

Abstract
N-acylethanolamine acid amidase (NAAA) is a cysteine amidase that hydrolyzes saturated or monounsaturated fatty acid ethanolamides, such as palmitoylethanolamide (PEA) and oleoylethanolamide (OEA). PEA has been shown to exert analgesic and anti-inflammatory effects by engaging peroxisome proliferator-activated receptor-α. Like other fatty acid ethanolamides, PEA is not stored in cells, but produced on demand from cell membrane precursors, and its actions are terminated by intracellular hydrolysis by either fatty acid amide hydrolase or NAAA. Endogenous levels of PEA and OEA have been shown to decrease during inflammation. Modulation of the tissue levels of PEA by inhibition of enzymes responsible for the breakdown of this lipid mediator may represent therefore a new therapeutic strategy for the treatment of pain and inflammation. While a large number of inhibitors of fatty acid amide hydrolase have been discovered, few compounds have been reported to inhibit NAAA activity. Here, we describe the most representative NAAA inhibitors and briefly highlight their pharmacological profile. A recent study has shown that a NAAA inhibitor attenuated heat hyperalgesia and mechanical allodynia caused by local inflammation or nerve damage in animal models of pain and inflammation. This finding encourages further exploration of the pharmacology of NAAA inhibitors.
Keywords: N-acylethanolamine acid amidase, fatty acid ethanolamides, palmitoylethanolamide, pain, inflammation, NAAA inhibitors
1. Introduction
The amides of long-chain fatty acids with ethanolamine, or fatty acid ethanolamides (FAEs), are a family of bioactive lipids that participate in the control of multiple physiological functions, including pain and inflammation.[1-4] Polyunsaturated FAEs such as arachidonoylethanolamide (anandamide, Fig. 1) are endogenous agonists for G protein-coupled cannabinoid receptors and participate in the control of stress-coping responses and pain initiation.[1,5] On the other hand, monounsaturated and saturated FAEs, such as oleoylethanolamide (OEA, Fig. 1) and palmitoylethanolamide (PEA, Fig. 1), are potent or moderately potent agonists of the peroxisome proliferator-activated receptor-α (PPAR-α), a member of the nuclear receptor superfamily, which is responsible for most of their analgesic and anti-inflammatory properties. [4,6,7]
Fig. 1.
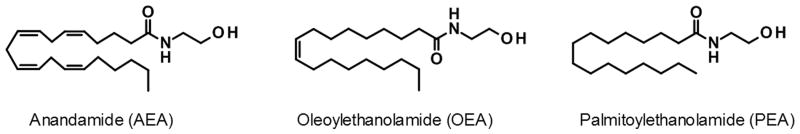
Chemical structures of anandamide, oleoylethanolamide, and palmitoylethanolamide.
FAEs are not stored in cells, but rather are produced on demand from cell membrane precursors.[8-10] OEA and PEA are generated in many mammalian tissues, including neurons[11] and innate immune cells,[12] where a selective phospholipase, N-acylphosphatidylethanolamine phospholipase D (NAPE-PLD) releases them by cleaving their membrane precursor, N-acylphosphatidylethanolamine.[13] The actions of these lipid messengers are terminated by enzyme-mediated hydrolysis, which is catalyzed by two known intracellular lipid amidases: N-acylethanolamine acid amidase (NAAA, previously referred to as N-acylethanolamine hydrolyzing acid amidase)[14-16] and fatty acid amide hydrolase (FAAH).[17,18] These enzymes share the ability to cleave lipid amide bonds, but differ in primary structure, substrate selectivity, and cellular localization. NAAA is a cysteine hydrolase that belongs to the N-terminal nucleophile (Ntn) family of enzymes,[15,16,19] and bears a significant degree of sequence homology with the choloylglycine hydrolases, which share the ability to cleave non-peptide amide bonds.[20] NAAA displays a strong preference for saturated FAEs such as PEA,[15] while FAAH, a member of the amidase signature family of serine hydrolases, displays broader substrate selectivity, but hydrolyzes preferentially monounsaturated and polyunsaturated FAEs such as anandamide and OEA.[17] Moreover, NAAA seems to be mainly localized to the lysosomal compartment of macrophages,[21] whereas FAAH is a membrane-bound enzyme that is found on the outer face of mitochondria and endoplasmic reticulum of most mammalian cells.[22]
Like other Ntn enzymes, such as acid ceramidase, a lysosomal enzyme that hydrolyses ceramide to sphingosine and fatty acid,[23,24] NAAA is activated by auto-proteolysis, which occurs at acidic pH and generates a catalytically competent form of the enzyme.[25] Comparison of the primary structure of NAAA with those of the other members of the choloylglycine hydrolase family followed by site-directed mutagenesis experiments have unequivocally identified cysteine 131 (Cys-131) in mice, or cysteine 126 (Cys-126) in humans, as the catalytic residue responsible for both auto-proteolysis and FAE hydrolysis.[26,27] The proposed mechanism of amide bond hydrolysis by Ntn enzymes consists in the attack of the catalytic N-terminal residue on the amide with formation of an acyl enzyme, followed by acyl enzyme hydrolysis with regeneration of the catalytically competent enzyme.[28,29] According to this mechanism, the thiol group of the catalytic cysteine of NAAA would react with substrate with the formation of a thioester bond. Acylation of Cys-126 of human NAAA by β-lactones, a class of NAAA inhibitors, was recently demonstrated by mass spectrometry experiments.[30,31]
