


Abstract
The challenge presented by high-throughput sequencing necessitates the development of novel tools for accurate alignment of reads to reference sequences. Current approaches focus on using heuristics to map reads quickly to large genomes, rather than generating highly accurate alignments in coding regions. Such approaches are, thus, unsuited for applications such as amplicon-based analysis and the realignment phase of exome sequencing and RNA-seq, where accurate and biologically relevant alignment of coding regions is critical. To facilitate such analyses, we have developed a novel tool, RAMICS, that is tailored to mapping large numbers of sequence reads to short lengths (<10 000 bp) of coding DNA. RAMICS utilizes profile hidden Markov models to discover the open reading frame of each sequence and aligns to the reference sequence in a biologically relevant manner, distinguishing between genuine codon-sized indels and frameshift mutations. This approach facilitates the generation of highly accurate alignments, accounting for the error biases of the sequencing machine used to generate reads, particularly at homopolymer regions. Performance improvements are gained through the use of graphics processing units, which increase the speed of mapping through parallelization. RAMICS substantially outperforms all other mapping approaches tested in terms of alignment quality while maintaining highly competitive speed performance.
INTRODUCTION
The issue of accurate pairwise sequence alignment is common to many fields in bioinformatics, whether as a core tool in fields such as reference-guided genome assembly (1–6) or as the seed for the generation of a progressive multiple sequence alignment (7–9). Ideally, analysis of a pairwise alignment should proceed in the knowledge that no inherent bias exists as a result of the alignment approach used. This is particularly challenging in the era of high-throughput sequencing, where every platform produces systematic errors (10–15) that should be considered in constructing an alignment.
The alignment of coding DNA, in particular, presents a unique challenge as it is critical that the final alignment takes into account the correct reading frame. Preservation of the reading frame ensures correct calling of gene structure (16–18) and SNPs, whether in, for example, the realignment phase of an exome sequencing pipeline (19), single-nucleotide polymorphism (SNP) calling from existing RNA-seq data (20,21) or amplicon-based analyses such as human immunodeficiency virus (HIV) drug resistance genotyping (22). When aligning coding DNA generated using high-throughput sequencing platforms, it is critical that codons present in the open reading frame remain intact and that codon-sized insertions and deletions are recognized and called correctly, as distinct from both genuine frameshifts and single indels created through sequencing error.
The landscape of pairwise alignment and reference mapping tools for high-throughput sequencing data is broad. Tools such as BOWTIE 2 (2) and BWA-MEM (3), while well-suited to mapping the location of query sequence reads within a complete reference genome, lack the subtle nuances required to correctly distinguish spurious indels from genuine codon-sized mutations in coding DNA. Other tools, such as MOSAIK (6) and SSAHA2 (5), perform full mapping and realignment using a Smith–Waterman approach, with a focus on correcting next-generation sequencing errors. However, basic Smith–Waterman alignment, even when taking into account quality scores as in MOSAIK and BWA-MEM, is not appropriate for reference mapping of coding DNA as it fails to maintain the intactness of codons. Finally, the Genome Analysis Toolkit (GATK) (23,24) performs the realignment step for tools such as BWA-MEM in, for example, the 1000 Genomes project pipeline (25), but does not consider the biological context (coding or non-coding) in its alignment.
One approach to ensure codons are maintained for downstream analysis, taken by the RevTrans program (26), is to initially translate query sequences into their corresponding amino acid sequences, align them to a translated reference sequence at the amino acid level and then ‘back-align’ the nucleotide sequences based on the amino acid alignment (26). While effective in some cases, this approach is useless in the presence of indels in the open reading frame, which results in mistranslation to amino acid space. Tools such as transAlign (27) repetitively translate, align, back-translate and correct multiple sequence alignments, which to some extent addresses frameshift errors. This approach is, however, time-consuming, as in order to align robustly it requires a full amino acid multiple sequence alignment to create a back-translated DNA profile to which low-scoring sequences (assumed to be frameshifted) are compared. An alternative is to use an approach that undertakes pairwise alignment in ‘codon space’—weighting the likelihood of each nucleotide substitution within the context of the codon to which it belongs. This, thus, facilitates the identification of indels, whether genuine frameshift mutations or the result of sequencing error.
Initial models of coding DNA such as those used by PairWise (16) and EST_Genome (28) were designed specifically for gene prediction, comparing sequences against all six translated frames simultaneously using a classical dynamic programming approach to discover exons. However, this approach was found to be time-consuming and overly specific (29). The HMMER tool (30) took a different approach, using profile hidden Markov models to align and compare protein sequences. GeneWise and GenomeWise (29) use a hidden Markov model approach to allow for the alignment of a nucleotide sequence, potentially including frameshifts, to an existing protein sequence, and thus represent the closest approximation to a codon–codon alignment tool. However, because the alignment is to a protein sequence, it is impossible to penalize synonymous mutations. Protein mismatches are all scored equally, as are single-nucleotide insertions resulting from sequencing error. This prevents the use of GeneWise and GenomeWise to correctly identify and account for homopolymer errors in next-generation sequencing platforms.
Here, we present a novel reference mapper (RAMICS: rapid amplicon mapping in codon space) that uses a hidden Markov model approach to align large numbers of coding DNA sequences to a coding region of a reference sequence in an accurate and biologically relevant manner, accounting for the inherent error biases of the sequencing platform used. RAMICS is developed to be executed on graphics processing units (GPUs) and, thus, provides significant speed improvements over other, similarly complex, mapping tools. RAMICS is, thus, ideal for any situation where a large number of sequence reads from a known, coding location in a genome must be mapped to a coding reference sequence.
MATERIALS AND METHODS
The open reading frame
RAMICS is designed specifically to align coding DNA. The tool first discerns the optimal open reading frame of the reference sequence by selecting a frame that would minimize the number of stop codons. RAMICS then performs a pairwise alignment to each query sequence in ‘codon space’ using a hidden Markov model (31) (Figure 1 and Supplementary information). Single- and double-nucleotide indels are identified and accounted for, thereby ensuring that biologically relevant complete codons are kept intact. This enables a correct and relevant comparison of a query sequence to the reference, as the concept of a codon-sized insertion that begins within one codon and ends within another (as would be output by most conventional alignment approaches) is structurally meaningless in a biological context (Figure 2A).
Figure 1.
