


Abstract
Conserved ribosomal proteins frequently harbor additional segments in eukaryotes not found in bacteria, which could facilitate eukaryotic-specific reactions in the initiation phase of protein synthesis. Here we provide evidence showing that truncation of the N-terminal domain (NTD) of yeast Rps5 (absent in bacterial ortholog S7) impairs translation initiation, cell growth and induction of GCN4 mRNA translation in a manner suggesting incomplete assembly of 48S preinitiation complexes (PICs) at upstream AUG codons in GCN4 mRNA. Rps5 mutations evoke accumulation of factors on native 40S subunits normally released on conversion of 48S PICs to 80S initiation complexes (ICs) and this abnormality and related phenotypes are mitigated by the SUI5 variant of eIF5. Remarkably, similar effects are observed by substitution of Lys45 in the Rps5-NTD, involved in contact with Rps16, and by eliminating the last two residues of the C-terminal tail (CTT) of Rps16, believed to contact initiator tRNA base-paired to AUG in the P site. We propose that Rps5-NTD-Rps16-NTD interaction modulates Rps16-CTT association with Met-tRNAiMet to promote a functional 48S PIC.
INTRODUCTION
Eukaryotic translation initiation is a complex process involving multiple steps (1). For the majority of cellular mRNAs it starts with the recruitment of Met-tRNAiMet to the 40S ribosomal subunit by eukaryotic-specific initiation factor eIF2 (1). In cooperation with initiation factors eIF3, 1/1A, the eIF2·GTP·Met-tRNAiMet ternary complex (TC), binds the 40S ribosomal subunit yielding the 43S preinitiation complex (PIC) (1). The 43S complex then binds to the 5′-end of mRNA and scans in search of the initiation codon to form the 48S PIC. Following recognition of the start codon and eIF5-induced irreversible hydrolysis of eIF2-bound Guanosine-5'-triphosphate (GTP), eIF5B promotes joining of the 40S and 60S subunits and the elongation process begins (1).
While recent studies have yielded detailed insights into the mechanism of translation initiation, many details of the process remain unknown. The exact placement and orientation of initiation factors on the ribosomal surface, structural rearrangements accompanying various steps of initiation, the role played by ribosomal proteins, the timing (and kinetics) of factor association and release and, finally, the exact architecture of the 43S and 48S PICs are either unknown or just beginning to emerge (2,3).
X-ray structures of the yeast Saccharomyces cerevisiae 80S ribosome (4,5) are opening up new opportunities to investigate the mechanism and regulation of translation initiation in eukaryotic cells. Eukaryotic ribosomes have evolved to be structurally more complex than those of prokaryotes and it is believed that this complexity is directly related to the evolution of the translation apparatus, appearance of new translation factors as well as appearance of multiple sophisticated translation control mechanisms, absent in prokaryotic cells (1–6). One of the key features differentiating eukaryotic (yeast) from prokaryotic ribosomes is the extent of protein–protein interactions on the ribosome surface (4,6); however, the significance of these interactions is unknown.
We aim to understand the evolutionary complexity of the eukaryotic (yeast) ribosome by studying the structure and function of yeast ribosomal protein S5, which belongs to the rpS7 ribosomal protein family that includes rpS7 in bacteria and Rps5 in eukaryotes (4,7,8). The protein forms part of the exit (E) site, is essential for cellular viability and recent data suggest that yeast Rps5 functions in translation initiation (9,10) as well as 40S head formation (11). Rps5/S7 proteins possess conserved central and C-terminal regions and exhibit variability at the N-terminus, with fungi and fruit flies exhibiting the longest N-terminal tail regions as compared to bacteria and metazoans.
Here we provide a detailed functional analysis of the N-terminal domain (NTD) of yeast Rps5 suggesting that it communicates with Rps16 to influence events surrounding recruitment of TC and assembly of functional 48S PICs. Our biochemical analysis suggests that truncation of the Rps5 N-terminal region, or mutation of an NTD residue (K45) that contacts Rps16, compromises hydrolysis of eIF2-bound GTP (in the 48S PIC), increasing accumulation of eIF1 and eIF5B (in addition to the eIF2 accumulation noted before (10)) while reducing association of eIF5, thereby delaying subunit joining and progression of the 80S ribosome to the elongation-competent state. Remarkably, similar effects were observed on eliminating the last two residues of the C-terminal tail (CTT) of Rps16, believed to contact initiator tRNA when base-paired to AUG in the P site (2,3,12). These defects can be rescued by introducing an eIF5 mutant (G31R), reported previously to possess elevated GTPase-activating-protein (GAP) function (13). We therefore hypothesize that communication of Rps5 with Rps16 has evolved to enhance recruitment of the eukaryotic-specific eIF2·GTP·Met-tRNAiMet ternary complex and regulated hydrolysis of eIF2-bound GTP, in a manner involving an altered location of the Rps16 CTT in the 40S decoding center.
MATERIALS AND METHODS
Yeast strains and growth methods
rps5-Δ0, rps5-Δ1-13, rps5-Δ1-24, rps5-Δ1-30 and rps5-Δ1-46 strains (Table 1) have been previously described (10), in which the chromosomal RPS5 gene is deleted and replaced with a kanMX cassette and mutant or wild type (WT) RPS5 alleles are present on high-copy plasmids and expressed from the strong S. cerevisiae TEF1 promoter. The following strains of a similar design, rps5-K41A Mata his3-1, leu2-0, ura3-0, rps5::kanMX, <yrps5-K41A; LEU2, 2μ>, rps5-F43G Mata his3-1, leu2-0, ura3-0, rps5::kanMX, <yrps5-F43G; LEU2, 2μ>, rps5-K45A Mata his3-1, leu2-0, ura3-0, rps5::kanMX, <yrps5-K45A; LEU2, 2μ>, expressing yeast rps5 variants with the indicated amino acid substitutions, were obtained as follows: point mutations were first introduced into the RPS5 gene in pTEF_yS5 plasmid (10) using side-directed mutagenesis and the following primers 5′-CAAACCGAGATTGCGTTGTTCAAC-3′ forward and 5′-GTTGAACAACGCAATCTCGGTTTG-3′ reverse (pTEF_yS5-K41A); 5′-GAGATTAAGTTGGGCAACAAATGGTC-3′ forward and 5′-GACCATTTGT TGCCCAACTTAATCTC-3′ reverse (pTEF_yS5-F43G); and 5′-AGTTGTTCAACGCATGGTCTTTTG-3′ forward and 5′-CAAAAGACCATGCGTTGAACAACT-3′ reverse (pTEF_yS5-K45A). The resultant pTEF_yS5 plasmids carrying mutant RPS5 were transformed into the heterozygous diploid BY4743 [4741/4742] MATa/MATα, his3-1/his3-1, leu2-0/leu2-0, lys2-0/+, met15-0/+, ura3-0/ura3-0, RPS5/rps5::kanMX. Transformants were allowed to sporulate using standard protocols (14) and tetrads were dissected. Haploid clones able to grow on YPED medium, resistant to G418 sulfate and expressing mutant RPS5 were selected. The rps5::kanMX genotype was further verified by PCR using 5′-CAGGTGCGACAATCTATCG-3′ and 5′-GAAACGTTACGTTTAGAGACAATG-3′ primers.
Table 1. Strains of S. cerevisiae.
| Strain | Genotype | Source/reference |
|---|---|---|
| rp5+ gcn2Δ | Mata his3-1, leu2-0, ura3-0, rps5::kanMX, <yrps5; LEU2, 2μ> gcn2::hisG | This work |
| rps5-Δ0 | Mata his3-1, leu2-0, ura3-0, rps5::kanMX, <yrps5; LEU2, 2μ> | (10) |
| rps5-Δ1-13 | Mata his3-1, leu2-0, ura3-0, rps5::kanMX, <yrps5-13; LEU2, 2μ> | (10) |
| rps5-Δ1-24 | Mata his3-1, leu2-0, ura3-0, rps5::kanMX, <yrps5-24; LEU2, 2μ> | (10) |
| rps5-Δ1-30 | Mata his3-1, leu2-0, ura3-0, rps5::kanMX, <yrps5-30; LEU2, 2μ> | (10) |
| rps5-Δ1-46 | Mata his3-1, leu2-0, ura3-0, rps5::kanMX, <yrps5-46; LEU2, 2μ> | (10) |
| rps5-K41A | Mata his3-1, leu2-0, ura3-0, rps5::kanMX, <yrps5-K41A; LEU2, 2μ> | This work |
| rps5-F43G | Mata his3-1, leu2-0, ura3-0, rps5::kanMX, <yrps5-F43G; LEU2, 2μ> | This work |
| rps5-K45A | Mata his3-1, leu2-0, ura3-0, rps5::kanMX, <yrps5-K45A; LEU2, 2μ> | This work |
| rps16+gcn2Δ | his3-1, leu2-0, met15-0, LYS, ura3-0, rps16B::kanMX4, rps16A::HIS3, <pGAL-RPS16A; LEU2, ARS1, CEN4> <pRPS28-RPS16; MET15, 2μ> gcn2::hisG | This work |
| rps16 WT | his3-1, leu2-0, met15-0, LYS, ura3-0, rps16B::kanMX4, rps16A::HIS3 <pGAL-RPS16A; LEU2, ARS1, CEN4> <pRPS28-RPS16A; URA3, 2μ> or his3-1, leu2-0, met15-0, LYS, ura3-0, rps16B::kanMX4, rps16A::HIS3 <pGAL-RPS16A; LEU2, ARS1, CEN4> <pRPS28-RPS16A; MET15, 2μ> | This work |
| rps16-R143G | his3-1, leu2-0, met15-0, LYS, ura3-0, rps16B::kanMX4, rps16A::HIS3 <pGAL-RPS16A; LEU2, ARS1, CEN4> <pRPS28-RPS16A; URA3, 2μ> | This work |
| rps16-RΔ | his3-1, leu2-0, met15-0, LYS, ura3-0, rps16B::kanMX4, rps16A::HIS3 <pGAL-RPS16A; LEU2, ARS1, CEN4> <pRPS28-RPS16A-R143Δ; URA3, 2μ> or his3-1, leu2-0, met15-0, LYS, ura3-0, rps16B::kanMX4, rps16A::HIS3 <pGAL-RPS16A; LEU2, ARS1, CEN4> <pRPS28-RPS16A-R143Δ; MET15, 2μ> | This work |
| rps16-YRΔΔ | his3-1, leu2-0, met15-0, LYS, ura3-0, rps16B::kanMX4, rps16A::HIS3 <pGAL-RPS16A; LEU2, ARS1, CEN4> <pRPS28-RPS16A-Y142ΔR143Δ; URA3, 2μ> or his3-1, leu2-0, met15-0, LYS, ura3-0, rps16B::kanMX4, rps16A::HIS3 <pGAL-RPS16A; LEU2, ARS1, CEN4> <pRPS28-RPS16A-Y142ΔR143Δ; MET15, 2μ> | This work |
| rps16-F46A | his3-1, leu2-0, met15-0, LYS, ura3-0, rps16B::kanMX4, rps16A::HIS3 <pGAL-RPS16A; LEU2, ARS1, CEN4> <pRPS28-RPS16A-F46A; MET15, 2μ> | This work |
| rps16-Y49G | his3-1, leu2-0, met15-0, LYS, ura3-0, rps16B::kanMX4, rps16A::HIS3 <pGAL-RPS16A; LEU2, ARS1, CEN4> <pRPS28-RPS16A-Y49G; MET15, 2μ> | This work |
RPS16-WT, rps16-R143G and rpS16-YRΔΔ strains were obtained as follows: the desired mutations were introduced into the RPS16A gene (expressed from RPS28 promoter) in high-copy plasmid K1005 (Yeplac195-pRPS28-FLAG-RPS16; URA3; 2μ) (a kind gift from Dr Philipp Milkereit, University of Regensberg, Germany) by site targeted mutagenesis using primers 5′-CCAAAAATCTTACGGTTAAGAAATTGTGG-3′ forward and 5′- CCACAATTTCTTAACCGTAAGATTTTTGG-3′ reverse (rps16-R143G); 5′-GATTCCAAAAATCTTAAGAAATTGTGG-3′ forward and 5′-CCACAATTTCTTAAGATTTTTGGAATC-3′ reverse (rpS16-YRΔΔ). Plasmids containing desired mutations were then transformed into strain Y-318 (pGAL-RPS16A) his3-1, leu2-0, met15-0, LYS, ura3-0, rps16B::kanMX4, rps16A::HIS3 <pGAL-RPS16A; LEU2, ARS1, CEN4>, lacking the chromosomal genes encoding the two isoforms of Rps16 and harboring a low-copy plasmid containing RPS16A under the glucose-repressible GAL promoter (15) (a kind gift from Dr Philipp Milkereit, University of Regensberg, Germany). The resulting strains were grown in glucose containing media to block the expression from pGAL-RPS16A; LEU2, ARS1, CEN4 and thus Rps16 expressed from Yeplac195-pRPS28-FLAG-RPS16 becomes the sole source of Rps16 protein expressed in these strains under glucose growth conditions. RPS16, rps16-RΔ and rps16-YRΔΔ yeast strains for GCN4-LacZ and SUI5 assays were obtained as follows: K1005 vector (Yeplac195-pRPS28-FLAG-RPS16; URA3; 2μ) harboring RPS16 wild-type or mutants was digested with PstI/NarI and the pRPS28-FLAG-RPS16 was inserted into PstI/ClaI digested pRS421 (2μ, MET15) vector. The resulting pRS421_RPS16 constructs were transformed into Y-318 strains already harboring K1005 plasmid with RPS16 wild type sequence. K1005 constructs were eliminated from the resulting strains by 5-FOA selection, thus obtaining yeast strains expressing wild-type or mutant Rps16 (expressed from pRS421 plasmids). Yeast cultures were grown as indicated using either synthetic media containing 0.67% Difco yeast nitrogen base, 1% ammonium sulfate, 2% glucose (or galactose) and supplemented with the appropriate amino acids or YEPD medium (14). Transformation was done using the lithium acetate method (16). For polysome analysis, yeast cells were grown in YEPD medium with 2% glucose.
