


Abstract
Despite growing consensus that long intergenic non-coding ribonucleic acids (lincRNAs) are modulators of cancer, the knowledge about the deoxyribonucleic acid (DNA) methylation patterns of lincRNAs in cancers remains limited. In this study, we constructed DNA methylation profiles for 4629 tumors and 705 normal tissue samples from 20 different types of human cancer by reannotating data of DNA methylation arrays. We found that lincRNAs had different promoter methylation patterns in cancers. We classified 2461 lincRNAs into two categories and three subcategories, according to their promoter methylation patterns in tumors. LincRNAs with resistant methylation patterns in tumors had conserved transcriptional regulation regions and were ubiquitously expressed across normal tissues. By integrating cancer subtype data and patient clinical information, we identified lincRNAs with promoter methylation patterns that were associated with cancer status, subtype or prognosis for several cancers. Network analysis of aberrantly methylated lincRNAs in cancers showed that lincRNAs with aberrant methylation patterns might be involved in cancer development and progression. The methylated and demethylated lincRNAs identified in this study provide novel insights for developing cancer biomarkers and potential therapeutic targets.
INTRODUCTION
Deep sequencing with new computational approaches for assembling transcriptome has identified tens of thousands of large intergenic transcripts across different tissues and cell types. These intergenic transcripts do not code for proteins and are named long intergenic non-coding ribonucleic acids (lincRNAs) (1,2). Many lincRNAs are dysregulated in human cancers and implicated in disease progression through modulating apoptosis, increasing cellular oncogenic potential or inhibiting tumor growth (3,4). Although several lincRNAs [lincRNA_p21 (5), HOTAIR (6), PCA3 (7) among others] have been depicted with relatively explicit molecular mechanisms in several cancers, little is known about the regulatory mechanisms of lincRNAs in tumors or normal tissues, especially on regulation by deoxyribonucleic acid (DNA) methylation.
DNA methylation at gene promoters is crucial for gene silencing and involved in many diseases (8). DNA methylation of lincRNA promoters might be an epigenetic regulator of lincRNAs expression (9), for instance, lincRNA Glt2 (MEG3), whose expression was indirectly regulated by mir-29a in hepatocellular carcinoma cells, which inhibited the activity of DNA methyltransferase and caused de-repression of MEG3 expression (10). Several lincRNAs were upregulated in the human colorectal cancer cell line HCT116 by treatment with a DNA-demethylating agent (11). However, systematically identifying cancer-related methylation patterns of human lincRNAs is still a challenge, partly because of a lack of global DNA methylation profiles for lincRNAs.
High-resolution next-generation sequencing and microarray technologies have been used for genome-scale mapping of DNA methylation (12). Illumina Infinium HumanMethylation450 BeadChip Array (Infinium 450k) has 485 577 probes that comprehensively cover most known CpG islands (CGIs) and 99% of NCBI Reference Sequence genes (13). The Cancer Genome Atlas (TCGA) Research Network contains a large number of data sets with Infinium 450k arrays for thousands of tumor samples with corresponding normal samples and matched clinical annotations that are all publicly available (14–16). Previous studies successfully extracted lincRNA expression information by repurposing the microarray data, which were originally designed to detect the expression of genes or exons (17–19). By reannotating the Infinium 450k array, we could obtain lincRNA methylation levels in a large number of samples.
We developed a computational strategy to reannotate the Infinium 450k array and observed that DNA methylation level of lincRNA promoters was tightly linked to lincRNA transcription. We constructed DNA methylation profiles for 20 distinct types of cancer according to lincRNA promoter methylation levels. We classified the lincRNAs into two categories and three subcategories and found that lincRNAs with resistant methylation patterns in tumors had conserved transcriptional regulation regions and were ubiquitously expressed across normal tissues. By analyzing the lincRNA methylation profiles together with clinical information for tumors in breast invasive cancer (BRCA), lung squamous cell cancer (LUSC) and uterine corpus endometrioid cancer (UCEC) among others, and subtype data of tumors in BRCA (20) and LUSC (21), we identified lincRNAs with promoter methylation patterns that were associated with cancer status, subtype or prognosis. These lincRNAs could be further evaluated for use as cancer biomarkers and potential cancer therapy targets. Some lincRNAs with aberrant methylation patterns in cancers might involve in cancer development and progression. Early detection of hypermethylated or hypomethylated lincRNAs could serve as cancer biomarkers for diagnosis or treatment.
MATERIALS AND METHODS
Data sources
DNA methylation data from Infinium 450k arrays and patients clinical data were downloaded from TCGA (https://tcga-data.nci.nih.gov/tcga/). RNA-seq data for 16 tissues were derived from Human Body Map 2 project (SRA, E-MTAB-513)(1). RNA-seq data of six cell lines were from Gene Expression Omnibus (GEO, GSE23316) (22). Corresponding lincRNA expression data from patients were calculated based on the RNA-seq V2 data from TCGA. CGI annotation and repetitive element (RE) annotation data were from UCSC Genome Browser (23). Annotation files for lincRNAs and protein-coding genes (PCGs) were downloaded from Human lincRNA Catalog (1).
Re-annotating data from the Infinium 450k array to construct lincRNA methylation profiles
We mapped 485 577 probe sequences (50 bp in length) to human genome (hg19) with BLAT (24). We treated BLAT output in two steps: first, we retained the probes uniquely mapped to a single location in human genome with a maximum of two mismatches. Second, we eliminated the sequences with gaps. A total of 485 512 probe sequences from Infinium 450k arrays were uniquely mapped. We then assigned the probe sequences into four lincRNA-associated regions according to the Human lincRNA Catalog annotation file (1): regions 10 kb upstream from the transcription start sites (TSSs), regions 10 kb downstream from the transcription termination sites (TTSs), exons and introns. We only retained the probe sequences exclusively mapped to a single region.
To estimate the methylation level of a given probe, we used the beta value: the ratio of intensities between methylated and unmethylated alleles. The beta value and corresponding P-value of each probe were obtained from the level 3 Infinium 450k data in TCGA. Beta value = Imeth/(Imeth+ Iunmeth), where Imeth is the intensity of methylation and Iunmeth is the intensity of unmethylation. We only used the beta values with significant detection P-values (P < 0.05) in calculations to avoid using the missing data.
For each type of cancer, we constructed lincRNA methylation profiles using the methylation levels of the probes mapped into 10 kb upstream from the TSSs of the lincRNAs. In research on a cis-regulatory element annotation system, Liu et al. specified the largest promoter size as 10 kb upstream from the TSS (25). Since the regulatory mechanism of lincRNAs transcription is similar to the regulation of genes (26), we used 10 kb upstream from the TSS for a relatively comprehensive range of lincRNA promoters. We used only the probes closest to each TSS to determine the DNA methylation status of lincRNA promoters.
Classification of lincRNAs with divergent methylation patterns in cancers
We classified lincRNAs into two categories: prone to methylation (PM) lincRNAs and resistant to methylation (RM) lincRNAs. LincRNA promoters with beta value ≤ 0.3 were considered as unmethylated promoters and those with beta value > 0.3 were considered as methylated ones. These cutoffs and strategies were similar to those in previous studies (27,28). LincRNAs with methylated promoters in more than 20% of all tumor samples were defined as PM lincRNAs. LincRNAs with unmethylated promoters in all tumors were named RM lincRNAs. PM lincRNAs that were methylated in more than 5% of tumors for each cancer were defined as consistently methylated (CM) lincRNAs. PM lincRNAs that were unmethylated in tumor samples for at least one cancer were classified as variable methylation (VM) lincRNAs.
Analysis of REs at lincRNA promoters
We obtained the position information of the REs from the Repeat Masker track (RMSK) in the UCSC Genome Browser (hg19) (29). We divided the region ±10 kb around lincRNA TSS into 20 equal-sized bins. The REs were considered to exist if they overlapped with the bins. We plotted the frequency of the REs in each bin for lincRNAs belonging to the three categories. We tested differences between the categories using Fisher's exact tests based on the density of the REs in an interval ± 2 kb around TSSs.