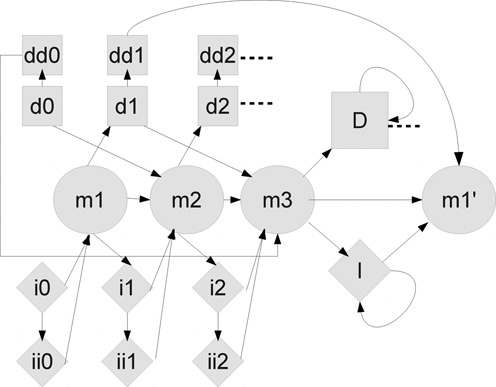
The RAMICS hidden Markov model. Match states are shown as circles, delete states as squares and insert states as diamonds. The states ‘I’ and ‘D’ are whole-codon insertions and deletions, and are the only self-transitioning states. One copy of this HMM exists for every codon in the reference sequence, and they are chained together to build a profile HMM. Dashed lines indicate an unshown transition to the next set of states in the chain..
Figure 2.
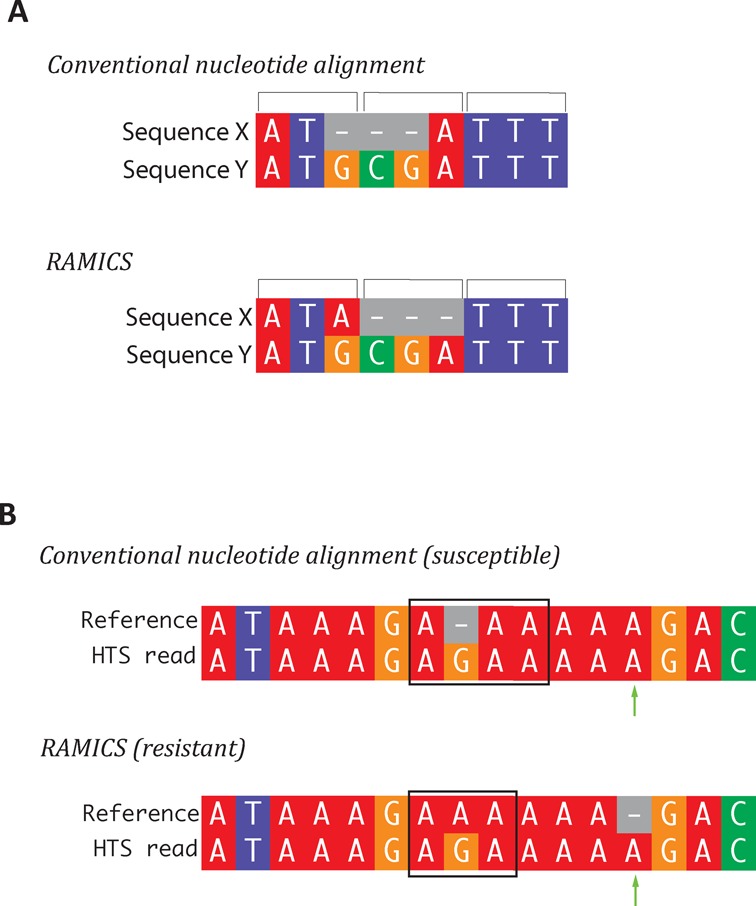
(A) ‘Biologically relevant alignments’. Sequences X and Y differ from each other by a three-nucleotide insertion. Conventional nucleotide-based approaches generate a statistically optimal pairwise alignment matching as many nucleotides as possible. RAMICS, on the other hand, considers the biological importance of codons and generates a biologically relevant alignment preserving the codon structure of the coding DNA being aligned. (B) ‘Correctly accounting for homopolymer errors’. Comparing alignments of an HIV pol gene sequence containing a homopolymer error (additional adenosine, green arrow) to a reference sequence shows that RAMICS correctly identifies and accounts for this error. The codon of interest in the reference sequence is marked with a black box. Conventional nucleotide alignment approaches would call this codon as AAA (flagging the G as an insertion) which encodes for lysine (sensitive form), while RAMICS would call the codon as AGA (arginine, the resistant form) correctly identifying and discounting the extra adenosine introduced as a result of the sequencing error at the homopolymer region.
RAMICS uses models of codon substitution (32–34) to give synonymous codon substitutions higher transition probabilities as well as favoring more likely non-synonymous substitutions. RAMICS can also be set to use a simple non-coding substitution model to align non-coding DNA to a non-coding reference sequence. However, RAMICS cannot concurrently align sequence reads to coding and non-coding regions of the same reference sequence (e.g. a reference sequence containing both introns and exons), and, as such, is unsuitable for use as the first-pass mapping tool in full-genome association studies. It can, however, be subsequently used for targeted realignment of coding regions as a second-pass mapping tool.
The sequencing platform
RAMICS uses reported average error rates for given sequencing platforms (10,12,13,15) to set the transition probabilities to single- and double-nucleotide indel states in the profile hidden Markov model. In addition, the model is more likely to place a single-nucleotide indel on either side of a homopolymer region when the sequencing platform is set to Roche/454 (35) or Ion Torrent Personal Genome Machine (36), as homopolymer miscalls are a common sequencing error on these platforms (13).
To illustrate the necessity of considering the sequencing platform used to generate the sequence data, we take an example from HIV-1 antiretroviral drug resistance testing. The drug resistance profile of a sequence is determined by characterizing mutations at various amino acid positions throughout the polymerase gene mapped in relation to the HXB2 HIV reference sequence (51). Drug resistant mutations include the mutation of lysine (K) to asparagine (N) at codon 103 (K103N), which confers drug resistance to certain non-nucleoside reverse transcriptase inhibitors (NNRTIs) (22).
The K103N codon is located in a homopolymer region and some high-throughput sequencing platforms such as Roche/454 (35) and Ion Torrent PGM (32), as well as to a lesser extent the Illumina platform (37), are prone to increased error rates in regions containing homopolymers, as they cannot successfully identify the correct number of nucleotides in these regions (12,13). Incorrect assignment of a homopolymer indel during the mapping step would result in a viral sequence being falsely called as susceptible when it is actually resistant (54).
A standard Needleman–Wunsch alignment to the HXB2 reference sequence of a sequence containing both the K103N mutation and a spurious extra adenosine directly following homopolymer region would result in the virus in question being falsely classified as susceptible to NNRTIs such as efavirenz (Figure 2B). Mapping of the same data using RAMICS, however, weighs the possibility of a false tyrosine (T) insertion in the center of a homopolymer region against a false adenosine (A) insertion at the end of the region, assigns the latter a higher probability and, thus, correctly calls the presence of the K103M mutation in spite of the hompolymer error (Figure 2B). Thus, a major strength of RAMICS is in its application to situations where the correctness of alignment in a coding region is crucial, such as alignment optimization in RNA-seq and exome sequencing (19–21,38,39).
Training
It can be a common case in reference mapping, particularly for amplicon-based analyses, that a set of query sequence reads will be more closely related to each other than they will to a given reference sequence—this is true in reference-guided assembly (40), drug-resistance genotyping (41) and applications in metagenomic function prediction (42). However, a comparison to the reference sequence is still required for these applications. RAMICS circumvents any potential bias introduced through a distantly related reference sequence by allowing the use of the reference sequence as a guide for the initial mapping and then iteratively training the underlying hidden Markov model to the resulting pairwise alignments. The result is a set of emission and transition probabilities that will correctly align outlier sequences (potentially with many sequencing errors) in the query set, based on the substitutions in sequences more similar to the reference. This approach generates a global reference alignment that approaches a reference-seeded multiple sequence alignment, and which can be used as such.