Reporter plasmids
Yeast p180, p196, p227, p226, p209 and pM226 reporter plasmids (17,18) derivatives of YCp50 (CEN, URA3) vector bearing GCN4-lacZ alleles were kindly provided by Drs Thomas Dever and Alan Hinnebusch (National Institutes of Health). pRS SUI3-S264Y-U plasmid (derivative of pRS316 (CEN, URA3)) harboring SUI3-S264Y allele and YCp SUI5-G31R-U plasmid (derivative of YCplac33 (CEN, URA3)) harboring TIF5-G31R allele (19) were kind gifts of Dr Leoš Valášek (Institute of Microbiology, Academy of Sciences of the Czech Republic). HIS4-LacZ reporter constructs with AUG or UUG initiation codons (p367 and p391, respectively) for assaying Sui phenotypes (20) were kind gifts of Dr Alan Hinnebusch (National Institutes of Health). Plasmids were transformed into wt and RPS5 or RPS16 mutant strains and grown on the minimal YNB medium. rps5+gcn2Δ and rps16+gcn2Δ strains were generated by transforming ancestor strains (rps5 and rps16, respectively) with an XbaI/EcoRI fragment derived from construct B2806 (18) containing the disruption cassette 5′-gcn2-hisG-URA3-hisG-gcn2-3′. Ura+ colonies were picked and were counter-selected on 5-FOA plates. The gcn2 deletion was confirmed by the strain's inability to grow under starvation conditions in the presence of sulfometuron methyl (SM) (an inhibitor of isoleucine-valine biosynthesis) and/or 3-amino-1,2,4-triazole (3-AT) (an inhibitor of histidine biosynthesis).
Fractionation of polyribosomes
Fractionation of polyribosomes was done essentially as described before (9,10) using 10–50% (17 000 rpm., 18 h) and/or 10–30% (20 000 rpm., 18 h) sucrose gradients and a Beckman SW32.1 rotor. All procedures were performed at 4°C. Yeast cells from 50 ml of log phase culture were pelleted, treated for 10 min with 100 μg/ml cycloheximide and repelleted. Cell extracts were made by glass bead cell disruption (3–5 cycles of 1 min each), with intermittent cooling on ice. The following buffer was used: 100 mM KCl, 2.5 mM magnesium acetate, 20 mM HEPES·KOH, pH 7.4, 14.4 mM β-mercaptoethanol, 100 μg/ml cycloheximide. Cell debris was removed by centrifugation at 7000 rpm for 8 min and polyribosomes were resolved by sucrose density gradient centrifugation as indicated. Gradients were collected using the ISCO Programmable Density Gradient System with continuous monitoring at 254 nm using an ISCO UA-6 absorbance detector. Analysis of ratios of 80S monosomes to polyribosomes was done essentially as before (10). Fractionation of cell extracts using formaldehyde cross-linking was done as described by Nielsen et al. (18). For western blotting, proteins collected from sucrose gradient fractions were solubilized in sodium dodecyl sulfate (SDS) polyacrylamide sample buffer for 10 min at 95°C, chilled on ice for 5 min and loaded onto polyacrylamide gel.
Western blotting
Western blotting was done following standard procedures (21). Anti-rpS5 antibodies derived against the AIKKKDELERVAKSNRC C-terminally conserved rpS5 peptide has been described previously (10). The anti-eIF2α (22), anti-eIF5B (23), anti-eIF1 (24) and anti-eIF5 antibodies were kindly provided by Drs Thomas Dever and Alan Hinnebusch (National Institutes of Health). Goat anti-rabbit HRP-conjugated antibodies and enhanced chemiluminescence detection kit (ECL™, GE Healthcare, Piscataway, NJ, USA) were used for detection.
β-Galactosidase assays
For GCN4-lacZ assays, cells were grown in a minimal synthetic (SD) medium supplemented with appropriate amino acids containing 2% galactose (for 2 h). To invoke amino acid starvation, sulfometuron methyl (SM) (final concentration 1 μg/ml), or 3-amino-triazole (3-AT) (final concentration 30 mM) was then added and the incubation was continued for additional 5 h. Cells were harvested, and extracts were prepared by subsequent cycles of cell freezing in liquid nitrogen and thawing at 37°C. For assaying Sui phenotypes, cells were grown in minimal synthetic media in presence of 2% galactose as inducer and β-galactosidase activity was assayed using the whole cell extract. β-Galactosidase activity was measured following the protocol described in Clontech Yeast Protocols Handbook using O-nitrophenyl β-d-galactopyranoside as a substrate.
Miscellaneous
Molecular cloning was performed following standard procedures. Deoxyribonucleic acid (DNA) sequencing was accomplished by the DNA Sequencing Core facility at Cleveland Clinic. Sodium dodecyl sulfate polyacrylamide gel electrophoresis was performed according to Laemmli (25). Yeast genomic DNA was isolated using the DNA-PureTM Yeast Genomic Kit (PureBiotech, Middlesex, NJ, USA) following the manufacturer's protocol.
RESULTS
Biochemical analysis of mutant yeast strains carrying truncated Rps5 variants suggested that the NTD of yeast Rps5 impacts the ability of 40S ribosomal subunits to function properly in translation initiation (10). Specifically, we reported increased eIF2 association with the mutant 40S subunits containing Rps5 lacking 30 or 46 N-terminal amino acid residues (in strains rps5-Δ1-30 and rps5-Δ1-46, respectively) compared to 40S subunits in the WT strain (10). We thus proposed that the N-terminal truncation of Rps5 either affects recruitment of eIF2-ternary complexes to 40S subunits or influences ribosomal association of eIF2 by modulating eIF5-stimulated hydrolysis of eIF2-bound GTP and/or subsequent dissociation of the eIF2·GDP complex (10).
To gain further insights into the exact step in the initiation process affected by Rps5-NTD truncations, we embarked on a more detailed analysis of translation initiation in Rps5 mutant cells by taking advantage of GCN4 translational control (26). Regulation of GCN4 translation is exerted via a reinitiation process involving four small upstream open reading frames (uORFs) preceding the GCN4 ORF and is known to be a sensitive indicator of translation initiation defects in vivo (for a review, see (26)). Following translation of the 5′-proximal uORF (uORF1) reinitiation depends on de novo recruitment of the eIF2 TC, which is required to recognize the next AUG codon, and is thus exquisitely sensitive to the eIF2·GTP level (for review, see (26)). Amino acid starvation derepresses GCN4 translation by provoking reduced availability of the TC (for review, see (26)).
Translation reinitiation is compromised in Rps5 mutants
To assess reinitiation in yeast strains expressing N-terminally truncated versions of Rps5, we assayed a set of GCN4-lacZ reporters with different arrangements of the uORFs (Figures 1 and 2 and Supplementary S1A). Plasmids were transformed into wild type strain rps5-Δ0 and its isogenic derivatives rps5-Δ1-13, rps5-Δ1-24, rps5-Δ1-30 and rps5-Δ1-46 carrying different truncated RPS5 genes. Cells were treated with sulfometuron methyl (SM), which inhibits isoleucine-valine biosynthesis, to derepress GCN4 expression (for review, see (26)). We found that truncation of 13 or 24 N-terminal amino acids in Rps5 does not substantially affect reinitiation in constructs containing all four uORFs (reporter plasmid p180, Figure 1A) or only uORFs 1 and 4 (p196, Figure 1B), as strains rps5-Δ1-13 and rps5-Δ1-24 exhibit basal levels of β-galactosidase activity under non-starvation conditions (−SM), and derepressed levels of activity under amino acid starvation conditions (+SM), that are nearly identical to those seen in the WT strain. Upstream ORFs 2 and 3 are known to be functionally redundant with uORF4 such that uORF1 and uORF4 are sufficient for nearly WT regulation of reinitiation on GCN4 mRNA (17). By contrast, GCN4-lacZ expression from p180 was reduced by ∼4-fold in rps5-Δ1-30 and by ∼40-fold in rps5-Δ1-46 in comparison with the WT strain (Figure 1A). Thus, while rps5-Δ1-30 retains a low-level induction of reinitiation (∼2-fold), both the basal level of reinitiation and its induction were almost completely abrogated in rps5-Δ1-46. The same holds true for the p196 construct that contains only uORFs 1 and 4 (Figure 1B). The GCN4-lacZ expression data were corroborated by growth assays (Figure 1C). Growth of rps5-Δ1-46 was compromised under amino acid starved conditions (Figure 1C, +SM), suggesting that GCN4 fails to efficiently activate the expression of genes encoding amino acid biosynthetic enzymes in this mutant (26). These observations led us to conclude that the rps5-Δ1-46 mutant exhibits a strong General control non-derepressible (Gcn−) phenotype (26).
Figure 1.
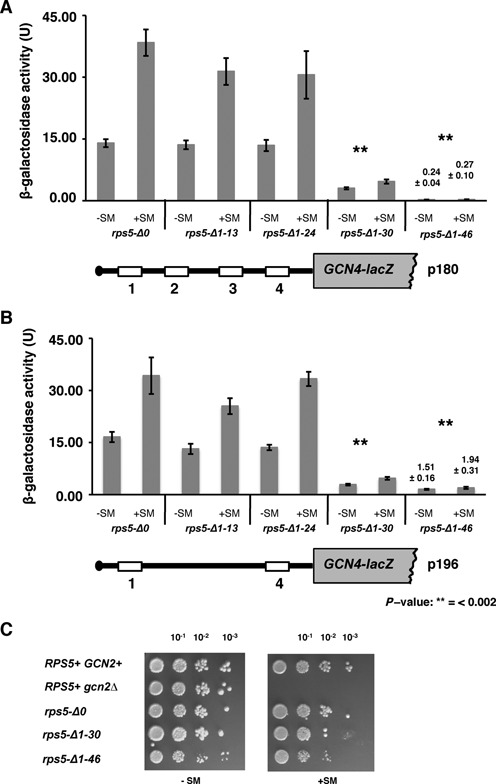
Rps5 N-terminal region is essential for reinitiation in yeast cells. (A, B) Expression of reporter GCN4-lacZ constructs. (A) p180 containing wild type GCN4 mRNA leader (four uORFs) and (B) p196 containing only uORFs 1 and 4, were transformed into wild type rps5-Δ0 and isogenic rps5-Δ1-13, rps5-Δ1-24, rps5-Δ1-30, rps5-Δ1-46 yeast strains. β-Galactosidase activity (units) are shown; measured under normal and amino acids starved conditions (+SM). (C) Yeast cell growth. Serial dilutions of strains spotted onto minimal media under non-starved (−SM), or amino acid (aa) starved conditions (+SM), respectively.
Figure 2.