Analysis of evolutionary conservation for lincRNA promoters
We used the measurements of base substitutions in 46 placental mammals (phastCons46way, UCSC) to analyze the evolutionary conservation for lincRNAs in different categories. We separated the region upstream and downstream 10 kb from the TSS of a lincRNA into 20 non-overlapping intervals, taking direction of transcription into account. We calculated the mean Phastcons scores for each interval. We tested the significance of differences between categories using scores calculated for intervals ± 2 kb around TSSs with Wilcoxon rank sum tests.
Predicting the functions of lincRNAs
We used two strategies to predict the functions of lincRNAs. According to lincRNA cis-regulatory mechanism (30), we used the PCGs adjacent to the target lincRNAs to infer the potential functions of lincRNAs with three rules: first, the PCGs must locate on the same strand with the target lincRNAs. Second, the PCGs were nearest to the target lincRNAs. Third, only PCGs within 10 kb from the target lincRNAs were retained. We also performed functional enrichment analysis of the PCGs co-expressed with the lincRNAs to predict the potential functions of lincRNAs in cancers.
Obtaining lincRNA expression values from the RNA-seq V2 data in TCGA
We recalculated the RPKM values for lincRNAs using the RNA-seq V2 data in TCGA (31). RPKM = (raw read counts × 109)/(total reads × length of lincRNA_X), where raw read counts = sum of raw read counts in all exons entirely mapped to the lincRNA locus; total reads = sum of raw read counts calculated for all transcripts of a sample; and length of lincRNA_X = sum of length of exons mapped to the lincRNA_X locus. To avoid ambiguous exons mapping, we merged the overlapping lincRNA transcripts into a single candidate lincRNA.
Identifying lincRNAs with prognosis- or cancer subtype-associated promoter methylation patterns
We calculated Kaplan–Meier log-rank P-values to identify lincRNAs with overall survival (OS)-associated methylation patterns. Tumors were separated according to the median methylation of each lincRNA. For each cancer, we divided tumors into a discovery set and a validation set. In the discovery phase, we retained lincRNAs with significant log-rank P-value < 0.05. We permuted the labels for tumors in the discovery set 5000 times to calculate the background distribution of log-rank P-values for each lincRNA. We then estimated the false discovery rate (FDR) for each lincRNA using its own background (32). We validated only lincRNAs under a threshold of FDR = 0.01 in the validation set. We identified lincRNAs with prognosis-associated (PA) methylation patterns in cancers with tumor sample size available for both clinical and methylation data ≥ 200 and a censoring (alive sample) rate ≤ 0.9; or the tumor sample size < 200 and a censoring rate ≤ 0.8 for an effective survival analysis (Supplementary Table S1) (33). We performed t-tests (one-tailed) to compare the lincRNA methylation pattern of patients in each subtype to those in other subtypes in each cancer. The lincRNAs that showed statistically higher or lower methylation (FDR < 0.05) in only one subtype were considered as having subtype-specific methylation patterns.
Statistical analyses
Functional enrichments of PCGs were consisted on the Fisher's exact test (two-tailed) implemented by DAVID v6.7 (http://david.abcc.ncifcrf.gov/) (34). Aberrantly methylated (AM) lincRNAs between tumors and corresponding normal samples were identified by t-test (two-tailed), when FDR < 0.05 and |average beta value of tumors − average beta value of normal samples| ≥ 0.3.
RESULTS
A reannotation strategy for constructing DNA methylation profiles of lincRNAs
To characterize DNA methylation patterns for lincRNAs, we designed a computational strategy to reannotate data of Infinium 450k arrays into four human lincRNA-associated regions (Figure 1A). In total, 3361 lincRNAs had at least one probe sequence uniquely mapped to one of the four regions (Supplementary Table S2). Most probe sequences corresponded to introns (6911, 47%) or exons (3447, 23%). Although a substantial set of probe sequences mapped to the regions 10 kb upstream from the TSSs, we retained only the probes closest to each TSS to determine the DNA methylation status of lincRNA promoters (2461, 13%). The remaining probe sequences (2001, 13%) were annotated closest to the regions 10 kb downstream from the TTSs (Figure 1B). To determine the validity of the reannotation strategy, we annotated the probe sequences to PCG loci using the same strategy. The results were consistent with the previous probe-PCG annotations provided by Illumina. To determine the reliability of lincRNA methylation status, we used data from Infinium 450k arrays and reduced representation bisulfite sequencing (RRBS) of nine cell lines from the ENCODE project (22). Methylation levels of probes annotated to lincRNAs were consistent with levels detected by RRBS (Supplementary Figure S1). In each cell line, all detected probe sites showed significant concordance between Infinium 450k array and RRBS for methylation, including probes annotated to the four lincRNA-associated regions (Supplementary Figure S2) and probes annotated only to lincRNA promoters (Supplementary Figure S3).
Figure 1.
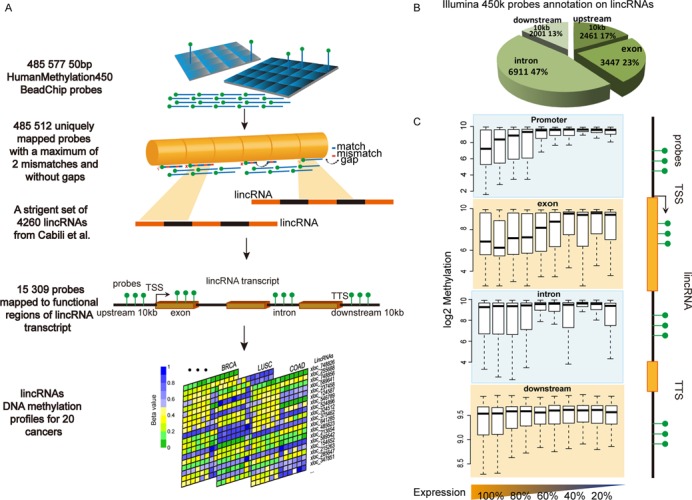
Computational strategy for reannotating Infinium 450k array data to construct lincRNA methylation profiles. (A) Probe reannotation pipeline for lincRNAs. (B) Pie chart with distribution and the number of probes annotated by functional region for all collected lincRNAs. (C) Methylation patterns of lincRNAs by 10 expression quantiles (lowest 10%–100%). Box plots show methylation levels in promoters, exons, introns and the regions 10 kb downstream from the TTS.
We compared the DNA methylation patterns for each region: promoters, exons, introns and the regions 10 kb downstream from the TTSs for lincRNAs within each of the 10 expression quantiles with the Infinium 450k array and RNA-seq data of an H1-hESC cell line from the ENCODE project (22) (Figure 1C). Compared with the other three regions, hypermethylation of promoters was more tightly linked to transcriptional silencing of lincRNAs (Supplementary Figure S4). Therefore, we constructed lincRNA promoter methylation profiles for 20 cancer types including 4629 tumors and 705 corresponding normal tissue samples (TCGA; Supplementary Table S3).
Dissecting lincRNAs promoter methylation patterns in cancers
For the lincRNAs in this study, the average DNA methylation levels showed significant differences between tumors and normal samples in 18 of the 20 cancer types (t-test, P < 0.05), with lower methylation levels in tumors than normal samples for 15 cancer types. Exceptions were kidney renal papillary cell cancer (KIRP), brain lower grade glioma (LGG) and prostate adeno cancer (PRAD) (Figure 2A). Since disrupting DNA methyltransferases may promote chromosome instability and tumor progression, cancer cells are usually less methylated at individual CpG dinucleotides than healthy cells (35–38). The lower average DNA methylation levels of lincRNAs in tumors than in corresponding normal samples for most cancer types were consistent with the global hypomethylation patterns of PCGs in cancer cells. In contrast, hypermethylation of lincRNAs might be involved in DNA repair, tumor cell invasion, cell cycle regulation and other events in which silencing might induce metastasis (38). Aberrant promoter methylation was frequently observed in cancer samples and might have contributed to tumor progression by silencing tumor suppressor genes or activating oncogenes. Therefore, we explored the methylation patterns at lincRNA promoters in each cancer type by dividing the 10-kb region upstream of the TSS into 10 equal-sized bins. We obtained three representative cancer type-specific methylation patterns for 20 cancer types, and examples were shown in bladder urothelial cancer (BLCA), head and neck squamous cell cancer (HNSC) and LGG (Supplementary Figure S5). We then assigned CGIs and CpG shores (±2-kb regions from CGI start or end sites) in the promoter regions and obtained two representative cancer type-specific methylation patterns according to the methylation levels of probes mapped to each region, and examples were shown in BLCA and LGG (Supplementary Figure S6).