The pharmacology of PEA has been extensively investigated.[32] The compound inhibits peripheral inflammation and mast cell degranulation,[33,34] and exerts strong antinociceptive effects in rat and mouse models of acute and chronic pain.[1,34-36] Moreover, in mice it suppresses pain behaviors induced by tissue injury, nerve damage or inflammation.[4] These properties are dependent on PPAR-α activation, since they are absent in PPAR-α-deficient mice, blocked by PPAR-α antagonists and mimicked by synthetic PPAR-α agonists.[4,37] The finding that PEA might attenuate skin inflammation and neuropathic pain in humans is highly significant and warrants additional clinical investigation.[38,39]
Endogenous levels of PEA and OEA (but not anandamide) undergo marked changes during inflammation. Stimulation with pro-inflammatory agents such as lipopolysaccharide (LPS) or carrageenan decreases PEA (and OEA) content in various cells and tissues of rodents.[3,27,40-42] The observation that synovial fluid from patients with rheumatoid arthritis and osteoarthritis contains lower amounts of PEA adds clinical relevance to previous findings.[43] The decrease in the cellular levels of PEA and OEA following an inflammatory stimulus is reversed by pharmacological blockade of NAAA-mediated FAE hydrolysis.[27,44,45] Moreover, protecting endogenous PEA and OEA from NAAA-catalyzed degradation in vivo has recently been reported to attenuate hyperalgesic and allodynic states elicited in mice and rats by local inflammation or nerve damage.[46]
The previous findings suggest that sustaining the levels of PEA by inhibition of intracellular NAAA activity might represent a novel approach to control inflammation and pain. The search for NAAA inhibitors is still in its infancy, and has developed mainly through two approaches. The first one relies on the modification of the structure of one of the natural substrates, PEA, with the goal of discovering derivatives that are able to inhibit enzyme activity while showing selectivity versus other lipid amide-hydrolyzing enzymes, such as FAAH. The second approach focuses on compounds containing chemical functionalities that are known to react with the thiol group of cysteine, referred to as “cysteine warheads”, in order to inactivate the catalytic cysteine in the catalytically-competent form of the enzyme, thus inhibiting its activity.
To date, a number of compounds originating from the two approaches have been discovered to inhibit NAAA activity with various potencies and selectivity. In this respect, it is important to point out that assay conditions should always be considered when comparing compounds' potencies. In fact, median inhibitory concentration (IC50) values may change depending on whether the compounds are tested in cell-free, homogenates, or whole-cell assays. This review will describe the most representative inhibitors and will briefly highlight their pharmacological profile.
2. Analogues of PEA
The search for inhibitors of NAAA activity relied at first on the structure of the natural substrate, PEA. Analogues of PEA were prepared by modification or replacement of the amidic function. Different amides and various retroamides, esters, and retroesters of palmitic acid (Fig. 2) were synthesized and evaluated for their ability to inhibit NAAA activity and their selectivity versus FAAH.[47,48]
Fig. 2.

(A) Chemical structures of amides, retro-amides, esters, and retro-esters of palmitic acid. (B) Chemical structures of selected NAAA inhibitors.
The palmitic acid retro-amides N-pentadecylbenzamide (1) and N-pentadecylcyclohexancarboxamide (2) were among the first potent and selective inhibitors of NAAA activity to be identified.[48] These compounds inhibited the activity of rat NAAA, from lung preparations, with IC50 values of 8.3 μM and 4.5 μM, respectively, and did not inhibit rat FAAH at concentrations up to 100 μM. The mechanism of NAAA inhibition by compound 2 was determined to be reversible and non-competitive, based on kinetic analyses. Compound 2 was shown to inhibit NAAA activity in rat alveolar macrophages, although preincubation of cells with high micromolar concentration of the inhibitor was required to achieve significant enzyme inhibition.
Amines with a long alkyl chain were reported to inhibit rat NAAA activity.[49] In particular, alkyl amines with a chain length of 14 or 15 carbon atoms showed the strongest activity, with the pentadecylamine 3 displaying IC50 of 5.7 μM. Esters of glycine with linear alkyl alcohols of 12 to 16 carbon atoms also inhibited NAAA activity. As observed with alkyl amines, the potency of glycine esters was modulated by the length of the alkyl chain, with highest inhibition of NAAA activity observed with the tridecyl ester 4 (IC50 = 11.8 μM). Compounds 3 and 4 inhibited NAAA with a competitive mechanism, as determined by kinetic studies. The amine 3 and the glycine ester 4 exist in solution as protonated or non-protonated species depending on the pH, and these species can interact differently with biomolecules. The selectivity of 3 and 4 against rat FAAH was therefore assessed at pH 5, the optimal pH of NAAA, and at pH 8.5, which is within the optimal range for FAAH. No inhibition of FAAH activity was observed at pH 8.5 for both compounds at 100 μM concentration, while at the same concentration only 25% and 7% decrease in FAAH activity was caused by compound 3 and 4, respectively, at pH 5.