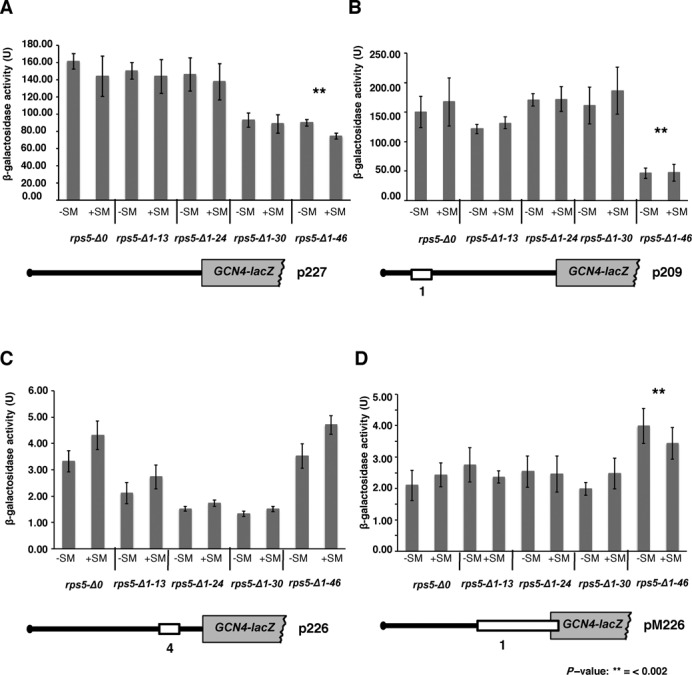
Truncation of Rps5 46 N-terminal amino acid residues confers leaky scanning phenotype. Expression of reporter GCN4-lacZ constructs. rps5-Δ0 and isogenic rps5-Δ1-13, rps5-Δ1-24, rps5-Δ1-30, rps5-Δ1-46 strains were transformed with (A) p227 containing uORFs less GCN4 mRNA leader; (B) p209 containing uORF1; (C) p226 containing only uORF4 and (D) pM226 containing uORF1 extended into GCN4 ORF. β-Galactosidase activity (units) are shown; measured under normal (−SM) and aa starved conditions (+SM) as in Figure 1.
We have previously found (using a reporter plasmid carrying lacZ with a short 5′UTR (∼50 nt) under control of the GAL1 promoter) that cap-dependent initiation was reduced in the rps5-Δ1-30 and rps5-Δ1-46 strains by ∼30% and ∼50%, respectively (10). We observed a similar reduction (∼2-fold) in cap-dependent initiation levels for a GCN4-lacZ reporter construct (p227) devoid of all four upstream open reading frames (uORFs) and thus possessing a long 5′UTR of ∼600 nt (Figure 2A and Supplementary S1A). This suggests that, independently of the 5′UTR length, initiation in rps5-Δ1-46 is reduced by only about 2-fold, while reinitiation is almost completely blocked. This finding led us to conduct a more thorough analysis of the role played by the Rps5 NTD in translation reinitiation.
The rps5-Δ1-46 mutant displays translation initiation defects downstream of 48S complex formation including a leaky scanning phenotype
The Gcn− phenotype observed in the rps5-Δ1-46 strain could arise for a number of reasons. Firstly, it is possible that the mutant ribosomal subunits are unable to resume scanning after they translate uORF1 and thus dissociate from the mRNA after terminating translation at the uORF1 stop codon. It is known that only those ribosomes that can successfully translate uORF1, terminate and resume scanning downstream, can bypass uORFs 2, 3, 4 and reinitiate translation at the GCN4 ORF (26). To test whether mutant ribosomes can resume scanning after translating uORF1, we assayed a GCN4-lacZ reporter (on p209) containing uORF1 at its original position (350 nt from the GCN4 AUG codon) as the only uORF in the leader (17) (Figure 2B and Supplementary Figure S1A). The uORF1 possesses specific surrounding sequences, both 5′ and 3′, which allow a large fraction of ribosomes to remain attached to the mRNA, resume scanning, and reinitiate downstream (26). Thus, the absence of uORFs 2–4, and the equivalent frequencies of reinitiation at GCN4 following uORF1 translation that occur in non-starvation and starvation conditions, confers high-level, constitutive expression from p209 (Figure 2B and Supplementary Figure S1A, rps5-Δ0, +SM versus –SM). A failure to resume scanning following uORF1 translation would reduce expression of the p209 construct. As expression of this construct in the rps5-Δ1-13, rps5-Δ1-24 and rps5-Δ1-30 strains were comparable to that in rps5-Δ0 (Figure 2B and Supplementary Figure S1A), it appears that none of these Rps5 truncations affect the ability of the mutant 40S ribosomal subunits to resume scanning after translating uORF1. This was not the case for the rps5-Δ1-46 mutant in which expression of p209 was reduced by a factor of ∼3 (Figure 2B and Supplementary Figure S1A). It is important to note, however, that the level of p209 expression in rps5-Δ1-46 remains more than 100-fold higher than that observed for the p180 construct in this mutant (Figure 1A). By contrast, in the WT strain, expression of the p209 construct is only ∼4-fold higher than that from p180 under starvation conditions, indicating that in WT cells a substantial fraction (∼25%) of the 40S subunits that resume scanning following uORF1 translation bypass uORFs 2–4 and reinitiate at GCN4 instead. The corresponding data for p209 and p180 in the rps5-Δ1-46 mutant indicate that <1% of the 40S subunits scanning downstream from uORF1 bypass uORFs 2–4 and reinitiate at GCN4. Accordingly, the failure to induce GCN4-lacZ expression from p180 in this mutant does not stem primarily from the reduced ability of the mutant 40S ribosomal subunits to resume scanning after translating uORF1. In WT cells, expression from p209 is expected to be ∼50% of that observed for p227 (17), owing to the fact that only ∼50% of the ribosomes that translate uORF1 can resume scanning and reinitiate at the GCN4 start codon (26). Because comparison of the results in Figure 2A and B for rps5-Δ0 cells did not reveal this difference, we made a side-by-side comparison of expression from p227 and p209 in the rps5-Δ0 strain and observed the predicted 2-fold higher expression for p227 versus p209 (data not shown). We presume that day to day variation in the measurement of β-galactosidase activities may have obscured this 2-fold difference in the results of Figure 2A and B.
We further considered a second possibility to account for the strong Gcn− phenotype of the rps5-Δ1-46 strain, wherein the mutant rps5-Δ1-46 40S subunits would exhibit a reduced rate of scanning that allows the TC to be acquired before the reinitiating ribosomal subunits bypass uORF4, thereby increasing reinitiation at uORF4 and producing an equivalent reduction in reinitiation at GCN4. To test this possibility, we assayed the p226 reporter containing a single uORF4 in its natural position (17). uORF4 is known to be the critical negative regulator of GCN4 expression (26). Indeed, GCN4-lacZ expression from the p226 construct in WT cells is ∼5-fold lower compared to that from the p180 or p196 constructs in which the presence of uORF1 allows a fraction of reinitiating ribosomes to bypass the uORF4 start codon (compare data for strain rps5-Δ0 in Figures 1A, B and 2C). Under normal circumstances, ribosomes that translate uORF4 reinitiate at the GCN4 start codon at a very low frequency partly because of the low propensity for resumption of scanning by post-termination ribosomes at the uORF4 stop codon, but also because of its proximity to the GCN4 AUG, as shown by the fact that GCN4 expression from a solitary uORF4 construct was partially derepressed by increasing the distance (scanning time) separating uORF4 from GCN4 (26,27). Thus, it can be predicted that increasing the time required to scan from the uORF4 stop codon to the GCN4 AUG by decreasing the rate of scanning by mutant 40S subunits would increase GCN4-lacZ expression from the p226 construct (28). This was not observed in the rps5-Δ1-46 mutant however, as expression of the p226 reporter was indistinguishable between this mutant and the WT strain (Figure 2C). Thus, it appears that a reduced rate of scanning by reinitiating 40S subunits does not contribute to the strong Gcn− phenotype of the rps5-Δ1-46 mutant. The other three Rps5 mutants display 2- to 3-fold reductions in expression of the p226 construct compared to the WT strain (Figure 2C and Supplementary Figure S1A), which might indicate that reinitiation following uORF4 translation is even less efficient in these mutants than in WT cells. Presumably, this defect is not seen for the p180 and p196 constructs (Figure 1A and B) because the great majority of GCN4-lacZ expression for these regulated constructs stems from reinitiating ribosomes that have bypassed uORF4.
We sought to confirm our conclusion that the rps5-Δ1-46 mutation does not decrease the rate of scanning by examining constructs pM199 and pG67 in which the distance between solitary uORF1 and the GCN4 AUG is smaller than normal (140 nt and 32 nt, respectively, Supplementary Figure S1B) versus the WT spacing of 350 nt present in construct p209 (Figure 2B). We observed a progressive decrease in β-galactosidase expression from these three constructs as the distance between uORF1 and GCN4 AUG decreased in both WT and rps5-Δ1-46 strains (compare expression levels from p209 in Figure 2B, Supplementary Figure S1A and pM199, pG67 in Supplementary Figure S1B for rps5-Δ0 and rps5-Δ1-46 strains). This reduction is expected from the progressive decrease in the amount of time available to reassemble an initiation complex by reinitiating 40S subunits before they reach the GCN4 AUG codon, diminishing the frequency of reinitiation (31). Importantly, expression from the pM199 and pG67 constructs was lower in the rps5-Δ1-46 mutant versus the rps5-Δ0 WT strain (Supplementary Figure S1B), whereas a decreased rate of scanning in the mutant should evoke higher expression by increasing the probability of reinitiation at the GCN4 AUG, especially for pG67 where the time required to scan the 32 nt uORF1-GCN4 interval is normally so small. The decreased expression of pM199 and pG67 in the rps5-Δ1-46 strain (Supplementary Figure S1B), also observed above for p209 (Figure 2B), supports our conclusion that the resumption of scanning following uORF1 translation is reduced in the mutant, decreasing the frequency of reinitiation at the GCN4 AUG codon.
A third possible defect underlying a Gcn− phenotype is the failure of scanning 40S subunits to recognize the uORF1 AUG codon, as translation of uORF1 is required to generate reinitiating 40S subunits that can bypass uORFs 2–4 and reinitiate at GCN4 when TC levels are reduced by starvation. To investigate this, we assayed GCN4-lacZ expression from the solitary-uORF1 reporter on pM226 in which uORF1 is extended to overlap the GCN4-lacZ coding region (Figure 2D and Supplementary Figure S1A), which destroys the ability of ribosomes to reinitiate at GCN4 after terminating at the elongated uORF1 stop codon. Hence, because initiation at uORF1 is normally so efficient and 40S subunits do not ‘leaky scan’ past the uORF1 AUG codon, this extension of uORF1 very effectively represses GCN4-lacZ expression (29) (compare in WT cells expression for pM226 in Figure 2D with p209 in Figure 2B). However, we observed only a slight ≤2-fold increase in GCN4-lacZ from pM226 in rps5-Δ1-46 compared to rps5-Δ0 (Figure 2D and Supplementary Figure S1A). While this degree of leaky scanning of the uORF1 AUG codon likely makes a small contribution to the strong reduction in expression of the p180/p196 constructs, it clearly cannot account for the virtually complete abrogation of GCN4-lacZ expression from these two regulated constructs (Figure 1A and B). Presumably, the small increase in leaky scanning of uAUG1 in the mutant is obscured for constructs p209, pM199 and pG67 by the decreased frequency of reinitiation following uORF1 translation deduced for these constructs.
