


Abstract
Escherichia coli DNA polymerase IV (Pol IV, also known as DinB) is a Y-family DNA polymerase capable of catalyzing translesion DNA synthesis (TLS) on certain DNA lesions, and accumulating data suggest that Pol IV may play an important role in copying various kinds of spontaneous DNA damage including N2-dG adducts and alkylated bases. Pol IV has a unique ability to coexist with Pol III on the same β clamp and to positively dissociate Pol III from β clamp in a concentration-dependent manner. Reconstituting the entire process of TLS in vitro using E. coli replication machinery and Pol IV, we observed that a replication fork stalled at (−)-trans-anti-benzo[a]pyrene-N2-dG lesion on the leading strand was efficiently and quickly recovered via two sequential switches from Pol III to Pol IV and back to Pol III. Our results suggest that TLS by Pol IV smoothes the way for the replication fork with minimal interruption.
INTRODUCTION
Genomic deoxyribonucleic acid (DNA) in all organisms is constantly exposed to endogenous and exogenous factors that can generate numerous types of DNA lesions. Although cells are equipped with various DNA repair mechanisms removing lesions, the replisome (the apparatus for DNA replication) inevitably encounters blocking lesions on the template DNA (1). The replication blocked by such lesion is rescued by damage tolerance mechanisms without which cells cannot survive (2–4).
The fate of a replication fork at blocking lesion depends on which strand contains the lesion. It was shown using an in vitro oriC-plasmid DNA replication system with the Escherichia coli replisome that a single abasic (AP) lesion on the lagging-strand, leaving a gap extending from the 5′-side of the lesion, had no effect on the progression of the replication fork and that one on the leading strand completely blocked the leading-strand synthesis and stalled the replication fork (5). In two thirds of the replication forks with blocked leading-strand synthesis, however, the lagging-strand synthesis continued until it stopped around 1 kb downstream of the lesion site. The lagging-strand synthesis uncoupled from the leading-strand synthesis thus left a single-stranded (ss) DNA segment on the leading strand. A similar uncoupling of leading- and lagging-strand synthesis was shown to occur in E. coli cells (6), and it seems that the leading-strand synthesis downstream of a cyclobutane pyrimidine dimer (CPD), which is strong blocking lesion, can be reinitiated through DnaG primase-directed repriming (7). In this case, the lesion would be left in the ssDNA gap on the leading strand. An ssDNA segment containing a lesion should therefore be formed whenever the replication fork encounters a blocking lesion, regardless of whether that lesion is in the lagging strand or the leading strand. The ssDNA segment with the blocking lesion, lethal if not repaired, is the target for the damage tolerance.
The damage tolerance mechanisms have two major branches: translesion DNA synthesis (TLS) and damage avoidance (DA). TLS branch involves a class of DNA polymerase (TLS polymerase) characterized by the ability to replicate damaged DNA (8,9). Not always mutagenic, the TLS branch is nonetheless inherently error-prone. The DA branch, on the other hand, has several pathways related to homologous recombination utilizing the information present in the undamaged sister chromatid, and the lesion bypass by the DA pathways is error-free (1). Although the molecular mechanisms of DA pathways are still poorly defined, it was clearly demonstrated that in normally growing E. coli cells, DA events greatly outnumbered TLS events when a single blocking lesion was grafted into the chromosome by a novel site-specific integration technique (10). A similar approach using mammalian cells, in contrast, showed more favorable usage of TLS events (11). This difference seems to be due to the proficiency of DA mechanisms and TLS polymerases differing between E. coli and mammalian cells. DNA polymerase V (Pol V), a major TLS polymerase in E. coli, is able to bypass common replication-blocking lesions, such as UV-induced photoproducts and chemical carcinogen-induced DNA adducts (12). However, the TLS action by Pol V absolutely requires RecA protein that is up-regulated and activated in the SOS response, and the upregulation and maturation of Pol V occur at the late stage of the SOS response (9). In normally growing cells, essentially no Pol V is present (8). TLS action by Pol V was observed with a delay of 50 min when plasmid DNA containing a single blocking lesion was introduced into SOS-induced E. coli cells (6). These data suggest that Pol V contributes only marginally to cellular damage tolerance mechanisms, and thus that DA pathways predominate in damage tolerance for the common blocking lesions in E. coli cells. The physiological role of Pol V can be viewed as a beneficial source of genetic diversity under genotoxic stress conditions (12).
Escherichia coli has another TLS polymerase, DNA polymerase IV (Pol IV), which belongs to the Y-family of DNA polymerase (13,14). Pol IV is present relatively abundantly, 250 molecules/cell, in normally growing cells and is up-regulated 10-fold in SOS-induced cells (15). Biochemical studies showed that the highly purified Pol IV has a catalytic proficiency in DNA synthesis and that its catalytic activity does not require RecA protein (13). Although the common blocking lesions are essentially not bypassed by Pol IV, a subset of minor groove lesions are efficiently and accurately bypassed by the enzyme. The major benzo[a]pyrene (BP) adduct to the N2 position of guanine, (+)-trans-anti-BP-N2-dG (abbreviated to dG-BP(+)), is bypassed by a mutual action of Pol IV and Pol V (16), while its stereo isomer, (−)-trans-anti-BP-N2-dG (abbreviated to dG-BP(−)) is bypassed by Pol IV alone (17,18). Another N2-dG adduct, N2-furfuryl-dG is also efficiently and accurately bypassed by Pol IV (19). Furthermore, the TLS action by Pol IV was observed with various spontaneous DNA lesions, including N2-N2-dG interstrand cross-link (20), acroline-mediated N2-dG peptide cross-links (21) and N2-(1-carboxyethyl)-dG adduct (22). A genetic study suggested an involvement of Pol IV in damage tolerance mechanisms for 3-methyldA and 3-methyldG (23). Therefore, as a prototype of Y-family TLS polymerase, Pol IV may be involved in proficient and accurate damage tolerance against various, but not all, spontaneous DNA lesions in normally growing cells.
Interestingly, a unique behavior of Pol IV toward Pol III was observed in in vitro DNA synthesis on SSB-coated singly primed ssDNA template (24,25). Association of Pol III with primer/template (P/T) DNA is strongly stabilized by binding of Pol III to β clamp, enabling the Pol III to remain bound to the P/T for more than 5 min. Indiani et al. found that Pol IV (12 nM) quickly takes control of the P/T from Pol III (4 nM) when it is idling on the P/T and that this polymerase exchange is less efficient when Pol III is moving in the presence of four dNTPs (24). They also showed that Pol III and Pol IV remain bound to the same β clamp during the polymerase switching. Pol IV has two distinct interfaces contacting with β clamp (26,27), both of which are required for the Pol III-Pol IV switching in vitro (28,29). On the other hand, Furukohri et al. reported that Pol IV inhibits DNA synthesis by Pol III in a concentration-dependent manner (25). Whereas no significant inhibition of processive DNA synthesis by Pol III was observed at low concentrations of Pol IV (22 and 45 nM), much higher concentrations (450 and 890 nM) actively promote rapid dissociation of the idling Pol III from β clamp (<15 s). This dislodging action does not require the association of Pol IV with β clamp but is stimulated by the β-clamp interaction. Taken together, these findings indicate that Pol IV affects Pol III through two distinct types of β-clamp interaction and a function dislodging Pol III from β clamp in a concentration-dependent manner: efficient polymerase switching on the same β clamp at lower concentration (12 nM) of Pol IV, and quick dissociation of Pol III from β clamp at higher concentration (450–890 nM) of Pol IV.
These in vitro studies of the nature of Pol IV may be of limited relevance to the intracellular concentration of Pol IV, 330 nM in normally growing cells and 3 μM in SOS-induced cells (8), but it was shown that Pol IV forms a multi-protein complex with RecA and UmuD proteins and that the complex formation modulates the function of Pol IV (30,31). The Pol IV concentration in normally growing cells may therefore be within a range suitable for a quick polymerase exchange rescuing the stalled Pol III. If so, DNA synthesis stalled at a subset of spontaneous DNA lesions such as N2-dG adducts would be quickly and accurately rescued by Pol IV without disruption of chain elongation.
In this work, we tried to reconstitute the entire TLS reaction in vitro by using the replication fork stalled at a typical N2-dG adduct so that we could investigate the rescuing action of Pol IV during DNA replication. We present, for the first time, experimental evidence that TLS by Pol IV can directly restart replication fork stalled at a DNA lesion. We also discuss a model of the TLS-mediated recovery of stalled replication forks.
MATERIALS AND METHODS
Nucleic acids
The 13-mer oligonucleotides containing a (−)-trans-anti-benzo[a]pyrene-N2-dG (dG-BP(−)) (5′-GAAGACCTGBP(−)CAGG-3′) were a generous gift from Dr N. Geacintov, NYU, New York. Parental plasmids 1 (P1) and 2 (P2) are derivatives of the oriC-terB plasmid pMOL7 (5). The sequence between the NsiI and ClaI site of pMOL7 is replaced by Cassette A, the 15-mer double-stranded oligonucleotides containing the EcoRV site (5′-ATTGAGATATCCTTA-3′) for P1, or Cassette B, the 30-mer double-stranded oligonucleotides containing the BsrGI site (5′-ATTGAGATGAAGACCTGTACAGGATCCTTA-3′) for P2.