Figure 2.
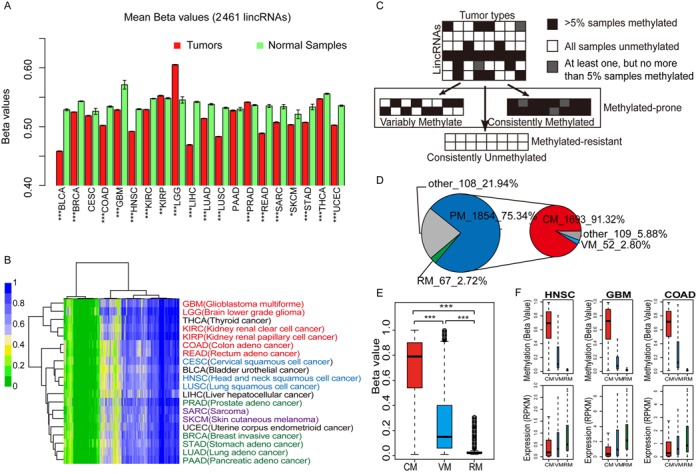
Dissecting lincRNA promoter methylation patterns in cancers. (A) Bar plots with average methylation levels of lincRNA promoters in tumors and corresponding normal samples (t-test, *P < 0.05, **P < 1.0e−3 and ***P < 1.0e−4). Error bars, mean ± SEM. (B) Unsupervised hierarchical clustering of average methylation profiles for 2461 lincRNAs in 20 cancer types. GBM-LGG, KIRC-KIRP and COAD-READ were three pairs of cancers with similar tissue of origin. PRAD, BRCA, STAD, LUAD and PAAD were cancers arising from adeno. CESC, HNSC and LUSC were cancers arising from squamous cells. SARC and SKCM were sarcomatoid carcinomas. (C) Strategy used to segregate lincRNAs into sets with distinct methylation patterns. (D) Pie chart with the number of lincRNAs in different categories. (E) Box plots show methylation level of lincRNAs in different categories. Differences between sets were tested using Wilcoxon rank sum tests. (F) Box plots of DNA methylation and expression levels of VM, CM and RM lincRNAs for three cancers. Methylation and the corresponding expression values were obtained from consistent sample sets.
We performed unsupervised hierarchical clustering on the average promoter methylation profiles of lincRNAs for 20 types of cancer. The results suggested that part of the lincRNAs had RM status in cancers, some lincRNAs had CM status in cancers and the others had VM patterns. Cancers with similar lincRNA methylation patterns were clustered together. Three pairs of cancers with adjacent tissue of origins showed similar lincRNA methylation patterns: glioblastoma multiforme (GBM) and LGG, colon adeno cancer (COAD) and rectum adeno cancer (READ) and kidney renal clear cell cancer (KIRC) and KIRP (Figure 2B).
To determine lincRNA methylation patterns in cancers, we classified the lincRNAs into two categories and three subcategories according to their methylation profiles of tumors (Figure 2C). We obtained 1854 PM lincRNAs and 67 RM lincRNAs. By subdividing the PM lincRNAs, we obtained 1693 (91.32%) CM lincRNAs and 52 (2.80%) VM lincRNAs (Figure 2D and Supplementary Table S4). Using our methodology, CM lincRNAs had the significantly highest median methylation levels and RM lincRNAs had the lowest levels in tumors (Figure 2E). As an important regulating factor of gene expression in cancers (39), DNA methylation might be involved in regulating lincRNA expression in cancers. We examined both methylation levels and expression levels of CM, VM and RM lincRNAs using the Infinium 450k array data and the RNA-seq V2 data for COAD, GBM and HNSC. In all three cancers, RM lincRNAs showed the overall lowest median methylation level and the highest median expression level (Figure 2F). Our results indicated that the three different lincRNA methylation patterns in cancers were related to lincRNA expression. Many lincRNAs have been identified as having regulatory functions in cancer-related pathways such as the MYC and p53 pathways (40). Therefore, we might be able to influence lincRNA expression by altering DNA methylation, thus disrupting the functions of lincRNAs in cancers. Further analysis of the three different DNA methylation patterns might help identify novel drug targets or cancer diagnostic biomarkers.
RM lincRNAs had the most conserved promoter regions and the least tissue-specific expression in normal tissues
Since REs are involved in reprogramming of DNA methylation (41,42), we investigated whether REs affected lincRNA methylation patterns. We quantified the REs around the TSSs of lincRNAs using the RMSK data from UCSC Genome Browser (43). All three major RE classes (LINEs, SINEs and LTRs) were depleted from lincRNA core promoters (2 kb upstream from the TSSs) (Figure 3A). Moreover, RM lincRNAs had significantly fewer REs than CM lincRNAs, possibly caused by activated DNA methylation of REs in lincRNA promoters. RE insertion close to a lincRNA promoter or RE hypermethylation might interrupt the transcription factors or other regulatory elements binding to lincRNA promoters, which could contribute to lincRNAs tissue-specific expression. We quantified the tissue specificity of lincRNA expression in 16 normal tissues (SRA, E-MTAB-513) and six cell lines (GEO, GSE23316) using an information theory method (Supplementary file) (44). CM lincRNAs had significantly higher tissue-specific expression than RM lincRNAs, which was consistent with our hypothesis (Figure 3B). To quantify the evolutionary conservation of lincRNA promoters, we used phastCons scores of placental mammals (45). The core promoters of lincRNAs showed the most conserved profiles. RM LincRNAs showed significantly greater conservation than CM lincRNAs at core promoters (Figure 3C).
Figure 3.
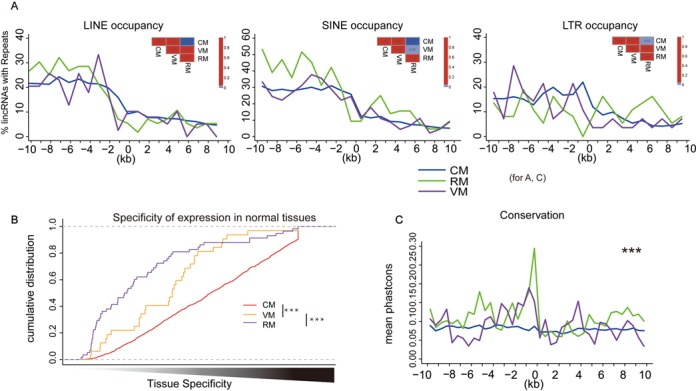
RM lincRNAs had conserved promoters. (A) RM lincRNAs were depleted of REs at promoters. Graphs show frequency of LINEs, SINEs and LTRs at 1-kb intervals around TSSs of CM, VM and RM lincRNAs. Significance of differences in densities was determined by Fisher's exact tests for repeat counts ± 2 kb from the TSSs. (B) RM lincRNAs had the lowest tissue-specific expression in normal tissues. Shown are cumulative distributions of tissue-specificity scores for CM, VM and RM lincRNAs. Differences between lincRNA sets were tested using Wilcoxon rank sum tests (***P < 0.001). (C) RM lincRNAs had evolutionarily conserved promoters. Shown are the graphs of conservation level in 500-bp intervals around the TSSs of CM, VM and RM lincRNAs. Conservation was determined by measuring the rate of base pair substitutions between species. Significance of observed differences between two categories was assessed using the Wilcoxon rank sum test for scores ± 2 kb around the TSSs (***P < 1.0e−3).