Output formats
RAMICS produces results in various formats with the choice of output format very much driven by the end-users requirements:
A global nucleotide alignment of all query sequences to the reference sequence, in FASTA format.
A derivation of the above global nucleotide alignment in which all single- and double-nucleotide insertions are widened to the width of a full codon using gaps (codons.fasta). This does not alter the sequences, but allows the open reading frame to be read clearly in alignment viewers such as SeaView (43).
A global alignment with all single- and double-nucleotide insertions removed (clean.fasta). This format does edit the sequences by assuming that all single- and double-indels are the result of sequencing/polymerase chain reaction (PCR) error. While this is extremely useful in some amplicon-based analyses where frameshift mutation can be safely treated as PCR/sequencing errors, it should not be used in cases where a frameshift mutation may have biological relevance.
A global alignment containing only one instance of each identical sequence with a count of the number of contributing sequence reads added to the sequence descriptor.
A set of SAM files describing mapped reads, with soft clipping used beyond the end of the reference sequence.
A file containing a set of pairwise gapped FASTA format alignments of each query sequence to the reference.
Evaluation of RAMICS
RAMICS is designed to map large numbers of high-throughput sequencing reads from a coding DNA region to a similar reference sequence, both quickly and accurately. RAMICS also has potential as an alignment refiner for the coding regions of whole-genome mapping or as the seed pairwise aligner for a profile HMM multiple sequence aligner in the style of the protein sequence aligner Clustal Omega (44). Both as a standalone amplicon mapper and as a component of these larger toolchains, it can align large numbers of reads quickly as well as aligning reads accurately in regions that have similar biological features but contain many synonymous or functionally similar substitutions. Although RAMICS has the ability to remove frameshifts in reads, under the assumption that they are sequencing errors, we did not remove any indels before evaluating the alignments.
We first compared the quality of alignments with RAMICS in non-coding mode, coding mode without training and coding mode with training, respectively. We then compared RAMICS for quality and speed against Bowtie-2 (2), BWA-MEM (3), BWA-MEM post-processed by the IndelRealigner from the GATK (23), MOSAIK (6), the basic Needleman–Wunsch aligners EMBOSS ‘needle’ and ‘water’ (45) (‘needle’ is used where a global alignment is more appropriate, ‘water’ for local alignment), NextGenMap (46), SSAHA-2 (5) and SHRiMP2 (1). These tools represent a mix of global, local and ‘glocal’ alignment techniques (47), as for the mapping of short reads to a relatively short amplicon all methods are potentially valid.
Quality performance
The RABEMA benchmark
Several benchmarks for the mapping of simulated next-generation sequencing data exist (48,49). The SEAL benchmark (49) is not easily extensible to new alignment tools, so we first performed a simple benchmark of RAMICS’ relative ability to correctly de-align spurious indels using the Rabema benchmark (48). Rabema uses the MASON read simulator (50) to simulate artificial short reads containing sequencing errors from a user-provided reference sequence and to output a ‘gold standard’ SAM files containing each of these reads mapped to the reference sequence. The alignments generated by a mapping tool are then compared to the ‘gold standard’ alignments.
The tool then bins reads by the number of errors found, ranging from perfect reads to highly erroneous outlier reads containing 4% or more errors. This allows us to test alignment performance not only of reads exhibiting the average error rates reported by manufacturers but also of rare outlier reads that exhibit a higher error rate than the reported average. The Rabema output gives the percentage of correctly mapped reads as a function of percentage sequencing error in the read. We ran each benchmark 10 times with 10 different random seeds to ensure statistical significance.
The AFAB benchmark
While Rabema is a good benchmark of a mapping tool's ability to detect sequencing errors such as spurious indels, it only determines whether an alignment is correct or incorrect and, thus, does not allow quantification of how incorrect the alignment is from the ‘gold standard’. Further, because all of the reads generated by Rabema originate from the same reference sequence, the levels of diversity of the query sequences being mapped are lower than what would be generated in many genuine sequencing experiments. Thus, we developed a flexible alignment benchmark (AFAB) that could evaluate not only whether a correct alignment was found, but also how similar this alignment was to a gold standard alignment, in the case where very divergent sequences made perfect mapping impossible. This was achieved using a combination of tools.
We began by selecting a ‘gold standard’ alignment of protein coding DNA which was the highly manually curated HIV-1 group M subtype reference alignment of the HIV envelope gene from the LANL HIV reference sequence database (51). This alignment was chosen for two reasons: (i) it encompasses the broad spectrum of diversity present within the HIV-1 group M subtypes (pairwise genetic diversity measured with the generalized time-reversible substitution model (52) ranged from 0.15 to 0.24 substitutions per site, median 0.218) and (ii) the envelope gene comprises both conserved and variable regions. This ensured that the benchmarking was performed on a complex example of ‘real-life’ data to which mapping tools are applied.
Using one representative sequence per subtype we extracted all possible pairwise alignments resulting in 55 unique alignments. For each alignment we used ART (48) to generate artificial Roche/454 and Illumina reads (1000 reads per pairwise alignment) from the second sequence of the pair. Further, ART generated SAM files containing the correct alignment of each simulated read to the reference sequence (the first sequence), which were used as the ‘gold standard’ for evaluation of the accuracy of the various mapping tools. The choice of ART has the added benefit of creating greater test coverage, ensuring that RAMICS is not just incidentally biased to MASON's model of read simulation. For both the Roche/454 and Illumina data the qscore tool (8) was used to evaluate each of the mapping tools, giving the Q Score and Cline shift similarity scores of each tool's generated alignment to the gold standard.
We then ranked the Q Score and Cline shift for each tool that chose to map each read, without penalizing tools that chose not to map the read as Rabema does. When multiple tools achieved the same score (most often 1.0 in the case of trivially mappable reads), we assigned each tool the average score they would receive, to avoid artificially inflating the scores of tools that chose to map only trivially mappable reads. This follows the ranking scheme used in the Friedman test (53) and removes the assumption of normality across easy and hard to map reads. We then report the average score obtained across all reads mapped, with a score closer to 1 indicating a consistently higher ranking of a mapping approach. We repeated each benchmark five times with five different random seeds to ensure statistical significance.
Speed performance
We benchmarked the wall clock time of RAMICS, and other tools, in mapping amplicon-style data. We mapped one million Roche GS Junior FASTQ reads (average length 237 base pairs) from the HIV reverse transcriptase (RT) gene to a short, 312 base pair region of the HXB2 HIV-1 reference sequence (54). All benchmarks were performed on an Intel® Core™ i5–3210M CPU @ 2.50 GHz and an NVIDIA® GeForce® GT 525M graphics card.