Proteins
N-terminal, His-tagged wild-type Pol IV (Pol IV) and polymerase-null mutant Pol IV (Pol IV D8A) proteins were purified as described previously (25). Enzymes required for the replication with oriC plasmid in vitro were also purified as described elsewhere (5,32). Pol III holoenzyme was purified as described previously (33,34) except that the final Superose 6 column purification step was omitted.
Construction of oriC plasmid containing a single dG-BP(−) adduct
Double-stranded, supercoiled oriC plasmids containing a single dG-BP(−) adduct were constructed by a method developed with the technique using gapped-duplex intermediates described previously (35,36). The approach is summarized in the legend of Supplementary Figure B.
Reconstitution of semi-bidirectional DNA replication of oriC plasmid in vitro
Reconstituted oriC plasmid DNA replication reactions were performed as described previously (5) but with minor modifications mentioned in the legend of Supplementary Figure A. Pol IV was added 5 min after the start of the replication reaction. The final concentrations of Pol IV and Pol III were 10 and 1.7 nM, respectively. To stop the reaction at each time point, an equal volume of stop solution containing 25 mM ethylenediaminetetraacetic acid (EDTA) pH 8.0 and 0.15% sodium dodecylsulphate was added. To monitor the DNA replication, the replication products were labeled by adding [α-32P] radiolabeled deoxynucleoside triphosphates to the reaction.
The radiolabeled replication products were extracted and analyzed by alkaline agarose gel electrophoresis, 8 M urea-denaturing 8% polyacrylamide-sequencing gel electrophoresis or two-dimensional (2D) gel electrophoresis followed by autoradiography as described previously (5) but with minor modifications. For the sequencing gel analysis, replication products were digested by MscI and NsiI. For the 2D gel electrophoresis, the replication products were digested by BglII. Phosphor-imaging and the measuring of each spot's intensities were performed using BAS2000 and Image Gauge (Fujifilm).
The reactions in which the results shown in Figures 5 and 6 were obtained were performed in the absence of radiolabeled nucleotides. The products were then digested by BglI, separated on alkaline agarose gel, and transferred onto a Hybond N+ membrane (GE healthcare). The products were detected by Southern blot analysis with each probes listed in Table 2 in an earlier study (5) or a probe with the complement sequence of probe D (probe D′, (5)). Phosphor-imaging and profiling were performed using BAS2000 and Image Gauge (Fujifilm).
Figure 5.
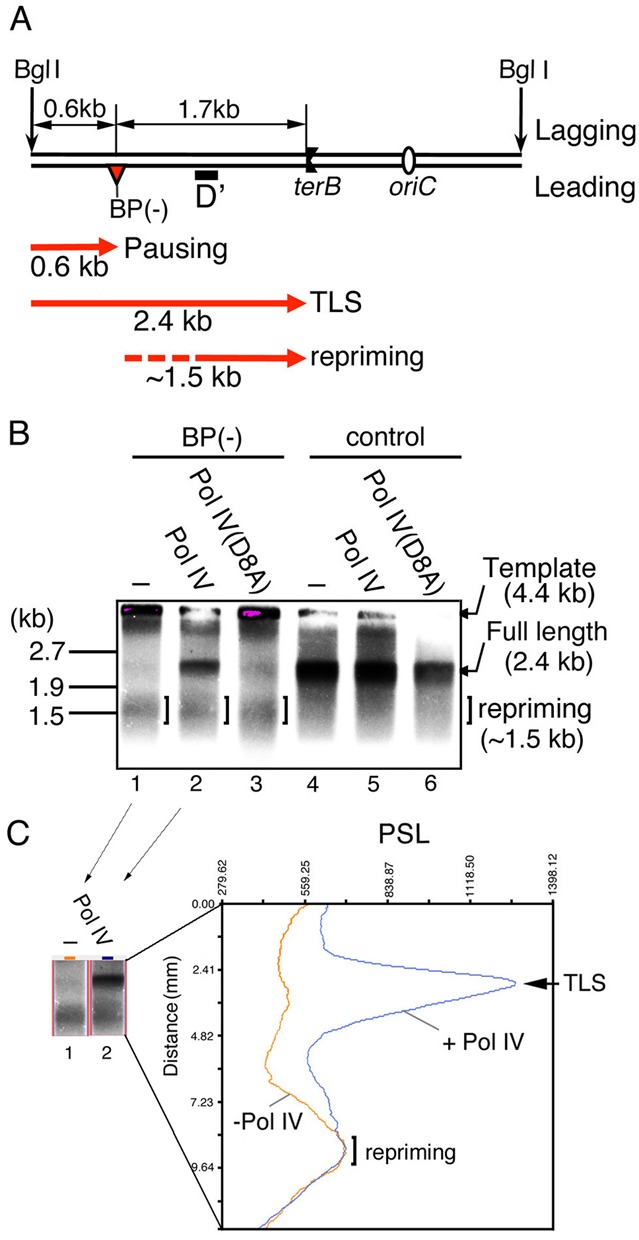
TLS and ‘downstream-lesion repriming’ events occur independently on the leading strand template. (A) Replication products were digested by BglI and subjected to Southern blot analysis using a leading-strand-products-specific probe (probe D′). Possible leading-strand products of counterclockwise forks are depicted as red lines with arrowheads. Pausing products would not be detected by probe D′ because probe D′ is 461 nt downstream of the lesion. (B) Replication reactions with pMOL7-BP(−) (lanes 1–3) or with pMOL7-control (lanes 4–6) were carried out in the absence (lanes 1 and 4) or presence of Pol IV (wild-type in lanes 2 and 5, D8A-mutant in lanes 3 and 6). The 4.4 kb BglI digested templates are barely detectable at the top of the gel because fragments with this size are hard to be transferred to a membrane. (C) Intensities of photostimulated luminescence (PSL) value of radioactive signals of the part of Figure 5B, lanes 1 and 2 (shown in the left panel), are shown with distances from the template signals (orange line, lane 1; blue line, lane 2).
Figure 6.
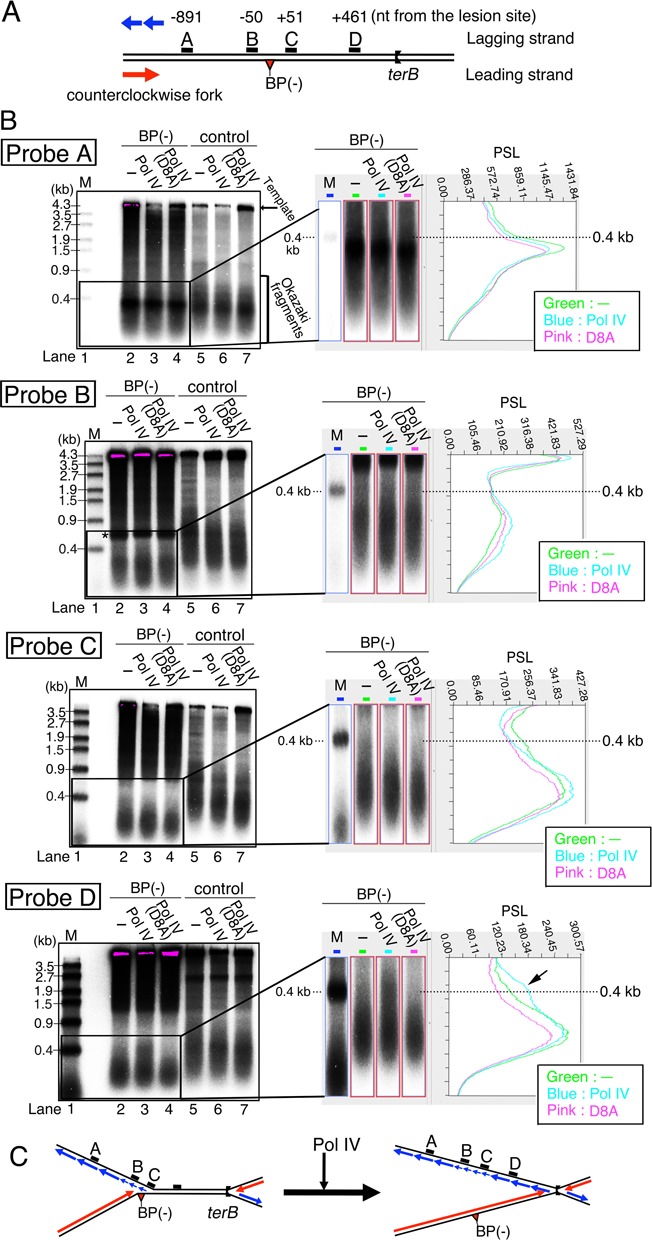
Restoration of normal fork speed is accompanied by concurrent synthesis of both leading- and lagging-strands. (A) The locations of each of the probes for Southern blot analysis on pMOL7-BP(−) are illustrated. Red and blue lines with arrowheads indicate leading- and lagging-strand synthesis of the counterclockwise fork. (B) Replication reactions were carried out with pMOL7-BP(−) (lanes 2–4) and with pMOL7-control (lanes 5–7). Okazaki fragments were detected by Southern blot analysis using specific probes located 891 nt upstream (probe A), 50 nt upstream (probe B), 51 nt downstream (probe C), and 461 nt downstream (probe D) of the lesion. Reactions without Pol IV (lanes 2 and 5) or with mutant Pol IV D8A (lanes 4 and 7) were carried out as controls. Size markers (M, 5′-32P-labeled λ/EcoT14I) are indicated in lane 1. Supercoiled pMOL7 purified from cells was used as a control in lanes 5–7. In the right panel, images enlarged in height nearby 0.4-kb size marker in lanes 1–4 of Figure 6B are shown. PSL intensities for these images are also shown (green line, lane 2; blue line, lane 3; pink line, lane 4). Longer-sized Okazaki fragments that accumulate as a result of Pol IV addition (probe D) are indicated by the black arrowhead in this PSL profile of probe D. (C) Schematic diagram of possible structure of replication forks before and after the addition of Pol IV. Red and blue lines with arrowheads indicate leading-strand and lagging-strand products.