We performed functional enrichment analysis of genes co-expressed with the RM, VM and CM lincRNAs (Pearson's correlation test, top 5% of P < 0.05) for BRCA, LUSC and GBM (46,47). RM genes (PCGs co-expressed with RM lincRNAs) in three cancers shared the GO terms ‘regulation of transcription’ and ‘transcription’. CM genes (PCGs co-expressed with CM lincRNAs) were enriched in GO terms ‘immune response’, ‘cell cycle’ and ‘chromatin modification’ among others (Supplementary Figure S7). For VM genes (PCGs co-expressed with VM lincRNAs), there were no significant functional enrichment results. In addition, 49 RM lincRNAs had homologous sequences in mice and 21 had homologs in zebrafish (48). RM lincRNAs had conserved transcriptional regulation regions and conserved sequences in multiple species, which suggested an evolutionary demand for correct regulation and expression of RM lincRNAs.
LincRNAs had promoter methylation patterns associated with cancer status, subtype and prognosis
Since aberrant promoter methylation silences tumor suppressor genes and activates oncogenes (49), we analyzed the different methylation patterns of lincRNA promoters between tumors and corresponding normal tissue samples. For example, 126 lincRNAs showed significantly aberrant methylation patterns in tumors compared to corresponding normal samples, including 24 hypermethylated and 28 hypomethylated lincRNAs for BRCA, and 14 hypermethylated and 60 hypomethylated lincRNAs for LUSC (Figure 4A and B and Supplementary Table S5). Most AM lincRNAs belonged to the CM category, indicating that these lincRNAs are consistently methylated in other types of tumors (Figure 4C and D). The hypomethylated CM lincRNAs in BRCA or LUSC showed a more common methylation pattern in normal samples than in tumors.
Figure 4.
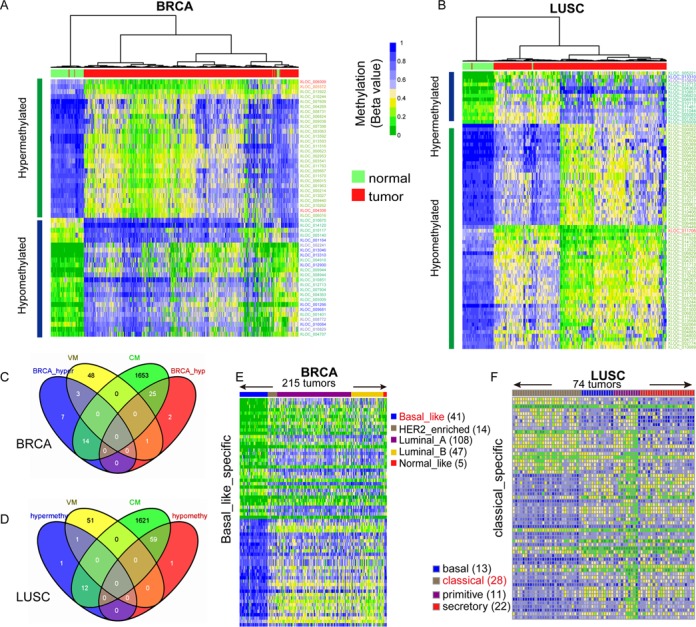
LincRNAs whose methylation patterns were associated with cancer status or subtypes. (A, B) Heat maps of bidirectional hierarchical clustering of lincRNAs with significantly different methylation levels between BRCA and normal breast (A) or LUSC and normal lung (B). (C, D) Venn diagrams showed that most of the AM lincRNAs in BRCA (C) or LUSC (D) belonged to the CM category. (E, F) Heat maps showing the methylation profiles of the top 5% lincRNAs with significantly different methylation levels (FDR < 0.05) in the basal-like subtype compared to the others for BRCA (E) and in the classical subtype compared to the others for LUSC (F). LincRNAs are ranked by ascending order of t-test FDR values.
We compared the methylation patterns of lincRNAs for different subtypes of BRCA (basal-like, HER2-enriched, luminal A, luminal B and normal-like) (20) and LUSC (basal, classical, primitive and secretory) (21). We identified the lincRNAs with subtype-specific methylation patterns in BRCA and LUSC (Figure 4E and F and Supplementary Table S6). Since tumors in each cancer molecular subtype had distinctive biological and clinical behaviors, lincRNAs with subtype-specific methylation patterns might have crucial functions in these subtypes. Several lincRNAs with subtype-specific methylation patterns have been functionally implicated in physiological or pathological processes through experimental validation. For instance, HOTAIR, a lincRNA hypomethylated in basal-like subtype BRCA, was highly expressed in metastatic breast cancers. Its high level of expression in primary breast tumors might predict subsequent metastasis and death (6). MEG3, which was hypomethylated in the luminal A subtype of BRCA, is an imprinted long non-coding RNA (50). MEG3 acted as a growth suppressor in tumor cells and activated p53 (51). In addition, HOTTIP, which binds the WDR5 protein and forms a complex with the histone methyltransferase protein MLL to target the WDR5-MLL complex to the HOXA region for transcriptional activation of HOXA (52), also showed subtype-specific methylation patterns in both BRCA (basal-like) and LUSC (classical).
We combined the lincRNA methylation profiles with clinical annotations and identified a subset of lincRNAs with methylation values showing a trend associated with OS in BRCA, LUSC and UCEC. We used a validation set as an independent data set to validate candidate reliability. For UCEC, we obtained 23 PA lincRNAs in the validation set from 30 lincRNAs in the discovery set (FDR < 0.01). For BRCA, we validated five lincRNAs associated with OS from the top 10 lincRNAs in the discovery set (FDR < 0.01). For LUSC, we obtained seven PA lincRNAs (Supplementary Tables S7–S9). For example, BRCA patients with lower methylation level of lincRNA XLOC_009284 had better prognosis (Figure 5A). LUSC patients with relatively lower methylation level of lincRNA XLOC_009367 showed poorer prognosis (Figure 5B). For UCEC, patients with the highest methylation level of lincRNA XLOC_007617 had a better prognosis than patients with lower methylation level (Figure 5C). GAS5, a lincRNA linked to apoptosis that is involved in progression of some types of cancers, was significantly correlated with prognosis for UCEC (53,54). The lincRNA MEG3 was associated with OS in the LUSC discovery set but not in the validation set, which inhibits proliferation of non-small cell lung cancer cells and induces apoptosis by affecting p53 expression (55). We hope to further validate candidates from the discovery set in the future, using a suitable tumor set. Besides, BRCA1, the ovarian cancer marker PCG, showed no correlation between methylation level and OS in a previous study (14). There were 42 lincRNAs showed a correlation between the methylation level and OS in BRCA, LUSC, UCEC, KIRC and LGG, some of which showed a negative correlation between methylation and expression in corresponding tumors (Pearson's correlation test; Supplementary Table S10), suggesting their potential as novel prognostic biomarkers.
Figure 5.