Having eliminated defects in the resumption of scanning, rate of scanning, or leaky scanning of uORF1 as the origin of the strong Gcn− phenotype in the rps5-Δ1-46 strains, it seems most likely that the reinitiating mutant 40S subunits are unable to bypass uORFs 2–4 at low TC concentrations (caused by amino acid starvation) because they are forced to queue up behind a mutant 40S subunit stalled at the AUG codon of uORF 2, 3 or 4 that is unable to complete the assembly of a 48S PIC competent for subsequent steps of initiation following AUG recognition, such as completion of GTP hydrolysis by eIF2, release of eIF2·GDP or other eIFs from the 40S subunit, or subunit joining. This delay would allow sufficient time to rebind TC to the 40S subunits queued up behind it, which would then go on to reinitiate at the uORF AUG when it is eventually cleared by the stalled 40S subunit, preventing them from reinitiating at GCN4. In fact, our finding that the elimination of uORFs 2 and 3 in construct p196 increased GCN4-lacZ in comparison to the WT p180 construct under starvation conditions specifically in the rps5-Δ1-46 mutant (Figure 1B and Supplementary Figure S2) supports this idea, as the absence of uAUGs 2 and 3 would eliminate two of the three sites where queuing would be imposed in the mutant. We note, however, that although we observed certain discrepancies in expression from p180 in rps5-Δ1-46 mutant (Supplementary Figure S1A versus S2; starved conditions) potentially due to overall very low levels of β-galactosidase activity in this strain, we have always observed a statistically significant increase in expression from p196 construct as compared to p180 in this strain (Supplementary Figure S1A and S2). Consistent with these data, a relatively smaller increase in expression under starvation conditions was observed in the rps5-Δ1-46 mutant on elimination of only uORF2 in construct p195 (Supplementary Figure S2, please compare 1.1 for p180, 1.64 for p195 and 2.01 for p196 at +SM). In addition, GCN4-lacZ expression from construct pG29, which lacks uORFs 2–3 as the result of a deletion between uORFs 1 and 4, also was elevated compared to the p180 construct in the rps5-Δ1-46 mutant under starvation conditions, whereas expression from pG29 is lower than that given by p180 in starved WT (rps5-Δ0) cells (Supplementary Figure S2). The reduced pG29 expression seen in WT cells under starvation conditions, as well as the higher expression of this construct compared to p180 observed in nonstarved WT cells, both are consistent with previous results (29); which were attributed to increased bypass of uORF4 in nonstarvation conditions, coupled with increased bypass of the GCN4 AUG in starvation conditions, by reinitiating 40S subunits. Our finding that expression from pG29 under starvation conditions is increased rather than decreased in the rps5-Δ1-46 mutant (Supplementary Figure S2) supports the idea that queuing of reinitiating 40S subunits (diminished by deletion of uORFs 2–3 in pG29) makes an important contribution to the Gcn− phenotype of the rps5-Δ1-46 mutant. We presume that expression of pG29 under starvation conditions remains lower in the rps5-Δ1-46 mutant versus WT cells (Supplementary Figure S2) because queuing of reinitiating subunits still occurs upstream of uORF4. Thus, our analysis of different GCN4-lacZ reporters leads us to predict that a step of initiation following AUG recognition by the scanning PIC is defective in rps5-Δ1-46 cells.
The rps5-Δ1-46 mutant exhibits altered association of eIF1, eIF5 and eIF5B with 48S complexes and does not exhibit a Sui− phenotype
Dynamic interaction of initiation factors with 40S subunits is important for translational control of GCN4 (for review, see (26)). In yeast, eIF1, the eIF2 TC, eIF3 and eIF5 comprise the multifactor complex (MFC), which may bind to the 40S ribosomal subunits as a preformed unit (30). Mutations that disrupt interactions of these initiation factors within the MFC or impair their recruitment to the ribosome affect reinitiation (for review, see (26)). We were thus interested to know whether the N-terminal Rps5 truncation evokes altered interactions of the mutant 40S subunits with MFC components, particularly eIF1 as this factor ensures accurate start codon recognition by blocking Pi release from the hydrolyzed eIF2·GDP·Pi complex and stabilizes the open, scanning-conducive conformation of the 40S subunit at non-AUG codons, while being released from the PIC on AUG recognition (for review, see (1,31)). Moreover, eIF1 is known to reside below the P-site and in the vicinity of the E-site (for review, see (1)) and hence in proximity to Rps5. Mutations in Rps5 might impede eIF1 release to decrease the efficiency of AUG recognition and confer the observed increase in leaky scanning of GCN4 uAUG1, and the postulated failure to complete the initiation process at uORFs 2–4 (with attendant queuing of scanning ribosomes) in the rps5-Δ1-46 mutant (Figure 2D and Supplementary Figure S1A).
Accordingly, we assessed the association of eIF1 with native 40S subunits in extracts of WT and rps5-Δ1-46 cells that were treated with formaldehyde to fix 43S/48S complexes in vivo and preserve their interactions with MFC components during separation of the whole cell extract (WCE) by sedimentation through sucrose density gradients. Western analysis of the gradient fractions with antibodies against eIF1 and the 40S subunit protein Rps5 revealed an increase in the eIF1:Rps5 ratio in the 40S fractions of the mutant versus WT cells (Figure 3A; top panel and 3B). We also checked the 40S-association of eIF5, as the eIF5 stimulates hydrolysis of eIF2-bound GTP and also plays an important role in eIF1 dissociation, Pi release and efficient AUG recognition through its interactions with eIF1, eIF2, eIF3 and eIF1A (for review, see (31)) (24,32). Moreover, there is evidence that eIF5 and eIF1 have antagonistic, positive and negative functions, respectively, in the efficiency and accuracy of start codon recognition ((33,34), reviewed in (35)). Western blotting of the gradient fractions revealed decreased association of eIF5 with the 43S/48S complexes formed in the rps5-Δ1-46 mutant compared to the wild type strain (Figure 3A; top panel and B). We further decided to check the 40S-association of eIF5B, as this factor ensures efficient 40S and 60S subunit joining in the last stage of the initiation pathway (for review, see (1)) and, thus, it was possible that the Rps5 truncation impairs eIF5B recruitment and thereby compromises formation of elongation-competent 80S ribosomes. We found that association of the mutant 40S ribosomal subunits with eIF5B was not reduced, and was even increased compared to its association with WT 40S subunits (Figure 3A; bottom panel and B).
Figure 3.
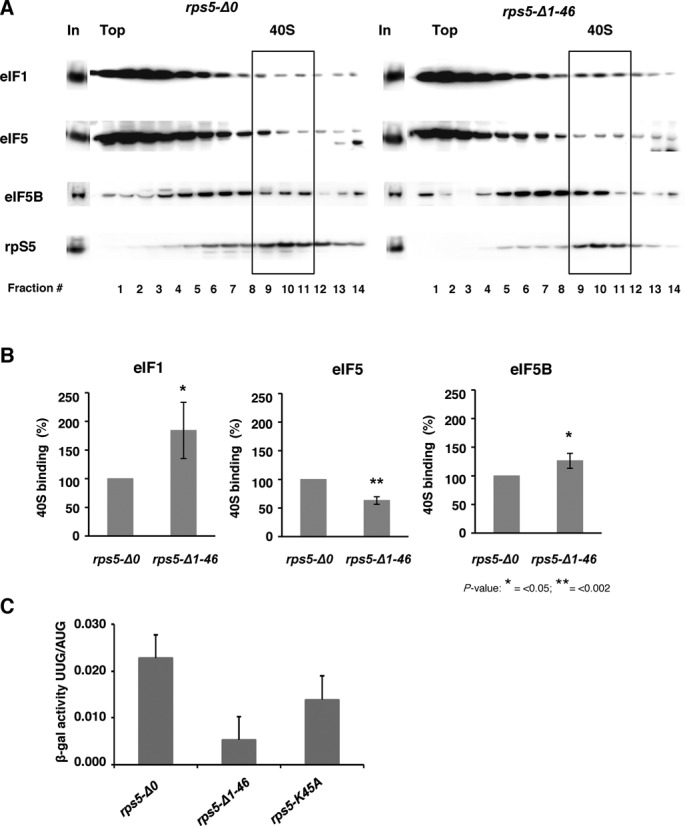
Association of eIF1, eIF5 and eIF5B with 40S ribosomal subunits in wt and mutant (rps5-Δ1-46) yeast strains and the stringency of AUG recognition on HIS4 mRNA (Sui phenotype). (A) Extracts from isogenic wt (rps5-Δ0) and Rps5 mutant (rps5-Δ1-46) strains were resolved by sucrose density gradient (10–30%) sedimentation. Western blot analyses were done using antibodies against eIF1, eIF5 and eIF5B and the ribosomal protein S5, respectively. ‘In’ for input - represents a 7% portion of each gradient fraction. Analysis of eIF1, eIF5 and eIF5B association was done using whole cell extract cross-linking with formaldehyde. (B) Association of eIF1, 5 and 5B with the 40S was quantified and expressed as percentage of 40S binding normalized against Rps5. (C) rps5-Δ0, rps5-Δ1-46 and rps5-K45A strains harboring reporter HIS4-LacZ constructs with either AUG or UUG initiation codons, respectively, were assayed for β-galactosidase activity. Mean ratio of expression from UUG to AUG reporter constructs are shown with standard errors from three experiments.
It is worth recalling here that we previously reported an accumulation of eIF2 in the 43S/48S complexes in the rps5-Δ1-46 mutant as compared to the WT strain (10). Taking these data together with the results in Figure 3AB and our analysis of GCN4 translational control, leads us to suggest that the initiation defect in the rps5-Δ1-46 mutant involves events occurring following AUG recognition by the scanning PIC. One interesting possibility is that release of eIF1 is impeded, with an attendant delay in both Pi release from eIF2·GDP·Pi and subsequent dissociation of eIF2·GDP from the 48S complex (36), which in turn delays the completion of subunit joining by 48S-associated eIF5B. In view of recent evidence that eIF1 and eIF5 compete for one or more binding sites on the 40S subunit (24,32), the accumulation of eIF1 in the PIC might be coupled to the apparent decrease in eIF5 association shown in Figure 3A for the rps5-Δ1-46 mutant.
Yeast eIF1 was originally identified by the isolation of mutations that reduce the stringency of AUG recognition on HIS4 mRNA (Sui− phenotype) (37). Sui− mutants increase initiation at the UUG codon encoding the third amino acid in the His4 protein, thus bypassing the requirement for the AUG start codon in HIS4 mRNA for yeast growth on medium lacking histidine (37). Extensive analysis of Sui− mutations in eIF1 has established that they elevate aberrant initiation at UUG codons by reducing the recruitment of eIF1, or enabling its inappropriate release from the 48S PIC at non-AUG codons, owing to weakened interactions of eIF1 mutants with the 40S subunit or MFC components (reviewed in (31) (38,39). It was further suggested that gain-of-function Sui− mutations in eIF2β or eIF5 that accelerate GTP hydrolysis by eIF2 and induce premature release of Met-tRNAiMet from eIF2·GDP on the scanning 40S subunit enable ribosomes to initiate translation from the UUG codon in HIS4 mRNA (13). We therefore also tested whether the rps5-Δ1-46 mutant is defective in rejecting UUG start codons by assaying two matched HIS4-lacZ reporter constructs containing AUG or UUG at the first codon of the HIS4 coding sequence and calculating the ratio of expression from the UUG versus AUG reporter. Interestingly, the rps5-Δ1-46 mutant exhibits a significant reduction in the UUG:AUG initiation ratio compared to the WT strain (Figure 3C and Supplementary Figure S3). This hyperaccuracy phenotype is similar to that conferred by so-called ssu mutations, which suppress the elevated UUG:AUG initiation ratios conferred by sui mutations in the same or other initiation factors (reviewed in (31)). The suppression of the UUG:AUG initiation ratio in rps5-Δ1-46 cells (Figure 3C and Supplementary Figure S3) would be compatible with a defect in start codon recognition resulting from a delay in eIF1 or Pi release if we assume that this defect has a relatively greater impact at UUG codons where mismatch with the anticodon of tRNAi already disfavors these events. This interpretation is supported by the results shown in (Supplementary Figure S3) indicating that the rps5-Δ1-46 mutation produces a greater reduction in expression of the HIS4-lacZ UUG reporter (∼10-fold) compared to that of the HIS4-lacZ AUG reporter (∼3-fold).
A gain-of-function variant of eIF5 rescues the translational defects and slow growth phenotype of rps5-Δ1-46 strain
The results obtained using GCN4-lacZ constructs (Figures 1A,B, 2, and Supplementary Figure S1, S2) combined with the biochemical data revealing elevated accumulation of eIF2 (10), eIF1 and eIF5B, along with decreased association of eIF5 with 43/48S complexes (Figure 3A and B) suggested that there might be a delay in eIF1 dissociation or Pi release from eIF2-GDP after scanning 48S PICs encounter the AUG codons at the GCN4 uORFs in rps5-Δ1-46 mutant cells. As explained above, this delay in assembling elongation-competent 48S PICs at the GCN4 uAUGs 2–4 would create a barrier for all reinitiating 40S subunits scanning the leader and increase the time available to re-acquire the TC before the uAUGs 2–4 are encountered, thereby restoring translation of the uORFs and reducing translation of GCN4 for the Gcn− phenotype. If this defect occurs during primary initiation events for most other mRNAs, it could also account for the Slg− phenotype of the rps5-Δ1-46 mutant. We reasoned that if this interpretation is correct, it might be possible to suppress both the Slg− and Gcn− phenotypes of rps5-Δ1-46 by introducing into this mutant plasmid-borne SUI3-S264Y or SUI5 mutant alleles encoding the gain-of-function Sui− variants eIF2β-S264Y and eIF5-G31R, respectively, reported to elevate the intrinsic rate of GTP hydrolysis by eIF2 (SUI3-S264Y) or GAP function of eIF5 (SUI5) (13). Subsequent biochemical analysis of SUI5 has revealed that it also stabilizes the closed conformation of the PIC, in which start codon recognition occurs, specifically at near-cognate start codons (including UUG), independent of any effect on rates of GTP hydrolysis (40).