RESULTS
Reconstitution of replication fork stalled at a benzo[a]pyrene DNA adduct that can be bypassed by DNA polymerase IV
To investigate whether Pol IV is able to rescue a stalled replication fork by resuming DNA synthesis over the dG-BP(−) lesion on the leading-strand template, we used the oriC-plasmid DNA-replication system in vitro (5) with 4.4 kb of plasmid pMOL7 DNA as a template (Supplementary Figure A) containing E. coli chromosomal replication origin oriC. Two replication forks are formed and start to proceed bi-directionally from the oriC. The replication termination sequence terB, inserted 0.9 kb away from oriC, blocks the progression of clockwise replication fork by forming a complex with the termination protein Tus. The counterclockwise replication fork travels a longer distance than the clockwise fork does, and it is stopped by the collision of the two forks. Consequently, the leading-strand replication products of each replication fork can be easily distinguished (clockwise fork, ∼0.9 kb; counterclockwise fork, ∼3.5 kb; Supplementary Figure A and Figure 1B, lanes 3 and 9). These products, especially 0.9-kb products, are detected as relatively broad bands because of the different priming position on the leading strand template. The progression of two forks on a plasmid produces two fully replicated plasmids catenated at or near the terB site (note that hypothetical decatenated products are shown in Supplementary Figure A and Figure 1A). A lesion-containing oriC-terB plasmid, called pMOL7-BP(−), was prepared as described in Supplementary Figure B. The dG-BP(−) adduct is located at the indicated dG residue within the following sequence context: 5′-GAAGACCTGBP(−)CAGG-3′. The damaged guanine is 1.7 kb away from the oriC region and was expected to block the leading-strand synthesis in the counterclockwise replication fork (Supplementary Figure B and Figure 1A).
Figure 1.
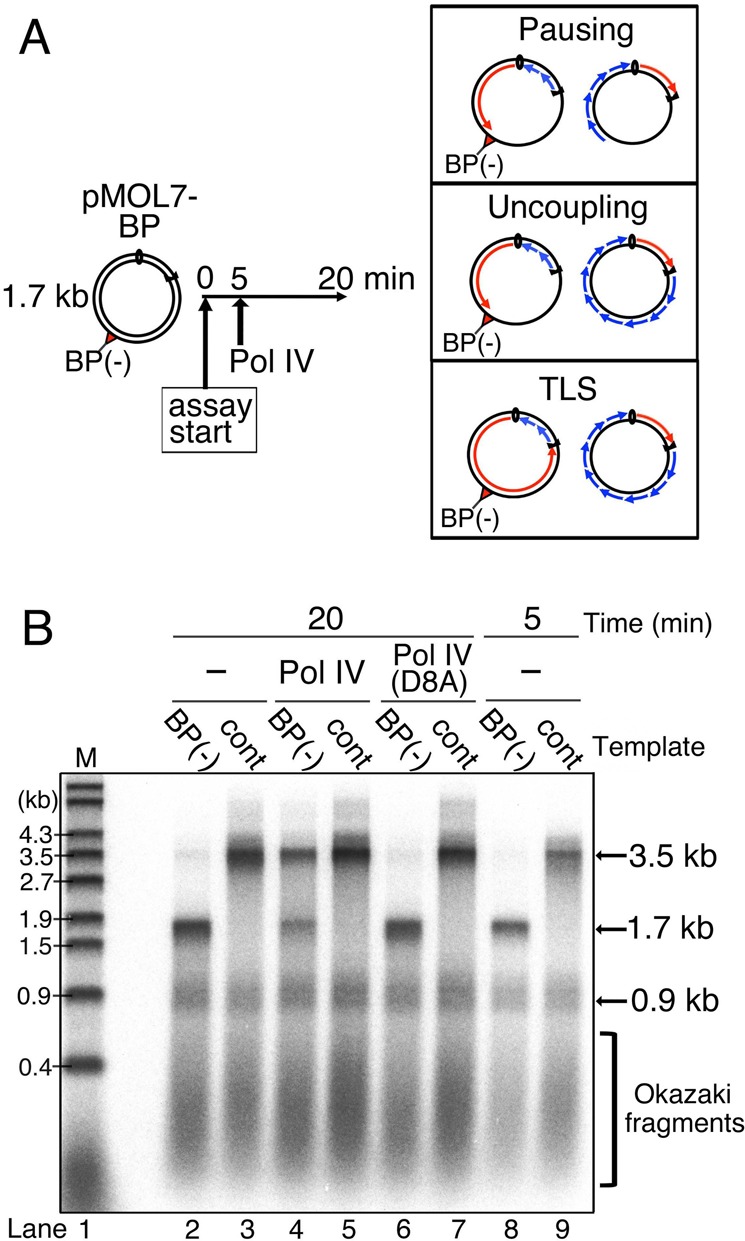
Leading-strand synthesis continues across the blocking lesion by Pol IV-mediated TLS. (A) Reconstitution of TLS in oriC replication system. Template pMOL7-BP(−) or pMOL7-control was incubated at 30°C with Pol III (1.7 nM) and other replicative proteins. Pol IV (10 nM) was added at 5 min after the start of the reaction. Replication products obtained at 20 min were purified, divided and analyzed by alkaline agarose gel electrophoresis (Figure 1), sequence gel electrophoresis (Figure 2) and 2D gel electrophoresis (Figure 4). Possible product structures are depicted in the panels on the right. (B) The 32P-labeled replication products of pMOL7-BP(−) (lanes 2, 4, 6 and 8) or pMOL7-control (lanes 3, 5, 7 and 9) in the absence (lanes 2, 3, 8 and 9) or presence of Pol IV (wild-type, in lanes 4 and 5; D8A-mutant, in lanes 6 and 7) were separated on an alkaline agarose gel. Size markers (M, 5′-32P-labeled λ/EcoT14I) are indicated in lane 1.
To reconstitute the stalled replication fork, this lesion-containing plasmid was first incubated with purified replication proteins. After 5-min incubation, paused products of the leading-strand synthesis (1.7 kb) were observed with the pMOL7-BP(−), while full-length products (3.5 kb) were detected with the lesion-free control plasmids in alkaline agarose gels (Figure 1B, lanes 8 and 9). Further incubation increased the amounts of each product because the initiation step of oriC-plasmid replication is not synchronized but undergoes randomly on each plasmid molecule during the incubation. Even after 20-min incubation, the full-length products were essentially not detected with pMOL7-BP(−). A very faint signal of the full-length products is seen at ∼3.5 kb (Figure 1B, lane 2), but this is likely due to DNA synthesis on a very small amount of parental lesion-free plasmid DNA. In our reaction condition, lagging-strand synthesis generated short products having lengths distributed with a peak at 0.3–0.4 kb because DNA ligase was omitted. Okazaki fragments and 0.9 kb of clockwise leading-strand synthesis products were not affected by the presence of a lesion (Figure 1B, compare lanes 2 and 3). We concluded that DNA-chain elongation by Pol III in the leading-strand synthesis is completely blocked by the dG-BP(−) adduct.
TLS efficiently restarts the leading-strand synthesis across the dG-BP(−) lesion
We tested whether the addition of Pol IV restarts the leading-strand synthesis stalled at the dG-BP(−) adduct (Figure 1A and B). When Pol IV was added 5 min after the incubation started and further incubated for 15 min, full-length leading-strand products were clearly detected (Figure 1B, lane 4). It is also clearly seen that the paused products (1.7 kb) were decreased coordinately with the appearance of full-length products, indicating that the addition of Pol IV rescued the leading-strand synthesis stalled at the lesion by elongating DNA chain across and beyond the lesion site. On the other hand, full-length products were not observed when a catalytically defective mutant Pol IV (Pol IV D8A) was added to the reaction (Figure 1B, lane 6). These data demonstrated that the TLS reaction proceeds efficiently within the replication fork stalled at the dG-BP(−) adduct and that the TLS depends on the polymerase activity of Pol IV.
