

Abstract
This review focuses on exogenous MRI contrast agents that are responsive to enzyme activity. Enzymes can catalyze a change in water access, rotational tumbling time, the proximity of a 19F-labeled ligand, the aggregation state, the proton chemical exchange rate between the agent and water, or the chemical shift of 19F, 31P, 13C or a labile 1H of an agent, which can be used to detect enzyme activity. The variety of agents attests to the creativity in developing enzyme-responsive MRI contrast agents.
Keywords: MRI contrast agent, enzyme activity, relaxation-based MRI, CEST MRI, MR
Introduction
The Problem: Detecting Enzyme Activity
Many outstanding chemistry methods have been developed to detect protein concentrations during in vitro studies or ex vivo analyses.[1] However, detecting the concentration of an enzyme does not necessarily reflect the importance of the enzyme to the pathology or biological process under study. In particular, enzymes can be expressed as inactive zymogens or pro-enzymes, and are subsequently activated only when their catalytic functions are required. Examples include pro-enzymes that are activated by binding cofactors or metal ions, through phosphorylation and dephosphorylation, or by cleaving a chemical bond in the enzyme.[2] We contend that evaluating enzymes based on their activity is a more direct and more accurate assessment of their importance in a pathological state or biological process, relative to grading attendance.
Developing a chemical assay that can detect or measure an enzyme activity is much more difficult than measuring enzyme concentration. As a major challenge, many enzymes are notoriously promiscuous and catalyze substrates of other enzymes.[3] Designing a chemical assay to detect the activity of one specific enzyme can prove challenging. Enzyme activities are inherently based on chemical kinetics, and therefore require a temporal assessment, which adds to the difficulty of the assay.[4] Finally, many enzymes are sensitive to their environmental conditions, including redox state, pH, and temperature in addition to concentrations of cofactors or metal ions. Assessing enzyme activity in an in vivo context is the ideal approach for ensuring that relevant environmental conditions are met.
Developing a chemical assay to study in vivo enzyme activity poses additional challenges. Perhaps most importantly, the pharmacokinetics of delivering chemical agents to the in vivo site of the enzyme is inherently based on stepwise transport kinetics, which is conceptually identical to stepwise chemical kinetics.[5] Accounting for multiple steps of dynamic kinetic processes can be overwhelming, and often requires additional measurements or assumptions to simplify the analysis. In addition, other in vivo conditions besides enzyme activity can influence a chemical assay, so that care must be taken to ensure that the assay is specifically reporting on enzyme activity. The chemical agent may directly or indirectly influence the enzyme by affecting biological homeostasis, especially if the agent is a substrate that is administered at concentrations that approach saturating conditions (which is typical for Michaelis-Menten enzyme kinetics studies).[6]
A Potential Solution Magnetic Resonance Imaging
Despite these challenges, non-invasive imaging methods using exogenous chemical contrast agents have been developed to assess in vivo enzyme activities. A large number of fluorescent dyes have been developed to detect enzyme activity during biochemical and in vitro assays.[7] Many of these dyes that are excited and/or emit in the red or near-infrared wavelength ranges have also been used for in vivo imaging, because these wavelength ranges have low absorbance in tissues. Bioluminescence has also been used to image in vivo enzyme activity using chemiluminescent substrates[8.9] and as reporter genes.[10,11] PET and SPECT imaging has also been used to detect in vivo enzyme activity by monitoring changes in pharmacokinetics of radiolabeled substrates. For example, 18F-fluorodeoxyglucose is trapped in cells by hexokinase, and therefore pharmacokinetic accumulation of the 18F radioisotope is a marker for hexokinase activity.[12] 18F-FLT (3’-deoxy-3’-fluorothymidine), can detect the activity of thymidine kinase 1,[13] whereas fluoromisonidazole (FMISO) gets reduced by nitroreductases in the cell and are used to image tumor hypoxia.[14] In vivo enzyme activity has not yet been detected with ultrasound imaging, although monitoring the proteolytic degradation of colloids and hydrogels with ultrasound imaging may possibly be translated to in vivo studies.[15] Similarly, detection of in vivo enzyme activity with electron paramagnetic resonance imaging has not yet been realized, although EPR spin probes that detect enzymes in vitro may eventually be translated to in vivo studies.[16] Each of these imaging modalities has advantages and disadvantages for in vivo studies, including in vivo assessments of enzyme activities.
Magnetic resonance imaging (MRI) with exogenous contrast agents also has advantages and disadvantages for in vivo imaging studies. Fortunately, the disadvantages of MRI are not as deleterious for enzyme detection relative to MRI studies of other biomarkers. In particular, MRI has low detection sensitivity relative to other imaging modalities, with minimum detection levels of ~1 μM to ~100 mM (depending on the MRI contrast agent),[17] which is greater than the ~10 pM to ~10 nM concentrations of an enzyme within in vivo tissues. Yet if an enzyme has high turn-over for a substrate that acts as a MRI contrast agent, then low concentration of enzyme can process a high concentration of agent that is above the detection sensitivity threshold. In effect, the MRI detection sensitivity is not ‘target-limited’ (which is the case for agents that directly bind an enzyme target) and instead the sensitivity is ‘agent-limited’. This agent-limited approach uses an agent that irreversibly changes through enzyme catalysis, such as an irreversible bond cleavage or bond formation. For this reason, few enzyme-responsive MRI contrast agents are designed to be reversible and return to their initial state after interaction with the enzyme, which is more common for agents that target other proteins, nucleic acids, metabolites, and metal ions, or measure pH, redox state, and temperature.[18]
The advantages of MRI are an excellent match for the requirements for detecting in vivo enzyme activity. MRI can obtain 3D images without concern for tissue depth and with good spatial resolution of 0.1-1 mm (depending on the size of the animal model or patient).[19] MRI can also acquire images with good temporal resolution of ~5-30 seconds (depending on the contrast mechanism and the MRI scanner hardware). These advantages can be used to dynamically track the specific location of an exogenous contrast agent, which improves the analyses of pharmacokinetics and enzyme kinetics.[20] As another advantage, these agents can change MR image contrast through a variety of mechanisms (as described below), which provides for creative chemistry approaches that may lead to improved detection specificity for a single enzyme. Some contrast mechanisms can selectively detect multiple contrast agents during the same scan session, and contrast mechanisms may possibly be combined to detect multiple agents. This advantage of multiplexing provides opportunity to simultaneously track a second “control” contrast agent that is unresponsive to enzyme activity, but which is sensitive to other environmental conditions that also affect an enzyme-responsive agent. The comparison of the enzyme-responsive and control agents can more specifically detect the enzyme activity relative to other environmental conditions.
This review describes each of the contrast mechanisms used by MRI contrast agents, and lists examples of enzyme-responsive agents that employ each type of contrast mechanism to detect enzyme activity. An emphasis is placed on approaches that constitute a platform technology that can be easily modified to detect diverse kinds of enzymes, such as approaches that can simply substitute a ligand that is an enzyme substrate without requiring redesign of the agent or its chemical synthesis. A common theme of these approaches is the need to use a multidisciplinary approach that combines chemistry and biochemistry, molecular and cell biology, physiology, radiology, and biomedical engineering.
T1 MRI contrast agents that detect enzyme activity
When net magnetization of water is perturbed from it’s preferred direction along a static magnetic field, the water’s magnetization “relaxes to equilibrium” and returns to this preferred direction through a first-order rate process known as T1 relaxation.[21] To relax to equilibrium, the water transfers the excited energy of its magnetic spins to the surrounding lattice through dipole-dipole interactions. The T1 relaxation time is quantified by measuring the exponential time constant of this relaxation process of the water, typically reported in units of seconds.