By comparing the growth rates of the WT strain and the rps5-Δ1-46 mutant harboring these plasmids (Figure 4A), we found that SUI5, but not SUI3-S264Y diminished the Slg− phenotype of the mutant strain. It is important to mention that the enhanced hydrolysis of eIF2-bound GTP conferred by the SUI3-S264Y product was observed in the absence of eIF5 (13) and hence the difference in the effects observed here for SUI3-S264Y and SUI5 might stem from the different mechanisms involved in their phenotypes. We further found that the SUI5 allele partially rescues the Gcn− phenotype of the rps5-Δ1-46 mutant, increasing growth under conditions of amino acid starvation (Figure 4B). These results led us also to address whether introduction of SUI5 would reverse the accumulation of eIF2 in 43S/48S complexes shown previously to occur in rps5-Δ1-46 (10). Indeed, we found that association of eIF2 with native 40S subunits in strain rps5-Δ1-46 was reduced by the introduction of SUI5 (Figure 4C and D). Together, these data support the possibility that the translation initiation defect in the rps5-Δ1-46 mutant involves delayed hydrolysis of eIF2-bound GTP or Pi release from eIF2·GDP·Pi in 48S PICs after encountering the AUG start codon.
Figure 4.
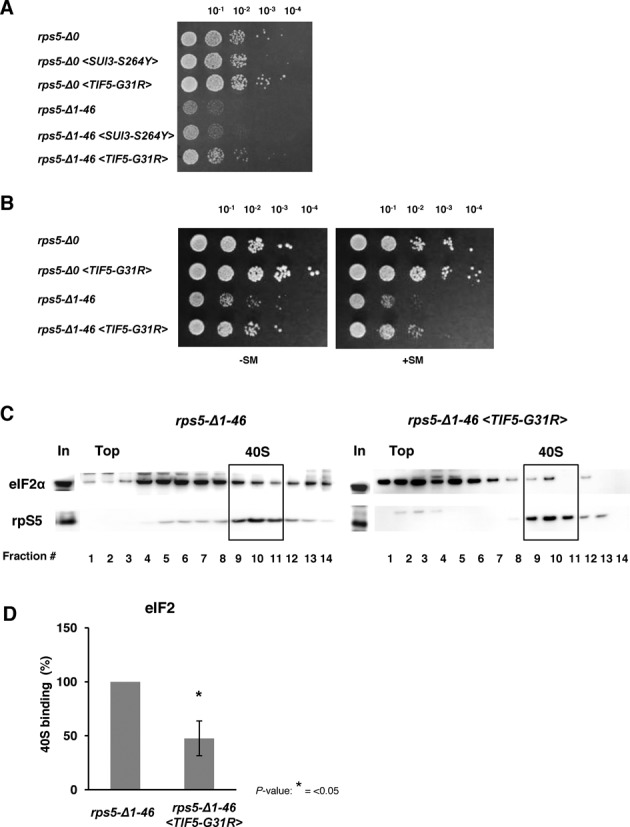
Increased GAP function of eIF5 rescues slow growth phenotype and initiation defects of rps5-Δ1-46 strain. (A) Yeast cell growth. Yeast rps5-Δ0 (Wt) and rps5-Δ1-46 strains (in particular harboring SUI3-S264Y and TIF5-G31R alleles) were spotted onto YNB agar plates with 2% glucose. (B) TIF5-G31R allele complements the Gcn− phenotype in rps5-Δ1-46. Serial dilutions of yeast strains were spotted onto YNB agar plates with 2% glucose and incubated under non-starved (−SM) conditions or aa starved conditions (+SM), respectively. (C) Association of initiation factor eIF2 with 40S subunits in rps5-Δ1-46 and rps5-Δ1-46 <TIF5-G31R> yeast strains. Western blot analysis of individual fractions with antibodies against eIF2α and rpS5 is shown. ‘In’ for input - represents a 7% portion of each gradient fraction. TIF5-G31R allele rescues enhanced accumulation of eIF2 on mutant 40S ribosomal subunits. (D) Association of eIF2 with the 40S subunits was quantified and expressed as percentage of 40S binding normalized against Rps5.
Disruption of Rps5-Rps16 interaction via Rps5 N-terminus confers a slow growth phenotype
We previously showed that eliminating all 50 N-terminal amino acids from Rps5 is lethal in yeast, and that global translation was compromised in rps5-Δ1-30 and severely impaired in the rps5-Δ1-46 mutant without a significant effect on ribosome biogenesis (10). These data, together with the results presented above, strongly suggest that the Rps5 N-terminal region (at least residues ∼40–50) is critical for translation and, hence, might have evolved in eukaryotes to perform an important function. Alignment of Rps5 from metazoans and fungi showed that this N-terminal region contains a number of highly conserved amino acids, including K41, F43 and K45 (Figure 5A). Interestingly, the crystal structure of the yeast 80S ribosome at 3.0 Å (3) revealed that the Rps5 N-terminal region interacts with the N-terminal region of Rps16 (belonging to the Rps9 family, which includes S9 in bacteria), and that conserved Rps5 residue K45 directly interacts with F46 in Rps16 (Figure 5B). Rps16 is located on the solvent side of the 40S head; however, it has a long protruding C-terminal tail (CTT) that reaches the mRNA cleft at the region occupied by the P-site tRNA (4,12). Thus, interaction of Rps5 and Rps16 via their respective N-terminal ends might affect the conformation of Rps16, including its CTT, and in turn perturb binding of TC to the PIC. It is also known that mammalian Rps5 and Rps16 mutually stabilize each other's binding to RNA and that the Rps5-Rps16 complex undergoes substantial structural rearrangement on RNA-binding (41).
Figure 5.
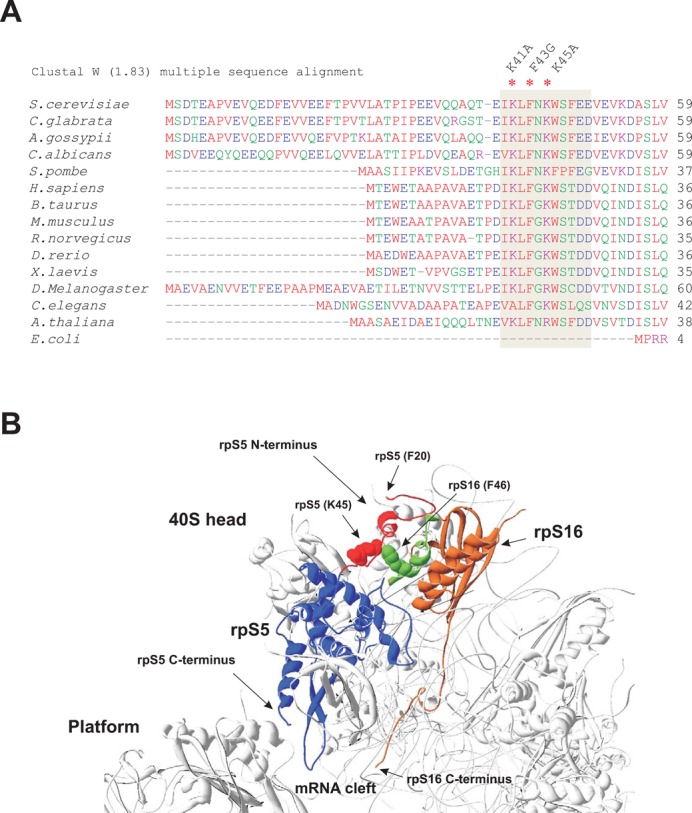
Sequence alignments of ribosomal protein S5/S7, the structure of yeast Rps5 and its ribosomal location relative to Rps16. (A) Sequence alignments of N-terminal amino acid regions of RpS5/S7 from various species. The amino acid residues, comprising residues 40–50 (in yeast Rps5) are boxed in gray. The three mutated residues are marked (*) and corresponding amino acid substitutions are shown. (B) Structures and ribosomal locations of Rps5 and Rps16. PDB files 3U5G and 3U5F were used and structures visualized using Swiss Pdbviewer (59). Rps5 is in blue with its eukaryote specific N-terminal extension in red. The Rps16 is shown in orange with part of it, which appears to interact with Rps5 N-terminus in green. Interacting Rps5 K45 (red) and F46 in Rps16 (green) are shown (van-der Waals radii of the side chain residues are shown). The view is from the solvent side of the 40S ribosomal subunit. The Rps5 N-terminus extends towards the solvent side and the Rps16 C-terminus is protruding towards the mRNA cleft.
To test this possibility, we mutated conserved Rps5 residues K41, F43 and K45, substituting positively charged K41 and K45 with alanines and bulky aromatic F43 with glycine and obtained yeast strains in which the wild type yeast RPS5 was replaced by the mutant RPS5 alleles. Interestingly, rps5-K41A (harboring lysine to alanine substitution at Rps5 position 41) and rps5-F43G (harboring phenylalanine to glycine substitution at position 43) exhibited growth rates very similar to that of the wild type strain, while rps5-K45A displayed a pronounced Slg− phenotype approaching that of the rps5-Δ1-46 strain (Figure 6A). Analysis of polysomes by sedimentation of whole cell extracts through sucrose density-gradients further revealed a marked reduction in polysome (P) to monosome (80S) ratio (P:M) in the rps5-K45A mutant as compared to the wild type strain, albeit not as severe as that seen for the rps5-Δ1-46 mutant (Figure 6B). A decrease in the P:M ratio is a characteristic phenotype of mutations that impair translation initiation without a commensurate reduction in the elongation or termination phases of translation, reducing the average number of ribosomes per mRNA. Importantly, we also found that the rps5-K45A mutant resembles the rps5-Δ1-46 strain in displaying a Gcn− phenotype, as indicated by increased sensitivity to SM (Supplementary Figure S4A) and reduced β-galactosidase activity expressed from the GCN4-lacZ reporter on p180 (Figure 6C and Supplementary Figure S4B); and also in displaying a reduced UUG:AUG ratio for the HIS4-lacZ reporters (Figure 3C and Supplementary Figure S3). Like the Slg− phenotype and reduction in P:M ratio (Figure 6A and B), both defects were less pronounced in rps5-K45A versus the rps5-Δ1-46 mutant. Interestingly, the reciprocal F46A mutation in RPS16 confers a slow growth phenotype (Supplementary Figure S5A) similar in degree to that observed for the rps5-K45A mutant (Figure 6A). In addition, Rps16 Tyrosine 49 also forms contacts with the Rps5 N-terminus, and the Y49G mutation in RPS16 (4,5) confers an even stronger Slg− phenotype (Supplementary Figure S5A). Importantly, F46A mutation causes a similar reduction in polysome (P) to monosome (80S) ratio (P:M) (as in the rps5-K45A mutant) as compared to the wild RPS16 type strain (Supplementary Figure S5B). Although further experiments are required to determine the exact consequences of these RPS16 mutations, these data support the possibility that interaction between the N-terminal regions of Rps5 and Rps16 is critical for efficient translation initiation at a step following AUG recognition. This further raises the question of how structural changes on the solvent side of the 40S ribosomal subunit may affect TC recruitment and GTP hydrolysis.
Figure 6.