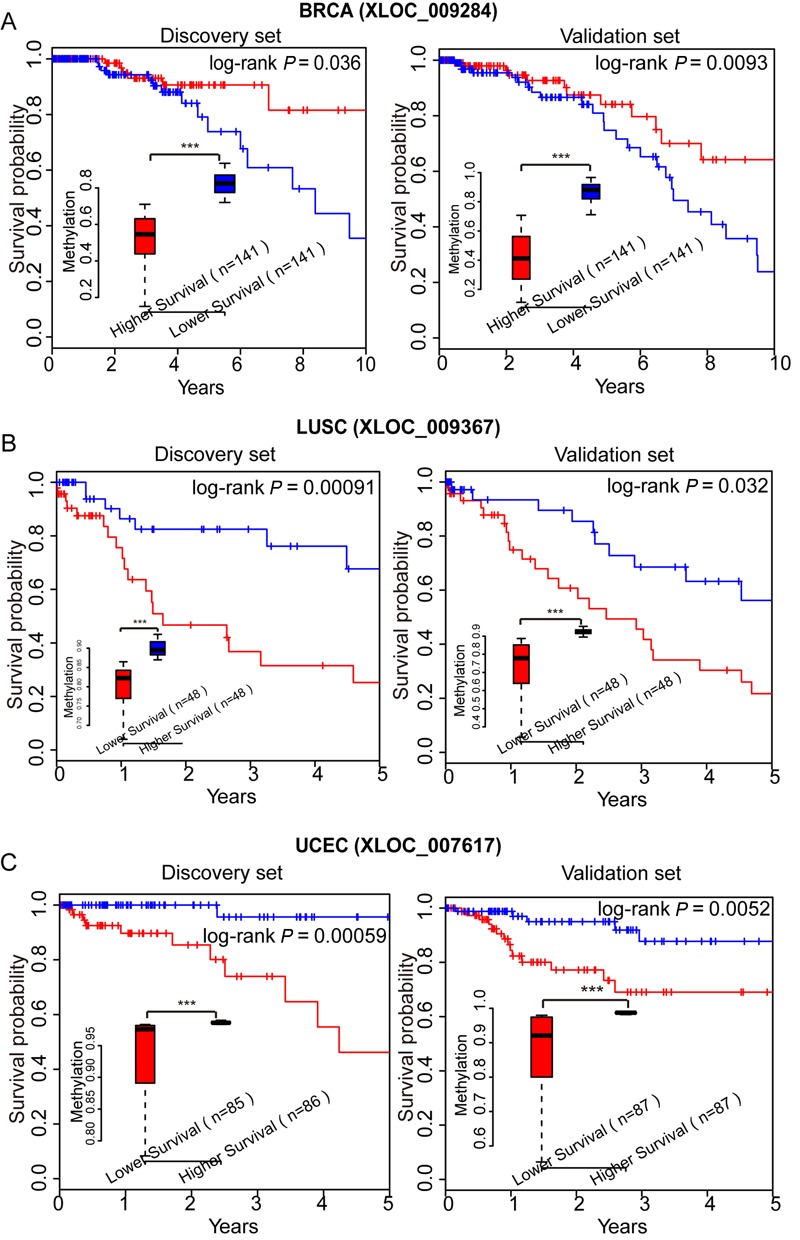
LincRNAs with PA methylation patterns in BRCA, LUSC or UCEC. (A) Kaplan–Meier curves for discovery-set patients (n = 282) with higher (top 50%; n = 141) or lower (bottom 50%; n = 141) methylation of XLOC_009284 in BRCA (left). Kaplan–Meier curves for validation-set patients as above (right). (B) Kaplan–Meier curves for discovery-set patients (n = 96) with higher (top 50%; n = 48) or lower (bottom 50%; n = 48) methylation of XLOC_009367 in LUSC (left). Kaplan–Meier curves for validation-set patients as above (right). (C) Kaplan–Meier curves for discovery-set patients (n = 171) with higher (top 50%; n = 86) or lower (bottom 50%; n = 85) methylation levels of XLOC_007617 in UCEC (left). Kaplan–Meier curves for validation-set patients as above (right). The methylation differences between patients sets were tested using Wilcoxon rank sum tests (***P < 1.0e−3).
We then used drug-free survival analysis to evaluate the possibility of promoter methylation of lincRNAs as drug targets. The drug-free interval was defined as from the end of chemotherapeutic drug treatment to the date of progression or recurrence or last contact (censored) (56). XLOC_007617 was a lincRNA that showed positive correlation between its methylation and drug-free survival in UCEC (log-rank P = 0.013; Supplementary Figure S8). In addition, we defined a methylation survival (MS) score using the 23 previously verified PA lincRNAs for UCEC. For each UCEC tumor, a point was given if the methylation level of a PA lincRNA was higher than the median methylation and was associated with longer OS or vice versa. The MS score of each tumor was assigned as the sum of the points. Patients were designated as sensitive for complete or partial response to platinum chemotherapy in the clinical data from TCGA (57) and as resistant for stable or progressive disease. Patients with higher MS scores were more sensitive to drugs (Supplementary Figure S9). Among patients whose tumors had MS scores higher than the median MS score, 87% were sensitive compared with 61% of patients with tumors with lower MS scores (P = 0.037, χ2 test). These results indicated that MS scores generated using lincRNA methylation levels might be used to predict patient sensitivity to chemotherapeutic drugs. Therefore, lincRNAs, whose promoter methylation patterns were associated with cancer status, subtype and prognosis, should be further studied as potential and novel cancer biomarkers.
Functional analyses of AM lincRNAs in cancers
Using the AM lincRNAs between tumors and corresponding normal samples identified from 14 types of cancers with at least seven normal samples, we constructed an AM lincRNA-cancer network (AMCN; Supplementary Figure S10A). The AMCN had two types of nodes: cancers and AM lincRNAs. Edges existed only between a lincRNA and cancer when the lincRNA was aberrantly methylated in the cancer. The AMCN illustrated that most lincRNAs were aberrantly methylated in a single cancer and a few lincRNAs were aberrantly methylated in multiple cancers (Supplementary Figure S10B). A total of 196 lincRNAs were aberrantly methylated in more than one cancer out of all 434 AM lincRNAs in the AMCN. Thirty one AM lincRNAs showed pairwise appearing in more than three types of cancer (Supplementary Figure S11). The lincRNA XLOC_013592, located in chromosome 20, co-occurred with other AM lincRNAs in six types of cancer. Chromosome 5 contained up to seven lincRNAs that co-occurred with other AM lincRNAs. By removing lincRNA nodes from the AMCN, we obtained a network of cancers (Figure 6A). Some pairs of cancers shared more AM lincRNAs than others. For instance, COAD and READ, two cancers that originate in the intestine, shared 69 AM lincRNAs, with 20 lincRNAs aberrantly methylated uniquely in these two cancers. Additionally, BLCA and UCEC shared 56 AM lincRNAs, indicating that these two cancers might share a common pathogenesis. Clinically, a high metastatic rate from UCEC to BLCA was seen (58). Furthermore, 191 AM lincRNAs showed consistent hypermethylated or hypomethylated status in diverse cancers. Five AM lincRNAs showed altered hypermethylation or hypomethylation status in five pairs of cancers (Figure 6B).
Figure 6.
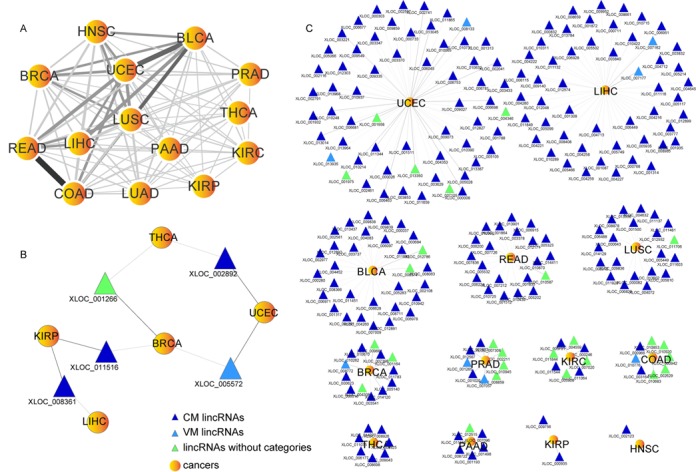
AM lincRNAs in cancers. (A) Network of cancers. The width and shades of color of the edges between two cancer types correlate with the numbers of shared AM lincRNAs. (B) AM lincRNAs with altered methylation status between cancers. (C) UAM LincRNAs. Dark gray, UAM lincRNA hypermethylation in tumors. Light gray, UAM lincRNA hypomethylated in tumors.
Except for lung adeno cancer, 238 lincRNAs aberrantly methylated in only one cancer were named uniquely aberrantly methylated (UAM) lincRNAs (Figure 6C). UCEC contained 60 UAM lincRNAs and liver hepatocellular cancer contained 51, amounting to nearly 47% of the total UAM lincRNAs. Theoretically, a lincRNA could intrinsically cis-regulate its neighbor PCGs by binding to its own locus. Thus, we used the PCGs neighbored to the target lincRNAs to infer the putative functions of the lincRNAs according to ‘guilt by association’ strategy (30,59). In UCEC, XLOC_013045 and XLOC_013050 were found to be adjacent to zinc-finger protein genes (ZNF181, ZNF30, ZNF404, ZNF45). The expression level of XLOC_013350 was negatively correlated with its methylation level (Pearson's correlation coefficient, PCC = −0.63, P < 0.05) and positively correlated with the expression of ZNF404 (PCC = 0.26, P < 0.05). We also observed a positive correlation between the expression of XLOC_013045 and ZNF181 (PCC = 0.18, P < 0.05), indicating that these two lincRNAs may be involved in cell growth and apoptosis. In PRAD, lincRNA XLOC_002726 was significantly hypermethylated in tumors and showed a negative correlation between its expression and methylation (PCC = −0.26, P < 0.05), which was a newly found susceptibility locus for prostate cancer in genome-wide association studies (1,60). CADM2, an upstream gene near XLOC_002726 on the same strand, is a prostate cancer suppressor gene (61). Furthermore, with the 24 AM lincRNAs found for PRAD, we built five classifiers based on Bayes network, naive Bayes, random forest, logistic regression and radial basis function network models to identify patient tumors from normal samples. All five classifiers showed good performance by 10-fold cross-validation (Supplementary Table S11). Therefore, lincRNAs with aberrant methylation patterns in cancers might be involved in cancer development and progression. Early detection of hypermethylation or hypomethylation of lincRNAs might serve as biomarkers for cancer diagnosis or treatment.