The interaction between water molecules and Gd(III) lanthanide metal ion is very efficient, creating a shorter T1 relaxation time fort he water when Gd(III) is present. These interactions between water molecules and Gd(III) are influenced by the rotational tumbling time of the Gd(III) complex, and the distance between the water proton and Gd(III). The distance can be altered by blocking water access to the Gd(III) ion, or by changing the rate at which the water molecule leaves a proximal position to the Gd(III) ion. The Gd(III) ion is typically chelated by an organic molecule, which reduces toxicity and provides opportunities to conjugate enzyme substrates to the chelate to develop enzyme-responsive MRI contrast agents.
The seminal example of an enzyme-responsive T1 MRI contrast agent consists of a Gd(III) chelate that includes a β-galactose ligand which blocks water access to the Gd(III) ion (Fig. 1).[22] Cleavage of this ligand by β-galactosidase improves water accessibility to Gd(III) which decreases the T1 relaxation time of the water. Only 4.3 μM of the enzyme was needed to cleave 0.5 mM of agent for sufficient detection with T1-weighted MRI, which demonstrates that detecting enzyme activity is more dependent on the concentration of the responsive agent than the concentration of the enzyme. The same approach was used to detect glutamic acid decarboxylase activity, which removed carboxylate ligands of the agent that blocked the access of water to the Gd(III) ion, resulting in a decrease in T1 relaxation time.[23] Serum anions can also block water access to the Gd(III) of a chelate. Esterase activity can cleave ethyl ester ligands of a Gd(III) chelate, and the post-cleaved ligand can displace the serum anions, which improves water access to the Gd(III) ion and decreases the T1 relaxation time.[24] This last example demonstrates that care should be taken to assess enzyme-responsive MRI contrast agents in the presence of endogenous metabolites, which may modulate expected behaviors of MRI contrast agents.
Figure 1.

Detecting enzyme activities through changes in T1 relaxation caused by alterations in water accessibility. Top panel: The cleavage of a galactopyranosyl ring of the T1 contrast agent, EgadMe, causes the inner sphere coordination site of the Gd3+ ion to become more accessible to water. Middle panel: A space-filling molecular model illustrates the increased accessibility of the Gd3+ ion (magenta) upon cleavage. Bottom Panel: Two living X. laevis embryos were injected with EgadMe, and the embryo shown on the right was also injected with β-galactosidase mRNA. The pseudocolor rendering of MR images shows that the signal strength is 45–65% greater in the embryo containing β-galactosidase mRNA, demonstrating the detection of β-galactosidase activity. Labeled anatomy: e, eye; c, cement gland; s, somite; b, brachial arches. Reproduced with permission from (22).
Well-established methods can measure the number of coordinated water molecules in a Gd(III) complex.[25] Other methods can measure the chemical exchange rates of water molecules that exchange between the Gd(III) complex and bulk water surrounding the agent.[26] These methods can be used to identify which of these two properties is most responsible for the shorter T1 relaxation time caused by the agent, and the change in T1 relaxation time after enzyme catalysis of the agent.
An understanding of these properties can be used to redesign the agent to improve the detection of enzyme activity. Yet some contrast agents rely on both properties, and therefore the distinction between both properties becomes less critical for these agents. For example, a PEG-peptide ligand of a Gd(III) chelate allows the chelate to have excellent solubility.[27] Cleavage of this ligand by MMP-2 causes the chelate to precipitate, which reduces the number of water molecules that can coordinate to the Gd(III) ion and severely reduces the chemical exchange rate of these water molecules with the water solvent, which results in an increase in T1 relaxation time. The same approach, but in the opposite sense, detects a decrease in T1 relaxation time when lipase activity cleaves a Gd(III) chelate’s aliphatic ligands, which allows the remaining Gd(III) chelate to solubilize, coordinate to water molecules and rapidly exchange coordinated water with the surrounding water solvent.[28] Similarly, a Gd(III) chelate in liposomes that are linked with protamine can experience greater water accessibility after trypsin cleaves the protamine linkers.[29] Gd(III)-based contrast agents can be conjugated to a variety of enzyme-sensitive ligands, so that this approach can be used as a platform technology.
The enzyme-catalyzed polymerization of monomeric agents can slow rotational tumbling time of a MRI contrast agent, which can lead to a decrease in T1 relaxation time. This approach has been used to detect the enzyme activities of several peroxidases that polymerize T1 MRI contrast agents, including myeloperoxidase and oxidoreductase (Fig. 2).[30] As another example, esterase activity can cleave a stearic acid ligand of a Gd(III) chelate, which leads to spontaneous polymerization of the cleaved chelate and a decrease in T1 relaxation time.[31] Detecting the combination of two enzyme activities has been accomplished by using β-galactosidase to convert Gd- DOTA-tyr-gal to GdDOTA-tyr, which is then polymerized by tyrosinase, causing the T1 relaxation time to decrease.[32] Because of this decrease in T1 relaxation time, it can be inferred that the polymerization of these agents did not cause precipitation that would reduce water accessibility to the Gd(III) chelate, which would cause an increase in T1 relaxation time. Conversely, enzyme-catalyzed degradation of a polymer containing MRI contrast agents can accelerate the rotational tumbling time of a contrast agent, increasing the T1 relaxation time caused by the agent. For example, a polymer with a glucuronide linker is cleaved by glucuronidase, which releases a Gd(III) chelate from the polymer and increases the T1 relaxation time.[33] The abilities to change the T1 relaxation time by polymerizing and depolymerizing agents doubles the flexibility of this platform technology.
Figure 2.

Detecting enzyme activities through changes in T1 relaxation time caused by alterations in rotational tumbling times. Top Panel: The contrast agent, hydroxytyraminyl-glycylmethylDOTA [D-DOTA(Gd)]; Bottom Panel: Polymerization of the contrast agent by perixodiase slows the rotational tumbling time of the agent, which causes a decrease in the T1 relaxation time of the agent on a per-Gd basis. (C) MR images of the contrast agent at various gadolinium concentrations in the presence of peroxidase (+ Px) or in the absence of peroxidase (− Px) and hydrogen peroxide, demonstrating the detection of peroxidase enzyme activity. Reproduced with permission from (31a).
Enzymes can also catalyze changes in T1 MRI contrast agents that cause the agent to bind to proteins, which slows rotational tumbling time of the agent and decreases the T1 relaxation time. β-galactosidase activity can cleave the galactopyranose ligand of a Gd(III) clelate, resulting in formation of a reactive phenolate anion that binds the contrast agent to a protein.[34] Myeloperoxidase activity can cause a hydroxytryamine ligand of a Gd(III) chelate to form a radical that binds the contrast agent to a protein.[35] Alkaline phosphatase activity can convert a phosphate monoester to an alcohol on a Gd(III) chelate, which allows the agent to non-covalently bind to a protein.[36] The enzyme activity of thrombin-activatable fibrinolysis inhibitor can cleave a lysine-containing ligand of a Gd(III) chelate, which enhances non-covalent binding of the agent to albumin.[37] These latter two examples represent a platform technology, because the masking group that prevents the agent from non-covalently binding to a protein may be conveniently substituted to detect other enzyme activities. A related example is the enzymatic cleavage of per-fluorinated dendrimers attched to silica nanoparticles by phosphate caged flurescien as a linker. The 19F NMR signal intensity increases as the dendrimeric portion is severed allowing for recovery of molecular rotation.[38]
A different approach exploits enzyme-catalyzed changes in the distance between 19F and Gd(III).[39] The close proximity of a 19F nucleus to a Gd(III) ion causes 19F to have a severely short T1 relaxation time, rendering the 19F signal to be undetectable. Enzyme-catalyzed separation of the 19F and Gd(III) increases the T1 relaxation time of the 19F nucleus, allowing the 19F signal to be detected. A 19F ligand has been tethered to a Gd(III) chelate using a peptide that is cleaved by caspase-3 activity,[40] or a lactam linker that is cleaved by β-lactamase.[41] A more complicated mechanism uses a two-step approach to release a 19F ligand from a Gd(III) chelate. β-galactosidase can cleave a galactose moiety of a Gd(III) chelate, which causes aromatic delocalization and subsequent release of a separate 19F-labeled ligand from a Gd(III) chelate.[42] The modular design of this agent may represent a platform technology for enzyme detection, because the galactose substrate may be easily replaced with a substrate for another enzyme.