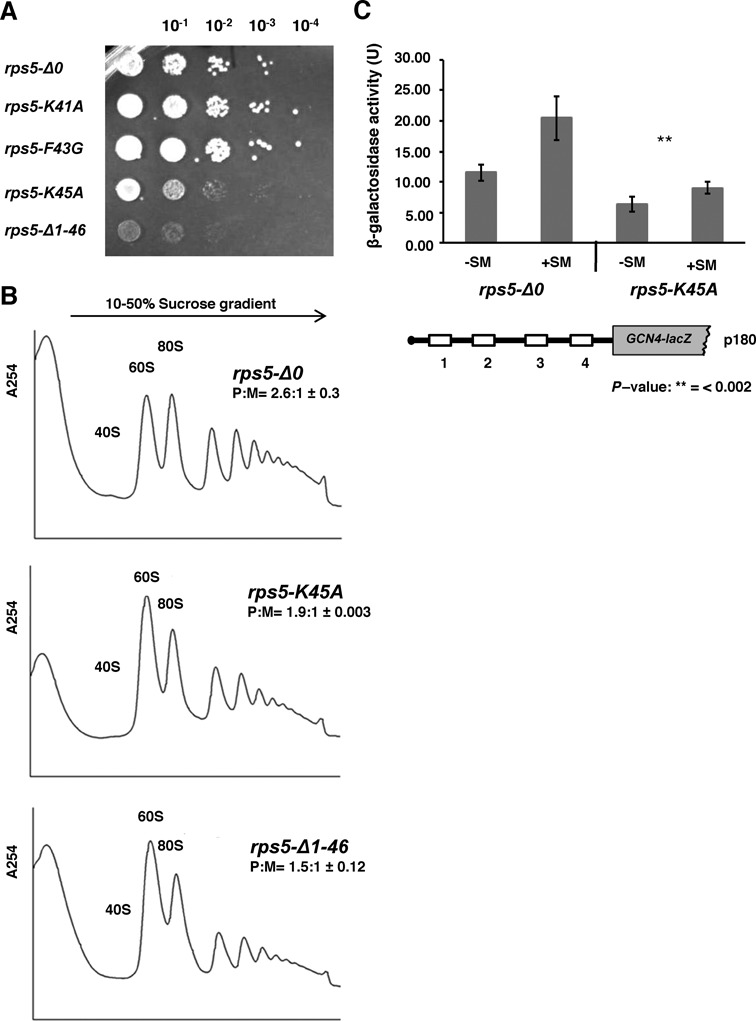
A single point K45A mutation in Rps5 confers phenotype as in rps5-Δ1-46 strain. (A) Yeast cell growth. Serial dilutions of yeast strains with the indicated genotypes (harboring point mutation in Rps5; K41A, F43G and K45A) and rps5-Δ1-46 were spotted onto YPD agar plates with 2% glucose. (B) Ribosome profiles of the wt, rps5-Δ1-46 and rps5-K45A mutant yeast strains. Extracts were resolved in 10–50% sucrose density gradients. The ratios of the area under the polysomal (P) and 80S (M) peaks (P:M) are shown with ± standard errors. rps5-Δ1-46 and rps5-K45A mutant yeast strains reveal very similar ribosome profiles. (C) rps5-Δ0 and rps5-K45A yeast strains were transformed with reporter construct p180 containing wild type GCN4 mRNA leader (with all four uORFs) upstream of GCN4-LacZ fusion gene. The strains were assayed for β-galactosidase activity under normal, nutrient rich, (−SM) and aa starved (+SM) conditions as in Figure 1.
Rps16 C-terminal residues are critical for ribosome function
It has been previously observed that the last two C-terminal residues of Rps16, which are highly conserved in all kingdoms of life (Figure 7A), interact with the P-site tRNA (12,42). Hence, the exact location of these residues in the PIC may be critical for efficient recruitment of the eIF2·GTP·Met-tRNAiMet TC, or for responding properly to an AUG codon in the P site during scanning. Accordingly, we asked whether mutations in these C-terminal residues in yeast Rps16 (Y142 and R143) confer phenotypes similar to those observed in the Rps5 (Δ1-46 and K45A) mutants described above. Indeed, we found that deletion of the last two residues of Rps16 (in yeast strain rps16-YRΔΔ) confers a Slg− phenotype (Figure 7C), a decrease in the P:M ratio (Figure 7B), and a Gcn− phenotype (Figure 7D and Supplementary Figure S6AB); note that GCN4-lacZ expression from the rps16 mutants was assayed using 3-AT, which blocks histidine biosynthesis, instead of SM (used for rps5 mutants) due to differences in the strain backgrounds of the rps16 and rps5 mutants. Replacement of the terminal Arg residue with Gly or its deletion conferred similar but less pronounced phenotypes (Figure 7B and D). Supporting the notion that the Rps16 C-terminal residues are important for the same step in initiation impacted by mutations in the Rps5 N-terminal region, we found that introduction of SUI5 eliminated the Slg− phenotype of the rps16-YRΔΔ mutant (Figure 7E). We further observed accumulation of eIF2 in the 43S/48S complex in the Rps16 mutant strains (Figure 7F) similar to that observed previously in strain rps5-Δ1-46 (10) (Figure 4C). The increase in association of eIF2 with 40S ribosomal subunits in the Rps16 mutant strains as compared to WT is statistically significant (P-values <0.05 in wt versus rps16-R143G and wt versus rps16-YRΔΔ), however, the difference in eIF2 association between the two mutants is not (P-values >0.2 in rps16-R143G versus rps16-YRΔΔ) (Figure 7F). We also observed an increased association of eIF1 along with a decreased association of eIF5 with 40S of the rps16-YRΔΔ mutant as compared to WT, similar to what has been observed in case of rps5 mutants (Supplementary Figure S6CD). These data support the idea that the Rps16 C-terminus cooperates with the N-terminal segments of Rps16 and Rps5 to promote hydrolysis of eIF2-bound GTP or Pi release from eIF2-GDP+Pi on AUG recognition, to ensure efficient general initiation and proper reinitiation on GCN4 mRNA.
Figure 7.
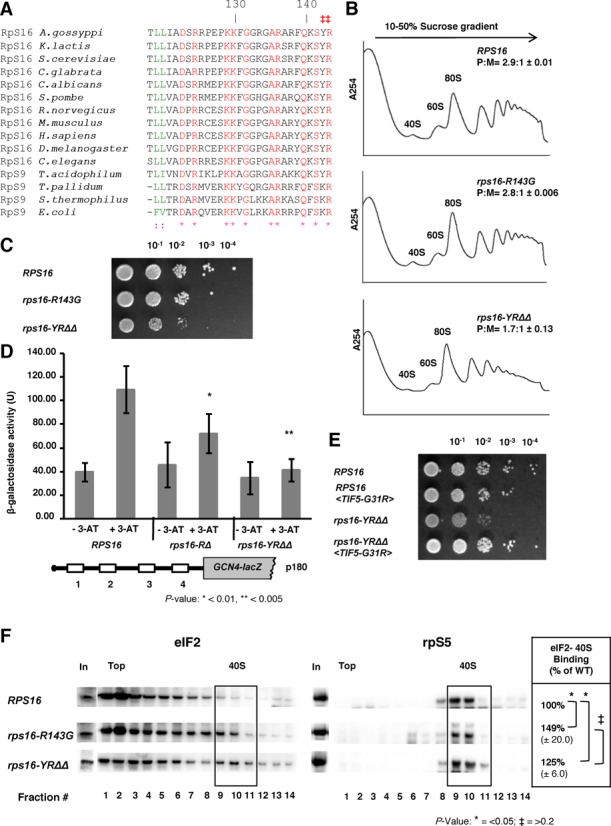
Rps16 C-terminus is important for efficient initiation. (A) Sequence alignments of the C-terminal region of ribosomal protein S16/S9 from various species. The last two amino acid residues (Y142 and R143 in budding yeast) which are extremely conserved are marked (‡). (B) Whole cell extracts from wild type and mutant yeast strains (rps16-R143G and rps16-YRΔΔ double deletion mutant) were resolved on a 10–50% sucrose density gradient. The ratio of the area under the polysomal (P) and 80S peaks are shown (P:M) with ± standard errors. (C) Wild type, rps16-R143G and rps16-YRΔΔ mutant yeast strains were grown in YEPD medium with 2% glucose till OD ∼0.7, serial diluted and spotted onto YEPD+2% glucose plates. (D) Wild type, rps16-RΔ and rps16-YRΔΔ mutants were transformed with p180 and assayed for GCN4 re-initiation efficiency using 3-AT. (E) RPS16, RPS16<TIF5-G31R>, rps16-YRΔΔ and rps16-YRΔΔ <TIF5-G31R> mutants were grown in YEPD medium containing 2% glucose till OD ∼0.5, serial diluted and spotted onto YEPD+2% glucose plates for growth assays. (F) Extracts from wild type, rps16-R143G and rps16-YRΔΔ double deletion mutant yeast strains after treatment with formaldehyde were resolved by velocity sedimentation on 10–30% sucrose gradients. Western blot analyses of individual fractions were performed using antibodies which recognize eIF2 and the ribosomal protein S5, respectively. ‘In’ for input - represents a 7% portion of each gradient fraction. Association of eIF2 with the 40S subunits was quantified and expressed as percentage of 40S binding normalized against Rps5.
DISCUSSION
Although ribosomal proteins are not involved in the catalysis of peptide bond formation (43), they do participate in a variety of activities in the translation process, such as the recruitment of tRNAs, translation factors and specific mRNAs (44–48). However, assignment of specific functions to individual eukaryotic ribosomal proteins has been very challenging, owing in part to the extreme functional cooperation between rRNA and ribosomal proteins and between ribosomal proteins themselves (49). In addition, the majority of eukaryotic ribosomal proteins are essential for cell viability, with many playing important roles in ribosome biogenesis (15,44,50,51), which impedes their extensive mutagenesis. As a result, only a few ribosomal proteins have been assigned specific functions in eukaryotic translation (52–54).
Rps5/S7 belongs to the extremely conserved family of ribosomal proteins that are present in all domains of life (45,47); however, eukaryotic Rps5 proteins contain an additional N-terminal segment (∼50–70 amino acid residues in lengths) absent in prokaryotes (Figure 5A). While the exact function of this N-terminal region was unclear, our finding that its complete removal is lethal in yeast (10) led us to hypothesize that it facilitates ribosome activities related to the presence of eukaryotic-specific translation factors (10). In particular, we argued that the N-terminal region of Rps5 modulates eIF2-bound GTP hydrolysis upon start codon recognition by the scanning 48S complex, and/or subsequent eIF2 dissociation (10).
In this report, we provide evidence that the eukaryotic specific N-terminal region of Rps5 plays an important role in start codon recognition during translation initiation and that this function involves its interaction with the eukaryotic-specific N-terminus of Rps16. Either truncating the N-terminal 46 residues of Rps5 (rps5-Δ1-46 mutant), or alanine substitution of Lys45 (rps5-K45A mutant), confers a Slg− phenotype and a reduced rate of bulk translation initiation, as manifested by reduced abundance of polyribosomes relative to 80S subunits. Both mutations also impair the derepression of GCN4 mRNA translation in response to amino acid limitation, which depends on efficient reinitiation at regulatory upstream AUGs in the GCN4 mRNA leader. We have further found that the F46A and Y49G substitutions of Rps16 residues involved in Rps16-NTD/Rps5-NTD interactions also confer slow growth phenotypes, supporting the possibility that interaction between the N-terminal regions of Rps5 and Rps16 is critical for efficient translation initiation. Examination of a panel of GCN4-lacZ reporters harboring different arrangements of uORFs revealed that the strong defect in the ability of scanning PICs to bypass the AUGs of the inhibitory uORFs 2–4 could not be explained by a failure of 40S subunits to resume scanning after terminating translation by uORF1, by a reduced rate of scanning between uORF1 and uORF4, or by a failure to recognize the AUG codon at uORF1. The most likely remaining explanation is that the scanning 40S subunits are delayed in reaching the AUGs at uORFs 2–4 by the presence of 48S PICs stalled at these start codons owing to a delay in completing a step of initiation following AUG recognition. This would evoke queuing of other scanning PICs behind the stalled 48S complexes and provide ample time for these queuing PICs to load the TC, overcoming the reduction in TC abundance produced in amino acid starved cells by eIF2α phosphorylation and attendant inhibition of GDP-GTP exchange on eIF2 catalyzed by eIF2B. As a result, virtually all the PICs scanning downstream from uORF1 would be forced to reinitiate at uORFs 2,3, or 4, and fail to reinitiate at the GCN4 AUG instead, blocking derepression of GCN4 translation.
The conclusion that initiation is impaired in the rps5-Δ1-46 mutant at a step following AUG recognition is supported by our previous findings that eIF2 accumulates on native 40S subunits in this strain (10), implying a failure in eIF5-stimulated GTP hydrolysis by TC in the scanning complex prior to AUG recognition, a defect in releasing Pi from eIF2·GDP at the AUG codon, or a failure to release eIF2·GDP from the 48S complex. Any of these defects are expected to block the joining of 60S subunits to assemble an 80S IC competent for elongation. Consistent with this, here we observed an accumulation in native PICs of eIF1, which is normally released on AUG recognition to allow Pi release from eIF2·GDP, and the subunit joining factor eIF5B, presumably because subunit joining cannot be completed.