A number of amides, retroamides, esters, retroesters and carbamates of palmitic acid were investigated to further explore PEA analogues as NAAA inhibitors and possibly obtain more potent compounds.[50] Among those compounds, only the esters of palmitic acid with cyclobutanol (5) and cyclopentanol (6) significantly inhibited the activity of human NAAA in membranes from HEK-293 cells stably overexpressing the enzyme (HEK-NAAA). When tested at 50 μM concentration, compounds 5 and 6 displayed 41% and 85% inhibition of NAAA activity, respectively. Kinetic analyses showed that cyclopentyl hexadecanoate 6 behaved as a competitive inhibitor of NAAA with an IC50 of 10 μM, and did not exhibit inhibitory activity on FAAH.[50] In intact HEK-NAAA cells, compound 6 increased the levels of PEA without influencing the levels of anandamide and OEA, thus suggesting that it inhibited NAAA activity.[51] Further characterization of compound 6 was done in a model of chronic inflammation in rats, carrageenan-induced granuloma,[41] in which PEA levels are significantly reduced during inflammation. Local administration of cyclopentyl hexadecanoate, together with carrageenan, led to a reduction in inflammation as observed with PEA given at a comparable dose.[51]
A series of PEA derivatives designed starting from N-pentadecylcyclohexancarboxamide, 2, led to the identification of 1-hexadecanoylpyrrolidine, 7, as a weak inhibitor of the activity of both recombinant rat NAAA (IC50 = 25 μM) and rat FAAH (IC50 = 21.8 μM) obtained from HEK-293 cells overexpressing those enzymes.[45] This compound was used as the starting point to discover additional inhibitors. Substitution of the pyrrolidine ring with other cyclic moieties resulted in a complete loss of inhibitory activity, while replacement of palmitic acid with 3-(p-biphenyl)propionic acid led to compound 8, which inhibited NAAA activity with an IC50 of 2.12 μM and showed only 30% inhibition of rat FAAH activity at 100 μM. Compound 8 had no inhibitory effects on the lipid-hydrolyzing enzymes monoacylglycerol lipase (MGL) and N-acylsphingosine amidohydrolase (ASAH). Kinetic analyses demonstrated that compound 8 acted through a competitive mechanism. The ability of compound 8 to inhibit NAAA activity in cells was evaluated in the RAW264.7 macrophage-like cell line. Exposure of these cells to LPS is known to result in a significant decrease in cellular PEA levels. Compound 8 was able to counteract the LPS-induced PEA reduction in RAW264.7 cells.
The compound 1-isothiocyanatopentadecane (9, AM9023) was recently reported as a potent and competitive inhibitor of purified human NAAA activity with IC50 of 0.35 μM, as measured with a fluorescent assay, and IC50 of 0.6 μM obtained using a radioactivity-based assay.[31] Despite the presence of an isothiocyanate group, which was expected to react irreversibly with the cysteine in the enzyme active site, AM9023 was shown to be a reversible inhibitor. AM9023 was selective for human NAAA as compared to human MGL and rat FAAH, showing IC50 > 10 μM on each of them.
3. Beta-lactone derivatives
The elucidation of the primary structure of NAAA and its classification as a cysteine hydrolase belonging to the N-terminal nucleophile (Ntn) family of enzymes oriented another line of research of NAAA inhibitors towards compounds containing chemical groups susceptible to nucleophilic attack by the thiol group of the catalytic cysteine. The screening of a series of molecules containing cysteine warheads led to the identification of the serine-derived β-lactone (S)-2-oxo-3-oxetanyl-carbamic acid benzyl ester (10, Fig. 3), an inhibitor of hepatitis A virus 3C protease,[52] as a weak inhibitor of native rat lung NAAA activity (IC50 = 2.96 μM). Structure–activity relationship (SAR) studies confirmed that the ability of 10 to inhibit NAAA depended on the β-lactone ring, rather than the carbamate fragment, because analogues lacking the β-lactone moiety were devoid of inhibitory activity.[27,44] Replacement of the carbamate group with an amide yielded the compound N-[(3S)-2-oxo-3-oxetanyl]-3-phenylpropanamide (11, (S)-OOPP, Fig. 3, IC50 = 0.42 M), which was significantly more potent than 10 at inhibiting NAAA. Kinetic analyses revealed that 11 blocked NAAA through a noncompetitive mechanism. In addition, compound 11 did not inhibit rat FAAH or other lipid hydrolases, such as MGL and diacylglycerol lipase type-α, and showed some selectivity vs. acid ceramidase (IC50 = 10.9 μM), a cysteine amidase that is both structurally and functionally related to NAAA.[16] The selectivity profile of 11 allowed to use this compound as a tool to investigate the effect of NAAA inhibition on inflammatory cells. When incubated with RAW264.7 macrophages stimulated with LPS, 11 inhibited NAAA activity in these cells, and blocked the LPS-induced reduction of PEA levels.[27] The presence of a chiral center in 11 allowed to explore the stereochemical requirements for enzyme recognition. The (S) stereochemistry at the α-carbon of the β-lactone ring turned out to be important for potent NAAA inhibition, as the enantiomer of compound 11, (R)-OOPP (12, Fig. 3, IC50 = 6.0 μM), showed a markedly diminished potency and did not inhibit NAAA activity when incubated with RAW264.7 macrophages stimulated with LPS.
Fig. 3.

(A) General structures of serine-derived β-lactones. (B) Chemical structures of selected serine-derived β-lactone NAAA inhibitors.