T2* MRI contrast agents that detect enzyme activity
A superparamagnetic iron oxide nanoparticle (SPION) can create a weak, local magnetic field gradient that changes the net magnetic field experienced by proximal water molecules.[18] This change in magnetic field causes the magnetization of the proximal water molecules to precess at different MR frequencies. The evolution of these different MR frequencies subsequently dephase through a process known as T2* relaxation. This dephasing reduces the total net MRI signal of water in the tissue that can be detected by the MRI scanner. The T2* relaxation time is quantified by measuring the exponential time constant of dephasing and relaxation processes oft he water, known as the T2* relaxation time, which is typically reported in units of seconds or milliseconds. The T2* relaxation time is primarily influenced by the SPIONs’ strength of superparamagnetism that is dependent on the aggregation state of the SPIONs, and the accessibility of water for the SPIONs.
Enzymes that catalyze the aggregation of SPIONs can increase the superparamagnetism of the nanoparticles, and therefore decrease the T2* relaxation time from the contrast agent. This mechanism has been described as an irreversible ‘magnetic relaxation switch’. For example, dextranase, MMP-2 and MMP-9 enzyme activities can cleave the hydrophilic coating of a SPION, causing the nanoparticles to aggregate (Fig. 3).[43] Protein kinase A phosphorylates the surface of ferritin (an endogenous SPION), which causes the ferritin to aggregate.[44] Peroxidase-induced polymerization of phenolic SPIONs can also facilitate the aggregation of the nanoparticles (25).[45] This aggregation can be sufficient to cause precipitation of the agent, which may potentially affect biodistribution and clearance during in vivo studies. β-galactosidase activity can cleave S-Gal (3,4-Cyclohexenoesculetin β-D-galactopyranoside), and the generated aglycone can chelate Fe3+ to form a superparamagnetic MRI contrast agent.[46] Interestingly, the Fe3+ of the S-Gal can shorten the T1 relaxation time before catalysis by β-galactosidase, which provides the opportunity to monitor the delivery of the agent to tissues in addition to monitoring β-galactosidase activity. S-Gal was originally developed as a stain for β-galactosidase activity during histological analyses, and therefore can be used as a multimodality imaging contrast agent. As with polymerizing T1 contrast agents, these many examples of aggregating T2* contrast agents demonstrate that this approach can be a platform technology.
Figure 3.

Detecting enzyme activities via changes in T2 relaxation. Top Panel: A nanoassembly of Gd-based contrast agents that are linked with DEVD peptides can be disaggregated following DEVD peptide cleavage by caspase-3. Bottom Panel: The incubation of the DEVD-linked nanoassembly with caspase-3 showed an increase in T2 relaxation time, caused by disassembly. Incubation of the nanoassembly with the enzyme and a caspase-3 inhibitor further confirmed that this approach detected caspase-3 enzyme activity. Reproduced with permission from (43a).
Enzymes can also catalyze the disaggregation of polymeric SPIONs, which can reduce superparamagnetism and increase the T2* relaxation time from the agent. For example, the protease activity of caspase-3 or MMP-2 can cleave short peptide linkers between SPIONs.[47] Other peptide linkers may be used to detect other proteases with this platform technology. The restriction enzyme EcoRV can cleave DNA duplexes that link SPIONs, which allows the SPIONs to irreversibly disaggregate.[48] A more complicated example detects the activity of secreted alkaline phosphatase, which can dephosphorylate 2’-AMP to create adenosine, which in turn can disrupt a DNA duplex that aggregates SPIONs, thereby separating the SPIONs and reduce their collective superparamagnetism.[49] As an exception to this behavior, a highly aggregated system of SPIONs in a hydrogel becomes less aggregated after trypsin digestion, which increased superparamagnetism and decreased the T2* relaxation time of the SPIONs.[50] Similarly, a highly aggregated bead with hyaluronan linkers becomes less aggregated when the linkers are cleaved by hyaluronidase, leading to decreased T2* relaxation time.[51] These last examples demonstrate that careful optimization of contrast agent formulations can be critical for generating an enzyme-dependent response, because other hydrogels or beads with less initial aggregation may follow the behavior of the other agents in this classification.
CEST MRI contrast agents that detect enzyme activity
Chemical Exchange Saturation Transfer is a relatively new mechanism for generating contrast in MR images.[52] The MR signal from a labile proton of a contrast agent can be selectively saturated, causing this MR signal to rapidly disappear. The labile proton can then exchange with a water proton, which transfers the saturation to the water molecule. This process can be repeated ~10 to ~10,000 times during the saturation period of a CEST MRI protocol, which causes many water molecules to be saturated and reduces the net MR signal from water. A CEST spectrum displays the net MR signal from water as a function of the selective saturation frequency, which can display both the amplitude and chemical shift of the CEST effect. A major advantage of CEST MRI is the ability to detect multiple CEST effects at different chemical shifts from multiple agents, which may be used to simultaneously interrogate multiple enzyme kinetics and/or pharmacokinetic events.
Many enzyme substrates have chemical groups with labile protons, such as amide, amine, and hydroxyl groups, providing many opportunities to monitor enzyme activities via changes in CEST MRI. Diamagnetic CEST (DIACEST) agents are metal-free organic molecules with chemical shifts in a limited range of 1 to 5 ppm (relative to water, which is defined to be 0 ppm for MRI studies).[53] Measuring enzyme-catalyzed changes in exogenous DIACEST agents can be difficult within this limited chemical shift range, especially because endogenous biomolecules also have chemical groups with labile protons that generate CEST within this range. Paramagnetic CEST (PARACEST) agents consist of a metal chelate that has labile protons with a greatly expanded chemical shift range due to their proximity to the metal ion of the chelate.[54] This facilitates the tracking of enzyme-catalyzed changes in PARACEST agents, with reduced interference from CEST generated from endogenous biomolecules. The greater chemical shift range also improves the specificity for saturating the MR frequency of the labile proton on the PARACEST agent without also directly saturating the MR frequency of water.
The seminal example of an enzyme-responsive CEST agent consists of a Tm(III) chelate with a peptidyl ligand (Fig. 4A).[55] Although the C-terminus of a peptide may be routinely coupled to lanthanide chelates, this example required coupling the peptide N-terminus to the chelate through a different synthesis strategy.[56] This ‘reverse’ coupling positioned a labile amide proton near the Tm(III) ion, which generated a unique chemical shift at −51 ppm. This amide was converted to an amine when caspase-3 cleaved this peptide at the amide bond that coupled the peptide to the chelate. This cleavage resulted in a change in the MR frequency of the CEST effect from −51 to +4 ppm, and a decrease in the amplitude of the CEST effect due to a change in the chemical exchange rate. Only 3.4 nM of caspase-3 enyme was required to cleave 20 mM of the agent in a practical time frame for CEST detection, which demonstrates that the limited detection sensitivity of CEST MRI can be overcome by exploiting the fast catalytic activity of an enzyme.
Figure 4.