Another phenotype of the rps5-Δ1-46 and rps5-K45A mutants consistent with a defect following start codon recognition is a reduction in the efficiency of utilizing near-cognate UUG start codons relative to AUG codons. The U:U mismatch between UUG and the anticodon of tRNAi destabilizes the closed, PIN state of the PIC from which eIF1 dissociates, Pi is released from eIF2·GDP·Pi and 60S subunit joining occurs (31). It can be expected that the delay in these reactions evoked by the U:U mismatch at UUG codons would be exacerbated by the defect postulated above engendered by alterations in the Rps5-NTD, to evoke a greater reduction in UUG versus AUG initiation in the rps5-Δ1-46 and K45A mutants.
Another line of evidence supporting our model is that the Slg− and Gcn− phenotypes and accumulation of eIF2 on native 40S subunits observed in the rps5-Δ1-46 mutant were all mitigated by introducing into the strain the dominant SUI5 allele of TIF5, encoding the eIF5-G31R variant. In contrast to the reduced UUG:AUG initiation ratio provoked by the Rps5 mutants, SUI5 confers an increased UUG:AUG ratio (Sui− phenotype). The dominance of the Sui− phenotype conferred by SUI5 implies an alteration of eIF5 function, and previous biochemical analysis suggested that eIF5-G31R increases eIF5 GAP function, elevating the rate of GTP hydrolysis by the eIF2 TC in a model assay containing these two factors, the 40S subunit and AUG triplet. In the current model of scanning, GTP hydrolysis and establishment of an internal equilibrium between GTP and GDP·Pi occurs in the scanning PIC prior to AUG recognition, with AUG recognition triggering Pi release rather than GTP hydrolysis per se (31,55). If the Rps5 NTD mutations impair GTP hydrolysis in the scanning complex, then it could be proposed that introducing eIF5-G31R into these mutants restores a more nearly WT rate of GTP hydrolysis, accounting for suppression of the Rps5 mutant phenotypes by SUI5.
Other studies in a more fully reconstituted yeast initiation system containing eIFs -1 and -1A and a model mRNA revealed that eIF5-G31R enhances rearrangement of the PIC from an open conformation thought to be compatible with scanning, to which eIF1A and TC are bound in a relatively unstable configuration (POUT), to a more closed state thought to be conducive to downstream steps of initiation, to which eIF1A and TC are bound more tightly (PIN) (31,39,40). In these latter studies, the ability of eIF5-G31R to promote the closed, PIN conformation was restricted to PICs reconstituted with mRNAs harboring a near-cognate start codon, including UUG, which is fully consistent with the Sui− phenotype of SUI5. As this altered activity of eIF5-G31R was observed in assays using a nonhydrolyzable GTP analog to form the eIF2 TC, they clearly occur independently of eIF5 GAP function. It seems difficult to account for the ability of SUI5 to mitigate the effects of the Rps5 NTD mutations on initiation at AUG codons by invoking the ability of eIF5-G31R to stabilize the closed, PIN state, as this activity is restricted to near-cognate codons. It is possible, however, that eIF5-G31R can stabilize the closed/PIN state at AUG codons in the context of the impaired PICs formed by the Rps5 NTD variants.
Our data suggest that Rps5 and specifically its NTD is (either directly or indirectly) involved in modulating critical activities in the initiation pathway. Because the Rps5-NTD interacts with the Rps16 NTD, with a direct contact involving the residues altered by the Rps5-NTD substitution K45A (Lys-45) and Rps16-NTD F46A (Phe-46), we consider it likely that the phenotypes of the Rps5-NTD mutations involve, at least in part, disruption of the interaction between these two eukaryotic-specific segments of Rps5 and Rps16. Furthermore, the fact that Rps16 contains a long unstructured CTT that appears to make direct contact with Met-tRNAiMet when the latter is base-paired with AUG in the P site (2,3,12) raises the intriguing possibility that impairing interaction between the NTDs of Rps5 and Rps16 will in turn impede productive interaction between the Rps16 CTT and Met-tRNAiMet in the P site of 48S PICs, thereby accounting for mutant phenotypes of the Rps5 NTD substitutions. Supporting this idea, we found that deleting the last two residues of the Rps16 CTT confers the same phenotypes we described for the Rps5 NTD mutations, namely, a Slg− phenotype, reduced rate of bulk translation initiation (decreased polysome content), impaired derepression of GCN4 mRNA translation (Gcn− phenotype) and accumulation of eIF2 on native 40S subunits. Moreover, the Slg− phenotype conferred by this rps16-YRΔΔ allele was suppressed by SUI5. This concordance of genetic and biochemical phenotypes between the Rps5-NTD mutations and the rps16-YRΔΔ substitution strongly suggests that they arise from a common molecular lesion, which we propose to be impaired Rps16-CTT interaction with Met-tRNAiMet base-paired with AUG in the P site. This defect would evoke reduced GTP hydrolysis, slower Pi release, or a delay in the conformational rearrangement from the open/POUT configuration to the closed/PIN state required for steps following AUG recognition.
There is recent evidence that domain 1 (D1) of eIF2α contacts the Rps5 C-terminal helix in mammalian PICs (2). This position of D1 is consistent with the reported interaction of eIF2α and the -3 position of the mRNA in 48S complexes (56). Moreover, it was very recently found that mammalian eIF2α crosslinks with Rps5 N-terminal (2TEWETAAPAVAETPDIK18), middle (72LTNSMMMHGRNNGK85) and C-terminal (165NIKTIAECLADELINAAK182) regions (57). It needs to be emphasized, however, that while the middle and C-terminal regions are extremely conserved among all Rps5 homologs, the N-terminal region mentioned above is rather dissimilar between yeast and mammalian proteins (Figure 5A). It thus remains possible that the mechanism of eIF2 recruitment in mammals and yeast differs slightly and/or that the Rps5 C-terminal region is primarily responsible for eIF2-Rps5 interaction (consistent with the study of Hashem et al. (2)) and the Rps5 N-terminal and middle regions only fine-tune the interaction. We thus hypothesize that interaction between eIF2 and Rps5 and subsequent Rps5-Rps16 association modulates the location of the Rps16 CTT to promote correct placement of TC in the P site and eIF5-stimulated GTP-hydrolysis or Pi release (Figure 8). The interaction between Rps5 and Rps16 may also indirectly influence the placement of eIF1, TC and eIF5 following start codon recognition. It is important to mention a very recent report indicating that positioning of the C-terminal tail of rpS9 (bacterial counterpart of Rps16) is important for fidelity of translation initiation in bacteria (58); although a molecular understanding of this effect is still lacking. Our study, however, provides some of the first evidence supporting the functional significance of protein–protein interactions within the ribosome that are absent in prokaryotes but represent a defining feature of eukaryotic ribosomes.
Figure 8.
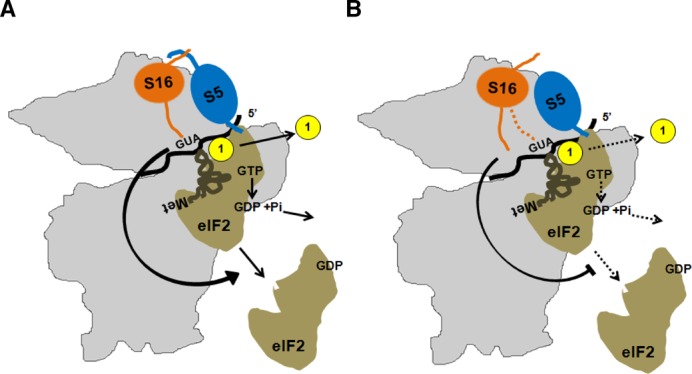
Proposed model for Rps5-Rps16 interaction affecting recruitment of eIF2·GTP·Met-tRNAiMet ternary complex and the eIF5 stimulated hydrolysis of eIF2-bound GTP. (A) Rps5 N-terminal region mediates critical interaction with Rps16, whose C-terminal region extends towards the mRNA cleft and the P-site and affects the dynamic events surrounding recruitment of TC, eIF1 stimulated recognition of AUG codon, subsequent eIF5 stimulated hydrolysis of eIF2-bound GTP and the release of eIF2. (B) Truncation of N-terminal 46 amino acids abolishes Rps5: Rps16 interaction, and affects regulated (via Rps16 C-terminal end) recruitment of TC and eIF5 stimulated hydrolysis of eIF2-bound GTP and the release of eIF2.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGMENTS
The authors would like to thank Drs Tom Dever, Leoš Valášek and Philipp Milkereit for generous gifts of antibodies, plasmids and strains used in this study. We are also grateful to Drs Tatyana Pestova and Christopher Hellen for many helpful discussions.
FUNDING
Human Frontiers Science Program (HFSP) [RGP0024 to A.A.K.]; Ohio Scholars Program [to A.A.K.]; Cleveland State University Doctoral Dissertation Research Expense Award [to A.G.]; Center for Gene Regulation in Health and Disease (GRHD) at Cleveland State University [to A.A.K.], and the Intramural Program of the National Institutes of Health [to A.G.H.]. Funding for open access charge: HFSP [RGP0024].
Conflict of interest statement. None declared.