Following the identification of the β-lactones 10 and 11 as NAAA inhibitors, this chemical class was extensively investigated to explore in more detail the SAR. The amidic chain turned out to be important for the modulation of NAAA inhibitory activity. Replacement of the 3-phenylpropanamide in 11 with a linear alkyl chain as in compound 13 (Fig. 3, IC50 = 0.46 μM), did not affect the potency, whereas the introduction of more lipophilic residues, such as a 2-naphthyl group, compound 14 (IC50 = 0.16 μM), or a p-biphenyl residue, compound 15 (IC50 = 0.115 μM), yielded potent NAAA inhibitors.[44] Pharmacological characterization of 15 showed that the compound inhibited NAAA with a rapid, noncompetitive, and reversible (upon overnight dialysis) mechanism. The in vivo activity of compounds 11 and 15 was tested in mice implanted subcutaneously with polyethylene sponges instilled with the pro-inflammatory polysaccharide carrageenan. These implants induced a localized inflammatory response characterized by leukocyte infiltration and reduction of PEA levels in these cells. When compounds 11 and 15 were added to the sponges, they prevented both leukocyte infiltration and reduction of PEA levels.[27,44] The ability of compound 11 to decrease inflammatory responses was not observed in PPAR-α deficient mice, and was mimicked by either exogenous PEA or the synthetic PPAR-α agonist GW7647.[27]
The β-lactone moiety, which is crucial for NAAA inhibitory activity, is known to display low chemical and plasma stability. In particular, serine-derived β-lactones, including compounds 11, 14 and 15, are all hydrolyzed in aqueous buffer at pH 7.4 with half-life of less than 20 minutes.[53] On the other hand, β-lactones deriving from threonine, which have a methyl substituent at the β-carbon of the lactone ring, were reported to display increased stability in aqueous media compared to serine-derived analogues.[52,54]
Threonine-derived β-lactones were therefore investigated for their ability to inhibit NAAA activity and their stability in aqueous media and plasma. The recognition of these compounds at the enzyme active site was dependent on the stereochemistry at the two chiral centers. The preference for the (S)-stereochemistry at the α-carbon of the lactone ring observed with serine-derived β-lactones appeared to be reversed, since the threonine-derived β-lactone analogue of 11, compound 16, lost the ability to inhibit NAAA (IC50 >100 μM), whereas its enantiomer, 17, having the (R)-configuration at the α-carbon, still inhibited NAAA activity, although with micromolar potency (IC50 = 3.2 μM).[44] The same trend in potency was reported for compounds 18 (IC50 = 10 μM), the threonine-derived β-lactone analogue of 10, and 19 (IC50 = 1 μM).[53] Consistent with previous observations,[52,54] these compounds displayed higher buffer stability than their serine-derived β-lactone analogues, with derivatives bearing a carbamate functionality at the α-carbon of the lactone ring being slightly more stable than the corresponding amides. However, no major improvement in plasma stability was observed.[53] A number of analogues were prepared bearing a carbamate functionality at the α-carbon of the lactone ring, and several potent NAAA inhibitors were identified within this chemical series. Among them, compound 20, also known as ARN077,[53] inhibited rat NAAA with IC50 of 50 nM and human NAAA with IC50 of 7 nM.[55] Modifications of the 5-phenylpentyl side chain of ARN077 led to potent inhibitors of both rat and human NAAA, such as compounds 21-23.[55] Within this series, compound 23 represented the first single-digit nanomolar inhibitor (IC50 = 7 nM) of both rat and human NAAA reported in the literature. As observed with compound 10, none of the compounds 20-23 inhibited rat FAAH activity at the concentration of 10 μM. At the same concentration, 23 showed 30% inhibition of rat acid ceramidase, while less than 10% inhibition was observed for compounds 21-23.[55]
Replacement of the methyl group at the β-carbon of the β-lactone ring with more sterically demanding alkyl groups such as ethyl, iso-propyl and tert-butyl has been recently reported to generally increase the hydrolytic stability of the compounds, but negatively affect the NAAA inhibitory potency, although still remaining in the nanomolar range.[56]
The compound ARN077 has been partially characterized for its pharmacological profile. Kinetic analyses with recombinant rat and human NAAA showed that ARN077 inhibited enzyme activity through a rapid and non-competitive mechanism.[30,46] Inhibition of rat NAAA was reversible upon overnight dialysis,[46] whereas only partial reversibility was observed with the human enzyme.[30] Further investigation of the mechanism of inhibition by high-resolution mass spectrometry indicated that ARN077 reacts with the catalytically active N-terminal cysteine (Cys 126) of human NAAA to form a thioester bond.[30] The formation of the covalent yet hydrolysable thioester bond is consistent with the non-competitive and partially reversible mechanism of inhibition of human NAAA. The selectivity of ARN077 was investigated by testing the compound on a panel of molecular targets including GPCRs, channels, transporters, and enzymes. At the concentration of 10 μM, the compound showed a clean profile on the targets of the panel.[57]
Like other β-lactone-based NAAA inhibitors, ARN077 is rapidly hydrolyzed in plasma.[53] This instability precludes in vivo studies by systemic administration, but not by topical application. When applied on mouse skin, ARN077 attenuated, in a dose-dependent manner, heat hyperalgesia and mechanical allodynia caused by carrageenan injection in the animal paw or by sciatic nerve ligation.[46] Topical application of ARN077 also dose-dependently reduced UVB-induced heat hyperalgesia in rats whose glabrous skin of the hind paw was exposed to UVB radiation. These studies also showed that ARN077 normalized the levels of PEA and OEA, but not anandamide, in inflamed tissues of mice, and that its antinociceptive effects were prevented by the selective PPAR-α antagonist GW6471 and were absent in PPAR-α-deficient mice. These findings suggest that ARN077 modulated nociceptive responses in mice and rats by blocking NAAA-mediated FAE degradation and restoring FAE signaling at PPAR-α.[46]
4. Conclusions
The discovery that FAEs exert analgesic and anti-inflammatory effects has stimulated a growing interest in the pharmacological modulation of their tissue levels. Since the action of these signaling molecules is terminated by enzyme-mediated hydrolysis, compounds that inhibit the enzymes responsible for FAE breakdown may represent new therapeutic agents for the treatment of pain and inflammatory states. NAAA is the primary hydrolyzing enzyme for the endogenous anti-inflammatory and analgesic molecule PEA. While a large number of inhibitors have been discovered for other hydrolases such as FAAH, relatively few compounds have been reported to inhibit NAAA activity.
Moderately potent inhibitors of NAAA have been obtained by structural modification of PEA. Most of these compounds inhibited enzyme activity in cell-free assays with IC50 values in the low micromolar or submicromolar range, and a few of them were reported to inhibit NAAA activity in cells. The therapeutic potential of these compounds, however, remains to be evaluated as only cyclopentyl hexadecanoate was reported to reduce carrageenan-induced inflammation in rats following local administration with carrageenan.