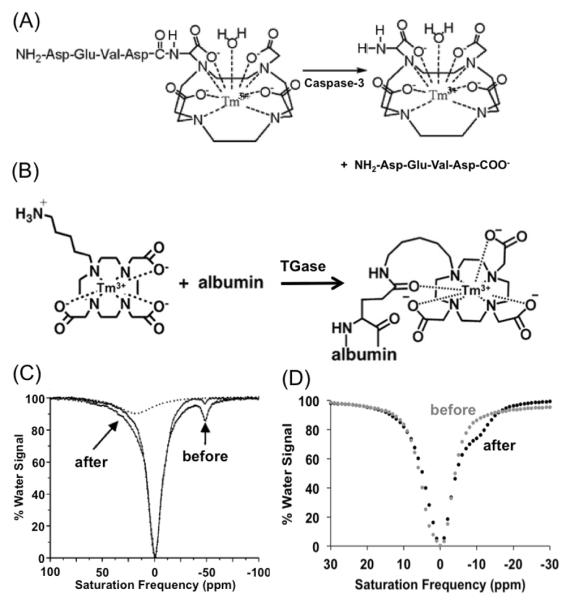
Detecting enzyme activities with CEST MR methods. (A): The cleavage of a peptidyl ligand of a CEST agent, DEVD-amido-(Tm-DOTA), by caspase-3 converts an amide to an amine. (B): The conjugation a CEST agent, Tm-DO3A-cadaverine, to a protein’s glutamine side chain by TGase converts an amine to an amide. (C): The CEST spectra of DEVD-amido-(Tm-DOTA) before (black) and after (gray) incubation with caspase-3 showed a decrease in CEST at μ̵11 ppm and the appearance of CEST at +8 ppm. This appearance of CEST was further confirmed by deconvoluting the CEST spectrum of the product (dashed line), which confirmed the detection of caspase-3 enzyme activity. (D): The CEST spectra of a mixture of albumin, Tm-DO3A-cadaverine and L-glutathione before (black) and after (gray) incubation with TGase showed a decrease in CEST at +4.6 ppm and the appearance of CEST at −9.2 ppm, which demonstrated detection of TGase activity. Reproduced with permission from (55) and (53).
This approach represents a platform technology that can detect many proteases by simply changing the peptide sequence of the chelate’s ligand. We refer to this platform technology as “catalyCEST MRI”. For example, similar PARACEST agents with a different peptide ligand can detect the protease activity of cathepsin-D[57] and urokinase Plasminogen Activator.[58] These examples used a Tm(III) chelate, which demonstrates that a variety of metal chelates may be used in this approach to expand this platform technology. More recently, a DIACEST agent has exploited a similar approach to detect the activity of cytosine deaminase. This enzyme removes an amine group from cytosine and causes a disappearance of CEST from cytosine during in vitro studies.[59] Because cytosine deaminase converts 5-fluorocytosine into the chemotherapeutic 5-fluorouracil, catalyCEST MRI may be able to track enzyme-activated prodrug therapy.
The detection of the enzyme activity shown by these examples uses an enzyme that “turns off” the CEST effect of the agent. This mechanism creates an absence of signal, and depending on the absence of data is a poor scientific design. To compensate for this pitfall, a “control” CEST agent that is unresponsive to enzyme activity can be included in the CEST MRI study to monitor pharmacokinetic delivery of the agents, and account for other physiological conditions that affect both agents. The ratio of the CEST effects from both agents can then be used to more specifically detect enzyme activity. This approach with two PARACEST agents has been demonstrated for the improved detection of uPA activity in chemical solutions and within an in vivo mouse model of pancreatic cancer.[60]
This platform technology has recently been expanded to detect transglutaminase enzyme activity, which is an “anti-protease” that forms an amide bond by coupling the side chain of glutamine to an amine group (Fig. 4B).[61] A PARACEST agent with an aliphatic amine ligand can generate a weak CEST effect, while coupling this agent to glutamine via transglutaminase catalysis creates a stronger CEST effect due to a change in the chemical exchange rate. This change is also accompanied by a small change in chemical shift from −15 ppm to −11 ppm, which can also be used to monitor transglutaminase activity. This detection method uses an enzyme that “turns on” the CEST effect of the agent. Although this method that creates a signa has advantages relative to the other examples that “turn off” the CEST effect, a control agent that is unresponsive to enzyme activity is still useful to include in the CEST MRI study to monitor pharmacokinetic delivery of the agents, and account for other physiological conditions that affect both agents.
As another example of monitoring enzymes that create covalent bonds, the DIACEST peptide (LRRASLG)8 generates lower CEST when Protein Kinase A forms a covalent bond between a serine side chain and a phosphate group, thereby detecting the kinase activity of this enzyme in vitro.[62] A unique 31P CEST study can detect bond formation by creatine kinase activity.[63] Saturation of the the 31P MR signal of γ-ATP phosphate, and subsequent enzyme-catalyzed exchange of the phosphate to creatine to form phosphocreatine, transferred the saturation to the 31P MR signal of the phosphocreatine. This 31P CEST mechanism was used to detect creatine kinase activity during both in vitro and in vivo studies, which was facilitated by the paucity of endogenous 31P MR signals that plague 1H catalyCEST MRI.
CatalyCEST MRI has been further expanded by employing modular designs that separate the substrate ligand that interacts with the enzyme from the portion of the agent that generates CEST. For example, the PARACEST agent Yb-(DOTA-aBz-bGal) can detect β-galactosidase activity through a multistep process: the enzyme cleaves the agent’s galactose ligand, which creates an electron donor group, which causes aromatic delocalization, which cleaves an amide bond to form an amine, which can generate CEST.[64] Similarly, the PARACEST agent Yb-(DO3A-oAA-TML-ester) can detect esterase activity through enzyme-catalyzed de-esterification, which triggers an intramolecular lactonization, which cleaves an amide bond to form an amine, which can generate CEST.[65] This separation of the substrate and CEST-generating chemical group may facilitate the synthesis of catalyCEST agents with new substrate ligands, thereby expanding this platform technology.
MRS contrast agents that detect enzyme activity
The chemical shift of an exogenous contrast agent can be detected with magnetic resonance spectroscopy (MRS). The chemical shift is extremely sensitive to the electronic distribution in a contrast agent, and therefore can be sensitive to changes in covalent bond structures caused by enzyme catalysis. The 1H signal of an exogenous contrast agent is difficult to detect in a crowded spectrum of 1H signals from water, proteins, and metabolites in tissues.[66] For comparison, the 19F and 31P signals from an exogenous agent are easier to detect because endogenous tissues are have no 19F MR signals and only few 31P signals.[67] As an example, fluorinated substrates of β-galactosidase experience a 6-10 ppm change in 19F chemical shift upon cleavage by the enzyme, which can be used to detect β-galactosidase activity in vitro and in vivo (Fig. 5).[68] As another example, vacuolar H+-ATPase activity adds inorganic phosphate to polyphosphate, which can be detected by recording a 24 ppm change in the 31P MR chemical shift.[69]
Figure 5.

Detecting enzyme activities with MR spectroscopy. A series of 19F-NMR spectra were acquired of OFPNPG in PBS (0.1M, pH 7.4, 600mL) before and after addition of β-galactosidase. Rapid hydrolysis of OFPNPG to form OFPNP was monitored by observing a new 19F-NMR signal for the aglycone, which was used to monitor the enzyme activity. The chemical structures of OFPNPG and OFPNP are shown. Reproduced with permission from (68).