REFERENCES
- 1.Jackson R.J., Hellen C.U., Pestova T.V. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 2010;11:113–127. doi: 10.1038/nrm2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hashem Y., des Georges A., Dhote V., Langlois R., Liao H.Y., Grassucci R.A., Hellen C.U., Pestova T.V., Frank J. Structure of the mammalian ribosomal 43S preinitiation complex bound to the scanning factor DHX29. Cell. 2013;153:1108–1119. doi: 10.1016/j.cell.2013.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lomakin I.B., Steitz T.A. The initiation of mammalian protein synthesis and mRNA scanning mechanism. Nature. 2013;500:307–311. doi: 10.1038/nature12355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ben-Shem A., Garreau de Loubresse N., Melnikov M., Jenner L., Yusupova G., Yusupov M. The structure of the eukaryotic ribosome at 3.0 Å resolution. Science. 2011;334:1524–1529. doi: 10.1126/science.1212642. [DOI] [PubMed] [Google Scholar]
- 5.Ben-Shem A., Jenner L., Yusupova G., Yusupov M. Crystal structure of the eukaryotic ribosome. Science. 2010;330:1203–1209. doi: 10.1126/science.1194294. [DOI] [PubMed] [Google Scholar]
- 6.Ramakrishnan V. The eukaryotic ribosome. Science. 2011;331:681–682. doi: 10.1126/science.1202093. [DOI] [PubMed] [Google Scholar]
- 7.Ignatovich O., Cooper M., Kulesza H.M., Beggs J.D. Cloning and characterisation of the gene encoding the ribosomal protein S5 (also known as rp14, S2, YS8) of Saccharomyces cerevisiae. Nucleic Acids Res. 1995;23:4616–4619. doi: 10.1093/nar/23.22.4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mager W.H., Planta R.J., Ballesta J.G., Lee J.C., Mizuta K., Suzuki K., Warner J.R., Woolford J. A new nomenclature for the cytoplasmic ribosomal proteins of Saccharomyces cerevisiae. Nucleic Acids Res. 1997;25:4872–4875. doi: 10.1093/nar/25.24.4872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Galkin O., Bentley A.A., Gupta S., Compton B.A., Mazumder B., Kinzy T.G., Merrick W.C., Hatzoglou M., Pestova T.V., Hellen C.U., et al. Roles of the negatively charged N-terminal extension of Saccharomyces cerevisiae ribosomal protein S5 revealed by characterization of a yeast strain containing human ribosomal protein S5. RNA. 2007;13:2116–2128. doi: 10.1261/rna.688207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lumsden T., Bentley A.A., Beutler W., Ghosh A., Galkin O., Komar A.A. Yeast strains with N-terminally truncated ribosomal protein S5: implications for the evolution, structure and function of the Rps5/RpS7 proteins. Nucleic Acids Res. 2010;38:1261–1272. doi: 10.1093/nar/gkp1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Neueder A., Jakob S., Pöll G., Linnemann J., Deutzmann R., Tschochner H., Milkereit P. A local role for the small ribosomal subunit primary binder Rps5 in final 18S rRNA processing in Yeast. PLoS One. 2010;5:e10194. doi: 10.1371/journal.pone.0010194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spahn C.M., Beckmann R., Eswar N., Penczek P.A., Sali A., Blobel G., Frank J. Structure of the 80S ribosome from Saccharomyces cerevisiae–tRNA-ribosome and subunit-subunit interactions. Cell. 2001;107:373–386. doi: 10.1016/s0092-8674(01)00539-6. [DOI] [PubMed] [Google Scholar]
- 13.Huang H., Yoon H., Hannig E.M., Donahue T.F. GTP hydrolysis controls stringent selection of the AUG start codon during translation initiation in Saccharomyces cerevisiae. Genes Dev. 1997;11:2396–2413. doi: 10.1101/gad.11.18.2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rose M.D., Winston F., Heiter P. Methods in Yeast Genetics: A Laboratory Course Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1990. [Google Scholar]
- 15.Ferreira-Cerca S., Pöll G., Gleizes P.E., Tschochner H., Milkereit P. Roles of eukaryotic ribosomal proteins in maturation and transport of pre-18S rRNA and ribosome function. Mol. Cell. 2005;20:263–275. doi: 10.1016/j.molcel.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 16.Ito H., Fukuda Y., Murata K., Kimura A. Transformation of intact yeast cells treated with alkali cations. J. Bacteriol. 1983;53:163–168. doi: 10.1128/jb.153.1.163-168.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mueller P.P., Hinnebusch A.G. Multiple upstream AUG codons mediate translational control of GCN4. Cell. 1986;45:201–207. doi: 10.1016/0092-8674(86)90384-3. [DOI] [PubMed] [Google Scholar]
- 18.Nielsen K.H., Szamecz B., Valášek L., Jivotovskaya A., Shin B.S., Hinnebusch A.G. Functions of eIF3 downstream of 48S assembly impact AUG recognition and GCN4 translational control. EMBO J. 2004;23:1166–1177. doi: 10.1038/sj.emboj.7600116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Valášek L., Nielsen K.H., Zhang F., Fekete C.A., Hinnebusch A.G. Interactions of Eukaryotic Translation Initiation Factor 3 (eIF3) subunit NIP1/c with eIF1 and eIF5 promote preinitiation complex assembly and regulate start codon selection. Mol. Cell. Biol. 2004;24:9437–9455. doi: 10.1128/MCB.24.21.9437-9455.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Donahue T.F., Cigan A.M. Genetic selection for mutations that reduce or abolish ribosomal recognition of the HIS4 translational initiator region. Mol. Cell. Biol. 1988;8:2955–2963. doi: 10.1128/mcb.8.7.2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Towbin H., Staehelin T., Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc. Natl. Acad. Sci. U.S.A. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dever T.E., Yang W., Aström S., Byström A.S., Hinnebusch A.G. Modulation of tRNA(iMet), eIF-2, and eIF-2B expression shows that GCN4 translation is inversely coupled to the level of eIF-2.GTP.Met-tRNA(iMet) ternary complexes. Mol. Cell. Biol. 1995;15:6351–6363. doi: 10.1128/mcb.15.11.6351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fringer J.M., Acker M.G., Fekete C.A., Lorsch J.R., Dever T.E. Coupled release of Eukaryotic translation initiation factors 5B and 1A from 80S ribosomes following subunit joining. Mol. Cell. Biol. 2007;27:2384–2397. doi: 10.1128/MCB.02254-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nanda J.S., Cheung Y.N., Takacs J.E., Martin-Marcos P., Saini A.K., Hinnebusch A.G., Lorsch J.R. eIF1 controls multiple steps in start codon recognition during eukaryotic translation initiation. J. Mol. Biol. 2009;394:268–285. doi: 10.1016/j.jmb.2009.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Laemmli U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 26.Hinnebusch A.G. Translational regulation of GCN4 and the general amino acid control of Yeast. Annu. Rev. Microbiol. 2005;59:407–450. doi: 10.1146/annurev.micro.59.031805.133833. [DOI] [PubMed] [Google Scholar]
- 27.Abastado J.P., Miller P.F., Jackson B.M., Hinnebusch A.G. Suppression of ribosomal reinitiation at upstream open reading frames in amino acid-starved cells forms the basis for GCN4 translational control. Mol. Cell. Biol. 1991;11:486–496. doi: 10.1128/mcb.11.1.486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Watanabe R., Murai M.J., Singh C.R., Fox S., Li M., Asano K. The eukaryotic initiation factor (eIF) 4G HEAT domain promotes translation re-initiation in yeast both dependent on and independent of eIF4A mRNA helicase. J. Biol. Chem. 2010;285:21922–21933. doi: 10.1074/jbc.M110.132027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grant C.M., Miller P.F., Hinnebusch A.G. Requirements for intercistronic distance and level of eIF-2 activity in reinitiation on GCN4 mRNA varies with the downstream cistron. Mol. Cell. Biol. 1994;14:2616–2628. doi: 10.1128/mcb.14.4.2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Asano K., Clayton J., Shalev A., Hinnebusch A.G. A multifactor complex of eukaryotic initiation factors eIF1, eIF2, eIF3, eIF5, and initiator tRNAMet is an important translation initiation intermediate in vivo. Genes Dev. 2000;14:2534–2546. doi: 10.1101/gad.831800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hinnebusch A.G. Molecular mechanism of scanning and start codon selection in eukaryotes. Microbiol. Mol. Biol. Rev. 2011;75:434–467. doi: 10.1128/MMBR.00008-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nanda J.S., Saini A.K., Munoz A.M., Hinnebusch A.G., Lorsch J.R. Coordinated movements of eukaryotic translation initiation factors eIF1, eIF1A and eIF5 trigger phosphate release from eIF2 in response to start codon recognition by the ribosomal preinitiation complex. J. Biol. Chem. 2013;288:5316–5329. doi: 10.1074/jbc.M112.440693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ivanov I.P., Loughran G., Sachs M.S., Atkins J.F. Initiation context modulates autoregulation of eukaryotic translation initiation factor 1 (eIF1) Proc. Natl. Acad. Sci. U.S.A. 2010;107:18056–18060. doi: 10.1073/pnas.1009269107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Loughran G., Sachs M.S., Atkins J.F., Ivanov I.P. Stringency of start codon selection modulates auto regulation of translation initiation factor eIF5. Nucleic Acids Res. 2012;40:2898–2906. doi: 10.1093/nar/gkr1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hinnebusch A.G., Lorsch J.R. The mechanism of eukaryotic translation initiation: new insights and challenges. Cold Spring Harb. Perspect. Biol. 2012 doi: 10.1101/cshperspect.a011544. 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paulin F.E., Campbell L.E., O'Brien K., Loughlin J., Proud C.G. Eukaryotic translation initiation factor 5 (eIF5) acts as a classical GTPase-activator protein. Curr. Biol. 2001;11:55–59. doi: 10.1016/s0960-9822(00)00025-7. [DOI] [PubMed] [Google Scholar]
- 37.Yoon H.J., Donahue T.F. The suil suppressor locus in Saccharomyces cerevisiae encodes a translation factor that functions during tRNA(iMet) recognition of the start codon. Mol. Cell. Biol. 1992;12:248–260. doi: 10.1128/mcb.12.1.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Karásková M., Gunišová S., Herrmannová A., Wagner S., Munzarová V., Valášek L. Functional characterization of the role of the N-terminal domain of the c/Nip1 subunit of eukaryotic initiation factor 3 (eIF3) in AUG recognition. J. Biol. Chem. 2012;287:28420–28434. doi: 10.1074/jbc.M112.386656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martin-Marcos P., Nanda J., Luna R.E., Wagner G., Lorsch J.R., Hinnebusch A.G. β-hairpin loop of eIF1 mediates 40S ribosome binding to regulate initiator tRNAMet recruitment and accuracy of AUG selection in vivo. J. Biol. Chem. 2013;288:27546–27562. doi: 10.1074/jbc.M113.498642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maag D., Algire M.A., Lorsch J.R. Communication between eukaryotic translation initiation factor 5 and 1A within the ribosomal pre-initiation complex plays a role in start site selection. J. Mol. Biol. 2006;356:724–737. doi: 10.1016/j.jmb.2005.11.083. [DOI] [PubMed] [Google Scholar]
- 41.Malygin A.A., Yanshina D.D., Karpova G.G. Interactions of human ribosomal proteins S16 and S5 with an 18S rRNA fragment containing their binding sites. Biochimie. 2009;91:1180–1186. doi: 10.1016/j.biochi.2009.06.013. [DOI] [PubMed] [Google Scholar]
- 42.Granneman S., Petfalski E., Swiatkowska A., Tollervey D. Cracking pre-40S ribosomal subunit structure by systematic analyses of RNA-protein crosslinking. EMBO J. 2010;29:2026–2036. doi: 10.1038/emboj.2010.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schmeing T.M., Ramakrishnan V. What recent ribosome structures have revealed about the mechanism of translation. Nature. 2009;461:1234–1242. doi: 10.1038/nature08403. [DOI] [PubMed] [Google Scholar]
- 44.Wool I.G., Chan Y.L., Gluck A. Structure and evolution of mammalian ribosomal proteins. Biochem. Cell Biol. 1995;73:933–947. doi: 10.1139/o95-101. [DOI] [PubMed] [Google Scholar]
- 45.Lecompte O., Ripp R., Thierry J.C., Moras D., Poch O. Comparative analysis of ribosomal proteins in complete genomes: an example of reductive evolution at the domain scale. Nucleic Acids Res. 2002;30:5382–5390. doi: 10.1093/nar/gkf693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dresios J., Panopoulos P., Synetos D. Eukaryotic ribosomal proteins lacking a eubacterial counterpart: important players in ribosomal function. Mol. Microbiol. 2006;59:1651–1663. doi: 10.1111/j.1365-2958.2006.05054.x. [DOI] [PubMed] [Google Scholar]
- 47.Roberts E., Sethi A., Montoya J., Woese C.R., Luthey-Schulten Z. Molecular signatures of ribosomal evolution. Proc. Natl. Acad. Sci. U.S.A. 2008;105:13953–13958. doi: 10.1073/pnas.0804861105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Landry D.M., Hertz M.I., Thompson S.R. RPS25 is essential for translation initiation by the Dicistroviridae and hepatitis C viral IRESs. Genes Dev. 2009;23:2753–2764. doi: 10.1101/gad.1832209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dinman J.D. The eukaryotic ribosomes: current status and challenges. J. Biol. Chem. 2009;284:11761–11765. doi: 10.1074/jbc.R800074200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vilardell J., Warner J.R. Ribosomal protein L32 of Saccharomyces cerevisiae influences both the splicing of its own transcript and the processing of rRNA. Mol. Cell. Biol. 1997;17:1959–1965. doi: 10.1128/mcb.17.4.1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Perreault A., Bellemer C., Bachand F. Nuclear export competence of pre-40S subunits in fission yeast requires the ribosomal protein Rps2. Nucleic Acids Res. 2008;36:6132–6142. doi: 10.1093/nar/gkn625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hershey J.W.B., Merrick W.C. The pathway and mechanism of eukaryotic protein synthesis. In: Sonenberg N., Hershey J.W.B., Mathews M.B., editors. Translational Control of Gene Expression. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2000. pp. 33–38. [Google Scholar]
- 53.Wilson D.N., Nierhaus K.H. Ribosomal proteins in the spotlight. Crit. Rev. Biochem. Mol. Biol. 2005;40:243–267. doi: 10.1080/10409230500256523. [DOI] [PubMed] [Google Scholar]
- 54.Brodersen D.E., Nissen P. The social life of ribosomal proteins. FEBS J. 2005;272:2098–2108. doi: 10.1111/j.1742-4658.2005.04651.x. [DOI] [PubMed] [Google Scholar]
- 55.Algire M.A., Maag D., Lorsch J.R. Pi release from eIF2, not GTP hydrolysis, is the step controlled by start-site selection during eukaryotic translation initiation. Mol. Cell. 2005;20:251–262. doi: 10.1016/j.molcel.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 56.Pisarev A.V., Kolupaeva V.G., Pisareva V.P., Merrick W.C., Hellen C.U., Pestova T.V. Specific functional interactions of nucleotides at key -3 and +4 positions flanking the initiation codon with components of the mammalian 48S translation initiation complex. Genes Dev. 2006;20:624–636. doi: 10.1101/gad.1397906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sharifulin D., Babaylova E., Kossinova O., Bartuli Y., Graifer D., Karpova G. Ribosomal protein S5e is implicated in translation initiation through its interaction with the N-Terminal domain of initiation factor eIF2α. Chembiochem. 2013;14:2136–2143. doi: 10.1002/cbic.201300318. [DOI] [PubMed] [Google Scholar]
- 58.Arora S., Bhamidimarri S.P., Bhattacharyya M., Govindan A., Weber M.H., Vishveshwara S., Varchney U. Distinctive contributions of the ribosomal P-site elements m2G966, m5C967 and the C-terminal tail of the S9 protein in the fidelity of the initiation of translation in Escherichia coli. Nucleic Acids Res. 2013;41:4963–4975. doi: 10.1093/nar/gkt175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Guex N., Peitsch M.C. SWISS-MODEL and the Swiss Pdbviewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.