We confirmed by sequencing gel analysis that the elongation by Pol III was indeed inhibited by the dG-BP(−) adduct (Figure 2). 32P-labeled replication products were digested with two restriction enzymes, separated by sequencing gel, and visualized by phosphor-imaging. When the control intact oriC-plasmid was used for the DNA synthesis, an 86-mer restriction fragment corresponding to the full-length leading-strand product was detected (Figure 2, lane 6). When pMOL7-BP(−) was used, the 86-mer fragment was not detected but a 40-mer product appeared (Figure 2, lane 5). This implies that the chain elongation by Pol III was arrested 1 nt before the dG-BP(−) adduct on the leading-strand template. The addition of wild-type Pol IV resulted in the appearance of a full-length 86-mer product and the concomitant reduction of the 40-mer paused product (Figure 2, lane 7). These data suggest that the polymerase exchange between Pol III and Pol IV (the first switch) occurred at the lesion site and that Pol IV then elongated the leading-strand DNA beyond the lesion site.
Figure 2.
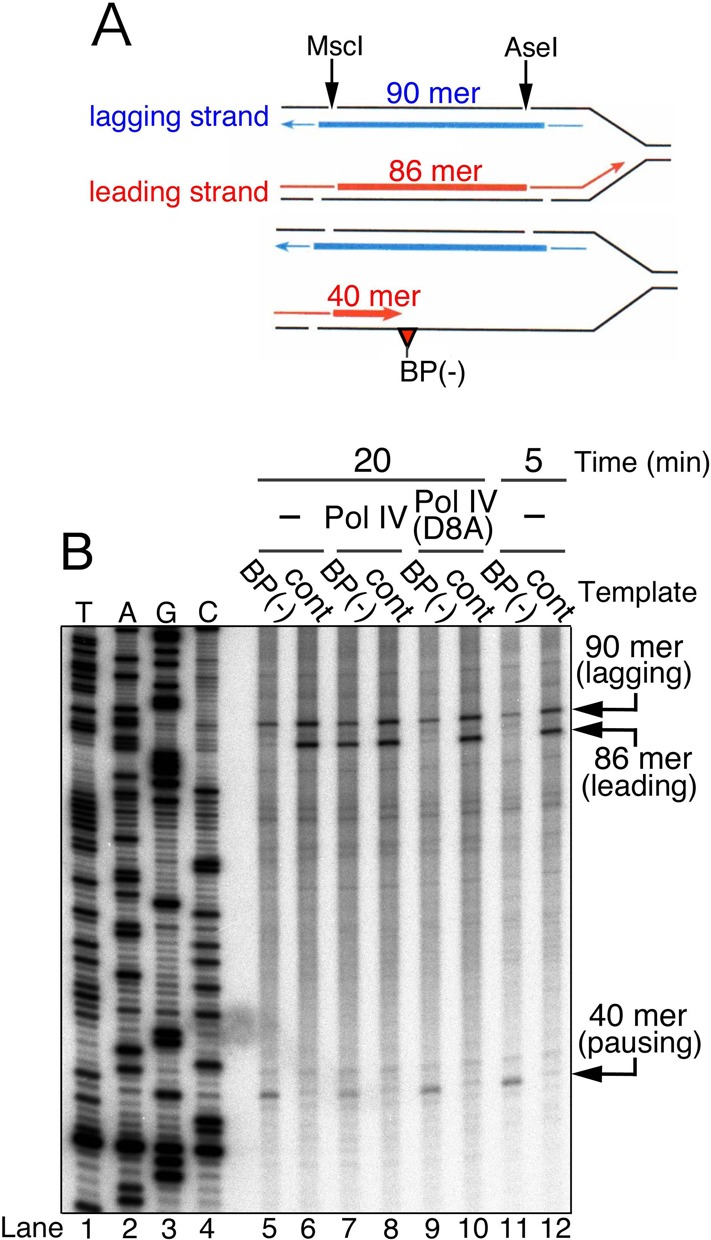
Nucleotide insertion at the lesion site by Pol IV restarts leading-strand synthesis across the blocking lesion. (A) Replication products were digested with MscI and AseI, yielding 32P-labeled products with the size of 86 mer for the leading strand and 90 mer for the lagging strand. If Pol III stops one-nucleotide before the BP(−) lesion, leading products would be 40 mer. (B) Digested replication products were subjected to sequencing gel analysis. The 32P-labeled products of pMOL7-BP(−) (lanes 5, 7, 9 and 11) or pMOL7-control (lanes 6, 8, 10 and 12) in the absence (lanes 5, 6, 11 and 12) or presence of Pol IV (wild-type in lanes 7 and 8, D8A-mutant in lanes 9 and 10) are shown. 5′-32P-labeled ladder markers are indicated in lanes 1–4.
The second polymerase switch occurs after completion of TLS across the lesion site
Although it is difficult to directly measure the length of the TLS patch produced by Pol IV in our in vitro system, another important question is does Pol III come back to the replication fork after completion of TLS by Pol IV? That is, is there a second switch? To answer this question, a time-course experiment was performed after the addition of Pol IV to the reaction (Figure 3). It is also difficult to determine how quickly forks restart leading-strand synthesis across the lesion, because during incubation the replication initiation events on each plasmid occur randomly. It was reported that Pol IV together with the β clamp slowly elongates DNA at ∼5 nt/s, while the Pol III holoenzyme catalyzes elongation at ∼900 nt/s (34,37). If Pol IV alone continues DNA-chain elongation from the lesion site all the way to terB site (∼1.7 kb), full-length products should appear no sooner than 5 min after the addition of Pol IV. A faint but detectable band representing 3.5-kb full-length products, however, was evident 1 min after the addition of Pol IV (Figure 3, lane 3). After 3-min incubation, more full-length products accumulated (Figure 3, lane 4). Without participation of Pol III, it would be difficult for forks to synthesize full-length products within 3 min, suggesting that Pol III came back to the primer terminus and elongated the leading-strand DNA after the completion of TLS by Pol IV.
Figure 3.
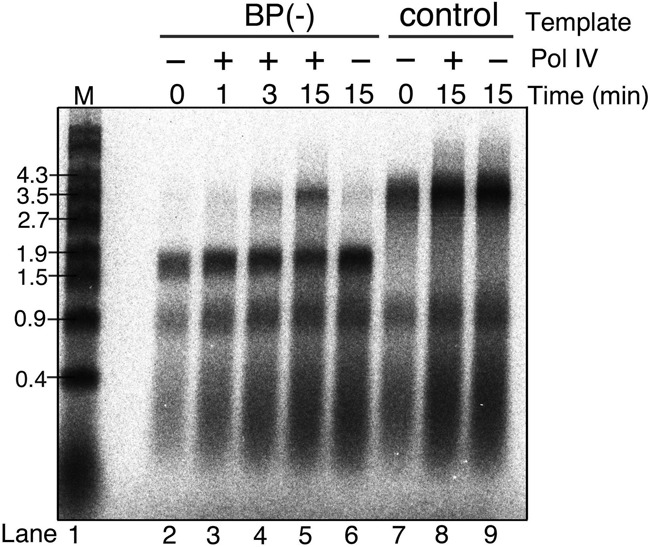
Pol III quickly resumes leading-strand synthesis after completion of TLS. Template pMOL7-BP(−) or pMOL7-control were incubated with replicative proteins including Pol III, and after 5 min of incubation, Pol IV was added. The 32P-labeled products of pMOL7-BP(−) (lanes 2–6) or pMOL7-control (lanes 7–9) obtained at the indicated times after addition of Pol IV were separated on an alkaline agarose gel. Reactions without Pol IV were carried out as controls (in lanes 2, 6, 7 and 9).
TLS can restart the progression of a stalled replication fork
Replication fork dynamics were assessed by 2D gel electrophoresis of the 32P-labeled replication products after digestion with a restriction enzyme, BglII (Figure 4). As shown in Figure 4A, analysis and interpretation of the gel pattern were performed as previously described (5,38). With intact control plasmid, the majority of replication products were detected as spot 1 (decatenated linear molecules) or spot 4 (catenated, asymmetric X-shape molecules) (Figure 4B, left panel). Spot 1 corresponds to fully replicated daughter molecules because the restriction enzyme digestion resolves the catenation of such molecules. Spot 4, on the other hand, corresponds to molecules almost fully replicated but not completed at the terB region (a predicted structure is shown in the lower part of Figure 4B). Since spot 1 comprises the vast majority of products in the oriC-plasmid replication without Tus protein (5), this incompleteness of DNA replication is likely due to a replication-blocking action of the Tus-terB complex. Spots 2 and 3 are due to a small amount of unidirectional replication.
Figure 4.
