


Abstract
We provide the first comprehensive analysis of any transcription factor family in Cryptosporidium, a basal-branching apicomplexan that is the second leading cause of infant diarrhea globally. AP2 domain-containing proteins have evolved to be the major regulatory family in the phylum to the exclusion of canonical regulators. We show that apicomplexan and perkinsid AP2 domains cluster distinctly from other chromalveolate AP2s. Protein-binding specificity assays of C. parvum AP2 domains combined with motif conservation upstream of co-regulated gene clusters allowed the construction of putative AP2 regulons across the in vitro life cycle. Orthologous Apicomplexan AP2 (ApiAP2) expression has been rearranged relative to the malaria parasite P. falciparum, suggesting ApiAP2 network rewiring during evolution. C. hominis orthologs of putative C. parvum ApiAP2 proteins and target genes show greater than average variation. C. parvum AP2 domains display reduced binding diversity relative to P. falciparum, with multiple domains binding the 5′-TGCAT-3′, 5′-CACACA-3′ and G-box motifs (5′-G[T/C]GGGG-3′). Many overrepresented motifs in C. parvum upstream regions are not AP2 binding motifs. We propose that C. parvum is less reliant on ApiAP2 regulators in part because it utilizes E2F/DP1 transcription factors. C. parvum may provide clues to the ancestral state of apicomplexan transcriptional regulation, pre-AP2 domination.
INTRODUCTION
Apicomplexan parasites are the causative agents of some of the world's most devastating infectious diseases, including malaria (caused by Plasmodium), toxoplasmosis (T. gondii) and cryptosporidiosis (Cryptosporidium). Cryptosporidium species, primarily C. parvum and C. hominis, have recently been revealed to be the second leading cause of infant diarrhea globally (1). Plasmodium and Cryptosporidium diverged between 824 and 350 mya (2) with more recent estimates of ∼420 mya (3,4), a distance comparable to that between humans and the ancestral chordate (5). While RNA polymerase-associated factors and basal transcription factors have been identified in the Apicomplexa (6), examination of apicomplexan proteomes yielded a surprising dearth of ‘typical’ eukaryotic enhancer proteins and their characteristic binding sites (7,8). These findings were highly unexpected, given the extensive evidence for transcriptional control (9–11). The lack of recognizable, specific transcription factors initially suggested that the specific transcription factors were likely so divergent from those found in other eukaryotes that they were unrecognizable. Balaji et al. (8) took a more sophisticated approach to tackle this issue by generating new Hidden Markov Models (HMMs) to perform sensitive sequence analyses of several apicomplexan genomes (Plasmodium, Cryptosporidium, Theileria) for all known DNA-binding domains (8). The team confirmed the dearth of ‘typical’ DNA-binding enhancer proteins, but they did identify Myb and zinc-finger proteins, as well as an unexpected family of proteins with multiple members present in all examined apicomplexan genomes. This Apicomplexan AP2 family of proteins (ApiAP2) bears resemblance to the AP2/ERF family of DNA-binding transcription factors first identified in plants (8). Subsequent work has indicated the near-complete domination of this acquired ApiAP2 family of transcription factors in apicomplexan transcriptional regulation (12–17). How the AP2 domain came to reside in the Apicomplexa and how radiation of this family led to the assumption of regulatory duties from traditional eukaryotic transcription factors, such as Myb and C2H2 zinc fingers, which are still found across Apicomplexa (18), or E2F/DP1, which is absent in all studied apicomplexans except for Cryptosporidium (19), is unresolved.
At the initial discovery of ApiAP2 proteins in 2005, the authors postulated that the apicomplexan AP2 domain is likely of plant origin. Given the evolutionary history of the Apicomplexa which includes the secondary endosymbiosis of an alga (recently shown to be rhodophyte in origin (20)) whose only remnants are the non-photosynthetic apicoplast organelle, it was highly plausible that the AP2 domain had been transferred from the algal endosymbiont to the host nuclear genome (8). Many cases of gene transfer from algae, cyanobacteria, viruses and even metazoa to the nuclear genome have been documented in the Apicomplexa (21–25). This hypothesis regarding the origin of AP2 domains in the Apicomplexa has had little follow-up. However, as more genome sequences have been generated, AP2 domains have been found throughout the tree of life, notably in several bacteria and their phages (8,26,27), and as noted at their initial discovery (8), sequence similarity between these domains does not link apicomplexan AP2 domains to plant AP2 domains to the exclusion of these other groups. Importantly, in most other AP2 families identified, the AP2 domain is associated with homing endonuclease or integrase domains of mobile elements. There is no evidence of mobile elements (active or otherwise) in the apicomplexan genomes examined here. Evidence of retrotransposable elements has been reported in another early branch of the Apicomplexa, the gregarines (28), and there is evidence that apicomplexan genomes which lack mobile elements used to contain them (29,30), or contain inactive elements as is the case in the apicomplexan coccidian parasite Eimeria tenella (31). More recently, non-integrase-associated bacterial AP2 proteins with architectures similar to AP2 proteins found in plants and alveolates have been reported (32). These AP2 proteins are also predicted to function as novel, specific transcription factors.
ApiAP2 proteins are highly divergent in both sequence and length (ranging from ∼200 to thousands of amino acids), and they generally display no discernable homology outside of the AP2 domain (which is ∼60 aa in length). AP2 domains are often the only globular domains in ApiAP2 proteins, and the domain can occur in architectures of one to four or more per protein (8,33). Since the discovery of the ApiAP2 proteins, much work has been done both computationally and experimentally to implicate these proteins in gene regulation. Five ApiAP2 proteins have been identified as key stage-specific regulators in Plasmodium (15–17,34,35). Another ApiAP2 protein (PFF0200c) has been implicated as a player in P. falciparum var gene regulation by binding the SPE2 DNA motif and acting as a DNA-tethering protein involved in formation and maintenance of heterochromatin (36). Campbell et al. (37) characterized the binding motifs for all 27 members of the ApiAP2 family in P. falciparum and used these data in conjunction with intra-erythrocytic expression data to predict putative regulatory targets of these proteins (37). The role of ApiAP2 proteins in gene regulation has also been investigated to a lesser degree in T. gondii, where several ApiAP2 proteins have been implicated in regulating progression through the cell cycle (38) as well as crucial virulence factors (14). Other studies have implicated ApiAP2s in regulating a developmental transition (13). Radke et al. (12) recently characterized a T. gondii AP2 that acts as a repressor of bradyzoite development. This is the first example of an ApiAP2 acting as a repressor and lends further support to the idea that members of the ApiAP2 family are multifaceted in their functional and regulatory capabilities.
The ApiAP2 literature focuses extensively on studies of regulation in Plasmodium and Toxoplasma spp., and there have been no extensive comparative studies of the AP2 domain-containing proteins across the phylum. The relatively low fraction of total gene content that is conserved across the phylum (orthologs) (39) and the widely variable size of total gene content (40) necessitate the evolution of the gene regulatory networks to facilitate these vastly different regulatory demands. Studies to date have not definitively addressed whether orthologous apicomplexan AP2 domains recognize the same DNA sequence motif and if these motifs are found upstream of orthologous sets of genes across apicomplexans. The sparse data that do exist suggest that putative ApiAP2 regulons may be quite different. For example, Campbell et al. found that though P. vivax, P. yoelli and P. falciparum AP2 domains are nearly perfectly conserved, and the timing of orthologous ApiAP2 protein expression is very similar, putative target gene sets of orthologous ApiAP2s are highly divergent (37). DeSilva et al. (41) investigated the binding specificity of a single C. parvum domain that was highly conserved with a Plasmodium AP2 domain. They found that the binding specificities were absolutely conserved; however, of the 127 putative P. falciparum targets of regulation, only 26 are conserved in C. parvum, suggesting the transcriptional network itself has evolved considerably since Plasmodium and Cryptosporidium diverged (41).
In this study, we have used HMMs and phylogenetic analysis to examine the distribution and evolutionary relationships of the AP2 DNA-binding domains across the Apicomplexa and an outgroup perkinsid oyster parasite, Perkinsus marinus. We also examine the relationship of these apicomplexan and perkinsid AP2 domains to the AP2 domains found throughout the chromalveolates. We used the insights gained from these comparisons to select AP2 protein domains representing most of the AP2 family from the basal-branching apicomplexan Cryptosporidium parvum for further study. We determined the binding specificities of these domains experimentally and searched for the identified binding motifs upstream of co-regulated C. parvum gene clusters to identify putative regulatory targets with the goal of identifying the putative ApiAP2 transcriptional regulatory network in this organism. Finally, we compare C. parvum results to the limited data available from C. hominis.
MATERIALS AND METHODS
Identification of AP2 and ApiAP2 domains
To identify ApiAP2 domains for phylogenetic analyses, we developed an HMM that appears to be more sensitive to the specific detection of ApiAP2s than the Pfam HMM designed for the detection of AP2 domains (www.pfam.org). We first ran the existing AP2 HMM on annotated protein sequences for apicomplexans T. gondii, Neospora caninum, P. falciparum, P. vivax, C. parvum, Theileria annulata and T. parva using HMMER (version 2.4i; http://hmmer.org/). We opted to use P. falciparum gene IDs from PlasmoDB version 6.0 (http://plasmodb.org) to facilitate comparisons with existing P. falciparum AP2 domain binding data, and we have provided a look-up table to the most recent gene IDs (Supplementary File S1, Table S1). We next constructed an alignment with the T-coffee package (42) of the most significant domain hits from this run (1e-4 or lower). The ApiAP2 HMM was built from this alignment using HMMER. We used this new HMM in conjunction with the Pfam AP2 HMM to search annotated protein sequences to examine the distribution of the AP2 domain across several chromalveolates, including apicomplexans P. falciparum, P. knowlesi, P. vivax, P. yoelli, T. parva, T. annulata, Babesia bovis, N. caninum, T. gondii, Cryptosporidium muris and C. parvum; the perkinsid oyster parasite Perkinsus marinus; dinoflagellates Karenia brevis and Alexandrium tamarense; ciliates Tetrahymena thermophila, Paramecium tetraurelia and Ichthyophthirius multifiliis and stramenopiles Thalassiosira pseudonana, Phaeodactylum tricornutum, Ectocarpus siliculosus, Phytophthora infestans and Phytophthora sojae. Extant representatives of purported algal endosymbionts Cyanidioschyzon merolae, Porphyra purpurea, P. yezeoensis (representative rhodophytes) and Chlamydomonas reinhardtii and Micromonas sp. RCC299 (representative chlorophytes) were also examined. All Plasmodium annotated proteins were obtained from PlasmoDB version 6.0. All T. gondii and N. caninum annotated proteins were obtained from ToxoDB version 5.2. C. parvum annotated proteins were obtained from CryptoDB version 4.6. T. parva data were obtained from TIGR Eukaryotic Genome Projects (ftp://ftp.tigr.org/pub/data/Eukaryotic_Projects/t_parva/annotation_dbs/). T. annulata data were obtained from the Wellcome Trust Sanger Institute (http://www.sanger.ac.uk/Projects/Pathogens/). As no annotated protein sequences were available at the time of our analyses, dinoflagellate analyses were run on six-frame translations of clustered Expressed Sequence Tags (ESTs) (Open Reading Frames [ORFs] > 75 AA) and perkinsid analyses were run on six-frame translations of the genome (ORFS > 50 AA). The sequences of all domains identified via six-frame translations are available in Supplementary File S2. All other organism annotated protein sequence data were downloaded from the National Center for Biotechnology Information, NCBI GenBank (http://www.ncbi.nlm.nih.gov). AP2 protein and domain counts for each organism were determined using a permissive domain e-value cutoff of 10.