RESULTS AND DISCUSSION
Evaluation of the RAMICS algorithm
Initially we evaluated the various components of the RAMICS algorithm to explore whether the addition of complexity improves the quality of the resulting alignment. We mapped both the Roche/454 and Illumina versions of the AFAB benchmarking data using the simplest incarnation of RAMICS (without codon alignment or training), RAMICS with codon alignment and the full RAMICS algorithm (with both the codon-based alignment and training parameters invoked). The addition of codon-based alignment increased the number of perfectly aligned sequence reads by 7.9 and 10.5% for the Roche/454 and Illumina data, respectively (Figure 3). Further, including three rounds of training of the HMM to the query data provided marginal improvements in the number of perfectly mapped reads by 1.5% (Roche/454) and 0.9% (Illumina). The number of discarded reads remained mostly unchanged regardless of the parameters used (Figure 3) showing that while alignment accuracy increases with algorithmic complexity, RAMICS is consistently capable of distinguishing between mappable and unmappable reads. The lesser impact of the training parameter can be explained by its intended use case: the proper alignment of extreme outlier reads with large numbers of sequencing errors. Thus, in most instances we suggest that RAMICS should be used with both the codon-based alignment and training parameters invoked. For all further benchmarking analysis we used the full RAMICS algorithm including both the codon-based alignment and training parameters.
Figure 3.
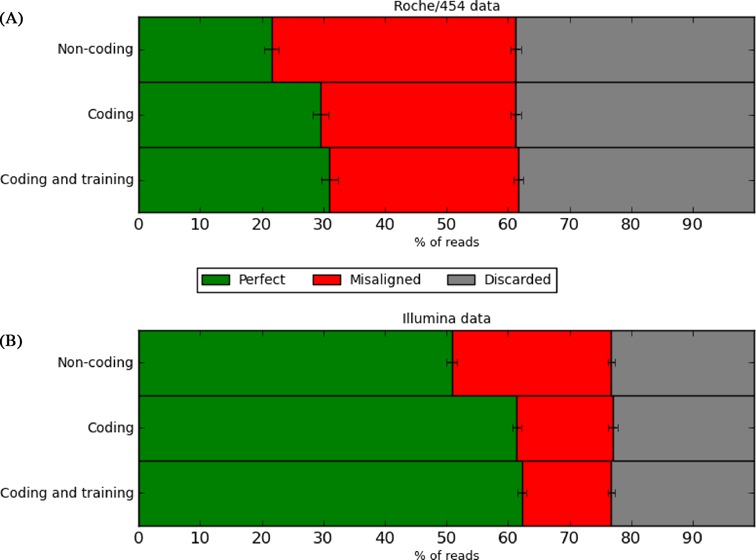
Alignment accuracy comparison of the components of the RAMICS algorithm. For each mapping approach, we show the percentage of simulated (A) Roche/454 and (B) Illumina reads that were misaligned (i.e. not discarded by the mapping tool but with an incorrect alignment when compared to the ‘gold standard’) as well as the percentage of reads that resulted in a perfect alignment when compared with the ‘gold standard’ alignment. For both of the data sets the most complex model (coding and training) performs best.
Comparison of RAMICS to other tools
The use case for which RAMICS is designed is rather unlike that for which most other mapping tools are tailored, because of its focus on coding DNA. Despite this, many studies have used various mapping tools to analyze sequence reads in ways similar to that which RAMICS was designed for (55–58) and, thus, we present comparisons of RAMICS to a number of commonly used mapping tools on the basis of alignment quality and speed.
Comparison on the basis of alignment quality
The Rabema benchmark
Initially we compared RAMICS’ mapping quality against other tools using the Rabema benchmark (39) for both Roche/454 and Illumina data. Reads were simulated from a single reference sequence using the reported error rates of the sequencing platforms as encoded by the Mason read simulator (50). These resulting sequence reads are then binned by their percentage difference from their seed sequence. For example, if the seed sequence used to generate a read was 100 nucleotides long and the resulting read had three differences relative to its seed, then that read would be added into the 3% error bin. This allowed the evaluation of the mapping tools using realistic error rates for both typical and outlier sequences. As would be expected, the observed error rate for vast majority of sequence reads for the Illumina data set fell around 1% with 88% of sequences exhibiting an error rate of ≤1%. For the 454 data, 49% of the sequence reads exhibited an error rate of ≤1% while the error rate of the remaining reads was in excess of 1%. The higher-than-expected number of sequences with an error rate of >1% most likely results from the fact that the single reference sequence used to seed the simulations contained a number of homopolymer regions, which are known to increase the overall observed error rate of the Roche/454 platform (12,13).
For the Roche/454 benchmark, the performance of all of the tools tested was comparable for reads containing zero errors, ranging from 100% to 97.9% of reads correctly mapped for RAMICS and SSAHA, respectively (Figure 4A). Such high performance is to be expected, as these sequence reads are identical to the reference sequence. As the percentage of erroneous nucleotides in a read increased, the accuracy of each of the mapping tools, with the exception of RAMICS, reduced (Figure 4A). SHRiMP (1) was excluded from the Roche/454 benchmark as it discarded over 70% of the reads containing 4% erroneous nucleotides with the recommended settings.
Figure 4.