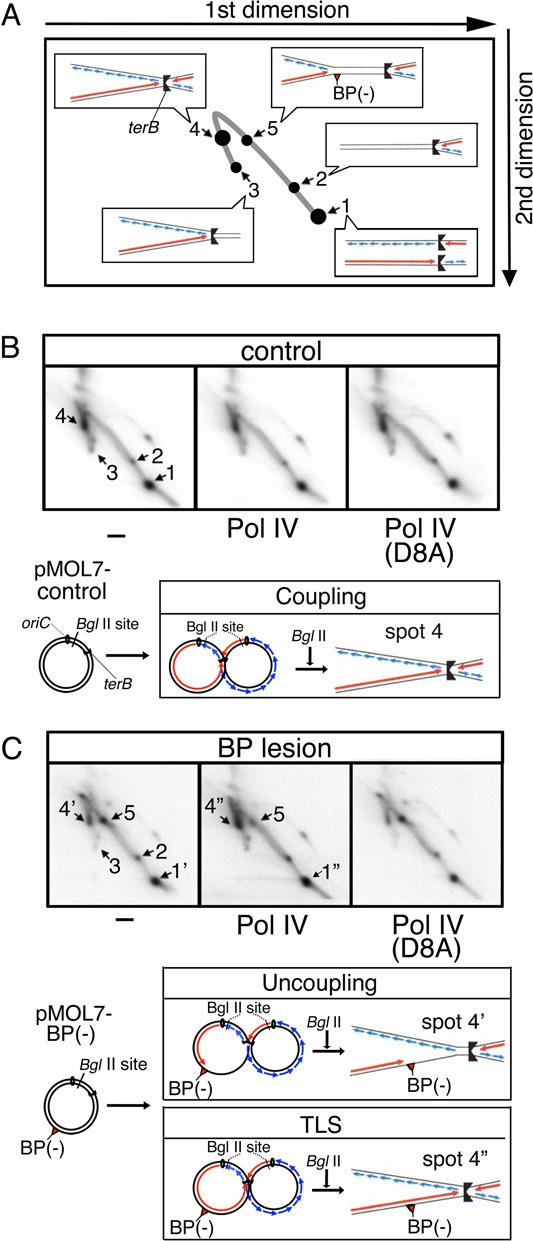
TLS by Pol IV restarts the progression of stalled replication forks. Replication products were treated with BglII and subjected to 2D gel analysis. Schematic diagrams of interpreted structures in each spots on the 2D gel are shown in the panel (A). The 32P-labeled products of pMOL7-control (B) or pMOL7-BP(−) (C) in the absence (left panel) or presence of Pol IV (wild-type, middle panel; D8A-mutant, right panel) are shown. Schematic diagram of possible products in spots 4, 4′, 4′′ are shown in the lower panel. Red and blue lines with arrowheads respectively indicate leading-strand and lagging-strand products.
When products of the in vitro replication with dG-BP(−)-containing plasmid DNA were analyzed, a new spot (spot 5) appeared within the double-Y arc (Figure 4C, left panel). The sizes of products at spot 5 were estimated from its migrating position to be 7.0–7.5 kb. Since the size of products having replication forks stalled only at the lesion site would be 7.0 kb, it is probable that the spot-5 products have stalled replication forks in which the leading-strand synthesis stopped at the lesion site and the lagging-strand synthesis ceased at the lesion site or some distance downstream of it. Spots that migrate to the same positions as spots 1 and 4 in Figure 4B, were also detected in the 2D gel analysis of replication products of pMOL7-BP(−) (shown as spots 1′ and 4′). Replication products corresponding to those spots were likely due to uncoupled lagging-strand DNA synthesis after the leading-strand synthesis ceased at the lesion (a predicted structure of spot 4′ is shown in the lower part of Figure 4C). This is because, as described later, we detected a significant amount of Okazaki fragments synthesized downstream of the lesion site. Quantification of each spot revealed that under our experimental conditions more than half of the replication forks encountering the BP(−) lesion on the leading strand continued to proceed toward the terB site in an uncoupled mode of replication (Figure 4C, left or right panel, the relative intensity ratios of spot 1′, 4′, 5 were respectively 50, 20 and 30%).
Addition of Pol IV decreased the amount of stalled replication products at spot 5, concomitantly increased the amount of products at the location of spot 4 (spot 4′′) and slightly increased the amount of products at the location of spot 1 (spot 1′′) (Figure 4C, middle panel, spots 1′′:4′′:5 = 55%:40%:5%), suggesting that a significant portion of stalled replication forks resumed activity and fully or almost fully completed DNA replication. In contrast, no such conversion of spot 5 was seen when the catalytically dead Pol IV was added to the in vitro system (Figure 4C, right panel). Taking this together with the findings in Figures 1 and 2, we concluded that the stalled replication fork resumed progression when the stalled leading-strand synthesis was restarted by the action of Pol IV. This means that TLS by Pol IV can restart the stalled replication fork.
Recovery of stalled fork by the TLS action of Pol IV is an event distinct from the restarting of leading-strand synthesis downstream of the lesion
Yeeles and Marians observed that the stalled replication fork reinitiated the leading-strand synthesis without TLS and proposed that DnaG primase catalyzes the synthesis of a new primer on the ssDNA segment downstream of the lesion site (7). They used an oriC-plasmid DNA containing a CPD lesion and an in vitro DNA replication system similar to ours. If the same phenomenon occurs in our system, the re-initiated leading-strand synthesis would produce DNA fragments with sizes up to 1.7 kb. We checked this possibility by Southern blot analysis using a specific probe for detecting leading-strand products downstream of the lesion site (Figure 5A). Together with BglI digested template (4.4 kb), as shown in Figure 5B, lane 1, we could detect a relatively broad band around 1.5 kb. This suggests that a restart of leading-strand DNA synthesis downstream of the lesion (we call this event ‘repriming’) occurred in our in vitro system with pMOL7-BP(−). In Figure 1B, lane 2, the repriming products probably overlapped with 1.7-kb stalled leading-strand replication products. Spots 1′′ and 4′′ in the middle panel of Figure 4C should also include the repriming products. Approximately 30% of the almost fully replicated products were repriming products, however, because the relative ratio of full-length TLS products and repriming products was 1.0:0.42 (Figure 5C). Interestingly, the amount of repriming products was not changed when Pol IV catalyzed TLS and yielded the full-length leading-strand products (Figure 5B, compare lanes 1 and 2, Figure 5C). It therefore seems that under the in vitro condition with 1.7 nM Pol III and 10 nM Pol IV, the resumption of leading-strand elongation interrupted at a lesion is an event distinct from the leading-strand repriming downstream of the lesion.
The second polymerase switching restores the speed of replication fork once slowed down around the lesion site
It has been widely accepted that a single Pol III holoenzyme, in which two or three catalytic subunits are connected to the DnaX complex, forms a complex with DnaB helicase moving on the lagging-strand template and that the Pol III–DnaB association ensures that the Pol III is always associating with the replication fork to replicate both strands concurrently (33,39,40). Therefore, another crucial question would be whether the Pol III holoenzyme switched back to the leading-strand synthesis is integrated in the replication fork to participate in Okazaki-fragment synthesis on the lagging strand at the same time. The results described below strongly suggest that after coming back to the leading-strand, Pol III holoenzyme can restart to promote concurrent DNA synthesis on both leading- and lagging-strands in a replication fork moving at a normal speed.
We analyzed lagging-strand synthesis during the whole TLS reaction by Southern blotting detection of Okazaki fragments synthesized at different locations. Replication products were digested by the restriction enzyme BglI, separated on an alkaline agarose gel, and detected by specific probes denoted as A–D on the lagging-strand template in Figure 6A. When the control plasmid pMOL7 was used, at the top of the gel 4.4-kb bands corresponding to digested template strands appeared (Figure 6B, lanes 5–7). Together with them, short smeared bands of Okazaki fragments appeared in the lower part of the gel, showing that in our system the lengths of Okazaki fragments were distributed with a peak at ∼0.3–0.4 kb. (Figure 6B, compare size marker in lane 1 and lanes 5–7). Supercoiled pMOL7 directly isolated from cells was used as a control template in this experiment, while in other experiments the pMOL7-control has been prepared via the gapped duplex procedure. In contrast to what it seen in this gel image of pMOL7, smeared signals and extra signals are seen in the upper part of the gel with pMOL7-BP(−) (lanes 2–4 in Figure 6B). These bands result from minor DNA contaminants carried over during the preparation of the lesion-containing template. The asterisk in the probe B panel indicates a band due to a small amount of contamination by linear plasmid. We confirmed that these bands unrelated to Okazaki fragments were also seen with pMOL7-control, but it was not shown here because the normal sizes of Okazaki fragment are masked by the signals of those contaminating fragments. The detection and interpretation of Okazaki fragments were unaffected by this contamination with pMOL7-BP(−).