The 13C signal of an exogenous contrast agent is also difficult to detect in a crowded spectrum of 13C signals from endogenous biomolecules.[70] Recent advances have developed hyperpolarized 13C contrast agents, which produce as much as 10,000-fold greater 13C signal than endogenous biomolecules, thereby facilitating their detection within tissues in vivo. MRS of several 13C-hyperpolarized agents have been used to detect enzyme activity by monitoring a change in 13C chemical shift. In particular, hyperpolarized [1-13C]pyruvate has been associated with the detection of pyruvate dehydrogenase activity, while [2-13C]pyruvate detects the activity of enzymes in the Krebs cycle.[71] In addition, hyperpolarized pyruvate has also been used to detect the activities of alanine transaminase, carbonic anhydrase, mitogen-activated protein kinase, lactate dehydrogenase, and choline kinase α.[72] Due to the ability to monitor multiple enzyme activities, 13C-hyperpolarized pyruvate should be considered to report on general metabolism rather than the activity of a specific enzyme. Similarly, the 13C-hyperpolarized metabolites lactate, alanine, fumarate, [5-13C]glutamine, 1-keto[1-13C]isocaproate, diethyl[1-13C]succinate, [2-13C]fructose, and 3,5-diflurorbenzoylglutamic acid potentially detect multiple enzyme activities, and therefore should be considered as methods to monitor biochemical pathways rather than specific enzymes.[73]
A pitfall of hyperpolarized 13C contrast agents is the transient nature of their hyperpolarization, typically lasting only a few minutes during in vivo studies. To address this pitfall, permanentily-labeled 13C- and 31P-labeled molecules have also been used to follow metabolism, and 2H-labeled metabolites have been used to determine rate limiting steps of enzyme catalysis. This approach has been used to study formaldehyde metabolism,[74,75] oxidation of methanol,[76] metabolism involving cannizzarase,[74] the citric acid cycle,[77-79] and the metabolism of phosphorylated metabolites.[80]
Future directions
MRI scanners are now available with “ultra-high” magnetic field strengths operating at 7 T for clinical imaging and 15.2 T for small animal imaging.[81] These stronger magnetic fields increase the net polarization of magnetic spins, which improves the net MR signal relative to the image’s noise level, as measured by the Signal-to-Noise Ratio (SNR). This improved SNR can be used to create MR images with finer spatial resolution and/or faster temporal resolution, which may indirectly improve the spatial and temporal detection of enzyme activity. However, a MR image with greater SNR does not directly lead to improved assessments of enzyme activity. Instead, the enzyme-mediated catalysis of a MRI contrast agent generates a difference in MR signal, which is measured as a Contrast-to-Noise Ratio (CNR). This CNR may be measured using the difference in MRI signals between two tissues with and without the enzyme, or the difference in MRI signals between an early time point and a latter time point during the dynamic process of enzyme catalysis. Therefore, future applications of ultra-high magnetic field strengths should exploit potential improvements in CNR rather than only rely on improvements in SNR.
MR contrast mechanisms that depend on chemical shift can benefit from ultra-high magnetic field strengths. For example, a higher magnetic field strength expands the MR chemical shift scale in units of Hz, which can improve the precision of measuring small changes in chemical shifts caused by enzyme catalysis. As another example, for a CEST agent to generate CEST, the chemical exchange rate must be less than the chemical shift in units of Hz. Because chemical shifts in Hz scale with magnetic field strengths, ultra-high magnetic field strengths provide opportunities to develop CEST agents with greater chemical exchange rates. In particular, the chemical exchange rate of amine, hydroxyl, phosphate, and sulfhydryl groups are often greater than their chemical shifts at current magnetic field strengths, but may generate CEST at higher magnetic field strengths. Many enzymes catalyze changes to these chemical groups, so that opportunities to detect changes to these chemical groups via CEST MRI may expand the armamentarium of CEST agents for enzyme detection.
Recent developments in imaging instrumentation and the chemistry of contrast agents have created strong interest in multimodality imaging. Many examples have incorporated an optical or photoacoustic imaging dye in an agent that can be detected by PET, SPECT, MRI, MRS or ultrasound imaging, perhaps because of the mature chemical synthesis methods developed for optical dyes.[82.83] Yet some multimodality imaging agents have been developed for combinations of PET, SPECT, MRI, and ultrasound imaging. developed for combinations of PET, SPECT, MRI, MRS and ultrasound imaging. The development of bionanotechnology has also bolstered this field by providing nanocarriers that package optical dyes and agents for other modalities.[84] These multimodality agents provide opportunities to combine an enzyme-responsive MRI contrast agent with a “control” agent for a different imaging modality that can account for pharmacokinetic delivery and other environmental conditions that may affect the signal from the MRI contrast agent. This comparison of the contrast changes from an enzyme-responsive agent and an unresponsive control agent is critical for improving the specificity of in vivo enzyme detection.
Reporter gene imaging has greatly contributed to our understanding of in vivo genetic expression in a variety of pathologies and biological processes.[85] Many examples of reporter gene imaging rely on the expression of enzymes. Indeed, the popularity of designing many responsive MRI contrast agents for detecting β-galactosidase and β-lactamase is largely driven by the popularity of using these enzymes for reporter gene imaging. Furthermore, the promiscuity of β-galactosidase provides flexibility in designing substrates of MRI contrast agents. Yet all of the enzymes that are detected with MRI contrast agents as presented in this review are capable of being used as reporter genes. Care should be taken that expression of the enzyme does not change the pathology or biological process, and that the MRI contrast agent has good specificity for detecting only the expressed enzyme (e.g., the MMP-2 protease would be a poor choice for reporter gene imaging due to these two conditions). Exploiting enzyme detection to explore gene expression is an outstanding example of how multidisciplinary approaches that combine chemistry and biochemistry, molecular and cell biology, physiology, radiology and biomedical engineering can improve biomedical studies.
Summary and Outlook
To summarize, the many examples of enzyme-responsive MRI contrast agents can be classified as T1 agents that change their water accessibility (7 agents); T1 agents that change their rotational tumbling time (10 agents); T1 agents that change 19F relaxation time (3 agents); T2* agents that change their aggregation state (11 agents); CEST agents that change their chemical shift and chemical exchange rate (8 agents); and MR spectroscopy agents that change their chemical shift (10) agents. Many of these contrast agents are designed to be a platform technology with ligands that can be easily modified to detect new enzymes while retaining the core structure of the agent. The designs and applications of many agents require a multidisciplinary approach, which attests to the creativity and future outlook of this research field.
Acknowledgements ((optional))
This work was supported by the Phoenix Friends of the Arizona Cancer Center, and NIH grants R01 CA169774-01 and P50 CA95060. DVH was sponsored by the TRIF Fellowship Program of the University of Arizona.
Biographies

Dina Hingorani received her B.S. degrees in Chemistry and Physics from the University of Pune, India in 2008. She is currently pursuing a PhD in chemistry at the University of Arizona, Tucson, AZ. Dina’s research interests include organic synthesis, medical imaging and cancer biology.

Dr. Byunghee Yoo earned B.S. degrees in Chemical Technology from Seoul National University, Republic of Korea, in 1994, and his Ph.D. in Biomedical Engineering from Case Western Reserve University, Cleveland, OH, in 2007. Byunghee is now a research fellow in MGH/MIT/HMS Athinoula A. Martinos Center for Biomedical Imaging. His research interests focus on the development of activatable molecular imaging probes and molecular sensors for monitoring gene expression and/or enzyme activity.

Adam Bernstein earned B.S. degrees in Biomedical Engineering and Physiology from the University of Arizona in 2013. Adam is currently pursuing a combined M.D./Ph.D. degree from the University of Arizona College of Medicine and plans to complete the Ph.D. portion of his training in Biomedical Engineering. Adam's research interests focus on the optimization of medical imaging protocols as well as image processing and analysis techniques.

Dr. Mark “Marty” Pagel earned B.A. degrees from Washington University, St. Louis, MO, in 1988, and his Ph.D. from the University of California, Berkeley, CA, in 1993. After managing research projects and facilities at Indiana University and Pharmacia Corp., Dr. Pagel joined Case Western Reserve University, Cleveland, OH, in 2003, and then moved his research program to the University of Arizona, Tucson, AZ, in 2008. Dr. Pagel's interests focus on the development and application of molecular imaging contrast agents that improve cancer diagnosis and evaluations of chemotherapies.