Compounds containing a properly substituted β-lactone moiety represent the most potent inhibitors of NAAA activity reported so far. Several β-lactone derivatives inhibited NAAA with IC50 in the low nanomolar range in cell-free assays. When tested in cells, compounds of this chemical class demonstrated NAAA inhibitory activity and restored PEA levels after challenge with proinflammatory stimuli. Although β-lactones display low plasma stability, thus preventing their systemic administration, topical application of the potent NAAA inhibitor ARN077 has been reported to dose-dependently attenuate heat hyperalgesia and mechanical allodynia elicited in mice by inflammation or nerve damage. This study suggests that β-lactone NAAA inhibitors could be envisaged as potential new anti-inflammatory and analgesic drugs for topical use. Under this clinical setting, the low plasma stability of these compounds can be considered as an advantage. In fact, NAAA inhibition would be restricted to the site of application of the drug, which upon reaching the systemic circulation would be converted into inactive metabolites thus strongly reducing their potential systemic side effects. The encouraging results in animal models of pain and inflammation observed with ARN077 applied topically support further exploration of the pharmacology of NAAA inhibitors. To this end, the discovery of compounds suitable for systemic administration would help better evaluate the therapeutic potential of NAAA inhibitors.
Fig. 4.

(A) General structure of threonine-derived β-lactones. (B) Chemical structures of selected threonine-derived β-lactones.
Footnotes
Chemical compounds studied in the article: Palmitoylethanolamide (PubChem CID: 4671); Oleoylethanolamide (PubChem CID: 5283454); N-Pentadecylbenzamide (PubChem CID: 44398681); N-Pentadecylcyclohexancarboxamide (PubChem CID: 44398718); Pentadecylamine (PubChem CID: 17386); 3-Phenyl-1-pyrrolidin-1-ylpropan-1-one (PubChem CID: 1533088); N-[(3S)-2-oxooxetan-3-yl]-3-phenylpropanamide (PubChem CID: 25227533); N-[(3S)-2-oxooxetan-3-yl]-4-phenylbenzamide (PubChem CID: 46899632); 5-phenylpentyl N-[(2S,3R)-2-methyl-4-oxooxetan-3-yl]carbamate (PubChem CID: 57523549); (4-phenylphenyl)methyl N-[(2S,3R)-2-methyl-4-oxooxetan-3-yl]carbamate (PubChem CID: 73291988)
Conflict of interest: The authors declare the following competing financial interests: T. Bandiera, S. Ponzano and D. Piomelli are inventors in patent applications protecting β-lactones reported in the present review.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Calignano A, La Rana G, Giuffrida A, Piomelli D. Control of pain initiation by endogenous cannabinoids. Nature. 1998;394:277–281. doi: 10.1038/28393. [DOI] [PubMed] [Google Scholar]
- 2.Rodriguez de Fonseca F, Navarro M, Gomez R, Escuredo L, Nava F, Fu J, Murillo-Rodriguez E, Giuffrida A, LoVerme J, Gaetani S, Kathuria S, Gall C, Piomelli D. An anorexic lipid mediator regulated by feeding. Nature. 2001;414:209–212. doi: 10.1038/35102582. [DOI] [PubMed] [Google Scholar]
- 3.Lo Verme J, Fu J, Astarita G, La Rana G, Russo R, Calignano A, Piomelli D. The nuclear receptor peroxisome proliferator-activated receptor-alpha mediates the anti-inflammatory actions of palmitoylethanolamide. Molecular Pharmacology. 2005;67:15–19. doi: 10.1124/mol.104.006353. [DOI] [PubMed] [Google Scholar]
- 4.LoVerme J, Russo R, La Rana G, Fu J, Farthing J, Mattace-Raso G, Meli R, Hohmann A, Calignano A, Piomelli D. Rapid broad-spectrum analgesia through activation of peroxisome proliferator-activated receptor-alpha. The Journal of Pharmacology and Experimental Therapeutics. 2006;319:1051–1061. doi: 10.1124/jpet.106.111385. [DOI] [PubMed] [Google Scholar]
- 5.Piomelli D. The molecular logic of endocannabinoid signalling. NatureReviews Neuroscience. 2003;4:873–884. doi: 10.1038/nrn1247. [DOI] [PubMed] [Google Scholar]
- 6.Fu J, Gaetani S, Oveisi F, Lo Verme J, Serrano A, Rodriguez De Fonseca F, Rosengarth A, Luecke H, Di Giacomo B, Tarzia G, Piomelli D. Oleylethanolamide regulates feeding and body weight through activation of the nuclear receptor PPAR-alpha. Nature. 2003;425:90–93. doi: 10.1038/nature01921. [DOI] [PubMed] [Google Scholar]
- 7.Suardiaz M, Estivill-Torrus G, Goicoechea C, Bilbao A, Rodriguez de Fonseca F. Analgesic properties of oleoylethanolamide (OEA) in visceral and inflammatory pain. Pain. 2007;133:99–110. doi: 10.1016/j.pain.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 8.Di Marzo V, Fontana A, Cadas H, Schinelli S, Cimino G, Schwartz JC, Piomelli D. Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature. 1994;372:686–691. doi: 10.1038/372686a0. [DOI] [PubMed] [Google Scholar]
- 9.Piomelli D. A fatty gut feeling. Trends in Endocrinology and Metabolism: TEM. 2013;24:332–341. doi: 10.1016/j.tem.2013.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ueda N, Tsuboi K, Uyama T. Metabolism of endocannabinoids and related N-acylethanolamines: Canonical and alternative pathways. The FEBS Journal. 2013;280:1874–1894. doi: 10.1111/febs.12152. [DOI] [PubMed] [Google Scholar]