We have found AP2 domain detection to be very sensitive to size of database searched, and some weakly scoring C. parvum domains can be detected when searching its proteins individually (35 domains are detected with the ApiAP2 HMM searching C. parvum only versus 21 domains when all organism proteins are searched concurrently; Supplementary File S1, Table S2); however, they are not significant enough when proteins from all included organisms are searched jointly. Due to these fluctuations, domain counts are only approximate. We tested the two most significant additional domains detected when C. parvum was searched alone (cgd2_2990 and cgd3_1980) on protein-binding microarrays (PBMs). Only one of these, cgd2_2990, had a detectable binding motif.
Phylogenetic analysis of AP2 domains
Determination of homolog groups
All phylogenetic analyses were carried out on AP2 domain sequences only, as full-length proteins are generally too divergent to be able to detect meaningful evolutionary relationships between them (as determined by multiple sequence alignment; data not shown). Alignments of AP2 domain sequences were performed using the HMMALIGN command of the HMMER package (version 2.4i; http://hmmer.org/) and edited using Jalview (43) (edited alignments can be found in Supplementary Files S3 and S4). Unrooted maximum likelihood trees were constructed from top-scoring (1e-3 or better) domain sequences across chromalveolates and green algae C. reinhardtii and Micromonas (Supplementary File S1, Table S3) using RAxML (version released 4/26/2012) with a gamma rate estimation and Dayhoff model of codon substitution (44). Taxa with very similar representatives were not included in the tree for purposes of simplification (P. knowlesi, P. yoelli, T. parva, N. caninum). Bootstrap support was obtained from 100 replicates. Trees were visualized using FigTree (v. 1.4.0; http://tree.bio.ed.ac.uk/software/figtree/). P. falciparum and C. parvum AP2 domains alone were analyzed and a tree was created using the same methods.
To identify homologous clusters of AP2 domains, a local install of the OrthoMCL algorithm (45) was run on all identified AP2 domains in apicomplexans and perkinsids using an e-value ranging from 1e-04 to 1e-11. Domains displaying similarity at these e-values were clustered into homolog groups. Homolog groups found at 1e-06 were used for subsequent analyses, as this is the highest stringency at which orthology could be detected between apicomplexan and P. marinus AP2 domains. 1e-11 is the highest stringency at which orthology between C. parvum and other apicomplexan AP2 domains can be detected. Relationships were visualized using Circos (46).
Comparisons between C. parvum and C. hominis
We focused on C. parvum in our phylogenetic analyses because of the better genome assembly (18 versus 1422 contigs for C. hominis) and availability of gene expression data. However, these species do exhibit differing host ranges and pathogenicity, and comparisons, where possible, are warranted. C. hominis is the predominant Cryptosporidium pathogen of humans, and the available genome sequence is 97% identical to C. parvum (47). The ApiAP2 HMM was run on C. hominis annotated proteins (downloaded from Cryptodb.org version 6.0). Orthologous C. parvum and C. hominis ApiAP2 proteins, as well as select upstream regions of orthologous predicted ApiAP2 target genes, were evaluated for conservation. C. parvum and C. hominis AP2 domains were compared to each other using the blastp package of NCBIblast (version 2.2.26). Upstream regions of select C. parvum AP2 target genes were compared against orthologous C. hominis upstream regions using NCBI blastn (version 2.2.26).
Determination of C. parvum ApiAP2 binding motifs
N-terminal GST fusion proteins were made using the pGEX4T-1 vector (GE Healthcare) and the 23 predicted C. parvum ApiAP2 domains and their flanking residues. Many flanking residues were included to ensure capture of each domain. Domain boundaries were determined using custom-built HMMs run on all annotated C. parvum proteins (downloaded from CryptoDB.org, version 4.6). The domains and flanking sequence were PCR-amplified and cloned into the BamHI restriction site in pGEX4T-1. Proteins were expressed and purified as previously described (41). Briefly, E. coli BL21 (RIL Codon PLUS, Stratagene) cells were induced with 200 mM isopropyl-beta-D-thiogalactopyranoside (IPTG) at 25°C. Proteins were then purified using Uniflow Glutathione Resin (Clontech) and eluted in 10 mM reduced glutathione, 50 mM Tris HCL, pH 8.0. Proteins were verified with western blots using an anti-GST antibody (Invitrogen) and purity was verified by silver stain.
A minimum of two PBM experiments were performed with each purified protein construct to determine their binding specificities as previously described (37,41). Motifs bound at a threshold of 0.45 or greater were considered significant. Similarity between C. parvum ApiAP2 binding sites was determined using the web-based STAMP tool (48). Comparisons between orthologous C. parvum and P. falciparum ApiAP2 binding sites (using P. falciparum ApiAP2 binding motif data from (37,41)), as well as comparisons between C. parvum ApiAP2 binding sites and C. parvum overrepresented upstream motifs (49) were also made using STAMP.
Predictions of putative ApiAP2 target genes
Definition of C. parvum upstream regions
Upstream regions were designated as in (49). Briefly, we downloaded the C. parvum genome (v 4.2) and nucleotide sequences for all protein-encoding genes from CryptoDB (http://cryptodb.org/cryptodb/, (50)). Custom Perl scripts were used to extract either (i) 1 kb of sequence upstream of each translation start site, or (ii) the upstream sequence until a gene was encountered on either strand. The translational start site was used because we do not have untranslated region (UTR) information for predicted genes. The C. parvum genome is only 9.1 Mb and is highly compact with very few introns and small intergenic spaces. To exclude the possibility of including coding regions in this set due to misannotation, a BLASTX was performed against the NCBI NR database using the set of upstream sequences as the query. Upstream sequences that contained significant portions of 100% identity to coding sequences were eliminated.
Target gene prediction
We modified the target prediction algorithm used in (37) for use with our data to identify putative AP2 target genes. This algorithm takes position weight matrices derived from PBM scores for each AP2 domain and searches for matches in the upstream sequence database. Each AP2 is assigned a score for each gene based on motifs found. The glmnet package in R (51) is then implemented to make a regression between this AP2 motif score and the expression pattern for each gene (C. parvum expression data from (52)) to determine how much the AP2 motif contributes to each gene's expression. An average expression pattern for genes possessing a particular AP2 motif upstream is then iteratively built, and genes that match this average expression pattern within a statistical threshold are designated as putative regulatory targets. P. falciparum regulatory targets were previously defined using a false discovery rate of 1% (37). As we have comparatively few time points over which we have expression information (seven for C. parvum versus 47 for P. falciparum) and thus have less statistical power, we considered genes falling within a false discovery rate of 25% as putative regulatory targets.
Evaluating evolutionary history of AP2 domains versus evolutionary history of putative target genes
Putative target genes of shared (‘ancestral’ or ‘pan-apicomplexan’) and lineage-specific ApiAP2 domains were compared against lists of three different evolutionary classes of apicomplexan genes as determined by OrthoMCL: (i) those shared between all of 12 apicomplexans (the 11 used for all other analyses, as well as P. berghei); (ii) genes shared between apicomplexans of at least two different genera and (iii) genus-specific genes (genes which have no orthologs outside of their respective genus). Putative targets were then classified as ‘shared’ or ‘lineage-specific’.
Comparisons between orthologous C. parvum and P. falciparum ApiAP2 networks
P. falciparum orthologs for C. parvum ApiAP2 targets were identified using the ‘transform by orthology’ tool at EupathDB (v. 2.17, http://EupathDB.org). Lists of putative targets for each orthologous P. falciparum ApiAP2 were then searched with the list of C. parvum ApiAP2 target orthologs to identify shared targets.
RESULTS
Perkinsid and apicomplexan AP2 domain families appear distinct from other chromalveolate AP2 domains
It is not known if AP2 domains were present in the chromalveolate ancestor, or if they arrived one or more times as a result of the multiple endosymbiotic and lateral transfer events that characterize the chromalveolates (21–24,53,54). Thus, we examined the distribution of AP2 domains across several chromalveolates including apicomplexans, a perkinsid, dinoflagellates, ciliates and stramenopiles, as well as in extant representatives of the endosymbiont donors to the chromalveolates, rhodophytes and chlorophytes using both a custom-built ApiAP2 HMM (Supplementary File S5) and an existing AP2 HMM available from Pfam. (Figure 1; Supplementary File S1, Tables S4 and S5). Phylogenies constructed from the identified domain sequences indicate that perkinsid Perkinsus marinus AP2 domains are closely related to apicomplexan AP2 domains, and both are more distantly related to other chromalveolate/endosymbiont AP2 domains (Figure 2). Deep evolutionary relationships are difficult, if not impossible to recover due to the short length (∼60 amino acids) of the domain and lack of discernable homology over the rest of the protein (as determined by multiple sequence alignment; data not shown); thus, most deep relationships within the tree are not well-supported. However, the bootstrap support for the divide between apicomplexan/perkinsid AP2 domains and the rest of the chromalveolates and algae is highly significant (Figure 2).
Figure 1.