DISCUSSION
Epigenetic factors tightly control expression patterns of lincRNAs (62). For example, DNA methylation disrupted a long non-coding RNA activity by affecting expression in a lethal lung developmental disorder (63). To determine the DNA methylation patterns for lincRNAs in human cancers, we developed a strategy to reannotate Infinium 450k array probes to lincRNA loci and constructed lincRNA methylation profiles for tumor patients. We investigated the patterns of lincRNA methylation in different cancer types and the functions of lincRNAs in cancers. By clustering analyses of lincRNA methylation levels in cancer, we revealed that some types of cancer had similar lincRNA methylation patterns and classified lincRNAs according to their methylation patterns in tumors. By integrating cancer subtype data and patients clinical information, we identified lincRNAs whose promoter methylation status was associated with cancer status, subtype and prognosis. By network analyses, we investigated the functions of AM lincRNAs in cancers. By literature mining, we validated that a few AM lincRNAs were efficacious in cancer development and progression. Experimentally validating the potential tumor-promoting functions of these candidate lincRNAs in cancers would be meaningful. LincRNAs whose promoter methylation status was associated with cancer status, subtype and prognosis could be investigated as disease signatures.
Two lincRNA catalogs were generated by Cabili et al.: a predicted catalog and a stringent catalog (1). During the reannotating process, we only considered the stringent lincRNA catalog with nine additional known lincRNAs, for these lincRNA transcripts might be more reliable (1). However, there were still some lincRNAs closed to the annotated PCGs. The probes used to evaluate PCGs might be mapped to nearby lincRNA-related regions. To avoid using these probes, we reannotated a subcatalog of 2167 lincRNAs that were more than 20 kb from the PCGs. Since we retained only probes annotated 10 kb upstream or downstream from lincRNAs or PCGs, probes annotated to the subcatalog of lincRNAs could not be related to PCGs. Methylation of the subcatalog of lincRNAs within each of the 10 expression quantiles showed that the lower the promoter methylation, the higher the expression of the corresponding lincRNAs. Methylation of the other three regions was not related to the expression (Supplementary Figure S12). Unsupervised hierarchical clustering of the average promoter methylation profiles of these lincRNAs showed CM, VM or RM patterns in 20 types of cancer (Supplementary Figure S13). Although the average promoter methylation levels of PRAD were higher than the corresponding normal samples, the trends of the other sample sets were consistent with previous analyses of the other 19 cancers (Supplementary Figure S14). Therefore, the methylation patterns of lincRNAs were maintained based on a small stringent set of lincRNAs.
The relationship between exonic and intronic methylation of lincRNAs in the H1-hESC cell line and their expression levels (Figure 1C) were not what we expect from PCGs, in which highly expressed genes have been shown to be more methylated in their gene bodies than genes expressed at low levels (64,65). However, the relationship between expression levels and gene-body methylation in PCGs has been shown to be complex in recent studies. For example, some tissue types showed a correlation between expression and gene-body methylation, whereas others showed no clear relationship (66,67). A relation between gene-body methylation and evolution has been suggested. For example, genes expressed at moderate levels had the highest methylation levels in some plants and invertebrates (68,69). Furthermore, the initiation and elongation of transcription showed different sensitivity to DNA methylation silencing in different genomic and cellular contexts (70). Although lincRNAs and PCGs both undergo the transcription process, lincRNAs have a much lower expression level and sequence conservation than PCGs (1), which could result in complex methylation patterns similar to those of PCGs. Therefore, the methylation patterns of lincRNA exons and introns in different species, cell types and phenotypes need to be further investigated.
The observed lower tissue-specific expression patterns and the higher promoter conservation of RM lincRNAs are consistent with the high conservation score of ubiquitously expressed lincRNAs (26). However, this pattern differed from that of PCGs, where PM promoters were more conserved and more depleted of REs (28), indicating that REs may play a different role in reprogramming the DNA methylation of lincRNA promoters. A recent study found that repetitive and transposable elements occurred in more than two-thirds of mature long non-coding RNA transcripts, particularly at their TSSs, whereas they seldom occurred in protein-coding transcripts (71), suggesting that they may play a role in the regulation of long non-coding RNA transcription (72). Since REs tend to be aberrantly methylated in human cancers (73,74), they might play a specific role in altering lincRNA promoter methylation levels and further affect lincRNA transcription in cancer. The depletion of REs observed at RM lincRNAs may reflect a need to preserve their stable methylation patterns in cancer.
In summary, we studied the functions and mechanisms of DNA methylation of lincRNAs in human cancers by reannotating publicly available data and integrating them with genomic analyses. The identified cancer-associated or clinically relevant lincRNAs could be further evaluated for use as cancer biomarkers and potential therapeutic targets.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online, including [1–4].
Acknowledgments
The authors gratefully thank the TCGA Research Network for providing data for this work.
Footnotes
The authors wish it to be known that, in their opinion, the first three authors should be regarded as Joint First Authors.
FUNDING
Funds for Creative Research Groups of the National Natural Science Foundation of China [81121003]; National Program on Key Basic Research Project [2014CB910504]; National Natural Science Foundation of China [91129710, 61073136 and 61170154]; Specialized Research Fund for the Doctoral Program of Higher Education of China [20102307110022].
Conflict of interest statement. None declared.