Okazaki fragments synthesized at about 1 kb upstream of the lesion site were detected by probe A, and in the presence and absence of Pol IV their size distributions were identical for lesion-containing and lesion-free plasmids (Figure 6B, probe A, compare lanes 2–4 to lanes 5–6). In the in vitro reaction with the lesion-free control plasmid, the size of Okazaki fragments was almost constant at every locations detected by probes A–D (Figure 6B, lanes 5–7). In the reaction with the plasmid DNA containing dG-BP(−) adduct on the leading strand, Okazaki fragments were readily detected at locations 51 nucleotides (probe C) and 461 nucleotides (probe D) downstream of the lesion site (Figure 6B, lanes 2–4). This indicated clearly that the lagging-strand DNA synthesis was uncoupled from the stalled leading-strand synthesis and continued up to about 500 nucleotides downstream of the lesion. Furthermore, the Okazaki fragment became significantly shorter at the location 50 nucleotides upstream of the lesion site (probe B) as well as the locations downstream of the lesion site (probes C and D) (Figure 6B, probes B, C and D, lanes 2–4). In the right panel, signals of Okazaki fragments of pMOL7-BP(−) were enlarged in height to highlight the difference of the size by the comparison with 0.4-kb size marker, that corresponds to the peak size of normal Okazaki fragments. Most Okazaki fragments detected by probe A correspond to the 0.4kb size marker, while Okazaki fragments detected by other probes are of shorter size (compare profiles of signal intensities in Figure 6B, right panel). Higuchi et al. previously observed a similar reduction in the size of Okazaki fragments with the pMOL7 plasmid containing an AP lesion on the leading strand (5). As discussed in their article, the shorter Okazaki fragments detected by probes B, C and D reflect a reduction of replication-fork speed when the leading- and lagging-strand syntheses are uncoupled. The length of the Okazaki fragments was previously shown to depend on two factors (41): the rate of primer formation, which is determined mostly by DnaG concentration, and the rate of replication fork movement. Because of the constant concentration of DnaG in the in vitro reaction, the size reduction of Okazaki fragments was likely due to the reduced speed of the replication fork that proceeds beyond the lesion site. It was reported that Pol III on the leading strand pushing DnaB helicase at a high rate generates a motive force for the replication fork (15). The speed of the replication fork was probably greatly reduced when Pol III on the leading strand stopped upon encountering the lesion on the pMOL7-BP(−).
When Pol IV was added to the reaction with pMOL7-BP(−) 5 min after the reaction started and incubated for further 15 min, a small but significant amount of longer Okazaki fragments appeared around 461 nucleotide downstream of the lesion site (Figure 6B, probe D, compare lanes 3 with 2, compare the blue line with the green one in the scanned profile, longer sized Okazaki fragments were indicated by the black arrowhead). The addition of catalytically dead Pol IV did not cause such a clear change in the size of Okazaki fragments (Figure 6B, probe D, lane 4). The size of longer products was ∼0.4 kb, which is close to that of the normal Okazaki fragment (Figure 6B, probe D, compare lanes 3 with 6, compare the blue line with 0.4-kb marker in the profile), indicating an almost full recovery of fork speed. On the other hand, in the same reaction with Pol IV, the Okazaki fragment remained shorter at the locations near the lesion site (probes B and C). A plausible scenario depicted by these data is the following (Figure 6C). When Pol III on the leading-strand stops at the lesion, the speed of replication fork immediately decreases and shorter Okazaki fragments are synthesized around the lesion site, both upstream and downstream of the lesion site. If Pol IV is present, TLS occurs on the first polymerase switching, and then Pol III takes back the leading-strand synthesis on the second polymerase switching. This reverse switch, from Pol IV back to Pol III, makes the replication fork resume normal speed and in turn leads to normal sized Okazaki fragment synthesis as seen in the Probe D panel. At the same time, lagging-strand synthesis probably regains its coupling with leading-strand synthesis. Although the speed of an uncoupled replication fork is unknown, the second polymerase switching seems to quickly occur, at least in some of the replication forks, in a time short enough for the replication fork to proceed about 500 nucleotides from the lesion site. It is conceivable that the sequential two times of polymerase switching between Pol III and Pol IV enables the replication fork to smoothly bypass the blocking lesion with minimum pausing and uncoupling.
DISCUSSION
In the work reported in this paper, we reconstituted the entire TLS reaction at the replication fork using an E. coli oriC-plasmid DNA-replication system, an E. coli TLS polymerase (Pol IV), and a model DNA lesion (dG-BP(−)) formed by covalent binding of the chemical carcinogen benzo[a]pyrene to the N2 position of guanine. The dG-BP(−) blocks DNA synthesis by Pol III in vitro and can be bypassed by Pol IV. We analyzed the replication-fork dynamics during the TLS reaction and found a quick recovery of the stalled fork by Pol IV. Analysis of the size distribution of Okazaki fragments revealed that the replication fork slows down when it encounters the lesion but quickly resumes its normal speed with the help of Pol IV. These results illuminate a possible important role of TLS by Pol IV in restarting the fork stalled on damaged DNA.
Lesion bypass by polymerase switching between replicative and TLS polymerases on the same β clamp
Undamaged single-stranded circular DNA was used as a template in previous in vitro studies examining functional interaction between Pol III and Pol IV. The idea that Pol IV takes control of the primer/template (P/T) from Pol III when Pol III is stalled on the P/T is based on different effects of Pol IV on Pol III in two situations: Pol IV quickly and efficiently takes over P/T from the Pol III idling on the P/T in a reaction mixture lacking one or two dNTPs, while Pol IV much less efficiently affects the Pol III moving on the P/T in the presence of four dNTPs (24,25). People considering the TLS capacity of Pol IV on a subset of N2-dG adducts as well as N2-dG-related interstrand DNA crosslinks and peptide crosslinks (19–22) have speculated that Pol IV would quickly rescue the Pol III stalled at such a DNA lesion in three sequential steps: (i) polymerase switching from Pol III to Pol IV, (ii) DNA chain elongation opposite the lesion by Pol IV, and (iii) polymerase switching from Pol IV to Pol III. The feasibility of efficient and coordinated action by the two polymerases, however, has never been demonstrated.
The present study showed clearly that Pol IV promotes a very efficient and quick recovery of a replication fork stalled at a typical N2-dG adduct, dG-BP(−), on the leading strand. We used 10 nM Pol IV and 1.7 nM Pol III in all the experiments, and in the present study this concentration of Pol IV showed no effect on the processive DNA synthesis by Pol III on either leading- or lagging-strand DNA synthesis in the oriC-plasmid replication reaction. Despite having no apparent inhibitory effect on the normal chain elongation by Pol III, 10 nM Pol IV appeared to be capable of bypassing the dG-BP(−) lesion on the leading strand when Pol III collides with the lesion, suggesting that the first polymerase switching from Pol III to Pol IV occurs only when Pol III is blocked by the lesion.
From the results of the time course experiment and the analysis of Okazaki fragments (Figures 3 and 6), we showed that Pol III regains control of P/T on the leading strand after the TLS by Pol IV. At the same time, the speed of replication fork also returns to its normal level (Figure 6). Because Pol III on the leading tightly docks to the DnaB helicase on the lagging strand, we speculate that Pol III holoenzyme resumes concurrent leading and lagging strand DNA synthesis. This occurs shortly after lesion bypass when Pol III regains access to the primer terminus freed from Pol IV on the leading strand and when fork speed is recovered. If this hypothesis is correct, the second polymerase switching from Pol IV to Pol III occurs quickly to restore the replisome to its former state, in which Pol IV does not affect the action of Pol III. Another possibility explaining the result in Figure 6 is that even if both strands are still uncoupled, the fork speed recovery increases the distance between primers formed by DnaG primase on the lagging strand and longer Okazaki fragments may be formed later on by free Pol III in the solution. In both cases, the result in Figure 6 suggests that the two sequential polymerase switches occur on the leading strand and consequently the fork recovers normal speed downstream of the lesion site.
For the following reasons it seems plausible that the same Pol III participates in DNA synthesis before and after the TLS by Pol IV. First, 22 nM Pol IV does not dissociate the idling Pol III from P/T and β clamp (25). Second, when Pol IV (12 nM) takes control of P/T from the idling Pol III, both Pol IV and Pol III coexist on the β clamp (24). And last, the replisome formed on oriC-plasmid DNA is stable enough to be isolable by a gel filtration (7). It thus seems that being triggered by the stall of Pol III at the lesion, two sequential polymerase switchings, the first from Pol III to Pol IV enabling the lesion bypass and the second from Pol IV to Pol III resuming the normal chain elongation on both strands, occur on the same β clamp in a quick process.
The undisturbed process of lesion bypass by Pol III and Pol IV, if the hypothesis mentioned above is the case, may be attributable to multiple and dynamic states of their interactions with β clamp (29,42). Recent biochemical analyses of the Pol III-β clamp complex revealed that both α and ϵ subunits of Pol III interact with different subunit of the dimeric β clamp in solution and strengthen the association between Pol III and β clamp (43,44). The data suggested that the interactions between β clamp and two subunits of Pol III would be dynamic, altering the binding states in different modes of Pol III action, such as an elongation mode, a proofreading mode and a stalling mode. It is apparently important to uncover how Pol IV recognizes and affects the different binding states between Pol III and β clamp through dynamic association of Pol IV with β clamp during the DNA synthesis.