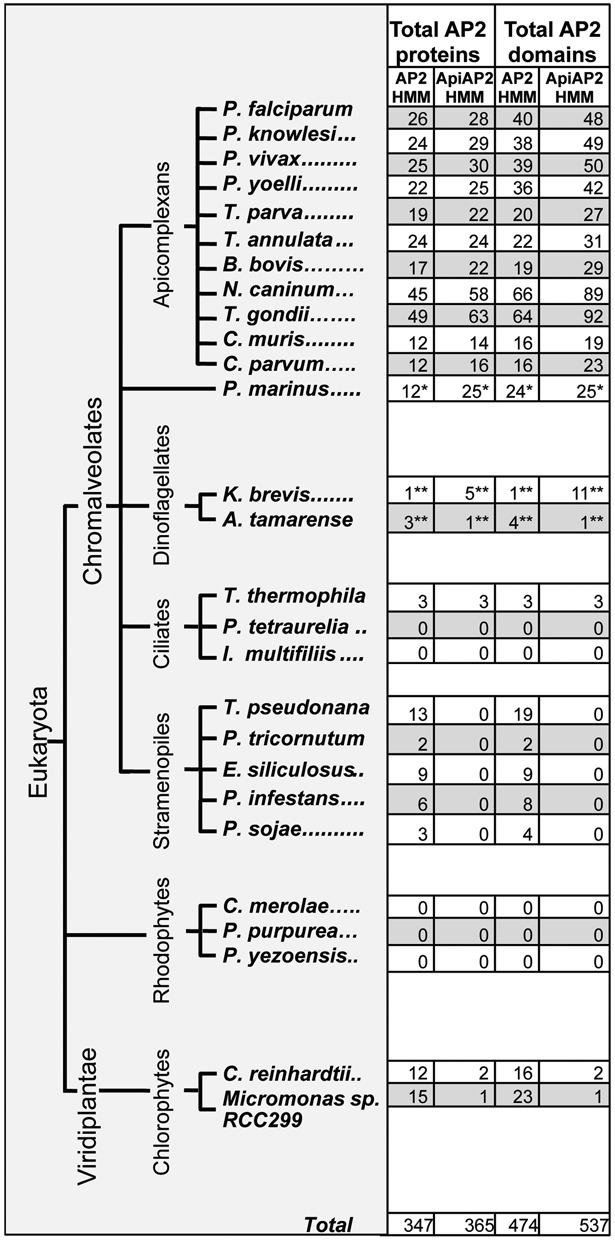
Distribution and quantification of AP2 proteins and domains across chromalveolates and algae. Counts of AP2 domain-containing proteins and the number of AP2 domains per species as determined by sensitive sequence profile analysis using either the AP2 HMM available from PFAM or our custom ApiAP2 HMM. Analyses on most species were run on fully annotated protein sets. **Dinoflagellate analyses were run on clustered EST data. *P. marinus analyses were run on clustered genome ORFs. These counts represent profile matches at or below a permissive e-value of 10. Approximately 85% of the hits were at or below 1e-3.
Figure 2.
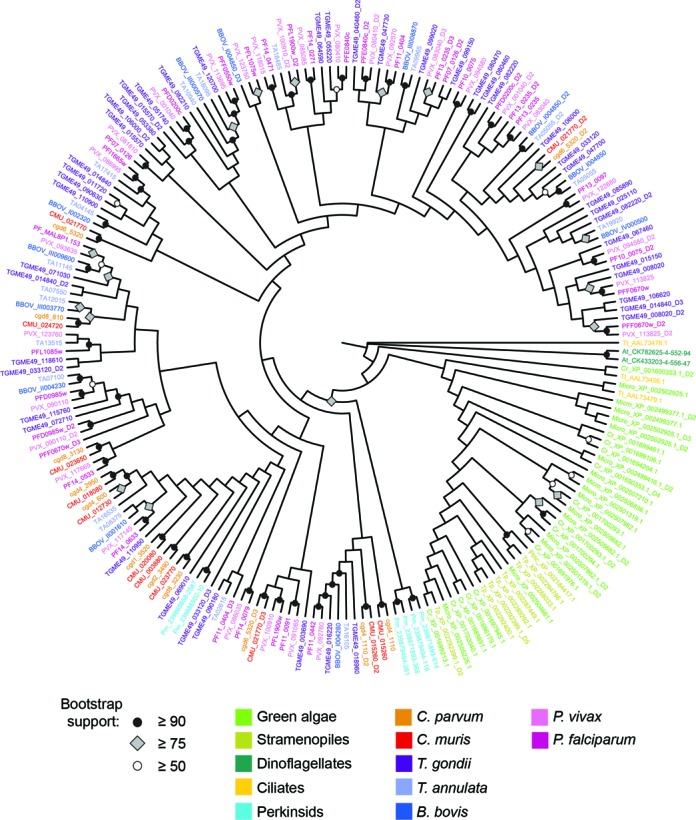
Evolutionary relationships of AP2 domains across chromalveolates and algae. Maximum likelihood tree constructed of top-scoring AP2 domains (hmmsearch domain e-value of 1e-3 or better using the AP2 HMM) from selected taxa. Bootstrap support obtained from 100 replicates are indicated on nodes where support = 50% or greater. Tree constructed with RAxML using a gamma rate estimation and a Dayhoff model of codon evolution, and visualized using FigTree (v. 1.4.0; http://tree.bio.ed.ac.uk/software/figtree/). Species abbreviation prefixes have been added to non-apicomplexan gene IDs for ease of understanding: At_, A. tamarense; Cr_, C. reinhardtii; K_brevis_, K. brevis; Micro_, Micromonas; Pm_, P. marinus; Ptric_, P. tricornutum; Tt_, T. thermophila; Tp_, T. pseudonana. Perkinsid and ApiAP2 domains group together outside of other chromalveolate and green algae AP2 domains with high bootstrap support, indicating possible independent origins for these domains.
Perkinsid and apicomplexan AP2 domains can be classified into evolutionary clades
There are two kinds of intraphylum domains: restricted lineage-specific domains and as many as 19 domains shared between all or most apicomplexans (Supplementary File S6, Figure S1; Table 1). Domain counts and composition of homolog groups vary depending on the stringency of e-value parameters used to assign orthologs to clusters; thus, we indicate ranges of domains determined by OrthoMCL clustering at 1e-4 to 1e-11 in Table 1 (Supplementary File S1, Tables S6–S9). We determined that 1e-6 is the most stringent e-value at which interphylum AP2 domains between apicomplexans and perkinsids can be detected, and we used ≤1e-6 for subsequent analyses. We used phyletic distribution data for all predicted perkinsid and apicomplexan AP2 domains to further subdivide them into evolutionary clades and classify the C. parvum domains. Twenty-three AP2 domains were detected in 18 different C. parvum proteins using these cutoffs.
Table 1. AP2 domain counts by evolutionary group.
| Domain classification | Number of domains in group (range) | Number of domains in group (1e-4) | Number of domains in group (1e-6) | Number of domains in group (1e-8) | Number of domains in group (1e-11) |
|---|---|---|---|---|---|
| Ancestral (shared with P. marinus) | 5 to 6 | 6 | 5 | – | – |
| Present in all or most apicomplexans | 10 to 18 | 18 | 18 | 16 | 10 |
| Plasmodium and piroplasms | 2 to 5 | 2 | 3 | 4 | 5 |
| Plasmodium and coccidians | 7 to 10 | 9 | 10 | 10 | 7 |
| Coccidians and piroplasms | 1 to 2 | 2 | 1 | 1 | 1 |
| Cryptosporidium and piroplasms | 0 to 1 | 1 | 0 | 1 | 1 |
| Cryptosporidium and coccidians | 0 to 2 | 2 | 1 | 2 | 0 |
| Plasmodium-specific | 14 to 29 | 14 | 16 | 22 | 29 |
| Piroplasm-specific | 4 to 9 | 4 | 6 | 6 | 9 |
| Theileria-specific | 2 to 10 | 2 | 2 | 10 | 6 |
| Coccidian-specific | 42 to 69 | 42 | 46 | 56 | 69 |
| Cryptosporidium-specific | 4 to 15 | 4 | 5 | 10 | 15 |
| P. marinus-specific | 0 to 2 | 0 | 2 | – | – |
Table 1. AP2 domain evolutionary classes across apicomplexans and P. marinus as determined by OrthoMCL clustering at e-values ranging from 1e-4 to 1e-11. There is no detectable orthology to P. marinus domains above 1e-6; the ‘ancestral’ and ‘P. marinus-specific’ categories were not determined above this cutoff. All identified apicomplexan and perkinsid AP2 domains were subjected to clustering. Refer to Supplementary File S1, Tables S6–S9 for IDs of domains falling into each classification at each e-value.
The 23 C. parvum AP2 domains were further classified as ancestral, pan-apicomplexan or lineage-specific based on their phyletic distribution with OrthoMCL clustering using an e-value cutoff of 1e-6 (Figure 3). Apicomplexan AP2 domains that clustered with a P. marinus AP2 domain were classified as ancestral; these domains likely predate the divergence between perkinsids and the Apicomplexa. Four C. parvum AP2 domains (cgd4_1110_D1, cgd4_1110_D3, cgd8_3130 and cgd8_3230) are ancestral (Figure 3). Domains are indicated by gene ID, and in the case of multidomain proteins, numbered D1–D4 starting from the N-terminus. Domains that span all or most apicomplexan lineages, but were absent in Perkinsus were classified as pan-apicomplexan (10 C. parvum domains are in this category). The remaining nine domains have no orthologs outside of Cryptosporidium and are classified as lineage-specific. It is necessarily true that some pan-apicomplexan domains may have been present in the perkinsid/apicomplexan ancestor as well, and were subsequently lost in Perkinsus. Because there is no extant evidence of these domains in Perkinsus and because there is ambiguity with respect to when these domains arose, we maintain separate ‘ancestral’ and ‘pan-apicomplexan’ designations. Lineage-specific domains have no identifiable orthologs outside their respective taxa, though again it is a formal possibility that these could also be true ‘ancestral’ domains that were lost in other lineages.
Figure 3.
