21 pathway by inducing DNA damage. DNA repair. 2012;11(2):146–156. doi: 10.1016/j.dnarep.2011.10.014. [DOI] [PubMed] [Google Scholar]
- Wang M, Medeiros BC, Erba HP, DeAngelo DJ, Giles FJ, Swords RT. Targeting protein neddylation: A novel therapeutic strategy for the treatment of cancer. Expert opinion on therapeutic targets. 2011;15(3):253–264. doi: 10.1517/14728222.2011.550877. [DOI] [PubMed] [Google Scholar]
- Wang Y, Fiskus W, Chong DG, Buckley KM, Natarajan K, Rao R, et al. Cotreatment with panobinostat and JAK2 inhibitor TG101209 attenuates JAK2V617F levels and signaling and exerts synergistic cytotoxic effects against human myeloproliferative neoplastic cells. Blood. 2009;114(24):5024–5033. doi: 10.1182/blood-2009-05-222133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warrener R, Beamish H, Burgess A, Waterhouse NJ, Giles N, Fairlie D, et al. Tumor cell-selective cytotoxicity by targeting cell cycle checkpoints. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2003;17(11):1550–1552. doi: 10.1096/fj.02-1003fje. [DOI] [PubMed] [Google Scholar]
- Watanabe T. Investigational histone deacetylase inhibitors for non-hodgkin lymphomas. Expert opinion on investigational drugs. 2010;19(9):1113–1127. doi: 10.1517/13543784.2010.504710. [DOI] [PubMed] [Google Scholar]
- Wei Y, Kadia T, Tong W, Zhang M, Jia Y, Yang H, et al. The combination of a histone deacetylase inhibitor with the bcl-2 homology domain-3 mimetic GX15-070 has synergistic antileukemia activity by activating both apoptosis and autophagy. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010;16(15):3923–3932. doi: 10.1158/1078-0432.CCR-10-0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegmans AP, Alsop AE, Bots M, Cluse LA, Williams SP, Banks KM, et al. Deciphering the molecular events necessary for synergistic tumor cell apoptosis mediated by the histone deacetylase inhibitor vorinostat and the BH3 mimetic ABT-737. Cancer research. 2011;71(10):3603–3615. doi: 10.1158/0008-5472.CAN-10-3289. [DOI] [PubMed] [Google Scholar]
- Willis SN, Chen L, Dewson G, Wei A, Naik E, Fletcher JI, et al. Proapoptotic bak is sequestered by mcl-1 and bcl-xL, but not bcl-2, until displaced by BH3-only proteins. Genes & development. 2005;19(11):1294–1305. doi: 10.1101/gad.1304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wissing MD, Mendonca J, Kortenhorst MS, Kaelber NS, Gonzalez M, Kim E, et al. Targeting prostate cancer cell lines with polo-like kinase 1 inhibitors as a single agent and in combination with histone deacetylase inhibitors. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2013;27(10):4279–4293. doi: 10.1096/fj.12-222893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xargay-Torrent S, Lopez-Guerra M, Saborit-Villarroya I, Rosich L, Campo E, Roue G, et al. Vorinostat-induced apoptosis in mantle cell lymphoma is mediated by acetylation of proapoptotic BH3-only gene promoters. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011;17(12):3956–3968. doi: 10.1158/1078-0432.CCR-10-3412. [DOI] [PubMed] [Google Scholar]
- Xu W, Ngo L, Perez G, Dokmanovic M, Marks PA. Intrinsic apoptotic and thioredoxin pathways in human prostate cancer cell response to histone deacetylase inhibitor. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(42):15540–15545. doi: 10.1073/pnas.0607518103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaseen A, Chen S, Hock S, Rosato R, Dent P, Dai Y, et al. Resveratrol sensitizes acute myelogenous leukemia cells to histone deacetylase inhibitors through reactive oxygen species-mediated activation of the extrinsic apoptotic pathway. Molecular pharmacology. 2012;82(6):1030–1041. doi: 10.1124/mol.112.079624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi C, Ma M, Ran L, Zheng J, Tong J, Zhu J, et al. Science. 6080. Vol. 336. New York, N.Y.: 2012. Function and molecular mechanism of acetylation in autophagy regulation; pp. 474–477. [DOI] [PubMed] [Google Scholar]
- Yu C, Rahmani M, Almenara J, Subler M, Krystal G, Conrad D, et al. Histone deacetylase inhibitors promote STI571-mediated apoptosis in STI571-sensitive and -resistant Bcr/Abl+ human myeloid leukemia cells. Cancer research. 2003;63(9):2118–2126. [PubMed] [Google Scholar]
- Yu C, Rahmani M, Conrad D, Subler M, Dent P, Grant S. The proteasome inhibitor bortezomib interacts synergistically with histone deacetylase inhibitors to induce apoptosis in Bcr/Abl+ cells sensitive and resistant to STI571. Blood. 2003;102(10):3765–3774. doi: 10.1182/blood-2003-03-0737. [DOI] [PubMed] [Google Scholar]
- Yu C, Rahmani M, Dent P, Grant S. The hierarchical relationship between MAPK signaling and ROS generation in human leukemia cells undergoing apoptosis in response to the proteasome inhibitor bortezomib. Experimental cell research. 2004;295(2):555–566. doi: 10.1016/j.yexcr.2004.02.001. [DOI] [PubMed] [Google Scholar]
- Yu X, Guo ZS, Marcu MG, Neckers L, Nguyen DM, Chen GA, et al. Modulation of p53, ErbB1, ErbB2, and raf-1 expression in lung cancer cells by depsipeptide FR901228. Journal of the National Cancer Institute. 2002;94(7):504–513. doi: 10.1093/jnci/94.7.504. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Tan J, Zhuang L, Jiang X, Liu ET, Yu Q. Inhibitors of histone deacetylases target the rb-E2F1 pathway for apoptosis induction through activation of proapoptotic protein bim. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(44):16090–16095. doi: 10.1073/pnas.0505585102. [DOI] [PMC free article] [PubMed] [Google Scholar]