Possible physiological role of Pol IV in DNA replication in normally growing E. coli cells
The above-mentioned hypothesis based on the present findings may pose a new insight into physiological roles of Pol IV. Accumulating data suggest that Pol IV efficiently and accurately bypasses a subset of minor groove N2-dG lesions and may play an important role in tolerating some types of spontaneous DNA damage in normally growing cells (19–23). Our in vitro observations suggest that smooth recovery of forks stalled at lesions that can be bypassed by Pol IV may occur in the cell. Although the effective concentration of Pol IV in E. coli cells is still unclear (30,31), it is possible that in normally growing cells, Pol IV always monitors the behavior of replisome in the chromosomal replication and quickly bypasses various, but not all, kinds of spontaneous DNA damage in an essentially error-free fashion when the replisome encounters such damage. Thus, the possible physiological role of Pol IV is to smooth the way for the replication fork by minimizing DNA replication interruptions caused by spontaneous DNA damages on chromosome. If this is the case, Pol IV would be recognized as a component of the replicative machinery that, like DnaG primase, is not physically integrated into the replisome but is always functioning. It is also possible that the positive action of Pol IV may be limited to the fork blockage by minor groove N2-dG adducts. Pol IV is known to be incapable of bypassing many other types of spontaneous lesions and UV- or chemical-induced lesions (45–47). It would be interesting to find out whether other TLS polymerases, such as Pol II or Pol V, play a role similar to that played by Pol IV in the smooth progress of the replication fork across lesions.
Previous studies showed that when Pol III collides with a blocking lesion on the lagging strand, the Pol III readily dissociates from β clamp and initiates the next Okazaki fragment synthesis (5,48). However, it seems possible that the undisturbed TLS process by Pol III and Pol IV on the same β clamp as seen here on the leading strand would also occur on the lagging strand. If the action of Pol IV rescues Pol III stalled at a subset of DNA damage on the leading or lagging strand, Pol IV may prevent the generation of ssDNA segments caused by replication stall at the lesion during chromosomal DNA synthesis. As a consequence, the spontaneous occurrence of DA pathways that are initiated by the ssDNA segment and involve several DNA transactions (2–4) may be reduced to some extent. The DA pathways are able to bypass common replication-blocking lesions in E. coli cells (10), but it has been thought that the replisome is temporally dissolved to initiate the DA events and re-loaded to resume the normal replication process after the DA events (3,4). In contrast to the undisturbed TLS process by Pol III and Pol IV, the DA process involves interruption of DNA replication, which may take a significant amount of time during cell propagation. Thus, depending on how frequently a spontaneous DNA lesion suitable for Pol IV is generated in normally growing cells, the TLS action of Pol IV may contribute to lightening the burden imposed by the DA pathway. Although in vitro data, including our findings, suggest that Pol IV plays a physiological role in the normal DNA replication process in cells, no direct evidence for that hypothesis has been provided by genetic studies. If Pol III is blocked by a lesion that cannot be bypassed by Pol IV, Pol V or DA pathways would solve the replication problem. In good agreement with this model, cells deficient in Pol IV do not exhibit any strong phenotype such as a growth defect, mutator or antimutator phenotype or constitutive induction of the SOS response. We must await a new approach if we want to know whether Pol IV is involved in smoothing the way for replisomes by replication-coupled TLS during chromosome replication in E. coli cells.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGMENT
We thank Dr N. Geacintov (NYU, New York) for generous gifts of oligonucleotides containing a dG-BP(−).
Footnotes
The authors wish it to be known that, in their opinion, the first two authors should be regarded as Joint First Authors.
FUNDING
Grants-in-Aid for Scientific Research KAKENHI [21870023, 25840009 to A.F., 25291077, 25131713 to H.M.] from the Japan Society for the Promotion of Science and the Ministry of Education, Culture, Sports, Science and Technology, Japan; LIGUE Contre le Cancer [labellisation 2011 to R.P.F.]; ANR ForkRepair [ANR 11 BSV8 017 01 to R.P.F.]; United States Public Health Services Grant [R01ES003775 to R.P.F. and E.L.]. Funding for open access charge: Grants-in-Aid for Scientific Research KAKENHI [to A.F.] from the Japan Society for the Promotion of Science and the Ministry of Education, Culture, Sports, Science and Technology, Japan.
REFERENCES
- 1.Friedberg E.C., Walker G.C., Siede W, Wood R.D., Schultz R.A., Ellenberger T. DNA Repair and Mutagenesis. 2nd edn. Washington, DC: ASM Press; 2006. [Google Scholar]
- 2.Michel B., Boubakri H., Baharoglu Z., LeMasson M., Lestini R. Recombination proteins and rescue of arrested replication forks. DNA Repair (Amst.) 2007;6:967–980. doi: 10.1016/j.dnarep.2007.02.016. [DOI] [PubMed] [Google Scholar]
- 3.Gabbai C.B., Marians K.J. Recruitment to stalled replication forks of the PriA DNA helicase and replisome-loading activities is essential for survival. DNA Repair (Amst.) 2010;9:202–209. doi: 10.1016/j.dnarep.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Costes A., Lambert S.A.E. Homologous recombination as a replication fork escort: fork-protection and recovery. Biomolecules. 2013;3:39–71. doi: 10.3390/biom3010039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Higuchi K., Katayama T., Iwai S., Hidaka M., Horiuchi T., Maki H. Fate of DNA replication fork encountering a single DNA lesion during oriC plasmid DNA replication in vitro. Genes Cells. 2003;8:437–449. doi: 10.1046/j.1365-2443.2003.00646.x. [DOI] [PubMed] [Google Scholar]
- 6.Pages V., Fuchs R.P. Uncoupling of leading- and lagging-strand DNA replication during lesion bypass in vivo. Science. 2003;300:1300–1303. doi: 10.1126/science.1083964. [DOI] [PubMed] [Google Scholar]
- 7.Yeeles J.T., Marians K.J. The Escherichia coli replisome is inherently DNA damage tolerant. Science. 2011;334:235–238. doi: 10.1126/science.1209111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sutton M.D. Coordinating DNA polymerase traffic during high and low fidelity synthesis. Biochim. Biophys. Acta. 2010;1804:1167–1179. doi: 10.1016/j.bbapap.2009.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sale J.E., Lehmann A.R., Woodgate R. Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat. Rev. Mol. Cell Biol. 2012;13:141–152. doi: 10.1038/nrm3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pages V., Mazon G., Naiman K., Philippin G., Fuchs R.P. Monitoring bypass of single replication-blocking lesions by damage avoidance in the escherichia coli chromosome. Nucleic Acids Res. 2012;40:9036–9043. doi: 10.1093/nar/gks675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Izhar L., Ziv O., Cohen I.S., Geacintov N.E., Livneh Z. Genomic assay reveals tolerance of DNA damage by both translesion DNA synthesis and homology-dependent repair in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 2013;110:E1462–E1469. doi: 10.1073/pnas.1216894110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fuchs R.P., Fujii S. Translesion synthesis and mutagenesis in prokaryotes. Cold Spring Harb. Perspect. Biol. 2013;5:a012682. doi: 10.1101/cshperspect.a012682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wagner J., Gruz P., Kim S.R., Yamada M., Matsui K., Fuchs R.P., Nohmi T. The dinB gene encodes a novel E. coli DNA polymerase, DNA pol IV, involved in mutagenesis. Mol. Cell. 1999;4:281–286. doi: 10.1016/s1097-2765(00)80376-7. [DOI] [PubMed] [Google Scholar]
- 14.Ohmori H., Friedberg E.C., Fuchs R.P., Goodman M.F., Hanaoka F., Hinkle D., Kunkel T.A., Lawrence C.W., Livneh Z., Nohmi T., et al. The Y-family of DNA polymerases. Mol. Cell. 2001;8:7–8. doi: 10.1016/s1097-2765(01)00278-7. [DOI] [PubMed] [Google Scholar]
- 15.Kim S., Dallmann H.G., McHenry C.S., Marians K.J. Coupling of a replicative polymerase and helicase: a tau-DnaB interaction mediates rapid replication fork movement. Cell. 1996;84:643–650. doi: 10.1016/s0092-8674(00)81039-9. [DOI] [PubMed] [Google Scholar]
- 16.Napolitano R., Janel-Bintz R., Wagner J., Fuchs R.P. All three SOS-inducible DNA polymerases (pol II, pol IV and pol V) are involved in induced mutagenesis. EMBO J. 2000;19:6259–6265. doi: 10.1093/emboj/19.22.6259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shen X., Sayer J.M., Kroth H., Ponten I., O'Donnell M., Woodgate R., Jerina D.M., Goodman M.F. Efficiency and accuracy of SOS-induced DNA polymerases replicating benzo[a]pyrene-7, 8-diol 9, 10-epoxide A and G adducts. J. Biol. Chem. 2002;277:5265–5274. doi: 10.1074/jbc.M109575200. [DOI] [PubMed] [Google Scholar]