References
- [1].Sittampalam GS. Assay Guidance Manual. Eli Lilly & Company and the National Center for Advancing Translational Sciences; Bethesda MD: 2004. [PubMed] [Google Scholar]
- [2].a Messerschmidt A, editor. Handbook of Metalloproteases. 2011. [Google Scholar]; b Adam JA. Chem. Rev. 2001,;101:2271–2290. doi: 10.1021/cr000230w. [DOI] [PubMed] [Google Scholar]; c Barrett AJ, Rawlings ND, Woessner JF, editors. Handbook of Proteolytic Enzymes. Academic Press; London: 1998. [Google Scholar]
- [3].Gatti-Lafranconi P, Hollfelder F. ChemBioChem. 2013;14:285–292. doi: 10.1002/cbic.201200628. [DOI] [PubMed] [Google Scholar]
- [4].Moffitt JR, Chemla YR, Bustamante C. Methods in Enzymology (Single Molecule Tools, Part B) 2010:221–257. doi: 10.1016/S0076-6879(10)75010-2. [DOI] [PubMed] [Google Scholar]
- [5].Maeng HJ, Chow ECY, Fan J, Pang KS. In: Encyclopedia of Drug Metabolism and Interactions. Lyubimov AV, Rodrigues AD, Sinz MA, editors. 2012. pp. 637–684. [Google Scholar]
- [6].a Michaelis L, Menten ML. Biochemische Zeitschrift. 1913;49:333–369. [Google Scholar]; b Johnson KA, Goody RS. Biochem. 2011;50:8264–8269. doi: 10.1021/bi201284u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a Razgulin A, Ma N, Rao J. Chem. Soc. Rev. 2011;40:4186–4216. doi: 10.1039/c1cs15035a. [DOI] [PubMed] [Google Scholar]; b Drake CR, Miller DC, Jones EF. Curr. Org. Synth. 2011;8:498–520. doi: 10.2174/157017911796117232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Paroo Z, Bollinger RA, Braasch DA, Richer E, Corey DR, Antich PP, Mason RP. Molec. Imaging. 2004;3:117–124. doi: 10.1162/15353500200403172. [DOI] [PubMed] [Google Scholar]
- [9].Contag CH, Jenkins D, Contag FR, Negrin RS. Neoplasia. 2000;2:41–52. doi: 10.1038/sj.neo.7900079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Liu L, Mason RP. PLoS One. 2010;5:e12024. doi: 10.1371/journal.pone.0012024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Gross S, Gammon ST, Moss BL, Rauch D, Harding J, Heinecke JW, Ratner L, Piwnica-Worms D. Nature Med. 2009;15:455–461. doi: 10.1038/nm.1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Smith TAD. Nucl. Med. Comm. 1998;19:97–105. doi: 10.1097/00006231-199802000-00002. [DOI] [PubMed] [Google Scholar]
- [13].Shields AF, Grierson JR, Dohmen B, Machula H-J, Stayanoff JC, Lawhorn-Crews JM, Obradovich-Muzik O, Manger TI. Nature Med. 1998;4:1334–1336. doi: 10.1038/3337. [DOI] [PubMed] [Google Scholar]
- [14].Krohn KA, Link JM, Mason RP. J. Nucl. Med. 2008;49:129S–148S. doi: 10.2967/jnumed.107.045914. [DOI] [PubMed] [Google Scholar]
- [15].a Ahvenainen R, Wirtanen G, Mattila-Sandholm T. Lebensmittel-Wissenschaft und Technologie. 1991;24:397–403. [Google Scholar]; b Shalaby WS, Blevins WE, Park K. Biomat. 1992;13:289–96. doi: 10.1016/0142-9612(92)90052-p. [DOI] [PubMed] [Google Scholar]
- [16].Khramtsov VV, Gorunova TE, Weiner LM. Biochem. Biophys. Res. Comm. 1991;179:520–527. doi: 10.1016/0006-291x(91)91402-x. [DOI] [PubMed] [Google Scholar]
- [17].a Ahrens ET, Rothbacher U, Jacobs RE, Fraser SE. Proc. Natl. Acad. Sci. USA. 1998;95:8443–8448. doi: 10.1073/pnas.95.15.8443. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Mills PH, Ahrens ET. Magn. Reson. Med. 2007;57:442–447. doi: 10.1002/mrm.21145. [DOI] [PubMed] [Google Scholar]
- [18].Yoo B, Pagel MD. Front. Biosci. 2008;13:1733–1752. doi: 10.2741/2796. [DOI] [PubMed] [Google Scholar]
- [19].Haake EM, Brown RW, Thompson MR, Venkatesan R. Magnetic Resonance Imaging: Physical Principles and Sequence Design. John Wiley & Sons; New York: 1999. [Google Scholar]
- [20].Yankeelov TE, Gore JC. Curr. Med. Imag. Rev. 2007;3:91–107. doi: 10.2174/157340507780619179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Merbach A, Toth E. The chemistry of contrast agents in medical magnetic resonance imaging. Wiley; New York: 2001. [Google Scholar]
- [22].Louie A, Huber HM, Ahrens ET, Rothbacher U, Moats R, Jacobs RE, Fraser SE, Meade TJ. Nature Biotech. 2000;18:321–325. doi: 10.1038/73780. [DOI] [PubMed] [Google Scholar]
- [23].Napolitano R, Pariani G, Fedeli F, Baranyai Z, Aswendt M, Aime S, Gianolio E. J. Med. Chem. 2013;56:2466–2477. doi: 10.1021/jm301831f. [DOI] [PubMed] [Google Scholar]
- [24].Giardiello M, Lowe MP, Botta M. Chem. Comm. 2007;44:4044–4046. doi: 10.1039/b711989e. [DOI] [PubMed] [Google Scholar]
- [25].Beeby A, Clarkson IM, Dickins RS, Faulkner S, Parker D, Royle L, de Sousa AS, Williams JAG, Woods M. J. Chem. Soc., Perkin Trans. 1999;2:493–504. [Google Scholar]
- [26].Randtke EA, Chen LQ, Corrales LR, Pagel MD. Magn. Reson. Med. 2013 in press. [Google Scholar]
- [27].a LePage M, Dow WC, Melchior M, You Y, Fingleton B, Quarles CC, Pépin C, Gore JC, Matrisian LM, McIntyre JO. Mol. Imaging. 2007;6:393–403. [PubMed] [Google Scholar]; b Jastrzebska B, Lebel R, Therriault H, McIntyre JO, Escher E, Guerin B, Paquette B, Neugebauer WA, Lepage M. J. Med. Chem. 2009;52:1576–1581. doi: 10.1021/jm801411h. [DOI] [PubMed] [Google Scholar]
- [28].Himmelreich U, Aime S, Hieronymus T, Justicia C, Uggeri F, Zenke M, Hoehn M. NeuroImage. 2006;32:1143–1149. doi: 10.1016/j.neuroimage.2006.05.009. [DOI] [PubMed] [Google Scholar]
- [29].Figueiredo S, Moreira JN, Geraldes CFGC, Aime S, Terreno E. Bioorg. Med. Chem. 2011;19:1131–1135. doi: 10.1016/j.bmc.2010.07.057. [DOI] [PubMed] [Google Scholar]
- [30].a Bogdanov A, Matuszewski L, Bremer C, Petrovski A, Weissleder R. Molecular Imaging. 2002;1:16–23. doi: 10.1162/15353500200200001. [DOI] [PubMed] [Google Scholar]; b Chen J, Pham W, Weissleder R, Bogdanov A. Magn. Reson. Med. 2004;52:1021–1028. doi: 10.1002/mrm.20270. [DOI] [PubMed] [Google Scholar]; c Chen J, Querol M, Bogdanov A, Weissleder R. Radiology. 2006;240:473–481. doi: 10.1148/radiol.2402050994. [DOI] [PubMed] [Google Scholar]; d Querol M, Chen JW, Weissleder R, Bogdanov A. Org. Lett. 2005;7:1719–1722. doi: 10.1021/ol050208v. [DOI] [PubMed] [Google Scholar]; e Querol M, Chen JW, Bogdanov A. Org. Biomol. Chem. 2006;4:1887–1895. doi: 10.1039/b601540a. [DOI] [PubMed] [Google Scholar]
- [31].Aime S, Cabella C, Colombatto S, Crich SG, Gianolio E, Maggioni F. J. Magn. Reson. Imaging. 2002;16:394–406. doi: 10.1002/jmri.10180. [DOI] [PubMed] [Google Scholar]
- [32].a Querol M, Bennett DG, Sotak C, Kang HW, Bogdanov A., Jr. Chembiochem. 2007;8:1637–1641. doi: 10.1002/cbic.200700157. [DOI] [PubMed] [Google Scholar]; b Arena F, Singh JB, Gianolio E, Stefanìa R, Aime S. Bioconjug. Chem. 2011;22:2625–2635. doi: 10.1021/bc200486j. [DOI] [PubMed] [Google Scholar]
- [33].Duimstra J, Meade TJ. WIPO Patent Application 05/115105. 2005.