- 11.Cadas H, Gaillet S, Beltramo M, Venance L, Piomelli D. Biosynthesis of an endogenous cannabinoid precursor in neurons and its control by calcium and cAMP. The Journal of Neuroscience: the official journal of the Society for Neuroscience. 1996;16:3934–3942. doi: 10.1523/JNEUROSCI.16-12-03934.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bisogno T, Maurelli S, Melck D, De Petrocellis L, Di Marzo V. Biosynthesis, uptake, and degradation of anandamide and palmitoylethanolamide in leukocytes. The Journal of Biological Chemistry. 1997;272:3315–3323. doi: 10.1074/jbc.272.6.3315. [DOI] [PubMed] [Google Scholar]
- 13.Okamoto Y, Morishita J, Tsuboi K, Tonai T, Ueda N. Molecular characterization of a phospholipase D generating anandamide and its congeners. The Journal of Biological Chemistry. 2004;279:5298–5305. doi: 10.1074/jbc.M306642200. [DOI] [PubMed] [Google Scholar]
- 14.Ueda N, Yamanaka K, Yamamoto S. Purification and characterization of an acid amidase selective for N-palmitoylethanolamine, a putative endogenous anti-inflammatory substance. The Journal of Biological Chemistry. 2001;276:35552–35557. doi: 10.1074/jbc.M106261200. [DOI] [PubMed] [Google Scholar]
- 15.Tsuboi K, Sun YX, Okamoto Y, Araki N, Tonai T, Ueda N. Molecular characterization of N-acylethanolamine-hydrolyzing acid amidase, a novel member of the choloylglycine hydrolase family with structural and functional similarity to acid ceramidase. The Journal of Biological Chemistry. 2005;280:11082–11092. doi: 10.1074/jbc.M413473200. [DOI] [PubMed] [Google Scholar]
- 16.Tsuboi K, Takezaki N, Ueda N. The N-acylethanolamine-hydrolyzing acid amidase (NAAA) Chemistry & Biodiversity. 2007;4:1914–1925. doi: 10.1002/cbdv.200790159. [DOI] [PubMed] [Google Scholar]
- 17.Desarnaud F, Cadas H, Piomelli D. Anandamide amidohydrolase activity in rat brain microsomes. Identification and partial characterization The Journal of Biological Chemistry. 1995;270:6030–6035. doi: 10.1074/jbc.270.11.6030. [DOI] [PubMed] [Google Scholar]
- 18.Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature. 1996;384:83–87. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- 19.Ueda N, Tsuboi K, Uyama T. N-acylethanolamine metabolism with special reference to Nacylethanolamine-hydrolyzing acid amidase (NAAA) Progress in Lipid Research. 2010;49:299–315. doi: 10.1016/j.plipres.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 20.Rossocha M, Schultz-Heienbrok R, von Moeller H, Coleman JP, Saenger W. Conjugated bile acid hydrolase is a tetrameric N-terminal thiol hydrolase with specific recognition of its cholyl but not of its tauryl product. Biochemistry. 2005;44:5739–5748. doi: 10.1021/bi0473206. [DOI] [PubMed] [Google Scholar]
- 21.Tsuboi K, Zhao LY, Okamoto Y, Araki N, Ueno M, Sakamoto H, Ueda N. Predominant expression of lysosomal N-acylethanolamine-hydrolyzing acid amidase in macrophages revealed by immunochemical studies. Biochimica et Biophysica Acta. 2007;1771:623–632. doi: 10.1016/j.bbalip.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 22.Gulyas AI, Cravatt BF, Bracey MH, Dinh TP, Piomelli D, Boscia F, Freund TF. Segregation of two endocannabinoid-hydrolyzing enzymes into pre- and postsynaptic compartments in the rat hippocampus, cerebellum and amygdala. The European Journal of Neuroscience. 2004;20:441–458. doi: 10.1111/j.1460-9568.2004.03428.x. [DOI] [PubMed] [Google Scholar]
- 23.Bernardo K, Hurwitz R, Zenk T, Desnick RJ, Ferlinz K, Schuchman EH, Sandhoff K. Purification, characterization, and biosynthesis of human acid ceramidase. The Journal of Biological Chemistry. 1995;270:11098–11102. doi: 10.1074/jbc.270.19.11098. [DOI] [PubMed] [Google Scholar]
- 24.Ferlinz K, Kopal G, Bernardo K, Linke T, Bar J, Breiden B, Neumann U, Lang F, Schuchman EH, Sandhoff K. Human acid ceramidase: Processing, glycosylation, and lysosomal targeting. The Journal of Biological Chemistry. 2001;276:35352–35360. doi: 10.1074/jbc.M103066200. [DOI] [PubMed] [Google Scholar]