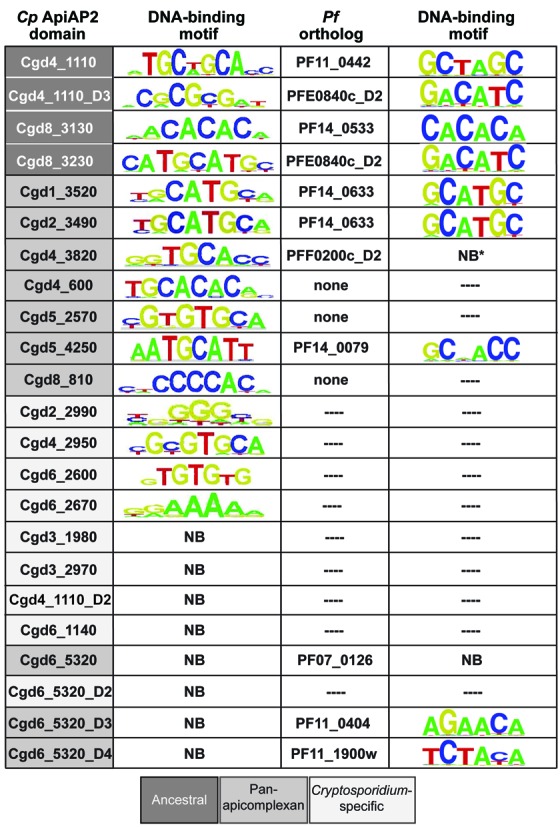
C. parvum and P. falciparum AP2 domain ortholog binding motifs as determined by PBM. C. parvum domains are color-coded according to evolutionary groups based on OrthoMCL clustering at 1e-6 as discussed in Materials and Methods. Data for core DNA motifs determined for P. falciparum AP2 domains obtained from (37).
C. parvum ApiAP2 domains bind diverse sequences
De Silva et al. (41) determined the DNA binding specificity of the C. parvum AP2 domain cgd2_3490, and we previously reported the DNA-binding specificity of cgd8_810 (41,49). To determine binding specificities for the remaining 21 predicted C. parvum AP2 domains, we created protein expression constructs and assayed binding using PBMs and cgd2_3490 as a control. Our results using PBMs agree with the previously reported 5′-TGCAT-3′ core binding motif for cgd2_3490, and we detect new binding specificities for 15 of the remaining predicted C. parvum AP2 domains (Figure 3).
We find that C. parvum AP2 domains bind a diversity of sequences similar to what is seen in P. falciparum. Although the C. parvum AP2 family can recognize a variety of sequences, we find that of the 15 domains for which we detected binding motifs, 10 of these bind one of three motif types: the 5′-TGCAT-3′ motif (recognized by four domains from four different proteins), the 5′-CACACA-3′ motif (recognized by four domains from four different proteins) or the G-box motif (5′-G[T/C]GGGG-3′, recognized by three domains from three different proteins). Cgd8_810 and cgd2_2990 bind the G-box as their primary motif, while cgd1_3520 binds the G-box motif secondarily (and is counted in each category; see below). Motifs and their PBM enrichment scores can be found in Supplementary File S6, Figures S2 and S3. P. falciparum also has four CACACA-binding AP2 domains, but this is the only markedly redundant P. falciparum AP2 binding motif (37).
Secondary and tertiary motif recognition
Secondary DNA binding motifs are thought to impart DNA-binding proteins with a broader range of binding interactions and thereby expand the repertoire of genes regulated by transcription factors (55). Multiple binding specificities above threshold were previously reported for several P. falciparum AP2 domains (37). Many of these secondary or tertiary binding sites had little similarity, indicating an additional layer of complexity to ApiAP2 regulation. C. parvum AP2s also display multiple motif recognition, though in the majority of cases secondary motifs are highly similar to, or are reverse complements of, the primary motif (Supplementary File S6, Figure S3). We find that only one C. parvum domain, cgd1_3520, is able to recognize two completely different motifs, both the 5′-TGCAT-3′ motif and the G-box.
Binding motif conservation between putative P. falciparum and C. parvum orthologs
It was noted previously that orthologous AP2 domains across P. falciparum, P. berghei and C. parvum (gene ids PF14_0633, PBANKA_132980 and cgd2_3490, respectively) have nearly identical binding specificities for the 5′-TGCATGCA-3′ motif (17,41). Our phylogenetic analyses support the orthology of this domain group, and we find an additional putative C. parvum ortholog to PF14_0633 (cgd1_3520) that also recognizes this motif (Figure 3). The putative orthologs cgd8_3130 and PF14_0533 bind highly similar motifs, as does putative ortholog pair cgd8_3230 and PFE0840c_D2.
A short, conserved linker region between AP2 domains is found in five P. falciparum ApiAP2 proteins (37). C. parvum proteins with multiple domains do not appear to contain this linker. The C. parvum multidomain protein cgd6_5320 has four predicted AP2 domains, and cgd4_1110 has three; Figure 3. Whether the C. parvum proteins utilize multiple DNA-binding regions simultaneously remains to be determined. Interestingly, C. parvum AP2 domain cgd4_3820 recognizes the sequence 5′-GGTGCACC-3′, while its putative P. falciparum ortholog PFF0200c_D2 (38% identity, with no conservation of residues predicted to be important for base-specific contacts (56)) failed to bind DNA as measured by PBMs. However, a construct of both AP2 domains PFF0200c_D1 (which does show binding) and PFF0200c_D2 joined by a short conserved linker region does bind the same motif as cgd4_3820. The D1 domain of PFF0200c shares only a single base-specific contact residue with cgd4_3820. These findings suggest that the binding interactions and specificities are complex.
Binding specificity is not conserved between the putative orthologs cgd4_1110_D3 and PFE0840c_D2, or putative orthologs cgd5_4250 and PF14_0079. There is no binding specificity above threshold in C. parvum for two domains (cgd6_5320_D3 and cgd6_5320_D4) whose putative orthologs (PF11_0404 and PFL1900w, respectively) do have binding motifs. These ill-conserved binding specificities may indicate that these domains are not true orthologs. Alternatively, the lack of conservation may be a true snapshot of evolving binding specificities, especially given the significant support of conserved binding specificities for the other putative ortholog groups.
Though putative orthologous apicomplexan AP2 domains often have similar binding specificities, evolutionary distance does not always predict binding specificity. We constructed a maximum likelihood tree of all predicted P. falciparum and C. parvum ApiAP2 domains and superimposed their binding motifs to examine the relationship between evolutionary distance and binding motif (Figure 4). AP2 domains that recognize similar motifs are interspersed throughout the tree. Putative orthologs PF14_0633, cgd2_3490, cgd1_3520 and cgd8_3230 all bind 5′-TGCAT-3′-like motifs, and are clustered together on the tree, though we also find TGCAT-binding AP2 domains that are more distantly related to this group. The G-box and CACACA-binding AP2 domains are more distantly related. These phyletic distributions could be explained by duplication of domains and divergence of their binding sites both within each species and as a consequence of speciation events. Determination of the families of AP2 binding motifs for intermediate taxa, such as T. gondii or the Piroplasmida, may further elucidate the relationship between AP2 binding sites and evolutionary descent.
Figure 4.
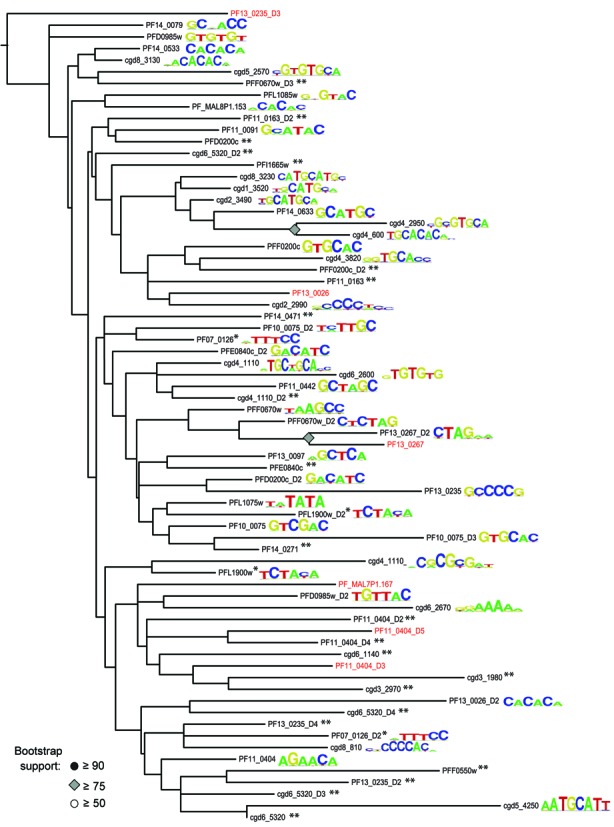
Maximum likelihood tree of P. falciparum and C. parvum AP2 domains and their DNA-binding motifs. Domain sequences were extracted from full-length proteins using HMM-defined coordinates, aligned and edited as described in Materials and Methods. A maximum likelihood tree was constructed from the edited alignment using RAxML with a Dayhoff protein evolution model, then visualized using FigTree. Domains in red were not identified in (37); as all domains were numbered from N-terminus to C-terminus, the numbering scheme therefore shifts slightly from (37). The previous domains PF11_0404_D3, PF13_025_D3, PF13_0267 and PF13_0026 correspond in this figure to domains PF11_0404_D4, PF13_0235_D4, PF13_0267_D2 and PF13_0026_D2, respectively. *Denotes domains that when tested alone, have no detectable binding specificity, but when tested with an adjacent domain, have the indicated binding specificity. **Denotes tested domains with no binding motifs over threshold.
Multiple ApiAP2 domains can bind C. parvum overrepresented upstream motifs
We previously reported 11 families of overrepresented motifs located upstream of C. parvum genes (49). Two of these motif families are known transcription factor motifs, E2F-like and CAAT-box-like. Another of these motif families, 5′-TGCAT-3′, which has a palindromic core, is an ApiAP2 binding site (designated ‘AP2_1’ in (49)). We found three additional C. parvum ApiAP2 proteins that bind this motif (Figure 5). While not constitutive, at least one cluster containing the 5′-TGCAT-3′ motif in their upstream regions is highly expressed at each of the surveyed life cycle time points (49). The same is true for transcripts representing each of the four TGCAT-binding ApiAP2 proteins; at least one transcript is maximally expressed at each of the surveyed time points (see cgd8_3230, cgd1_3520, cgd5_4250 and cgd2_3490 in Supplementary File S6, Figure S5). We additionally reported that ApiAP2 cgd8_810 binds the overrepresented G-box motif (49), and we find that cgd2_2990 and cgd1_3520 also recognize the G-box. Cgd2_2990 has a bimodal expression pattern, peaking at 6 and (to a lesser degree) 24 h post-infection, cgd1_3520 has peak expression at 12 h post-infection, while cgd8_810 is expressed at multiple later time points. Clusters containing overrepresented G-box motifs in the upstream regions of their genes are also maximally expressed, individually, at any of the surveyed time points across the life cycle. These results suggest that regulation of these differentially expressed gene clusters might be handled by the respective coexpressed ApiAP2 (Figure 6).