- 18.Seo K.Y., Nagalingam A., Miri S., Yin J., Chandani S., Kolbanovskiy A., Shastry A., Loechler E.L. Mirror image stereoisomers of the major benzo[a]pyrene N2-dG adduct are bypassed by different lesion-bypass DNA polymerases in E. coli. DNA Repair (Amst.) 2006;5:515–522. doi: 10.1016/j.dnarep.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 19.Jarosz D.F., Godoy V.G., Delaney J.C., Essigmann J.M., Walker G.C. A single amino acid governs enhanced activity of DinB DNA polymerases on damaged templates. Nature. 2006;439:225–228. doi: 10.1038/nature04318. [DOI] [PubMed] [Google Scholar]
- 20.Kumari A., Minko I.G., Harbut M.B., Finkel S.E., Goodman M.F., Lloyd R.S. Replication bypass of interstrand cross-link intermediates by Escherichia coli DNA polymerase IV. J. Biol. Chem. 2008;283:27433–27437. doi: 10.1074/jbc.M801237200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Minko I.G., Yamanaka K., Kozekov I.D., Kozekova A., Indiani C., O'Donnell M.E., Jiang Q., Goodman M.F., Rizzo C.J., Lloyd R.S. Replication bypass of the acrolein-mediated deoxyguanine DNA-peptide cross-links by DNA polymerases of the DinB family. Chem. Res. Toxicol. 2008;21:1983–1990. doi: 10.1021/tx800174a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yuan B., Cao H., Jiang Y., Hong H., Wang Y. Efficient and accurate bypass of N2-(1-carboxyethyl)-2’-deoxyguanosine by DinB DNA polymerase in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A. 2008;105:8679–8684. doi: 10.1073/pnas.0711546105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bjedov I., Dasgupta C.N., Slade D., Le Blastier S., Selva M., Matic I. Involvement of Escherichia coli DNA polymerase IV in tolerance of cytotoxic alkylating DNA lesions in vivo. Genetics. 2007;176:1431–1440. doi: 10.1534/genetics.107.072405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Indiani C., McInerney P., Georgescu R., Goodman M.F., O'Donnell M. A sliding-clamp toolbelt binds high- and low-fidelity DNA polymerases simultaneously. Mol. Cell. 2005;19:805–815. doi: 10.1016/j.molcel.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 25.Furukohri A., Goodman M.F., Maki H. A dynamic polymerase exchange with Escherichia coli DNA polymerase IV replacing DNA polymerase III on the sliding clamp. J. Biol. Chem. 2008;283:11260–11269. doi: 10.1074/jbc.M709689200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bunting K.A., Roe S.M., Pearl L.H. Structural basis for recruitment of translesion DNA polymerase pol IV/DinB to the beta-clamp. EMBO J. 2003;22:5883–5892. doi: 10.1093/emboj/cdg568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burnouf D.Y., Olieric V., Wagner J., Fujii S., Reinbolt J., Fuchs R.P., Dumas P. Structural and biochemical analysis of sliding clamp/ligand interactions suggest a competition between replicative and translesion DNA polymerases. J. Mol. Biol. 2004;335:1187–1197. doi: 10.1016/j.jmb.2003.11.049. [DOI] [PubMed] [Google Scholar]
- 28.Heltzel J.M., Maul R.W., Wolff D.W., Sutton M.D. Escherichia coli DNA polymerase IV (pol IV), but not pol II, dynamically switches with a stalled pol III* replicase. J. Bacteriol. 2012;194:3589–3600. doi: 10.1128/JB.00520-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wagner J., Etienne H., Fuchs R.P., Cordonnier A., Burnouf D. Distinct beta-clamp interactions govern the activities of the Y family PolIV DNA polymerase. Mol. Microbiol. 2009;74:1143–1151. doi: 10.1111/j.1365-2958.2009.06920.x. [DOI] [PubMed] [Google Scholar]
- 30.Godoy V.G., Jarosz D.F., Simon S.M., Abyzov A., Ilyin V., Walker G.C. UmuD and RecA directly modulate the mutagenic potential of the Y family DNA polymerase DinB. Mol. Cell. 2007;28:1058–1070. doi: 10.1016/j.molcel.2007.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cafarelli T.M., Rands T.J., Benson R.W., Rudnicki P.A., Lin I., Godoy V.G. A single residue unique to DinB-like proteins limits formation of the polymerase IV multiprotein complex in Escherichia coli. J. Bacteriol. 2013;195:1179–1193. doi: 10.1128/JB.01349-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Funnell B.E., Baker T.A., Kornberg A. Complete enzymatic replication of plasmids containing the origin of the Escherichia coli chromosome. J. Biol. Chem. 1986;261:5616–5624. [PubMed] [Google Scholar]
- 33.Maki H., Maki S., Kornberg A. DNA polymerase III holoenzyme of Escherichia coli. IV. the holoenzyme is an asymmetric dimer with twin active sites. J. Biol. Chem. 1988;263:6570–6578. [PubMed] [Google Scholar]
- 34.Sugaya Y., Ihara K., Masuda Y., Ohtsubo E., Maki H. Hyper-processive and slower DNA chain elongation catalysed by DNA polymerase III holoenzyme purified from the dnaE173 mutator mutant of Escherichia coli. Genes Cells. 2002;7:385–399. doi: 10.1046/j.1365-2443.2002.00527.x. [DOI] [PubMed] [Google Scholar]
- 35.Koehl P., Burnouf D., Fuchs R.P. Construction of plasmids containing a unique acetylaminofluorene adduct located within a mutation hot spot. A new probe for frameshift mutagenesis. J. Mol. Biol. 1989;207:355–364. doi: 10.1016/0022-2836(89)90259-3. [DOI] [PubMed] [Google Scholar]
- 36.Lenne-Samuel N., Janel-Bintz R., Kolbanovskiy A., Geacintov N.E., Fuchs R.P. The processing of a benzo(a)pyrene adduct into a frameshift or a base substitution mutation requires a different set of genes in Escherichia coli. Mol. Microbiol. 2000;38:299–307. doi: 10.1046/j.1365-2958.2000.02116.x. [DOI] [PubMed] [Google Scholar]
- 37.Wagner J., Fujii S., Gruz P., Nohmi T., Fuchs R.P. The beta clamp targets DNA polymerase IV to DNA and strongly increases its processivity. EMBO Rep. 2000;1:484–488. doi: 10.1093/embo-reports/kvd109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Friedman K.L., Brewer B.J. Analysis of replication intermediates by two-dimensional agarose gel electrophoresis. Methods Enzymol. 1995;262:613–627. doi: 10.1016/0076-6879(95)62048-6. [DOI] [PubMed] [Google Scholar]
- 39.Kurth I., O'Donnell M. New insights into replisome fluidity during chromosome replication. Trends Biochem. Sci. 2013;38:195–203. doi: 10.1016/j.tibs.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maki H., Furukohri A. DNA polymerase III, bacterial. In: Lennarz W.J., Lane M.D., editors. The Encyclopedia of Biological Chemistry. Vol. 2. Waltham, MA: Academic Press; 2013. pp. 92–95. Vol. [Google Scholar]
- 41.Marians K.J. Prokaryotic DNA replication. Annu. Rev. Biochem. 1992;61:673–719. doi: 10.1146/annurev.bi.61.070192.003325. [DOI] [PubMed] [Google Scholar]
- 42.Heltzel J.M., Maul R.W., Scouten Ponticelli S.K., Sutton M.D. A model for DNA polymerase switching involving a single cleft and the rim of the sliding clamp. Proc. Natl. Acad. Sci. U.S.A. 2009;106:12664–12669. doi: 10.1073/pnas.0903460106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ozawa K., Horan N.P., Robinson A., Yagi H., Hill F.R., Jergic S., Xu Z.Q., Loscha K.V., Li N., Tehei M., et al. Proofreading exonuclease on a tether: the complex between the E. coli DNA polymerase III subunits alpha, epsilon, theta and beta reveals a highly flexible arrangement of the proofreading domain. Nucleic Acids Res. 2013;41:5354–5367. doi: 10.1093/nar/gkt162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Toste Rego A., Holding A.N., Kent H., Lamers M.H. Architecture of the pol III-clamp-exonuclease complex reveals key roles of the exonuclease subunit in processive DNA synthesis and repair. EMBO J. 2013;32:1334–1343. doi: 10.1038/emboj.2013.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jasti V.P., Das R.S., Hilton B.A., Weerasooriya S., Zou Y., Basu A.K. (5'S)-8, 5’-cyclo-2’-deoxyguanosine is a strong block to replication, a potent pol V-dependent mutagenic lesion, and is inefficiently repaired in escherichia coli. Biochemistry. 2011;50:3862–3865. doi: 10.1021/bi2004944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yuan B., Wang J., Cao H., Sun R., Wang Y. High-throughput analysis of the mutagenic and cytotoxic properties of DNA lesions by next-generation sequencing. Nucleic Acids Res. 2011;39:5945–5954. doi: 10.1093/nar/gkr159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ippoliti P.J., Delateur N.A., Jones K.M., Beuning P.J. Multiple strategies for translesion synthesis in bacteria. Cells. 2012;1:799–831. doi: 10.3390/cells1040799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McInerney P., O'Donnell M. Functional uncoupling of twin polymerases: Mechanism of polymerase dissociation from a lagging-strand block. J. Biol. Chem. 2004;279:21543–21551. doi: 10.1074/jbc.M401649200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.