- 25.Zhao LY, Tsuboi K, Okamoto Y, Nagahata S, Ueda N. Proteolytic activation and glycosylation of N-acylethanolamine-hydrolyzing acid amidase, a lysosomal enzyme involved in the endocannabinoid metabolism. Biochimica et Biophysica Acta. 2007;1771:1397–1405. doi: 10.1016/j.bbalip.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 26.Wang J, Zhao LY, Uyama T, Tsuboi K, Tonai T, Ueda N. Amino acid residues crucial in pH regulation and proteolytic activation of N-acylethanolamine-hydrolyzing acid amidase. Biochimica et Biophysica Acta. 2008;1781:710–717. doi: 10.1016/j.bbalip.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 27.Solorzano C, Zhu C, Battista N, Astarita G, Lodola A, Rivara S, Mor M, Russo R, Maccarrone M, Antonietti F, Duranti A, Tontini A, Cuzzocrea S, Tarzia G, Piomelli D. Selective N-acylethanolamine-hydrolyzing acid amidase inhibition reveals a key role for endogenous palmitoylethanolamide in inflammation. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:20966–20971. doi: 10.1073/pnas.0907417106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oinonen C, Rouvinen J. Structural comparison of Ntn-hydrolases. Protein Science: a publication of the Protein Society. 2000;9:2329–2337. doi: 10.1110/ps.9.12.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lodola A, Branduardi D, De Vivo M, Capoferri L, Mor M, Piomelli D, Cavalli A. A catalytic mechanism for cysteine n-terminal nucleophile hydrolases, as revealed by free energy simulations. PloS One. 2012;7:e32397. doi: 10.1371/journal.pone.0032397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Armirotti A, Romeo E, Ponzano S, Mengatto L, Dionisi M, Karacsonyi C, Bertozzi F, Garau G, Tarozzo G, Reggiani A, Bandiera T, Tarzia G, Mor M, Piomelli D. Beta-lactones inhibit N-acylethanolamine acid amidase by S-acylation of the catalytic N-terminal cysteine. ACS Medicinal Chemistry Letters. 2012;3:422–426. doi: 10.1021/ml300056y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.West JM, Zvonok N, Whitten KM, Vadivel SK, Bowman AL, Makriyannis A. Biochemical and mass spectrometric characterization of human N-acylethanolamine-hydrolyzing acid amidase inhibition. PloS One. 2012;7:e43877. doi: 10.1371/journal.pone.0043877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.LoVerme J, La Rana G, Russo R, Calignano A, Piomelli D. The search for the palmitoylethanolamide receptor. Life Sciences. 2005;77:1685–1698. doi: 10.1016/j.lfs.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 33.Berdyshev E, Boichot E, Corbel M, Germain N, Lagente V. Effects of cannabinoid receptor ligands on LPS-induced pulmonary inflammation in mice. Life Sciences. 1998;63:PL125–129. doi: 10.1016/s0024-3205(98)00324-5. [DOI] [PubMed] [Google Scholar]
- 34.D'Agostino G, La Rana G, Russo R, Sasso O, Iacono A, Esposito E, Raso GM, Cuzzocrea S, Lo Verme J, Piomelli D, Meli R, Calignano A. Acute intracerebroventricular administration of palmitoylethanolamide, an endogenous peroxisome proliferator-activated receptor-alpha agonist, modulates carrageenan-induced paw edema in mice. The Journal of Pharmacology and Experimental Therapeutics. 2007;322:1137–1143. doi: 10.1124/jpet.107.123265. [DOI] [PubMed] [Google Scholar]
- 35.Mazzari S, Canella R, Petrelli L, Marcolongo G, Leon A. N-(2-hydroxyethyl)hexadecanamide is orally active in reducing edema formation and inflammatory hyperalgesia by down-modulating mast cell activation. European Journal of Pharmacology. 1996;300:227–236. doi: 10.1016/0014-2999(96)00015-5. [DOI] [PubMed] [Google Scholar]
- 36.Calignano A, La Rana G, Piomelli D. Antinociceptive activity of the endogenous fatty acid amide, palmitylethanolamide. European Journal of Pharmacology. 2001;419:191–198. doi: 10.1016/s0014-2999(01)00988-8. [DOI] [PubMed] [Google Scholar]
- 37.Khasabova IA, Xiong Y, Coicou LG, Piomelli D, Seybold V. Peroxisome proliferator-activated receptor alpha mediates acute effects of palmitoylethanolamide on sensory neurons. The Journal of Neuroscience: the official journal of the Society for Neuroscience. 2012;32:12735–12743. doi: 10.1523/JNEUROSCI.0130-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kemeny L, Koreck A, Kis K, Kenderessy-Szabo A, Bodai L, Cimpean A, Paunescu V, Raica M, Ghyczy M. Endogenous phospholipid metabolite containing topical product inhibits ultraviolet light-induced inflammation and DNA damage in human skin. Skin Pharmacology and Physiology. 2007;20:155–161. doi: 10.1159/000098702. [DOI] [PubMed] [Google Scholar]
- 39.Keppel Hesselink JM, Hekker TA. Therapeutic utility of palmitoylethanolamide in the treatment of neuropathic pain associated with various pathological conditions: A case series. Journal of Pain Research. 2012;5:437–442. doi: 10.2147/JPR.S32143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Capasso R, Izzo AA, Fezza F, Pinto A, Capasso F, Mascolo N, Di Marzo V. Inhibitory effect of palmitoylethanolamide on gastrointestinal motility in mice. British Journal of Pharmacology. 2001;134:945–950. doi: 10.1038/sj.bjp.0704339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.De Filippis D, D'Amico A, Cipriano M, Petrosino S, Orlando P, Di Marzo V, Iuvone T, Grp ER. Levels of endocannabinoids and palmitoylethanolamide and their pharmacological manipulation in chronic granulomatous inflammation in rats. Pharmacological Research. 2010;61:321–328. doi: 10.1016/j.phrs.2009.11.005. [DOI] [PubMed] [Google Scholar]