REFERENCES
- 1.Cabili M.N., Trapnell C., Goff L., Koziol M., Tazon-Vega B., Regev A., Rinn J.L. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011;25:1915–1927. doi: 10.1101/gad.17446611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guttman M., Garber M., Levin J.Z., Donaghey J., Robinson J., Adiconis X., Fan L., Koziol M.J., Gnirke A., Nusbaum C., et al. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat. Biotechnol. 2010;28:503–510. doi: 10.1038/nbt.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prensner J.R., Chinnaiyan A.M. The emergence of lncRNAs in cancer biology. Cancer Discov. 2011;1:391–407. doi: 10.1158/2159-8290.CD-11-0209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tsai M.C., Spitale R.C., Chang H.Y. Long intergenic noncoding RNAs: new links in cancer progression. Cancer Res. 2011;71:3–7. doi: 10.1158/0008-5472.CAN-10-2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yoon J.H., Abdelmohsen K., Srikantan S., Yang X., Martindale J.L., De S., Huarte M., Zhan M., Becker K.G., Gorospe M. LincRNA-p21 suppresses target mRNA translation. Mol. Cell. 2012;47:648–655. doi: 10.1016/j.molcel.2012.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gupta R.A., Shah N., Wang K.C., Kim J., Horlings H.M., Wong D.J., Tsai M.C., Hung T., Argani P., Rinn J.L., et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 2010;464:1071–1076. doi: 10.1038/nature08975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee G.L., Dobi A., Srivastava S. Prostate cancer: diagnostic performance of the PCA3 urine test. Nat. Rev. Urol. 2011;8:123–124. doi: 10.1038/nrurol.2011.10. [DOI] [PubMed] [Google Scholar]
- 8.Jones P.A. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012;13 doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 9.Wu W., Bhagat T.D., Yang X., Song J.H., Cheng Y., Agarwal R., Abraham J.M., Ibrahim S., Bartenstein M., Hussain Z., et al. Hypomethylation of noncoding DNA regions and overexpression of the long noncoding RNA, AFAP1-AS1, in Barrett's esophagus and esophageal adenocarcinoma. Gastroenterology. 2013;144:956–966. doi: 10.1053/j.gastro.2013.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Braconi C., Kogure T., Valeri N., Huang N., Nuovo G., Costinean S., Negrini M., Miotto E., Croce C.M., Patel T. microRNA-29 can regulate expression of the long non-coding RNA gene MEG3 in hepatocellular cancer. Oncogene. 2011;30:4750–4756. doi: 10.1038/onc.2011.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lujambio A., Portela A., Liz J., Melo S.A., Rossi S., Spizzo R., Croce C.M., Calin G.A., Esteller M. CpG island hypermethylation-associated silencing of non-coding RNAs transcribed from ultraconserved regions in human cancer. Oncogene. 2010;29:6390–6401. doi: 10.1038/onc.2010.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laird P.W. Principles and challenges of genomewide DNA methylation analysis. Nat. Rev. Genet. 2010;11:191–203. doi: 10.1038/nrg2732. [DOI] [PubMed] [Google Scholar]
- 13.Sandoval J., Heyn H., Moran S., Serra-Musach J., Pujana M.A., Bibikova M., Esteller M. Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome. Epigenetics. 2011;6:692–702. doi: 10.4161/epi.6.6.16196. [DOI] [PubMed] [Google Scholar]
- 14.The Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Du Z., Fei T., Verhaak R.G., Su Z., Zhang Y., Brown M., Chen Y., Liu X.S. Integrative genomic analyses reveal clinically relevant long noncoding RNAs in human cancer. Nat. Struct. Mol. Biol. 2013;20:908–913. doi: 10.1038/nsmb.2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo X., Gao L., Liao Q., Xiao H., Ma X., Yang X., Luo H., Zhao G., Bu D., Jiao F., et al. Long non-coding RNAs function annotation: a global prediction method based on bi-colored networks. Nucleic Acids Res. 2013;41:e35. doi: 10.1093/nar/gks967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bu D., Yu K., Sun S., Xie C., Skogerbo G., Miao R., Xiao H., Liao Q., Luo H., Zhao G., et al. NONCODE v3.0: integrative annotation of long noncoding RNAs. Nucleic Acids Res. 2012;40:D210–D215. doi: 10.1093/nar/gkr1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.The Cancer Genome Atlas Research Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.The Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–525. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bernstein B.E., Birney E., Dunham I., Green E.D., Gunter C., Snyder M. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karolchik D., Barber G.P., Casper J., Clawson H., Cline M.S., Diekhans M., Dreszer T.R., Fujita P.A., Guruvadoo L., Haeussler M., et al. The UCSC Genome Browser database: 2014 update. Nucleic Acids Res. 2014;42:D764–D770. doi: 10.1093/nar/gkt1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kent W.J. BLAT–the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shin H., Liu T., Manrai A.K., Liu X.S. CEAS: cis-regulatory element annotation system. Bioinformatics. 2009;25:2605–2606. doi: 10.1093/bioinformatics/btp479. [DOI] [PubMed] [Google Scholar]
- 26.Popadin K., Gutierrez-Arcelus M., Dermitzakis E.T., Antonarakis S.E. Genetic and epigenetic regulation of human lincRNA gene expression. Am. J. Hum. Genet. 2013;93:1015–1026. doi: 10.1016/j.ajhg.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sproul D., Nestor C., Culley J., Dickson J.H., Dixon J.M., Harrison D.J., Meehan R.R., Sims A.H., Ramsahoye B.H. Transcriptionally repressed genes become aberrantly methylated and distinguish tumors of different lineages in breast cancer. Proc. Natl. Acad. Sci. U.S.A. 2011;108:4364–4369. doi: 10.1073/pnas.1013224108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sproul D., Kitchen R.R., Nestor C.E., Dixon J.M., Sims A.H., Harrison D.J., Ramsahoye B.H., Meehan R.R. Tissue of origin determines cancer-associated CpG island promoter hypermethylation patterns. Genome Biol. 2012;13:R84–R99. doi: 10.1186/gb-2012-13-10-r84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karolchik D., Hinrichs A.S., Furey T.S., Roskin K.M., Sugnet C.W., Haussler D., Kent W.J. The UCSC Table Browser dataretrieval tool. NucleicAcids Res. 2012;32:D493–496. doi: 10.1093/nar/gkh103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guttman M., Rinn J.L. Modular regulatory principles of large non-coding RNAs. Nature. 2012;482:339–346. doi: 10.1038/nature10887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wagner G.P., Kin K., Lynch V.J. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci. 2012;131:281–285. doi: 10.1007/s12064-012-0162-3. [DOI] [PubMed] [Google Scholar]
- 32.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ranstam J., Karrholm J., Pulkkinen P., Makela K., Espehaug B., Pedersen A.B., Mehnert F., Furnes O. Statistical analysis of arthroplasty data. II. Guidelines. Acta Orthop. 2011;82:258–267. doi: 10.3109/17453674.2011.588863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sherman B.T., Huang da W., Tan Q., Guo Y., Bour S., Liu D., Stephens R., Baseler M.W., Lane H.C., Lempicki R.A. DAVID Knowledgebase: a gene-centered database integrating heterogeneous gene annotation resources to facilitate high-throughput gene functional analysis. BMC Bioinformatics. 2007;8:426–436. doi: 10.1186/1471-2105-8-426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Herman J.G., Baylin S.B. Gene silencing in cancer in association with promoter hypermethylation. N. Engl. J. Med. 2003;349:2042–2054. doi: 10.1056/NEJMra023075. [DOI] [PubMed] [Google Scholar]
- 36.Feinberg A.P., Tycko B. The history of cancer epigenetics. Nat. Rev. Cancer. 2004;4:143–153. doi: 10.1038/nrc1279. [DOI] [PubMed] [Google Scholar]
- 37.Egger G., Liang G., Aparicio A., Jones P.A. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004;429:457–463. doi: 10.1038/nature02625. [DOI] [PubMed] [Google Scholar]
- 38.Robertson K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005;6:597–610. doi: 10.1038/nrg1655. [DOI] [PubMed] [Google Scholar]
- 39.Baylin S.B. DNA methylation and gene silencing in cancer. Nat. Clin. Pract. Oncol. 2005;2(Suppl. 1):S4–S11. doi: 10.1038/ncponc0354. [DOI] [PubMed] [Google Scholar]
- 40.Huarte M., Guttman M., Feldser D., Garber M., Koziol M.J., Kenzelmann-Broz D., Khalil A.M., Zuk O., Amit I., Rabani M., et al. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell. 2010;142:409–419. doi: 10.1016/j.cell.2010.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Su J., Shao X., Liu H., Liu S., Wu Q., Zhang Y. Genome-wide dynamic changes of DNA methylation of repetitive elements in human embryonic stem cells and fetal fibroblasts. Genomics. 2012;99:10–17. doi: 10.1016/j.ygeno.2011.10.004. [DOI] [PubMed] [Google Scholar]