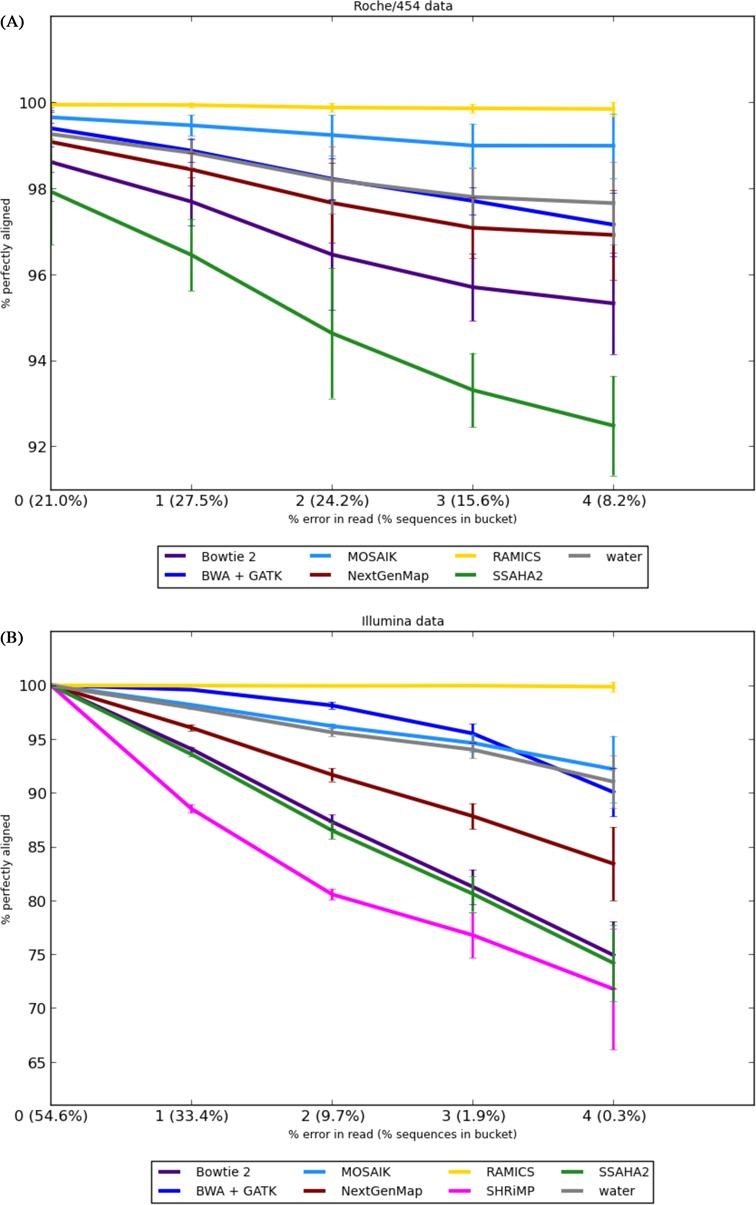
Alignment accuracy using the Rabema benchmarks. The percentage of reads mapped correctly as a function of the percentage error in simulated sequence reads for both the Roche/454 (A) and Illumina (B) sequencing platforms. Reads are binned together by the Rabema tool according to the percentage of erroneous nucleotides in the simulated read relative to its seed sequence. The numbers in parentheses following the percentage error represent the mean percentage of simulated reads (across the five Rabema runs) that were binned by Rabema into that error category. We have excluded BWA-MEM without GATK as it performed identically to BWA-MEM + GATK. SHRiMP was excluded from the Roche/454 benchmark as it performed extremely poorly on this test.
A similar result was observed for the Illumina benchmarking data with all methods correctly mapping 100% of reads at zero error rate and the accuracy of the various approaches, with the exception of RAMICS, reducing as the percentage error rate increased (Figure 4B). For the Illumina benchmark data we observed that compared to the Roche/454 data, the rate of reduction of accuracy is much sharper as the error rate increases. We suggest this effect is due to the shorter lengths of Illumina reads, which leave fewer correct nucleotides on either side of an error with which to anchor an alignment.
RAMICS outperforms all of the other approaches for both the Roche/454 and Illumina benchmarking data sets. This is because RAMICS renders sequencing error in reads when aligning to a reference sequence almost irrelevant, even in outlier reads with a large number of errors, by correctly identifying likely sequencing errors. MOSAIK was the next best performing tool (albeit with a reduction of 8% in accuracy at an error rate of 4% in the Illumina data), while SHRiMP and SSAHA2, both of which conservatively choose not to align reads, quickly deteriorate in their mapping ability with increasing error rate (Figure 4). The GATK IndelRealigner (23) made no statistically significant difference to the test results, so we have removed it from Figure 4 for clarity.
The AFAB benchmark
The Rabema benchmark clearly shows that RAMICS outperforms all other mapping approaches on the outlier sequences with a higher-than-average error rate. However, these data are not a true reflection of the type of data for which RAMICS has been optimized, namely mapping a diverse set of sequence reads to a related reference sequence. Rabema data only differ from the reference sequence through the insertion of sequencing errors and, thus, we developed a benchmarking tool to enable the assessment of mapping tools at aligning coding sequence reads containing sequencing errors that are diverse from the reference sequence. In contrast to the Rabema benchmarking tool, this approach enabled the use of two robust methods for evaluating relative alignment accuracy enabling the identification of perfectly aligned reads and misaligned reads. Reads discarded during mapping were not penalized in the final ranking thereby allowing tools to discard ambiguous reads without incurring unnecessary bias in the benchmarking process.
Extraction of all possible pairwise alignments of subtypes A through G from the ‘gold standard’ HIV alignment resulted in 55 sequence pairs, each of which were evaluated using this approach. Firstly, we considered the percentage of reads that each mapping approach was capable of aligning regardless of the quality of the alignment. Needle (45) does not possess the capability to discard any reads, resulting in 100% of reads being mapped for both the Illumina and Roche/454 data (Figure 5). The percentage of reads retained by the other approaches ranges from 38% (SHRiMP) to 94% (MOSAIK) for the Roche/454 data and 20% (BWA-MEM) and 62% (RAMICS) for the Illumina data (Figure 5).
Figure 5.
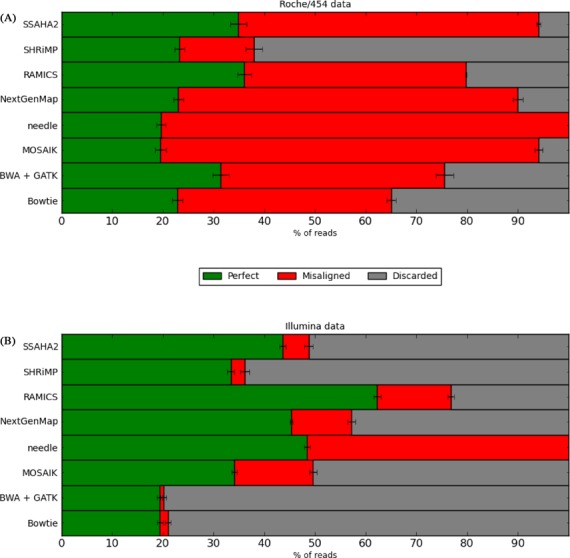
Alignment accuracy for coding data. For each mapping approach evaluated we show the percentage of simulated (A) Roche/454 and (B) Illumina reads that were misaligned (i.e. not discarded by the mapping tool but with an incorrect alignment when compared to the ‘gold standard’) as well as the percentage of reads that resulted in a perfect alignment when compared with the ‘gold standard’ alignment. We have excluded BWA-MEM without GATK as it performed identically to BWA-MEM + GATK.
As expected, because these data contained sequencing errors and were diverse from the reference sequence, the percentage of perfectly mapped reads was lower for all approaches than was observed for the Rabema benchmarking data. For the Illumina data, RAMICS significantly outperforms all of the other mapping approaches by aligning 62% of the total number of reads perfectly, with the next best approach (needle) mapping 48% of all reads correctly (Figure 5). For the Roche/454 data RAMICS performed best, correctly mapping 36% of reads perfectly, while the performance of the other methods ranges between 19% (MOSAIK) and 34% (SSAHA2). The GATK IndelRealigner made no difference to the outcome for BWA-MEM, so we have removed standalone BWA-MEM from Figure 5 for clarity.