Figure 5.
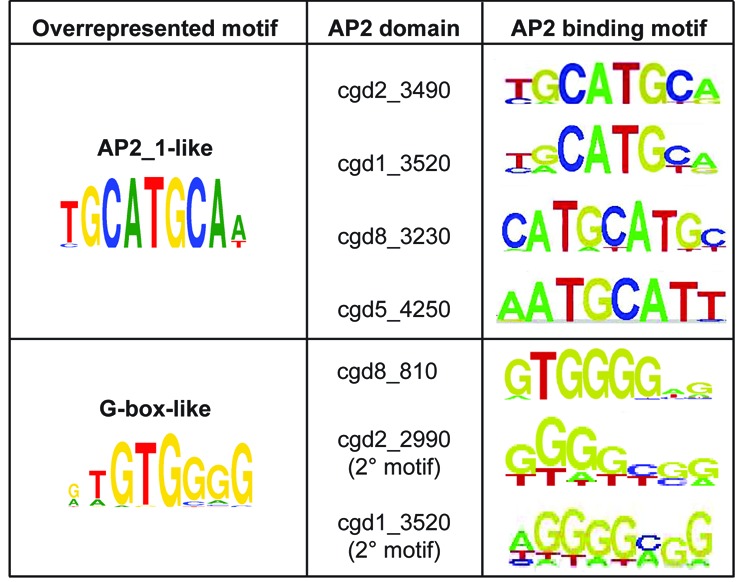
Overrepresented C. parvum motifs bound by AP2 domains. Overrepresented motifs as determined by (49). Overrepresented motifs that are (i) recognized by known non-AP2 proteins (E2F; CAAT-box) or (ii) are not AP2 binding motifs (GAGA-like; Unknown Set 1, Unknown Set2 and Unknown Motifs 14, 21, 22 and 25) are not included.
Figure 6.
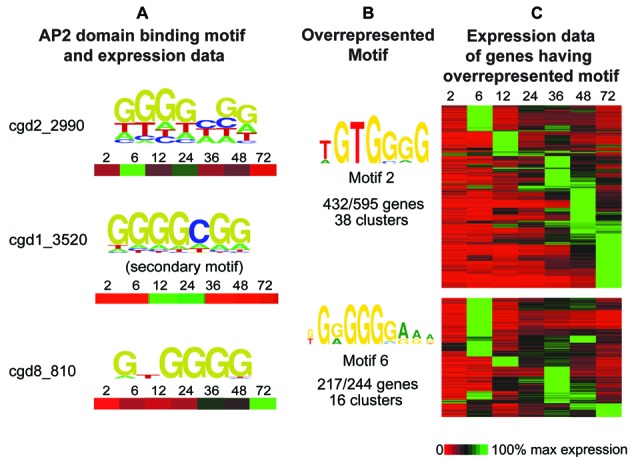
Expression patterns of genes containing overrepresented C. parvum G-box motifs and potential G-box-binding ApiAP2 regulators. (A) The PBM-determined binding motif as well as expression data for each G-box-binding AP2 domain is displayed for seven time points from 2 to 72 h across the in vitro life cycle (expression data from (52)). (B) Two G-box motifs were identified as overrepresented in upstream regions of C. parvum genes clustered by expression profile (49). The number of clusters, and the number of genes having the motif out of those clusters is shown. (C) Expression data for all genes in clusters identified as having overrepresented G-box motifs. Genes with overrepresented G-box motifs are expressed across the life cycle; there is a G-box-binding ApiAP2 protein expressed at each of those time points, suggesting ApiAP2s could be driving expression of these genes.
We did not detect ApiAP2 protein-DNA interactions for nine additional previously predicted overrepresented upstream motifs (49). Interestingly, we identified four different AP2 domains that can bind the palindromic 5′-CACACA-3′ motif, yet the motif is not overrepresented upstream of the 200 coregulated C. parvum gene clusters we previously identified. We were able to predict putative regulatory targets for two of these CACACA-binding AP2 domains, cgd8_3130 and cgd4_600. The other CACACA-binding AP2 domains, cgd5_2570 and cgd6_2600, have no predicted targets below statistical threshold. Most of these putative targets have a bimodal expression pattern, peaking at 12 and 36 h post-infection (data not shown). ApiAP2 proteins cgd8_3130 and cgd4_600 are expressed during these time points, and thus could plausibly be involved in regulation of these genes.
ApiAP2 network evolution: comparisons of orthologous and lineage-specific ApiAP2s and their regulatory targets
Behnke et al. (38) found that genes expressed throughout the T. gondii cell cycle define subtranscriptomes expressed in two separate waves: genes responsible for basal processes, such as DNA replication, protein translation and glycolysis; and genes specific to apicomplexan processes, such as those involved in invasion or immune evasion (38). They noted that 24 ApiAP2 proteins are expressed in a cascade across the cell cycle. These findings raise the intriguing possibility that the evolutionary history of AP2 domains is somehow correlated with the evolutionary history of their regulatory targets—i.e. that ancestral or pan-apicomplexan AP2 domains might be responsible for regulating basal housekeeping processes, while lineage-specific AP2 domains might regulate apicomplexan-specific processes. To further investigate this possibility, we used a modified version of the algorithm Campbell et al. (37) developed to predict regulatory targets (which incorporates genome-wide expression data and presence of AP2 binding motifs in upstream regions) for a number of C. parvum AP2 domains (37). We selected lineage-specific and orthologous AP2 domains from both C. parvum and P. falciparum and evaluated the category composition of their predicted target genes (see Materials and Methods). However, we did not find a significant correlation between evolutionary class of AP2 domain and putative targets in either organism (Supplementary File S6, Figure S4). Comparison of putative targets between ancestral or pan-apicomplexan C. parvum AP2 domains and their P. falciparum orthologs revealed very little overlap between them (Supplementary File S1, Table S10). P. falciparum ApiAP2 proteins on average are predicted to regulate a much higher percentage of genes.
Conservation between C. parvum and C. hominis ApiAP2s and their putative targets
We find that although the C. parvum and C. hominis genome sequences are 97% identical (47), the ApiAP2s themselves do not display this average similarity. 18/35 C. parvum domains are 100% identical to domains in C. hominis; another eight domains have 80–99% aa identity; three domains do not appear to have orthologs in C. hominis and the remaining six have similarities as low as 43%. While some of these differences can probably be explained by assembly status of the genomes, the results are intriguing.
Few C. hominis expression data exist and no protein binding data exist; thus, regulatory targets for C. hominis ApiAP2 proteins could not be predicted. Instead, we compared the upstream regions of the putative targets of two C. parvum ApiAP2's that share 100% identity with C. hominis orthologs cgd8_3230 and cdg5_4250. Cgd8_3230 has 15 predicted targets with orthologs in C. hominis. Of these 15 orthologous pairs of upstream regions, seven are >90% identical across 90% or more of their length; five are >90% identical across 50% or more of their length and the remaining three are >90% identical across <50% of their length. Cgd5_4250 has 23 predicted targets with orthologs in C. hominis. Of these 23 orthologous pairs of upstream regions, nine are >90% identical across 90% or more of their length; one is > 90% identical across >50% of their length and the remaining 13 are >90% identical over <50% of their length.
The ApiAP2 expression cascade is conserved in C. parvum
Unlike Plasmodium spp. and Toxoplasma, a number of other possible sequence-specific transcription factor families have been detected in the C. parvum genome (summarized in (49)), some of which are absent in other apicomplexans (E2F, for example). The ratio of available putative C. parvum transcription factors to regulate target genes (∼1:340) is much higher than the P. falciparum ratio (∼1:800), due both to the lower gene count in C. parvum and a higher absolute number of possible transcription factors (19). We have also determined that the E2F binding motif is one of the most overrepresented motifs in the upstream regions of C. parvum genes (49). Given these observations, it might be expected that C. parvum is less reliant on the ApiAP2 family for transcriptional regulation than P. falciparum and other apicomplexans (see Supplementary File S7 for C. parvum gene cluster upstream motif co-occurrence (49) and clusters with E2F motifs, but no known AP2 binding motif). However, expression data for each predicted C. parvum ApiAP2 protein indicate that the expression cascade observed across the P. falciparum blood stage (37) and across the T. gondii cell cycle (38) is conserved in C. parvum (Supplementary File S6, Figure S5), though putative orthologous ApiAP2s do not necessarily appear at similar temporal/developmental windows in the cascades (to the extent they can be correlated).
DISCUSSION
We identified and characterized the family of C. parvum AP2 domains by experimentally determining their DNA sequence targets. We then used this information to examine ApiAP2 regulation in a kingdom-wide context by performing evolutionary analyses of the distribution of, and relationships between, AP2 domains, many of which also have experimental binding-specificity data. Phylogenies constructed from AP2 domains spanning chromalveolates and extant representatives of their endosymbionts indicate a distinct divide between AP2s found in the plants, stramenopiles, deep alveolates, such as ciliates and dinoflagellates, and those found in the Apicomplexa. The perkinsid AP2 domains group confidently with the apicomplexans to the exclusion of other chromalveolates. Some domains are orthologous, spanning several apicomplexan taxa and thus, must predate speciation events.
Our results suggest that by whichever manner AP2 domains came to reside in the apicomplexan/perkinsid ancestor (mobile element invasion, transfer from an algal endosymbiont or some mixture of these events), perkinsid and apicomplexan AP2 domains share a common origin. Based on our homology analyses, we propose that there were five to six progenitor domains arising from the acquisition event which occurred sometime between the split from dinoflagellates and the appearance of the latest perkinsid/apicomplexan ancestor. The domains in the perkinsid and apicomplexan lineages then amplified independently. The apicomplexan ancestor likely possessed 10–18 domains (or a maximum of 10–18 ApiAP2 proteins, an estimate in approximate agreement with the maximum of nine proteins proposed in (8)). A more precise estimate will require additional, diverse sampling across the phylum as well as additional structural analysis of the domain. Though some domains present in both apicomplexans and perkinsids are ancestral, domains spanning other combinations of taxa may be either ancestral or have been lost in a few of the extant lineages, or they may have arisen as a result of recent amplification. The most striking amplifications have occurred in the coccidian and Plasmodium lineages, with anywhere from 42 to 69 of the ∼90 coccidian domains and 14 to 29 of the ∼50 Plasmodium domains being lineage-specific. Due to the thresholds used, it is a possibility that we have not detected all AP2 domains, or that we have designated some weakly homologous domains as AP2s when they are not functional DNA-binding domains. Interestingly, several high-scoring predicted domains had no detected DNA binding motifs (such as cgd6_5320_D1 through D4, cgd4_1110_D2 and cgd6_1140, Figure 3; Supplementary File S1, Table S4). It is also possible that we have not detected all binding motifs for C. parvum AP2s, whether through failure to capture critical binding residues in our constructs or other experimental shortcoming. However, other AP2 domains from Plasmodium also have no detected DNA binding (37), one of which is orthologous to cgd6_5320_D1. These results continue to suggest that some predicted apicomplexan AP2 domains may function outside the context of binding DNA. Alternatively, it has been suggested that AP2 domains may be discriminators of methylated DNA (57); it is possible that these domains are DNA-binding, but our assay did not reflect subtle DNA modifications necessary for binding to occur.