- [34].Chang YT, Cheng CM, Su YZ, Lee WT, Hsu JS, Liu GC, Cheng TL, Wang YM. Bioconjug. Chem. 2007;18:1716–1727. doi: 10.1021/bc070019s. [DOI] [PubMed] [Google Scholar]
- [35].Rodriguez E, Nilges M, Weissleder R, Chen JW. J. Am. Chem. Soc. 2010;132:168–177. doi: 10.1021/ja905274f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lauffer R, McMurry TJ, Dunham SO, Scott DM, Parmelee DJ, Dumas S. Bioactivated diagnostic imaging contrast agents. WIPO Patent Application 97/36619. 1997.
- [37].Nivorozhkin A, Kolodziej AF, Caraban P, Greenfield MT, Lauffer RB, McMurry TJ. Angew. Chem. Int. Ed. 2001;40:2903–2906. [PubMed] [Google Scholar]
- [38].Tanaka K, Kitamura N, Chujo Y. Bioconj. Chem. 2011;22:1484–1490. doi: 10.1021/bc100381x. [DOI] [PubMed] [Google Scholar]
- [39].Harvey P, Kuprov I, Parker D. Lanthanide Complexes as Paramagnetic Probes for 19F Magnetic Resonance. Eur. J. Inorg. Chem. 2012:2015–2022. [Google Scholar]
- [40].Mizukami S, Takikawa R, Sugihara F, Hori Y, Tochio H, Wälchli M, Shirakawa M, Kikuchi K. J. Am. Chem. Soc. 2008;130:794–795. doi: 10.1021/ja077058z. [DOI] [PubMed] [Google Scholar]
- [41].Matsushita H, Mizukami S, Mori Y, Sugihara F, Shirakawa M, Yoshioka Y, Kikuchi K. ChemBioChem. 2012;13:1579–1583. doi: 10.1002/cbic.201200331. [DOI] [PubMed] [Google Scholar]
- [42].Keliris A, Mamedov I, Hagberg GE, Logothetis NK, Scheffler K, Engelmann J. Contrast Media Molec. Imaging. 2012;7:478–483. doi: 10.1002/cmmi.1470. [DOI] [PubMed] [Google Scholar]
- [43].a Granot D, Shapiro EM. Magn. Reson. Med. 2011;65:1253–1259. doi: 10.1002/mrm.22839. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Matsumura S, Aoki I, Saga T, Shiba K. Molec. Pharmaceutics. 2011;8:1970–1974. doi: 10.1021/mp2001999. [DOI] [PubMed] [Google Scholar]; c Schellenberger E, Rudloff F, Warmuth C, Taupitz M, Hamm B, Schnorr J. Bioconjug. Chem. 2008;19:2440–2445. doi: 10.1021/bc800330k. [DOI] [PubMed] [Google Scholar]
- [44].Shapiro MG, Szablowski JO, Langer R, Jasanoff A. J. Am. Chem. Soc. 2009;131:2484–2486. doi: 10.1021/ja8086938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Perez J, Simeone FJ, Tsourkas A, Josephson L, Weissleder R. Nanoletters. 2004;4:119–122. [Google Scholar]
- [46].Cui W, Liu L, Kodibagkar VD, Mason RP. Magn. Reson. Med. 2010;64:65–71. doi: 10.1002/mrm.22400. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Bengtsson NE, Brown G, Scott EW, Walter GA. Magn. Reson. Med. 2010;63:745–753. doi: 10.1002/mrm.22235. [DOI] [PubMed] [Google Scholar]
- [47].a Perez J, Josephson L, O’Loughlin T, Hogemann D, Weissleder R. Nature Biotechnol. 2002;20:816–820. doi: 10.1038/nbt720. [DOI] [PubMed] [Google Scholar]; b Zhao M, Josephson L, Tang Y, Weissleder R. Angew. Chem. Int. Ed. 2003;43:1375–1378. doi: 10.1002/anie.200390352. [DOI] [PubMed] [Google Scholar]
- [48].Yu SS, Scherer RL, Ortega RA, Bell CS, O’Neil CP, Hubbell JA, Giorgio TD. J. Nanobiotech. 2011;9:1–10. doi: 10.1186/1477-3155-9-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Westmeyer GG, Durocher Y, Jasanoff A. Angew. Chem. Int. Ed. 2010;49:3909–3911. doi: 10.1002/anie.200906712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Colomb J, Louie K, Masia SP, Bennett KM. Magn. Reson. Med. 2010;64:1792–1799. doi: 10.1002/mrm.22570. [DOI] [PubMed] [Google Scholar]
- [51].a Shiftan L, Israely T, Cohen M, Frydman V, Dafni H, Stern R, Neeman M. Cancer Res. 2005;65:10316–10323. doi: 10.1158/0008-5472.CAN-04-3947. [DOI] [PubMed] [Google Scholar]; b Shiftan L, Neeman M. Contrast Media Molec. Imaging. 2006;1:106–112. doi: 10.1002/cmmi.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Ward KM, Aletras AH, Balaban RS. J. Magn. Reson. 2000;143:79–87. doi: 10.1006/jmre.1999.1956. [DOI] [PubMed] [Google Scholar]
- [53].McMahon MT, Gilad AA, DeLiso MA, Berman SM, Bulte JW, van Zijl PC. Magn. Reson. Med. 2008;60:803–812. doi: 10.1002/mrm.21683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Zhang S, Merritt M, Woessner DE, Lenkinski RE, Sherry AD. Acc. Chem. Res. 2003;36:783–790. doi: 10.1021/ar020228m. [DOI] [PubMed] [Google Scholar]
- [55].Yoo B, Pagel MD. J. Am. Chem. Soc. 2006;128:14302–14303. doi: 10.1021/ja063874f. [DOI] [PubMed] [Google Scholar]
- [56].Yoo B, Pagel MD. Bioconjugate Chem. 2007;18:903–911. doi: 10.1021/bc060250q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Suchy M, Ta R, Li A, Wojciechowski F, Pasternak SH, Bartha R, Hudson RHE. Org. Biomolec. Chem. 2010;8:2560–2566. doi: 10.1039/b926639a. [DOI] [PubMed] [Google Scholar]
- [58].Yoo B, Sheth V, Pagel MD. Tet. Lett. 2009;50:4459–4462. doi: 10.1016/j.tetlet.2009.05.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Liu G, Liang Y, Bar-Shir A, Chan KWY, Galpoththawela CS, Bernard SM, Tse T. J. Am. Chem. Soc. 2011;133:16326–16329. doi: 10.1021/ja204701x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Yoo B, Sheth VR, Howison CM, Douglas MJK, Pineda CT, Maine EA, Baker AF, Pagel MD. Mag. Reson. Med. 2013;71:1221–1230. doi: 10.1002/mrm.24763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Hingorani DV, Randtke EA, Pagel MD. J. Am. Chem. Soc. 2013;135:6396–6398. doi: 10.1021/ja400254e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Airan RD, Bar-Shir A, Liu G, Pelled G, McMahon MT, van Zijl PCM, Bulte JWM, Gilad AA. Magn. Reson. Med. 2012;68:1919–1923. doi: 10.1002/mrm.24483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Li Z, Qiao H, Lebherz C, Choi SR, Zhou X, Gao G, Kung HF, Rader DJ, Wilson JM, Glickson JD, Zhou R. Hum. Gene Ther. 2005;16:1429–1438. doi: 10.1089/hum.2005.16.1429. [DOI] [PubMed] [Google Scholar]