- 42.Estecio M.R., Gallegos J., Vallot C., Castoro R.J., Chung W., Maegawa S., Oki Y., Kondo Y., Jelinek J., Shen L., et al. Genome architecture marked by retrotransposons modulates predisposition to DNA methylation in cancer. Genome Res. 2010;20:1369–1382. doi: 10.1101/gr.107318.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meyer L.R., Zweig A.S., Hinrichs A.S., Karolchik D., Kuhn R.M., Wong M., Sloan C.A., Rosenbloom K.R., Roe G., Rhead B., et al. The UCSC Genome Browser database: extensions and updates 2013. Nucleic Acids Res. 2013;41:D64–D69. doi: 10.1093/nar/gks1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Martinez O., Reyes-Valdes M.H. Defining diversity, specialization, and gene specificity in transcriptomes through information theory. Proc. Natl. Acad. Sci. U.S.A. 2008;105:9709–9714. doi: 10.1073/pnas.0803479105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Siepel A., Bejerano G., Pedersen J.S., Hinrichs A.S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L.W., Richards S., et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liao Q., Liu C., Yuan X., Kang S., Miao R., Xiao H., Zhao G., Luo H., Bu D., Zhao H., et al. Large-scale prediction of long non-coding RNA functions in a coding-non-coding gene co-expression network. Nucleic Acids Res. 2011;39:3864–3878. doi: 10.1093/nar/gkq1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liao Q., Xiao H., Bu D., Xie C., Miao R., Luo H., Zhao G., Yu K., Zhao H., Skogerbo G., et al. ncFANs: a web server for functional annotation of long non-coding RNAs. Nucleic Acids Res. 2011;39:W118–W124. doi: 10.1093/nar/gkr432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Volders P.J., Helsens K., Wang X., Menten B., Martens L., Gevaert K., Vandesompele J., Mestdagh P. LNCipedia: a database for annotated human lncRNA transcript sequences and structures. Nucleic Acids Res. 2013;41:D246–D251. doi: 10.1093/nar/gks915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Esteller M. CpG island hypermethylation and tumor suppressor genes: a booming present, a brighter future. Oncogene. 2002;21:5427–5440. doi: 10.1038/sj.onc.1205600. [DOI] [PubMed] [Google Scholar]
- 50.Zhou Y., Zhang X., Klibanski A. MEG3 noncoding RNA: a tumor suppressor. J. Mol. Endocrinol. 2012;48:R45–R53. doi: 10.1530/JME-12-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhou Y., Zhong Y., Wang Y., Zhang X., Batista D.L., Gejman R., Ansell P.J., Zhao J., Weng C., Klibanski A. Activation of p53 by MEG3 non-coding RNA. J. Biol. Chem. 2007;282:24731–24742. doi: 10.1074/jbc.M702029200. [DOI] [PubMed] [Google Scholar]
- 52.Wang K.C., Yang Y.W., Liu B., Sanyal A., Corces-Zimmerman R., Chen Y., Lajoie B.R., Protacio A., Flynn R.A., Gupta R.A., et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature. 2011;472:120–124. doi: 10.1038/nature09819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pickard M.R., Mourtada-Maarabouni M., Williams G.T. Long non-coding RNA GAS5 regulates apoptosis in prostate cancer cell lines. Biochim. Biophys. Acta. 2013;1832:1613–1623. doi: 10.1016/j.bbadis.2013.05.005. [DOI] [PubMed] [Google Scholar]
- 54.Qiao H.P., Gao W.S., Huo J.X., Yang Z.S. Long non-coding RNA GAS5 functions as a tumor suppressor in renal cell carcinoma. Asian Pac. J. Cancer Prev. 2013;14:1077–1082. doi: 10.7314/apjcp.2013.14.2.1077. [DOI] [PubMed] [Google Scholar]
- 55.Lu K.H., Li W., Liu X.H., Sun M., Zhang M.L., Wu W.Q., Xie W.P., Hou Y.Y. Long non-coding RNA MEG3 inhibits NSCLC cells proliferation and induces apoptosis by affecting p53 expression. BMC Cancer. 2013;13:461–471. doi: 10.1186/1471-2407-13-461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang D., Khan S., Sun Y., Hess K., Shmulevich I., Sood A.K., Zhang W. Association of BRCA1 and BRCA2 mutations with survival, chemotherapy sensitivity, and gene mutator phenotype in patients with ovarian cancer. JAMA. 2011;306:1557–1565. doi: 10.1001/jama.2011.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kandoth C., Schultz N., Cherniack A.D., Akbani R., Liu Y., Shen H., Robertson A.G., Pashtan I., Shen R., Benz C.C., et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67–73. doi: 10.1038/nature12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ali-El-Dein B., Mosbah A., Osman Y., El-Tabey N., Abdel-Latif M., Eraky I., Shaaban A.A. Preservation of the internal genital organs during radical cystectomy in selected women with bladder cancer: a report on 15 cases with long term follow-up. Eur. J. Surg. Oncol. 2013;39:358–364. doi: 10.1016/j.ejso.2013.02.004. [DOI] [PubMed] [Google Scholar]
- 59.Lee J.T. Lessons from X-chromosome inactivation: long ncRNA as guides and tethers to the epigenome. Genes Dev. 2009;23:1831–1842. doi: 10.1101/gad.1811209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Takata R., Akamatsu S., Kubo M., Takahashi A., Hosono N., Kawaguchi T., Tsunoda T., Inazawa J., Kamatani N., Ogawa O., et al. Genome-wide association study identifies five new susceptibility loci for prostate cancer in the Japanese population. Nat. Genet. 2010;42:751–754. doi: 10.1038/ng.635. [DOI] [PubMed] [Google Scholar]
- 61.Chang G., Xu S., Dhir R., Chandran U., O'Keefe D.S., Greenberg N.M., Gingrich J.R. Hypoexpression and epigenetic regulation of candidate tumor suppressor gene CADM-2 in human prostate cancer. Clin. Cancer Res. 2010;16:5390–5401. doi: 10.1158/1078-0432.CCR-10-1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Guttman M., Amit I., Garber M., French C., Lin M.F., Feldser D., Huarte M., Zuk O., Carey B.W., Cassady J.P., et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458:223–227. doi: 10.1038/nature07672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Szafranski P., Dharmadhikari A.V., Brosens E., Gurha P., Kolodziejska K.E., Zhishuo O., Dittwald P., Majewski T., Mohan K.N., Chen B., et al. Small noncoding differentially methylated copy-number variants, including lncRNA genes, cause a lethal lung developmental disorder. Genome Res. 2013;23:23–33. doi: 10.1101/gr.141887.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ball M.P., Li J.B., Gao Y., Lee J.H., LeProust E.M., Park I.H., Xie B., Daley G.Q., Church G.M. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat. Biotechnol. 2009;27:361–368. doi: 10.1038/nbt.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hellman A., Chess A. Gene body-specific methylation on the active X chromosome. Science. 2007;315:1141–1143. doi: 10.1126/science.1136352. [DOI] [PubMed] [Google Scholar]
- 66.Aran D., Toperoff G., Rosenberg M., Hellman A. Replication timing-related and gene body-specific methylation of active human genes. Hum. Mol. Genet. 2011;20:670–680. doi: 10.1093/hmg/ddq513. [DOI] [PubMed] [Google Scholar]
- 67.Jjingo D., Conley A.B., Yi S.V., Lunyak V.V., Jordan I.K. On the presence and role of human gene-body DNA methylation. Oncotarget. 2012;3:462–474. doi: 10.18632/oncotarget.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zemach A., McDaniel I.E., Silva P., Zilberman D. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science. 2010;328:916–919. doi: 10.1126/science.1186366. [DOI] [PubMed] [Google Scholar]
- 69.Zilberman D., Gehring M., Tran R.K., Ballinger T., Henikoff S. Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat. Genet. 2007;39:61–69. doi: 10.1038/ng1929. [DOI] [PubMed] [Google Scholar]
- 70.Rountree M.R., Selker E.U. DNA methylation inhibits elongation but not initiation of transcription in Neurospora crassa. Genes Dev. 1997;11:2383–2395. doi: 10.1101/gad.11.18.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kapusta A., Kronenberg Z., Lynch V.J., Zhuo X., Ramsay L., Bourque G., Yandell M., Feschotte C. Transposable elements are major contributors to the origin, diversification, and regulation of vertebrate long noncoding RNAs. PLoS Genet. 2013;9:e1003470. doi: 10.1371/journal.pgen.1003470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fatica A., Bozzoni I. Long non-coding RNAs: new players in cell differentiation and development. Nat. Rev. Genet. 2014;15:7–21. doi: 10.1038/nrg3606. [DOI] [PubMed] [Google Scholar]
- 73.Wild L., Flanagan J.M. Genome-wide hypomethylation in cancer may be a passive consequence of transformation. Biochim. Biophys. Acta. 2010;1806:50–57. doi: 10.1016/j.bbcan.2010.03.003. [DOI] [PubMed] [Google Scholar]
- 74.Wu H.C., Delgado-Cruzata L., Flom J.D., Perrin M., Liao Y., Ferris J.S., Santella R.M., Terry M.B. Repetitive element DNA methylation levels in white blood cell DNA from sisters discordant for breast cancer from the New York site of the Breast Cancer Family Registry. Carcinogenesis. 2012;33:1946–1952. doi: 10.1093/carcin/bgs201. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.