The current lack of continuous in vitro propagation and molecular genetic tools in Cryptosporidium imposes a critical barrier to further functional characterization of predicted ApiAP2 transcription factors and their putative regulatory targets. Our target predictions are based on gene expression data from the limited in vitro life cycle. Additional clusters and upstream patterns will undoubtedly appear when in vivo data are available. It is also important to note that although proteomics data from the very early stages of the C. parvum life cycle are available (58,59), there are no proteomics data for the majority of the life cycle. Thus, we do not know how closely mRNA expression indicates protein expression in C. parvum, and the expectation that ApiAP2 mRNA expression profiles should correlate highly with those of predicted target genes may be flawed. The correlation between mRNA and protein expression in P. falciparum was found to be moderately positive, though a delay has been observed for several genes, indicating post-transcriptional regulatory mechanisms in Plasmodium (10,60). Unlike what has previously been indicated for P. falciparum ApiAP2s (37), C. parvum ApiAP2 mRNA expression does not correlate well with predicted target gene expression profiles in many cases (data not shown). The upstream sequence database used to mine for putative transcription factor binding sites is greatly affected by the status of the annotation, and untranslated regions are largely undefined in C. parvum. Though it has been suggested that UTRs may overlap with coding regions in highly compact genomes, such as that of C. parvum (61), the prevalence of this phenomenon has not been established, and we did not search any coding regions in the construction of our upstream sequence database. It should also be noted that we have far fewer time points over which expression data were measured (seven time points post-infection spread over 72 h versus 48 hourly time points for P. falciparum), and we do not have the resolution nor as much statistical power in target prediction as has been achieved in P. falciparum (37).
Our ApiAP2 network analysis, based on a combination of in vitro (PBMs) and computational data, lays the foundation for further exploration of transcriptional regulation in the absence of molecular genetic tools. Even when considering model organisms for which there are a myriad of genetic tools, few large transcription factor family networks have been characterized in depth (37,55,62–66). Here, we have presented evidence that ApiAP2s are likely major players in C. parvum transcriptional regulation, namely: (1) An ApiAP2 regulatory cascade is conserved in C. parvum, and (2) C. parvum ApiAP2s bind a diverse set of motifs, many of which are conserved with P. falciparum and overrepresented upstream of many co-expressed gene clusters. The conservation of a putative ApiAP2 regulatory cascade despite complete reordering of orthologous ApiAP2 expression between C. parvum and P. falciparum further suggests extensive ApiAP2 network rewiring over its evolutionary history. Intriguingly, even with the much smaller time scale between C. parvum and C. hominis (genome sequences 97% identical), we find that orthologous ApiAP2 proteins themselves, as well as orthologous upstream regions of predicted target genes are not absolutely conserved. Some of these differences may be due to differences in assembly status, but others likely indicate divergences that may play a role in the differing host range and pathogenicity between the two species. Questions regarding divergence between C. parvum and C. hominis can be more fully addressed as more C. hominis genome sequence and expression data become available. In conjunction with our phylogenetic analyses, these results contribute to the beginnings of a framework for understanding ApiAP2 regulation in other apicomplexans. Binding motifs have been identified for several members of a single AP2 domain ortholog group (PF14_0633 in P. falciparum, cgd2_3490 in C. parvum, TGME49_110950 in T. gondii and AP2-Sp in P. berghei; (16,38,41)), all of which bind the 5′-TGCAT-3′ motif. Our results build on these previous observations. Putatively orthologous domains have conserved binding specificities between two of the most distantly related apicomplexans, P. falciparum and C. parvum, indicating that binding specificities can be inferred by orthology.
We have noted that orthologous AP2 domains often have conserved DNA-binding motifs, yet the putative networks of target genes are vastly different between orthologous AP2s, with very few shared targets. Our broadscale comparisons of ApiAP2 network composition between P. falciparum and C. parvum suggest that there is no relationship between evolutionary class of AP2 domain and evolutionary class of predicted targets. If network divergence does not appear to be driven by evolution of the ApiAP2 protein binding specificities, divergence could instead be driven by genome rearrangements, through shuffling, ablation and creation of cognate cis elements upstream of completely different sets of genes. Apicomplexa have undergone a striking degree of genome rearrangement, with no three genes found together, in the same order, across the phylum; even in closely related lineages, such as Plasmodium and the Piroplasmida (∼300 my divergence time, (67)) synteny is rare (40). Regulatory network evolution by way of transcription factor binding site turnover has been documented in several cases in yeast as well as animals (reviewed in (68)).
Transcription factor substitution may also play a role in ApiAP2 network divergence. We previously reported evidence of substitution in the ribosomal protein regulon between a P. falciparum G-box-binding AP2 and C. parvum E2F (49), and further evidence suggests multiple transcription factor handoffs, as yet another AP2 binding site, 5′-TGCAT-3′, is conserved upstream of T. gondii and N. caninum ribosomal genes (69). Ribosomal gene regulon transcription factor substitution has also been noted in yeast (70,71). Campbell et al. (37) reported extensive divergence between predicted orthologous ApiAP2 regulons in P. falciparum, P. vivax and P. yoelli, indicating that there is extensive network divergence even on relatively small evolutionary time scales (∼120 million years). Conservation of transcription factor binding in the face of extensive regulon divergence has been noted across several organisms (72–75).
The function of ApiAP2 proteins outside of the AP2 domain(s) itself is unclear, though the rest of the protein presumably has some involvement in facilitating protein–protein interactions. Yeast-two-hybrid studies have indicated that some P. falciparum ApiAP2 proteins interact with each other, as well as with other regulatory proteins, such as the histone acetyltransferase Gcn5 (76). Structural studies of a P. falcparum ApiAP2 (PF14_0633) demonstrate that AP2 domains can dimerize to bind DNA (56). We previously reported that clustered C. parvum gene expression patterns cannot be attributed to the presence of any one type of upstream motif (49). Further protein-interaction studies on ApiAP2 proteins are needed to establish the degree to which ApiAP2 trans regulatory environments are conserved. Rewiring of transcriptional regulatory networks via evolving combinatorial interactions has also been reported in yeast (reviewed in (68)).
Many C. parvum AP2 domains bind redundant motifs, and the majority of C. parvum AP2 domains bind only one motif. Thus, C. parvum ApiAP2 regulation does not appear to be as multifaceted as is suggested in P. falciparum (37). The presence of additional non-ApiAP2 transcription factors in the C. parvum genome may explain the decreased diversity of ApiAP2 binding motifs. We noted previously that the E2F motif is the most abundantly overrepresented motif in the upstream regions of the C. parvum genome, being found upstream of 161 of 200 predicted co-regulated gene clusters (49). E2Fs are notably absent in Plasmodium and other apicomplexans (19), and they are also among the most ancient transcription factor families that can be traced back to the last eukaryotic common ancestor (as well as Myb, C2H2 zinc finger, bZIP and AT-hook domains, most of which are present across the Apicomplexa) (18). It is possible that the three predicted E2F transcription factors and their two DP1 dimerization partners are responsible for a disproportionate amount of the transcriptional regulation, such that C. parvum is less reliant on ApiAP2s. The apparent redundancy in C. parvum ApiAP2 binding motifs may also be important for stage-specific transcriptional regulation, as ApiAP2s binding the same or similar motifs are expressed at various points across the life cycle. While we identified several AP2 domains that can potentially bind two predicted C. parvum regulatory motif families (49), the function of seven of the remaining overrepresented motif families is still unknown. Several players in C. parvum transcriptional regulation have yet to be identified. The mechanisms by which AP2 domain-containing proteins came to regulate the vast majority of genes in many apicomplexans beginning from just a few, or perhaps a single, vertically inherited factor are likely varied, involving a combination of modalities. C. parvum, with its more diverse complement of transcription factor families and possible reduced reliance on ApiAP2 proteins, offers clues to the ancestral state of apicomplexan transcriptional regulation, pre AP2-domination.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We would like to thank our colleagues Jeremy DeBarry (UGA) for discussions that strengthened the quality of this work and Sandeep J. Joseph (Emory) for assistance with the Figure S5 heat map. This study was supported in part by resources and technical expertise from the Georgia Advanced Computing Resource Center, a partnership between the University of Georgia's Office of the Vice President for Research and Office of the Vice President for Information Technology.
Footnotes
Present addresses:
Jenna Oberstaller, Department of Global Health, University of South Florida, Tampa, FL 33612, USA.
Manuel Llinás, Department of Biochemistry and Molecular Biology and Center for Infectious Disease Dynamics, The Pennsylvania State University, University Park, PA 16802, USA.
FUNDING
National Institutes of Health (NIH) [R01-AI065246 to Mitch Abrahamsen (transferred to Mark Rutherford) PI; J.C.K. Co-PI]; [R01-AI076276 to M.L.]. Centre for Quantitative Biology [NIH P50 GM071508 to M.L.]. NIH Training Grant T32 [AI060546 to the UGA Center for Tropical and Emerging Global Diseases to J.O.]. Source of open access funding: University of Georgia funds allocated to the Kissinger Research Group were used to finance open access funding charges.
Conflict of interest statement. None declared.