It is also interesting to consider the read discarding strategies employed by the various approaches. Ideally, a mapping tool would discard all reads that it could not align perfectly, while still aligning as many reads as possible. Considering that all reads in this benchmark data set are simulated from a gold standard alignment, Bowtie and BWA-MEM's strategy of discarding almost 80% of the reads leads to a high rate of perfect alignment, but an unacceptable loss of coverage depth in situations where many variants are being aligned or deep coverage is necessary.
For all of the reads that were aligned by one or more mapping tools we used two approaches for evaluating alignment quality, the Q Score (59) and Cline shift score (60), to rank each of the mapping tools for each of the 5500 pairs of sequences reads that were tested.
The perfect mapping tool would rank first or joint first compared to all other tools for every single comparison. When two or more tools received equal ranking ties were broken according to the Friedman test (53) by assigning, for example, a ranking of 1.5 to each tool if two tools were equally first, preventing a bias toward tools that aligned more reads. The average Q Score ranks for the methods ranged from 2.512 to 4.846 and 2.978 to 4.562 for the Illumina and Roche/454 data, respectively (Table 1). The average Cline shift ranks range from 2.522 to 4.565 and 3.039 to 4.817 for the Illumina and Roche/454 data, respectively. For both benchmarking data sets RAMICS was the highest ranked mapping approach tested, with BWA-MEM performing worst over both data sets (Table 1). The addition of the GATK (23) to the BWA-MEM toolchain, the approach used in the 1000 Genomes Project (25), had a negligible effect on the quality of alignment for this use case.
Table 1.
The average rank over aligned reads only for the given mapping tools
| Tool | Illumina rank | Roche/454 rank |
|---|---|---|
| Bowtie 2 | 4.00 | 3.76 |
| BWA-MEM | 4.13 | 4.00 |
| MOSAIK | 3.98 | 3.98 |
| needle | 2.97 | 3.70 |
| NextGenMap | 3.45 | 3.80 |
| RAMICS | 2.43 | 2.46 |
| SHRiMP | 3.71 | 3.82 |
| SSAHA2 | 3.37 | 3.43 |
For both test data sets, the highest ranked mapping approach is marked in bold.
Thus, for both benchmarking approaches used across both Illumina and Roche/454 data, RAMICS outperforms all other methods tested here in terms of alignment quality. We must reiterate, however, that all approaches here are being tested against data that have been simulated to test the use case for which RAMICS has been specifically designed. While RAMICS outperforms all of the mapping approaches tested here, it is context based (focused on coding DNA that is divergent from the reference sequence) and, thus, it should not be assumed that RAMICS outperforms all methods in all scenarios, e.g. whole-genome mapping.
Speed performance
While alignment accuracy for mapping tools is essential, the scale of the data sets being generated by high-throughput sequencing approaches means that mapping speed is also a critical factor for an optimal method. With this in mind we have designed RAMICS to harness the computing power of graphic processing units (GPUs) for optimal speed. We have also developed a CPU-based version for researchers who do not have access to GPU hardware. Here, we have compared the mapping speeds of both the CPU and GPU versions of RAMICS against the other mapping approaches in mapping one million Roche/454 reads from the HIV RT gene to a reference sequence. We found that, on average, the speeds for all approaches scale linearly as the data set size increases (data not shown) and find that in most instances NextGenMap is the fastest of the tools tested while MOSAIK is the slowest (Figure 6). Not surprisingly, we find that the GPU version of RAMICS outperforms the CPU version with as much as a 3-fold speedup. Nonetheless we find that RAMICS performs in line with most of the read mappers tested.
Figure 6.
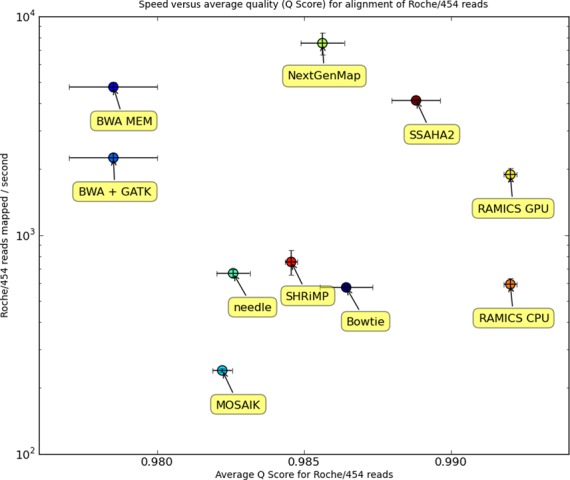
Comparing mapping tools in terms of both speed (reads mapped per second) and accuracy (average Q Score of the Roche/454 reads generated using the AFAB benchmarking tool). The error bars on each data point reflect the range for both the speed (y-axis) and accuracy (x-axis). RAMICS’ speed performance remains competitive with some of the fastest mappers, while its quality performance outstrips them significantly.
The majority of the other mapping tools are built to align reads quickly to a large genome using various heuristics and, thus, much of the overhead necessary to hash a large genome is redundant in these analyses. RAMICS, on the other hand, is designed to perform a full dynamic programming alignment to a shorter reference sequence quickly and accurately.
Comparing the RAMICS GPU version to the EMBOSS tool needle (45), which performs a full dynamic programming alignment, shows that RAMICS is roughly twice as fast (Figure 6). Further, the efficiency of RAMICS is evident in that the codon-based approach implemented means it must perform eight comparisons to needle's three, and makes extensive use of floating point arithmetic to achieve the fine-grained distinctions required for its more complicated alignment model. Indeed, RAMICS non-coding, which performs a Needleman–Wunsch style alignment but takes into account homopolymer errors, is itself twice as fast as the full RAMICS algorithm (data not shown).
There should, however, be no trade-off in quality for speed or vice versa and, thus, the optimal mapping approach is one that can map the greatest number of reads with the highest accuracy in the shortest time. By evaluating each mapping approach in terms of speed and accuracy together (Figure 6), we find that the GPU-based version of the full RAMICS codon-based algorithm outperforms all other approaches suggesting that, for amplicon-style analyses at least, many of the other approaches exhibit such a trade-off.
To our knowledge, RAMICS is the first mapping tool that undertakes accurate mapping of coding DNA to a reference sequence at speed, while concurrently identifying and correcting sequencing platform-induced errors. RAMICS’ accuracy in generating biologically correct alignments of coding DNA means that it can be applied wherever the generation of high-quality alignments for sensitive detection of SNPs and novel variants are required.