- [64].Chauvin T, Durand P, Bernier M, Meudal H, Doan BT, Noury F, Badet B, Beloeil JC, Tóth E. Angew. Chemie Int. Ed. 2008;47:4370–4372. doi: 10.1002/anie.200800809. [DOI] [PubMed] [Google Scholar]
- [65].Li Y, Sheth VR, Liu G, Pagel MD. Contrast Media Molec. Imaging. 2011;6:219–228. doi: 10.1002/cmmi.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].a Holloway C, ten Hove M, Clarke K, Neubauer S. Front. Biosci. (Schol Ed) 2011;3:331–340. doi: 10.2741/s154. [DOI] [PubMed] [Google Scholar]; b Marino S, Ciurleo R, Bramanti P, Federico A, De Stefano N. Neurocrit. Care. 2011;14:127–33. doi: 10.1007/s12028-010-9406-6. [DOI] [PubMed] [Google Scholar]; c Bolan PJ, Nelson MT, Yee D, Garwood M. BreastCancerRes. 2005;7:149–52. doi: 10.1186/bcr1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].a Yu JX, Kodibagkar VD, Cui W, Mason RP. Curr. Med. Chem. 2005;12:819–48. doi: 10.2174/0929867053507342. [DOI] [PubMed] [Google Scholar]; b Arias-Mendoza F, Payne GS, Zakian KL, Schwarz AJ, Stubbs M, Stoyanova R, Ballon D, Howe FA, Koutcher JA, Leach MO, et al. NMR Biomed. 2006;19:504–512. doi: 10.1002/nbm.1057. [DOI] [PubMed] [Google Scholar]
- [68].Yu JX, Kodibagkar VD, Liu L, Mason RP. NMR Biomed. 2008;21:704–712. doi: 10.1002/nbm.1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Ki S, Sugihara F, Kasahara K, Tochio H, Okada-Marubayashi A, Tomita S, Morita M, Ikeguchi M, Shirakawa M, Kokubo T. Nucleic Acids Res. 2006;34:e51. doi: 10.1093/nar/gkl135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Kurhanewicz J, Bok R, Nelson SJ, Vigneron DB. J. Nucl. Med. 2008;49:341–344. doi: 10.2967/jnumed.107.045112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Chen AP, Hurd RE, Schroeder MA, Lau AZ, Gu Y, Lam WW, Barry J, Tropp J, Cunningham CH. NMR Biomed. 2012;25:305–311. doi: 10.1002/nbm.1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].a Barb AW, Hekmatyar SK, Glushka JN, Prestegard JH. J. Magn. Reson. 2013;228:59–65. doi: 10.1016/j.jmr.2012.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Schroeder MA, Ali MA, Hulikova A, Supuran CT, Clarke K, Vaughn-Jones RD, Tyler DJ, Swietach P. Proc. Nat. Acad. Sci. USA. 2013;110:E958–E967. doi: 10.1073/pnas.1213471110. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Lodi A, Woods SM, Ronen SM. NMR Biomed. 2013;26:299–306. doi: 10.1002/nbm.2848. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Laustsen C, Ostergaard JA, Lauritzen MH, Norregaard R, Bowen S, Sogaard LV, Flybjerg A, Pedersen M, Ardenkjaer-Larsen JH. Diabetes/Met Res Reviews. 2013;29:125–129. doi: 10.1002/dmrr.2370. [DOI] [PubMed] [Google Scholar]; e Venkatesh HS, Chaumeil MM, Ward CS, Haas-Kogan DA, David JC, Ronen SM. Neuro-Oncology. 2012;14:315–325. doi: 10.1093/neuonc/nor209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].a Gallagher FA, Kettunen MI, Day SE, Lerche M, Brindle KM. Magn. Reson. Med. 2008;60:253–257. doi: 10.1002/mrm.21650. [DOI] [PubMed] [Google Scholar]; b Karlsson M, Jensen PR, in’t Zandt R, Gisselsson A, Hansson G, Duus JO, Meier S, Lerche MH. Int. J. Cancer. 2010;127:729–736. doi: 10.1002/ijc.25072. [DOI] [PubMed] [Google Scholar]; c Zacharias NM, Chan HR, Sailasuta N, Ross BD, Bhattacharya P. J. Am. Chem. Soc. 2012;134:934–943. doi: 10.1021/ja2040865. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Keshari KR, Wilson DM, Chen AP, Bok R, Larson PEZ, Hu S, Van Criekinge M, MacDonald JM, Vigneron DB, Kurhanewicz J. J. Am. Chem. Soc. 2009;131:17591–17596. doi: 10.1021/ja9049355. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Jamin Y, Gabellieri C, Smythe L, Reynolds S, Robinson SP, Springer CJ, Leach MO, Payne GS, Eykyn TR. Magn. Reson. Med. 2009;62:1300–1304. doi: 10.1002/mrm.22049. [DOI] [PubMed] [Google Scholar]
- [74].Mason RP, Sanders JKM. Biochemistry. 1989;28:2160–2168. doi: 10.1021/bi00431a030. [DOI] [PubMed] [Google Scholar]
- [75].Mason RP, Sanders JKM, Crawford A, Hunter BK. Biochemistry. 1986;25:4504–4507. doi: 10.1021/bi00364a008. [DOI] [PubMed] [Google Scholar]
- [76].Mason RP, Sanders JKM, Cornish A. FEBS Lett. 1987;216:4–6. [Google Scholar]
- [77].Jeffrey FMH, Rajagopal A, Malloy CR, Sherry AD. Biochem. Sci. 1991;16:5–10. doi: 10.1016/0968-0004(91)90004-f. [DOI] [PubMed] [Google Scholar]
- [78].Cohen SM, Ogawa S, Shulman RG. Proc. Nat. Acad. Sci. USA. 1979;76:1603–1607. doi: 10.1073/pnas.76.4.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Cohen SM, Ogawa S, Shulman RG. Fed. Proc. 1979;38:828–828. [Google Scholar]
- [80].Jeffrey FMHJ, Malloy CR. Biochem. J. 1992;287:117–123. doi: 10.1042/bj2870117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Vaughan JT, Snyder CJ, DelaBarre LJ, Bolan PJ, Tian J, Bolinger L, Adriany G, Andersen P, Strupp J, Ugurbil K. Magn. Reson. Med. 2009;61:244–248. doi: 10.1002/mrm.21751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Swierczewska M, Lee S, Chen X. Molec. Imaging. 2011;10:3–16. [PMC free article] [PubMed] [Google Scholar]
- [83].Yu JX, Kodibagkar VD, Liu L, Zhang ZW, Liu L, Magnusson J, Liu YT. Chem. Sci. 2013;4:2132–2142. [Google Scholar]
- [84].Gao J, Xie J, Xu B, Chen X. Nanoplatform-Based Molecular Imaging. 2011:25–45. [Google Scholar]
- [85].Pagel MD, Basilion JP. In: Molecular Imaging with Reporter Genes. Gambhir SS, Yaghoubi SS, editors. Cambridge University Press; 2011. [Google Scholar]