REFERENCES
- 1.Kotloff K.L., Nataro J.P., Blackwelder W.C., Nasrin D., Farag T.H., Panchalingam S., Wu Y., Sow S.O., Sur D., Breiman R.F., et al. Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): a prospective, case-control study. Lancet. 2013;382:209–222. doi: 10.1016/S0140-6736(13)60844-2. [DOI] [PubMed] [Google Scholar]
- 2.Escalante A.A., Ayala F.J. Evolutionary origin of Plasmodium and other Apicomplexa based on rRNA genes. Proc. Natl. Acad. Sci. U.S.A. 1995;92:5793–5797. doi: 10.1073/pnas.92.13.5793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Okamoto N., McFadden G.I. The mother of all parasites. Future Microbiol. 2008;3:391–395. [Google Scholar]
- 4.Berney C., Pawlowski J. A molecular time-scale for eukaryote evolution recalibrated with the continuous microfossil record. Proc. Biol. Sci. 2006;273:1867–1872. doi: 10.1098/rspb.2006.3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Putnam N.H., Butts T., Ferrier D.E., Furlong R.F., Hellsten U., Kawashima T., Robinson-Rechavi M., Shoguchi E., Terry A., Yu J.K., et al. The amphioxus genome and the evolution of the chordate karyotype. Nature. 2008;453:1064–1071. doi: 10.1038/nature06967. [DOI] [PubMed] [Google Scholar]
- 6.Meissner M., Soldati D. The transcription machinery and the molecular toolbox to control gene expression in Toxoplasma gondii and other protozoan parasites. Microbes Infect. 2005;7:1376–1384. doi: 10.1016/j.micinf.2005.04.019. [DOI] [PubMed] [Google Scholar]
- 7.Hakimi M.A., Deitsch K.W. Epigenetics in Apicomplexa: control of gene expression during cell cycle progression, differentiation and antigenic variation. Curr. Opin. Microbiol. 2007;10:357–362. doi: 10.1016/j.mib.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 8.Balaji S., Babu M.M., Iyer L.M., Aravind L. Discovery of the principal specific transcription factors of Apicomplexa and their implication for the evolution of the AP2-integrase DNA binding domains. Nucleic Acids Res. 2005;33:3994–4006. doi: 10.1093/nar/gki709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Llinas M., DeRisi J.L. Pernicious plans revealed: Plasmodium falciparum genome wide expression analysis. Curr. Opin. Microbiol. 2004;7:382–387. doi: 10.1016/j.mib.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 10.Hall N., Karras M., Raine J.D., Carlton J.M., Kooij T.W.A., Berriman M., Florens L., Janssen C.S., Pain A., Christophides G.K., et al. A comprehensive survey of the Plasmodium life cycle by genomic, transcriptomic, and proteomic analyses. Science. 2005;307:82–86. doi: 10.1126/science.1103717. [DOI] [PubMed] [Google Scholar]
- 11.Radke J.R., Behnke M.S., Mackey A.J., Radke J.B., Roos D.S., White M.W. The transcriptome of Toxoplasma gondii. BMC Biol. 2005;3:26. doi: 10.1186/1741-7007-3-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Radke J.B., Lucas O., De Silva E.K., Ma Y., Sullivan W.J., Weiss L.M., Llinas M., White M.W. ApiAP2 transcription factor restricts development of the Toxoplasma tissue cyst. Proc. Natl. Acad. Sci. 2013;110:6871–6876. doi: 10.1073/pnas.1300059110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Walker R., Gissot M., Croken M.M., Huot L., Hot D., Kim K., Tomavo S. The Toxoplasma nuclear factor TgAP2XI-4 controls bradyzoite gene expression and cyst formation. Mol. Microbiol. 2013;87:641–655. doi: 10.1111/mmi.12121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walker R., Gissot M., Huot L., Alayi T.D., Hot D., Marot G., Schaeffer-Reiss C., Van Dorsselaer A., Kim K., Tomavo S. Toxoplasma transcription factor TgAP2XI-5 regulates the expression of genes involved in parasite virulence and host invasion. J. Biol. Chem. 2013;288:31127–31138. doi: 10.1074/jbc.M113.486589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iwanaga S., Kaneko I., Kato T., Yuda M. Identification of an AP2-family protein that is critical for malaria liver stage development. PloS ONE. 2012;7:e47557. doi: 10.1371/journal.pone.0047557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yuda M., Iwanaga S., Shigenobu S., Kato T., Kaneko I. Transcription factor AP2-Sp and its target genes in malarial sporozoites. Mol. Microbiol. 2010;75:854–863. doi: 10.1111/j.1365-2958.2009.07005.x. [DOI] [PubMed] [Google Scholar]
- 17.Yuda M., Iwanaga S., Shigenobu S., Mair G.R., Janse C.J., Waters A.P., Kato T., Kaneko I. Identification of a transcription factor in the mosquito-invasive stage of malaria parasites. Mol. Microbiol. 2009;71:1402–1414. doi: 10.1111/j.1365-2958.2009.06609.x. [DOI] [PubMed] [Google Scholar]
- 18.Iyer L.M., Anantharaman V., Wolf M.Y., Aravind L. Comparative genomics of transcription factors and chromatin proteins in parasitic protists and other eukaryotes. Intl. J. Parasitol. 2008;38:1–31. doi: 10.1016/j.ijpara.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 19.Templeton T.J., Iyer L.M., Anantharaman V., Enomoto S., Abrahante J.E., Subramanian G.M., Hoffman S.L., Abrahamsen M.S., Aravind L. Comparative analysis of apicomplexa and genomic diversity in eukaryotes. Genome Res. 2004;14:1686–1695. doi: 10.1101/gr.2615304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Janouskovec J., Horak A., Obornik M., Lukes J., Keeling P.J. A common red algal origin of the apicomplexan, dinoflagellate, and heterokont plastids. Proc. Natl. Acad. Sci. U.S.A. 2010;107:10949–10954. doi: 10.1073/pnas.1003335107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang J., Mullapudi N., Lancto C.A., Scott M., Abrahamsen M.S., Kissinger J.C. Phylogenomic evidence supports past endosymbiosis, intracellular and horizontal gene transfer in Cryptosporidium parvum. Genome Biol. 2004;5:R88. doi: 10.1186/gb-2004-5-11-r88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kishore S., Stiller J., Deitsch K. Horizontal gene transfer of epigenetic machinery and evolution of parasitism in the malaria parasite Plasmodium falciparum and other apicomplexans. BMC Evol. Biol. 2013;13:1–12. doi: 10.1186/1471-2148-13-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schoenfeld T.W., Murugapiran S., Dodsworth J.A., Floyd S., Lodes M., Mead D.A., Hedlund B.P. Lateral gene transfer of Family A DNA polymerases between thermophilic viruses, Aquificae, and Apicomplexa. Mol. Biol. Evol. 2013;30 doi: 10.1093/molbev/mst078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Striepen B., Pruijssers A.J.P., Huang J.L., Li C., Gubbels M.J., Umejiego N.N., Hedstrom L., Kissinger J.C. Gene transfer in the evolution of parasite nucleotide biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 2004;101:3154–3159. doi: 10.1073/pnas.0304686101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang J.L., Mullapudi N., Sicheritz-Ponten T., Kissinger J.C. A first glimpse into the pattern and scale of gene transfer in the Apicomplexa. Intl. J. Parasitol. 2004;34:265–274. doi: 10.1016/j.ijpara.2003.11.025. [DOI] [PubMed] [Google Scholar]
- 26.Magnani E., Sjolander K., Hake S. From endonucleases to transcription factors: evolution of the AP2 DNA binding domain in plants. Plant Cell. 2004;16:2265–2277. doi: 10.1105/tpc.104.023135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wuitschick J.D., Lindstrom P.R., Meyer A.E., Karrer K.M. Homing endonucleases encoded by germ line-limited genes in Tetrahymena thermophila have APETELA2 DNA binding domains. Eukaryot. Cell. 2004;3:685–694. doi: 10.1128/EC.3.3.685-694.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Templeton T.J., Enomoto S., Chen W.J., Huang C.G., Lancto C.A., Abrahamsen M.S., Zhu G. A genome-sequence survey for Ascogregarina taiwanensis supports evolutionary affiliation but metabolic diversity between a Gregarine and Cryptosporidium. Mol. Biol. Evol. 2010;27:235–248. doi: 10.1093/molbev/msp226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roy S.W., Penny D. Large-scale intron conservation and order-of-magnitude variation in intron loss/gain rates in apicomplexan evolution. Genome Res. 2006;16:1270–1275. doi: 10.1101/gr.5410606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roy S.W., Penny D. Widespread intron loss suggests retrotransposon activity in ancient apicomplexans. Mol.Biol. Evol. 2007;24:1926–1933. doi: 10.1093/molbev/msm102. [DOI] [PubMed] [Google Scholar]
- 31.Ling K.H., Rajandream M.A., Rivailler P., Ivens A., Yap S.J., Madeira A.M., Mungall K., Billington K., Yee W.Y., Bankier A.T., et al. Sequencing and analysis of chromosome 1 of Eimeria tenella reveals a unique segmental organization. Genome Res. 2007;17:311–319. doi: 10.1101/gr.5823007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iyer L.M., Aravind L. Insights from the architecture of the bacterial transcription apparatus. J. Struct. Biol. 2012;179:299–319. doi: 10.1016/j.jsb.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Altschul S.F., Wootton J.C., Zaslavsky E., Yu Y.K. The construction and use of log-odds substitution scores for multiple sequence alignment. PLoS Comput. Biol. 2010;6:e1000852. doi: 10.1371/journal.pcbi.1000852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kafsack B.F.C., Rovira-Graells N., Clark T.G., Bancells C., Crowley V.M., Campino S.G., Williams A.E., Drought L.G., Kwiatkowski D.P., Baker D.A., et al. A transcriptional switch underlies commitment to sexual development in malaria parasites. Nature. 2014;507:248–252. doi: 10.1038/nature12920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sinha A., Hughes K.R., Modrzynska K.K., Otto T.D., Pfander C., Dickens N.J., Religa A.A., Bushell E., Graham A.L., Cameron R., et al. A cascade of DNA-binding proteins for sexual commitment and development in Plasmodium. Nature. 2014 doi: 10.1038/nature12970. doi:10.1038/nature12970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Flueck C., Bartfai R., Niederwieser I., Witmer K., Alako B.T.F., Moes S., Bozdech Z., Jenoe P., Stunnenberg H.G., Voss T.S. A Major Role for the Plasmodium falciparum ApiAP2 Protein PfSIP2 in Chromosome End Biology. Plos Pathog. 