AVAILABILITY
The RAMICS software (University of the Western Cape, Copyright Reserved, 2013) is available under license from the University of the Western Cape, South Africa at http://hiv.sanbi.ac.za/tools#/ramics. Terms of the license will include but are not limited to the following: an annual up-front licensing fee for use only will be applicable for any use of the software for commercial purposes (to be defined), and a ‘free use’ only license will be available to verified and approved academic institutions and public not-for-profit research organizations for non-commercial use and/or application only.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online, including [1–19].
ACKNOWLEDGMENT
Thanks are due to Hannah Ajoge, Baruch Lubinsky, Ram Krishna Shrestha and Natasha Wood for useful discussions.
FUNDING
South African Department of Science and Technology [to S.A.T.]; South African National Research Foundation DAAD Study Bursary (11/4/1) [to I.A.W.]; South African Medical Research Council [to SANBI]. Funding for open access charge: South African Department of Science and Technology.
Conflict of interest statement. None declared.
REFERENCES
- 1.David M., Dzamba M., Lister D., Ilie L., Brudno M. SHRiMP2: sensitive yet practical short read mapping. Bioinformatics. 2011;27:1011–1012. doi: 10.1093/bioinformatics/btr046. [DOI] [PubMed] [Google Scholar]
- 2.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li H., Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ning Z. SSAHA: a fast search method for large DNA databases. Genome Res. 2001;11:1725–1729. doi: 10.1101/gr.194201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith D.R., Quinlan A.R., Peckham H.E., Makowsky K., Tao W., Woolf B., Shen L., Donahue W.F., Tusneem N., Stromberg M.P. Rapid whole-genome mutational profiling using next-generation sequencing technologies. 2008;18:1638–1642. doi: 10.1101/gr.077776.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Notredame C., Higgins D.G., Heringa J. T-coffee: a novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000;302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- 8.Edgar R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thompson J.D., Higgins D.G., Gibson T.J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Balzer S., Malde K., Lanzén A., Sharma A., Jonassen I. Characteristics of 454 pyrosequencing data—enabling realistic simulation with flowsim. Bioinformatics. 2010;26:i420–i425. doi: 10.1093/bioinformatics/btq365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harismendy O., Ng P.C., Strausberg R.L., Wang X., Stockwell T.B., Beeson K.Y., Schork N.J., Murray S.S., Topol E.J., Levy S., et al. Evaluation of next generation sequencing platforms for population targeted sequencing studies. Genome Biol. 2009;10:R32. doi: 10.1186/gb-2009-10-3-r32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huse S.M., Huber J.A., Morrison H.G., Sogin M.L., Welch D.M. Accuracy and quality of massively parallel DNA pyrosequencing. Genome Biol. 2007;8:R143. doi: 10.1186/gb-2007-8-7-r143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Quail M.A., Smith M., Coupland P., Otto T.D., Harris S.R., Connor T.R., Bertoni A., Swerdlow H.P., Gu Y. A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genomics. 2012;13:341. doi: 10.1186/1471-2164-13-341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang X., Chockalingam S.P., Aluru S. A survey of error-correction methods for next-generation sequencing. Brief. Bioinform. 2013;14:56–66. doi: 10.1093/bib/bbs015. [DOI] [PubMed] [Google Scholar]
- 15.Loman N.J., Misra R.V., Dallman T.J., Constantinidou C., Gharbia S.E., Wain J., Pallen M.J. Performance comparison of benchtop high-throughput sequencing platforms. Nat. Biotechnol. 2012;30:434–439. doi: 10.1038/nbt.2198. [DOI] [PubMed] [Google Scholar]
- 16.Birney E., Thompson J.D., Gibson T.J. PairWise and SearchWise: finding the optimal alignment in a simultaneous comparison of a protein profile against All DNA translation frames. Nucleic Acids Res. 1996;24:2730–2739. doi: 10.1093/nar/24.14.2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guan X., Uberbacher E.C. Alignments of DNA and protein sequences containing frameshift errors. Comput. Appl. Biosci. CABIOS. 1996;12:31–40. doi: 10.1093/bioinformatics/12.1.31. [DOI] [PubMed] [Google Scholar]
- 18.Ranwez V., Harispe S., Delsuc F., Douzery E.J.P. MACSE: Multiple Alignment of Coding SEquences accounting for frameshifts and stop codons. PLoS ONE. 2011;6:e22594. doi: 10.1371/journal.pone.0022594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang T., Yang L., Jiang H., Tian G., Zhang X. High-performance single-chip exon capture allows accurate whole exome sequencing using the Illumina Genome Analyzer. Sci. China Life Sci. 2011;54:945–952. doi: 10.1007/s11427-011-4232-4. [DOI] [PubMed] [Google Scholar]
- 20.Piskol R., Ramaswami G., Li J.B. Reliable identification of genomic variants from RNA-seq data. Am. J. Hum. Genet. 2013;93:641–651. doi: 10.1016/j.ajhg.2013.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Quinn E.M., Cormican P., Kenny E.M., Hill M., Anney R., Gill M., Corvin A.P., Morris D.W. Development of strategies for SNP detection in RNA-seq data: application to lymphoblastoid cell lines and evaluation using 1000 Genomes data. PLoS ONE. 2013;8:e58815. doi: 10.1371/journal.pone.0058815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clavel F., Hance A.J. HIV drug resistance. N. Engl. J. Med. 2004;350:1023–1035. doi: 10.1056/NEJMra025195. [DOI] [PubMed] [Google Scholar]
- 23.McKenna A.H., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M., et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.The 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wernersson R., Pedersen A.G. RevTrans: multiple alignment of coding DNA from aligned amino acid sequences. Nucleic Acids Res. 2003;31:3537–3539. doi: 10.1093/nar/gkg609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bininda-Emonds O.R. transAlign: using amino acids to facilitate the multiple alignment of protein-coding DNA sequences. BMC Bioinformatics. 2005;6:156. doi: 10.1186/1471-2105-6-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mott R. EST_GENOME: a program to align spliced DNA sequences to unspliced genomic DNA. Comput. Appl. Biosci. CABIOS. 1997;13:477–478. doi: 10.1093/bioinformatics/13.4.477. [DOI] [PubMed] [Google Scholar]
- 29.Birney E., Clamp M., Durbin R. GeneWise and Genomewise. Genome Res. 2004;14:988–995. doi: 10.1101/gr.1865504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eddy S.R. Profile hidden Markov models. Bioinformatics. 1998;14:755–763. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- 31.Baum L.E., Petrie T. Statistical inference for probabilistic functions of finite state Markov chains. Ann. Math. Stat. 1966;37:1554–1563. [Google Scholar]