- 42.Zhu C, Solorzano C, Sahar S, Realini N, Fung E, Sassone-Corsi P, Piomelli D. Proinflammatory stimuli control N-acylphosphatidylethanolamine-specific phospholipase D expression in macrophages. Molecular Pharmacology. 2011;79:786–792. doi: 10.1124/mol.110.070201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Richardson D, Pearson RG, Kurian N, Latif ML, Garle MJ, Barrett DA, Kendall DA, Scammell BE, Reeve AJ, Chapman V. Characterisation of the cannabinoid receptor system in synovial tissue and fluid in patients with osteoarthritis and rheumatoid arthritis. Arthritis Research & Therapy. 2008;10:R43. doi: 10.1186/ar2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Solorzano C, Antonietti F, Duranti A, Tontini A, Rivara S, Lodola A, Vacondio F, Tarzia G, Piomelli D, Mor M. Synthesis and structure-activity relationships of N-(2-oxo-3-oxetanyl)amides as N-acylethanolamine-hydrolyzing acid amidase inhibitors. Journal of Medicinal Chemistry. 2010;53:5770–5781. doi: 10.1021/jm100582w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li Y, Yang L, Chen L, Zhu C, Huang R, Zheng X, Qiu Y, Fu J. Design and synthesis of potent N-acylethanolamine-hydrolyzing acid amidase (NAAA) inhibitor as anti-inflammatory compounds. PloS One. 2012;7:e43023. doi: 10.1371/journal.pone.0043023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sasso O, Moreno-Sanz G, Martucci C, Realini N, Dionisi M, Mengatto L, Duranti A, Tarozzo G, Tarzia G, Mor M, Bertorelli R, Reggiani A, Piomelli D. Antinociceptive effects of the N-acylethanolamine acid amidase inhibitor ARN077 in rodent pain models. Pain. 2013;154:350–360. doi: 10.1016/j.pain.2012.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vandevoorde S, Tsuboi K, Ueda N, Jonsson KO, Fowler CJ, Lambert DM. Esters, retroesters, and a retroamide of palmitic acid: Pool for the first selective inhibitors of N-palmitoylethanolamine-selective acid amidase. Journal of Medicinal Chemistry. 2003;46:4373–4376. doi: 10.1021/jm0340795. [DOI] [PubMed] [Google Scholar]
- 48.Tsuboi K, Hilligsmann C, Vandevoorde S, Lambert DM, Ueda N. N-cyclohexanecarbonylpentadecylamine: A selective inhibitor of the acid amidase hydrolysing N-acylethanolamines, as a tool to distinguish acid amidase from fatty acid amide hydrolase. The Biochemical Journal. 2004;379:99–106. doi: 10.1042/BJ20031695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yamano Y, Tsuboi K, Hozaki Y, Takahashi K, Jin XH, Ueda N, Wada A. Lipophilic amines as potent inhibitors of N-acylethanolamine-hydrolyzing acid amidase. Bioorganic and Medicinal Chemistry. 2012;20:3658–3665. doi: 10.1016/j.bmc.2012.03.065. [DOI] [PubMed] [Google Scholar]
- 50.Saturnino C, Petrosino S, Ligresti A, Palladino C, De Martino G, Bisogno T, Di Marzo V. Synthesis and biological evaluation of new potential inhibitors of N-acylethanolamine hydrolyzing acid amidase. Bioorganic and Medicinal Chemistry Letters. 2010;20:1210–1213. doi: 10.1016/j.bmcl.2009.11.134. [DOI] [PubMed] [Google Scholar]
- 51.Petrosino S, Iuvone T, Di Marzo V. N-palmitoyl-ethanolamine: Biochemistry and new therapeutic opportunities. Biochimie. 2010;92:724–727. doi: 10.1016/j.biochi.2010.01.006. [DOI] [PubMed] [Google Scholar]
- 52.Lall MS, Karvellas C, Vederas JC. Beta-lactones as a new class of cysteine proteinase inhibitors: Inhibition of hepatitis a virus 3C proteinase by N-Cbz-serine beta-lactone. Organic Letters. 1999;1:803–806. doi: 10.1021/ol990148r. [DOI] [PubMed] [Google Scholar]
- 53.Duranti A, Tontini A, Antonietti F, Vacondio F, Fioni A, Silva C, Lodola A, Rivara S, Solorzano C, Piomelli D, Tarzia G, Mor M. N-(2-oxo-3-oxetanyl)carbamic acid esters as N-acylethanolamine acid amidase inhibitors: Synthesis and structure-activity and structure-property relationships. Journal of Medicinal Chemistry. 2012;55:4824–4836. doi: 10.1021/jm300349j. [DOI] [PubMed] [Google Scholar]
- 54.Lall MS, Ramtohul YK, James MNG, Vederas JC. Serine and threonine beta-lactones: A new class of hepatitis a virus 3C cysteine proteinase inhibitors. Journal of Organic Chemistry. 2002;67:1536–1547. doi: 10.1021/jo0109016. [DOI] [PubMed] [Google Scholar]
- 55.Ponzano S, Bertozzi F, Mengatto L, Dionisi M, Armirotti A, Romeo E, Berteotti A, Fiorelli C, Tarozzo G, Reggiani A, Duranti A, Tarzia G, Mor M, Cavalli A, Piomelli D, Bandiera T. Synthesis and structure-activity relationship (SAR) of 2-methyl-4-oxo-3-oxetanylcarbamic acid esters, a class of potent N-acylethanolamine acid amidase (NAAA) inhibitors. Journal of Medicinal Chemistry. 2013;56:6917–6934. doi: 10.1021/jm400739u. [DOI] [PubMed] [Google Scholar]
- 56.Vitale R, Ottonello G, Petracca R, Bertozzi SM, Ponzano S, Armirotti A, Berteotti A, Dionisi M, Cavalli A, Piomelli D, Bandiera T, Bertozzi F. Synthesis, structure-activity, and structure-stability relationships of 2-substituted-n-(4-oxo-3-oxetanyl) N-acylethanolamine acid amidase (NAAA) inhibitors. ChemMedChem. 2014;9:323–336. doi: 10.1002/cmdc.201300416. [DOI] [PubMed] [Google Scholar]
- 57.Unpublished results. The compound was tested at CEREP (France) in a panel of ca. 100 targets.