2010;6:e1000784. doi: 10.1371/journal.ppat.1000784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Campbell T.L., De Silva E.K., Olszewski K.L., Elemento O., Llinas M. Identification and genome-wide prediction of DNA binding specificities for the ApiAP2 family of regulators from the malaria parasite. Plos Pathog. 2010;6:e1001165. doi: 10.1371/journal.ppat.1001165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Behnke M.S., Wootton J.C., Lehmann M.M., Radke J.B., Lucas O., Nawas J., Sibley L.D., White M.W. Coordinated progression through two subtranscriptomes underlies the tachyzoite cycle of Toxoplasma gondii. PloS ONE. 2010;5:e12354. doi: 10.1371/journal.pone.0012354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuo C.H., Kissinger J.C. Consistent and contrasting properties of lineage-specific genes in the apicomplexan parasites Plasmodium and Theileria. BMC Evol. Biol. 2008;8:108. doi: 10.1186/1471-2148-8-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.DeBarry J.D., Kissinger J.C. Jumbled genomes: missing Apicomplexan synteny. Mol. Biol. Evol. 2011;28:2855–2871. doi: 10.1093/molbev/msr103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.De Silva E.K., Gehrke A.R., Olszewski K., Leon I., Chahal J.S., Bulyk M.L., Llinas M. Specific DNA-binding by Apicomplexan AP2 transcription factors. Proc. Natl. Acad. Sci. U.S.A. 2008;105:8393–8398. doi: 10.1073/pnas.0801993105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Notredame C., Higgins D.G., Heringa J. T-Coffee: a novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000;302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- 43.Waterhouse A.M., Procter J.B., Martin D.M.A., Clamp M., Barton G.J. Jalview Version 2, a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 45.Li L., Stoeckert C.J., Jr, Roos D.S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krzywinski M.I., Schein J.E., Birol I., Connors J., Gascoyne R., Horsman D., Jones S.J., Marra M.A. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19 doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xu P., Widmer G., Wang Y., Ozaki L.S., Alves J.M., Serrano M.G., Puiu D., Manque P., Akiyoshi D., Mackey A.J., et al. The genome of Cryptosporidium hominis. Nature. 2004;431:1107–1112. doi: 10.1038/nature02977. [DOI] [PubMed] [Google Scholar]
- 48.Mahony S., Benos P.V. STAMP: a web tool for exploring DNA-binding motif similarities. Nucleic Acids Res. 2007;35:W253–W258. doi: 10.1093/nar/gkm272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Oberstaller J., Joseph S.J., Kissinger J.C. Genome-wide upstream motif analysis of Cryptosporidium parvum genes clustered by expression profile. BMC Genom. 2013;14:516. doi: 10.1186/1471-2164-14-516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Puiu D., Enomoto S., Buck G.A., Abrahamsen M.S., Kissinger J.C. CryptoDB: the Cryptosporidium genome resource. Nucleic Acids Res. 2004;32:D329–D331. doi: 10.1093/nar/gkh050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.R Development Core Team. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2012. [Google Scholar]
- 52.Mauzy M.J., Enomoto S., Lancto C.A., Abrahamsen M.S., Rutherford M.S. The Cryptosporidium parvum transcriptome during in vitro development. PloS ONE. 2012;7 doi: 10.1371/journal.pone.0031715. doi:10.1371/journal.pone.0031715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Keeling P.J. Chromalveolates and the evolution of plastids by secondary endosymbiosis. J. Eukaryot. Microbiol. 2009;56:1–8. doi: 10.1111/j.1550-7408.2008.00371.x. [DOI] [PubMed] [Google Scholar]
- 54.Woehle C., Dagan T., Martin W.F., Gould S.B. Red and problematic green phylogenetic signals among thousands of nuclear genes from the photosynthetic and apicomplexa-related Chromera velia. Genome Biol. Evol. 2011;3:1220–1230. doi: 10.1093/gbe/evr100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Badis G., Berger M.F., Philippakis A.A., Talukder S., Gehrke A.R., Jaeger S.A., Chan E.T., Metzler G., Vedenko A., Chen X., et al. Diversity and complexity in DNA recognition by transcription factors. Science. 2009;324:1720–1723. doi: 10.1126/science.1162327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lindner S.E., De Silva E.K., Keck J.L., Llinas M. Structural determinants of DNA binding by a P. falciparum ApiAP2 transcriptional regulator. J. Mol. Biol. 2010;395:558–567. doi: 10.1016/j.jmb.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Iyer L.M., Abhiman S., Aravind L. Natural history of eukaryotic DNA methylation systems. Progress Mol. Biol. Transl. Sci. 2011;101:25–104. doi: 10.1016/B978-0-12-387685-0.00002-0. [DOI] [PubMed] [Google Scholar]
- 58.Sanderson S.J., Xia D., Prieto H., Yates J., Heiges M., Kissinger J.C., Bromley E., Lal K., Sinden R.E., Tomley F., et al. Determining the protein repertoire of Cryptosporidium parvum sporozoites. Proteomics. 2008;8:1398–1414. doi: 10.1002/pmic.200700804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Snelling W.J., Lin Q., Moore J.E., Millar B.C., Tosini F., Pozio E., Dooley J.S., Lowery C.J. Proteomics analysis and protein expression during sporozoite excystation of Cryptosporidium parvum (Coccidia, Apicomplexa) Mol. Cell. Proteom. 2007;6:346–355. doi: 10.1074/mcp.M600372-MCP200. [DOI] [PubMed] [Google Scholar]
- 60.Le Roch K.G., Johnson J.R., Florens L., Zhou Y., Santrosyan A., Grainger M., Yan S.F., Williamson K.C., Holder A.A., Carucci D.J., et al. Global analysis of transcript and protein levels across the Plasmodium falciparum life cycle. Genome Res. 2004;14:2308–2318. doi: 10.1101/gr.2523904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Keeling P.J., Slamovits C.H. Causes and effects of nuclear genome reduction. Curr. Opin. Genet. Dev. 2005;15:601–608. doi: 10.1016/j.gde.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 62.Berger M.F., Badis G., Gehrke A.R., Talukder S., Philippakis A.A., Peña-Castillo L., Alleyne T.M., Mnaimneh S., Botvinnik O.B., Chan E.T., et al. Variation in homeodomain DNA binding revealed by high-resolution analysis of sequence preferences. Cell. 2008;133:1266–1276. doi: 10.1016/j.cell.2008.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Badis G., Chan E.T., van Bakel H., Pena-Castillo L., Tillo D., Tsui K., Carlson C.D., Gossett A.J., Hasinoff M.J., Warren C.L., et al. A library of yeast transcription factor motifs reveals a widespread function for Rsc3 in targeting nucleosome exclusion at promoters. Mol. Cell. 2008;32:878–887. doi: 10.1016/j.molcel.2008.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Noyes M.B., Christensen R.G., Wakabayashi A., Stormo G.D., Brodsky M.H., Wolfe S.A. Analysis of homeodomain specificities allows the family-wide prediction of preferred recognition sites. Cell. 2008;133:1277–1289. doi: 10.1016/j.cell.2008.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhu C., Byers K.J.R.P., McCord R.P., Shi Z., Berger M.F., Newburger D.E., Saulrieta K., Smith Z., Shah M.V., Radhakrishnan M., et al. High-resolution DNA-binding specificity analysis of yeast transcription factors. Genome Res. 2009;19:556–566. doi: 10.1101/gr.090233.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Harbison C.T., Gordon D.B., Lee T.I., Rinaldi N.J., Macisaac K.D., Danford T.W., Hannett N.M., Tagne J.-B., Reynolds D.B., Yoo J., et al. Transcriptional regulatory code of a eukaryotic genome. Nature. 2004;431:99–104. doi: 10.1038/nature02800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Silva J.C., Egan A., Friedman R., Munro J.B., Carlton J.M., Hughes A.L. Genome sequences reveal divergence times of malaria parasite lineages. Parasitology. 2011;138:1737–1749. doi: 10.1017/S0031182010001575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li H., Johnson A.D. Evolution of Transcription Networks Lessons from Yeasts. Curr. Biol. 2010;20:R746–R753. doi: 10.1016/j.cub.2010.06.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Essien K., Stoeckert C.J., Jr Conservation and divergence of known apicomplexan transcriptional regulons. BMC Genom. 2010;11:147. doi: 10.1186/1471-2164-11-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Planta R.J., Goncalves P.M., Mager W.H. Global regulators of ribosome biosynthesis in yeast. Biochem. Cell. Biol. 1995;73:825–834. doi: 10.1139/o95-090. [DOI] [PubMed] [Google Scholar]
- 71.Tanay A., Regev A., Shamir R. Conservation and evolvability in regulatory networks: the evolution of ribosomal regulation in yeast. Proc. Natl. Acad. Sci. U.S.A. 2005;102:7203–7208. doi: 10.1073/pnas.0502521102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Borneman A.R., Gianoulis T.A., Zhang Z.D., Yu H., Rozowsky J., Seringhaus M.R., Wang L.Y., Gerstein M., Snyder M. Divergence of transcription factor binding sites across related yeast species. Science. 2007;317:815–819. doi: 10.1126/science.1140748. [DOI] [PubMed] [Google Scholar]
- 73.Moses A.M., Pollard D.A., Nix D.A., Iyer V.N., Li X.-Y., Biggin M.D., Eisen M.B. Large-scale turnover of functional transcription factor binding sites in Drosophila. PLoS Comput. Biol. 2006;2:e130. doi: 10.1371/journal.pcbi.0020130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Odom D.T., Dowell R.D., Jacobsen E.S., Gordon W., Danford T.W., MacIsaac K.D., Rolfe P.A., Conboy C.M., Gifford D.K., Fraenkel E. Tissue-specific transcriptional regulation has diverged significantly between human and mouse. Nat. Genet. 2007;39:730–732. doi: 10.1038/ng2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schmidt D., Wilson M.D., Ballester B., Schwalie P.C., Brown G.D., Marshall A., Kutter C., Watt S., Martinez-Jimenez C.P., Mackay S., et al. Five-vertebrate ChIP-seq reveals the evolutionary dynamics of transcription factor binding. Science. 2010;328:1036–1040. doi: 10.1126/science.1186176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.LaCount D.J., Vignali M., Chettier R., Phansalkar A., Bell R., Hesselberth J.R., Schoenfeld L.W., Ota I., Sahasrabudhe S., Kurschner C., et al. A protein interaction network of the malaria parasite Plasmodium falciparum. Nature. 2005;438:103–107. doi: 10.1038/nature04104. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.