- 32.Doron-Faigenboim A., Pupko T. A combined empirical and mechanistic codon model. Mol. Biol. Evol. 2007;24:388–397. doi: 10.1093/molbev/msl175. [DOI] [PubMed] [Google Scholar]
- 33.Goldman N., Yang Z. A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol. Biol. Evol. 1994;11:725–736. doi: 10.1093/oxfordjournals.molbev.a040153. [DOI] [PubMed] [Google Scholar]
- 34.Kosiol C., Holmes I., Goldman N. An empirical codon model for protein sequence evolution. Mol. Biol. Evol. 2007;24:1464–1479. doi: 10.1093/molbev/msm064. [DOI] [PubMed] [Google Scholar]
- 35.Margulies M., Egholm M., Altman W.E., Attiya S., Bader J.S., Bemben L.A., Berka J., Braverman M.S., Chen Y.-J., Chen Z., et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rothberg J.M., Hinz W., Rearick T.M., Schultz J., Mileski W., Davey M., Leamon J.H., Johnson K., Milgrew M.J., Edwards M., et al. An integrated semiconductor device enabling non-optical genome sequencing. Nature. 2011;475:348–352. doi: 10.1038/nature10242. [DOI] [PubMed] [Google Scholar]
- 37.Bennett S. Solexa Ltd. Pharmacogenomics. 2004;5:433–438. doi: 10.1517/14622416.5.4.433. [DOI] [PubMed] [Google Scholar]
- 38.Choi M., Scholl U.I., Ji W., Liu T., Tikhonova I.R., Zumbo P., Nayir A., Bakkaloğlu A., Özen S., Sanjad S., et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc. Natl. Acad. Sci. U.S.A. 2009;106:19096–19101. doi: 10.1073/pnas.0910672106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ng S.B., Turner E.H., Robertson P.D., Flygare S.D., Bigham A.W., Lee C., Shaffer T., Wong M., Bhattacharjee A., Eichler E.E., et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461:272–276. doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Trapnell C., Salzberg S.L. How to map billions of short reads onto genomes. Nat. Biotechnol. 2009;27:455–457. doi: 10.1038/nbt0509-455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.de Oliveira T., Deforche K., Cassol S., Salminen M., Paraskevis D., Seebregts C., Snoeck J., van Rensburg E.J., Wensing A.M.J., van de Vijver D.A., et al. An automated genotyping system for analysis of HIV-1 and other microbial sequences. Bioinformatics. 2005;21:3797–3800. doi: 10.1093/bioinformatics/bti607. [DOI] [PubMed] [Google Scholar]
- 42.Mende D.R., Waller A.S., Sunagawa S., Jarvelin A.I., Chan M.M., Arumugam M., Raes J., Bork P. Assessment of metagenomic assembly using simulated next generation sequencing data. PLoS ONE. 2012;7:e31386. doi: 10.1371/journal.pone.0031386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gouy M., Guindon S., Gascuel O. SeaView Version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 2010;27:221–224. doi: 10.1093/molbev/msp259. [DOI] [PubMed] [Google Scholar]
- 44.Sievers F., Wilm A., Dineen D., Gibson T.J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rice P., Longden I., Bleasby A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 2000;16:276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 46.Sedlazeck F.J., Rescheneder P., von Haeseler A. NextGenMap: fast and accurate read mapping in highly polymorphic genomes. 2013;29:2790–2791. doi: 10.1093/bioinformatics/btt468. [DOI] [PubMed] [Google Scholar]
- 47.Brudno M., Malde S., Poliakov A., Do C.B., Couronne O., Dubchak I., Batzoglou S. Glocal alignment: finding rearrangements during alignment. Bioinformatics. 2003;19:i54–i62. doi: 10.1093/bioinformatics/btg1005. [DOI] [PubMed] [Google Scholar]
- 48.Holtgrewe M., Emde A.-K., Weese D., Reinert K. A novel and well-defined benchmarking method for second generation read mapping. BMC Bioinformatics. 2011;12:210. doi: 10.1186/1471-2105-12-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ruffalo M., LaFramboise T., Koyutürk M. Comparative analysis of algorithms for next-generation sequencing read alignment. Bioinformatics. 2011;27:2790–2796. doi: 10.1093/bioinformatics/btr477. [DOI] [PubMed] [Google Scholar]
- 50.Holtgrewe M. Mason—a read simulator for second generation sequencing data. 2010. Technical Report, FU Berlin.
- 51.Frahm N., Baker B., Brander C. Identification and optimal definition of HIV-derived cytotoxic T lymphocyte (CTL) epitopes for the study of CTL escape, functional avidity and viral evolution. HIV Mol. Immunol. 2008;2008:3–24. [Google Scholar]
- 52.Tavaré S. Some probabilistic and statistical problems in the analysis of DNA sequences. Lect. Math. Life Sci. 1986;17:57–86. [Google Scholar]
- 53.Friedman M. The use of ranks to avoid the assumption of normality implicit in the analysis of variance. J. Am. Stat. Assoc. 1937;32:675–701. [Google Scholar]
- 54.Ratner L., Haseltine W., Patarca R., Livak K.J., Starcich B., Josephs S.F., Doran E.R., Rafalski J.A., Whitehorn E.A., Baumeister K., et al. Complete nucleotide sequence of the AIDS virus, HTLV-III. Nature. 1985;313:277–284. doi: 10.1038/313277a0. [DOI] [PubMed] [Google Scholar]
- 55.Chang S.T., Sova P., Peng X., Weiss J., Law G.L., Palermo R.E., Katze M.G. Next-generation sequencing reveals HIV-1-mediated suppression of T cell activation and RNA processing and regulation of noncoding RNA expression in a CD4+ T cell line. mBio. 2011;2:e00134-11. doi: 10.1128/mBio.00134-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Henn M.R., Boutwell C.L., Charlebois P., Lennon N.J., Power K.A., Macalalad A.R., Berlin A.M., Malboeuf C.M., Ryan E.M., Gnerre S., et al. Whole genome deep sequencing of HIV-1 reveals the impact of early minor variants upon immune recognition during acute infection. PLoS Pathog. 2012;8:e1002529. doi: 10.1371/journal.ppat.1002529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Radford A.D., Chapman D., Dixon L., Chantrey J., Darby A.C., Hall N. Application of next-generation sequencing technologies in virology. J. Gen. Virol. 2012;93:1853–1868. doi: 10.1099/vir.0.043182-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Watson S.J., Welkers M.R.A., Depledge D.P., Coulter E., Breuer J.M., de Jong M.D., Kellam P. Viral population analysis and minority-variant detection using short read next-generation sequencing. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013;368:1614. doi: 10.1098/rstb.2012.0205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang G., Dunbrack R.L. Jr. Scoring profile-to-profile sequence alignments. Protein Sci. 2004;13:1612–1626. doi: 10.1110/ps.03601504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cline M., Hughey R., Karplus K. Predicting reliable regions in protein sequence alignments. Bioinformatics. 2002;18:306–314. doi: 10.1093/bioinformatics/18.2.306. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.