

Background: Molecular basis for the dephosphorylation step in biosynthesis of methyl glucose lipopolysaccharides (MGLPs) of Mycobacterium tuberculosis is unknown.
Results: Structures of unliganded, vanadate-bound, and phosphate-bound glycosyl-3-phosphoglycerate phosphatase (GpgP) reveal pivotal conformational changes in enzyme during dephosphorylation.
Conclusion: Dimerization and maneuvers of Loop2 play essential roles during dephosphorylation.
Significance: We present first structures of histidine phosphatase-type GpgP and explain the mechanism of catalysis.
Keywords: Bacterial Protein Phosphatase, Crystal Structure, Enzyme Mechanism, Mutagenesis, Mycobacterium Tuberculosis, Enzymatic Mechanism
Abstract
Mycobacterium tuberculosis (Mtb) synthesizes polymethylated polysaccharides that form complexes with long chain fatty acids. These complexes, referred to as methylglucose lipopolysaccharides (MGLPs), regulate fatty acid biosynthesis in vivo, including biosynthesis of mycolic acids that are essential for building the cell wall. Glucosyl-3-phosphoglycerate phosphatase (GpgP, EC 5.4.2.1), encoded by Rv2419c gene, catalyzes the second step of the pathway for the biosynthesis of MGLPs. The molecular basis for this dephosphorylation is currently not understood. Here, we describe the crystal structures of apo-, vanadate-bound, and phosphate-bound MtbGpgP, depicting unliganded, reaction intermediate mimic, and product-bound views of MtbGpgP, respectively. The enzyme consists of a single domain made up of a central β-sheet flanked by α-helices on either side. The active site is located in a positively charged cleft situated above the central β-sheet. Unambiguous electron density for vanadate covalently bound to His11, mimicking the phosphohistidine intermediate, was observed. The role of residues interacting with the ligands in catalysis was probed by site-directed mutagenesis. Arg10, His11, Asn17, Gln23, Arg60, Glu84, His159, and Leu209 are important for enzymatic activity. Comparison of the structures of MtbGpgP revealed conformational changes in a key loop region connecting β1 with α1. This loop regulates access to the active site. MtbGpgP functions as dimer. L209E mutation resulted in monomeric GpgP, rendering the enzyme incapable of dephosphorylation. The structures of GpgP reported here are the first crystal structures for histidine-phosphatase-type GpgPs. These structures shed light on a key step in biosynthesis of MGLPs that could be targeted for development of anti-tuberculosis therapeutics.
Introduction
The resilience and pathogenicity of Mycobacterium tuberculosis (Mtb)4 has been partly ascribed to its unique cell envelope that is made up of a remarkable mixture of polysaccharides and lipidic moieties, some of which are found exclusively in mycobacteria (1–4). Among the various components that make up the cell wall, mycolic acids impart structural integrity to the cell wall. The synthesis of mycolic acids is regulated by methylglucose lipopolysaccharides (MGLPs) via their association with and modulation of activity of the fatty acid synthase I complex (5). In this context, MGLPs have been studied vigorously and have been shown to be physiologically important. The significance of MGLPs in Mtb is further underscored by the fact that many enzymes involved in the biosynthesis of MGLPs are essential for the survival of the bacterium (6).
MGLPs constitute complexes of 6-O-methyl glucose lipopolysaccharides and long chain fatty acid molecules mixed in 1:1 stoichiometry. The biosynthesis of MGLPs requires the concerted action of many enzymes (Fig. 1A) (7–10). These include enzymes necessary for the transfer of glucosyl, methyl, and acetyl groups, as well as accessory enzymes like glucoside hydrolases and phosphatases. Although some of these enzymatic activities have been characterized, the identity of a number of enzymes catalyzing discrete steps in the biosynthesis of MGLPs is currently unknown. The first enzymatic activity proposed for the pathway leading to the biosynthesis of MGLP is the glucosyl-3-phosphoglycerate synthase activity that fuses the glucosyl moiety of UDP-glucose with d-3-phosphoglyceric acid. The resultant glucosyl-3-phosphoglycerate (GPG) is subjected to dephosphorylation by a recently identified GPG phosphatase (GpgP) to produce glucosyl glycerate (GG) (Fig. 1A) (11). In the next step, two molecules of GG are linked together by di-glucosyl glycerate synthase to produce di-glucosyl glycerate. A glucosyl transferase extends the di-glucosyl glycerate moiety further via formation of α-(1→4) linkages. The hexose units of the polymer are modified with position specific methyl and acetyl groups (12, 13). However, the identity of many of these enzymes catalyzing methyl and acetyl transfers is currently unknown.
FIGURE 1.
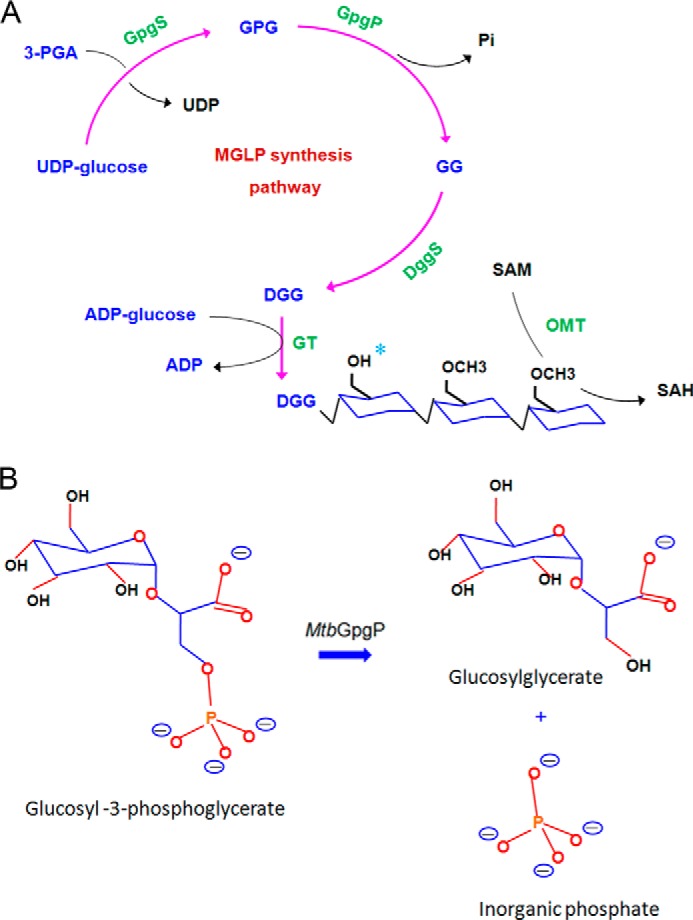
Biosynthesis of MGLPs. A, schematic diagram of enzymatic activities involved in the biosynthesis of MGLPs. The hexose units are modified by O-methyl transferases and acetyl transferases (possible acetylation site marked with a cyan asterisk). B, reaction catalyzed by MtbGpgP.
Recently, Rv2419c was shown to dephosphorylate GPG with high specificity and hence was annotated as the GpgP catalyzing the second step in the pathway for the biosynthesis of MGLPs (Fig. 1B) (14). Unlike halo acid dehalogenase-like phosphatases that require a metal ion for catalysis (11, 15–17), MtbGpgP carries out metal ion-independent dephosphorylation. The enzyme harbors a characteristic RHG motif and therefore belongs to the histidine phosphatase superfamily (18). Catalysis by members belonging to this family proceeds via the formation of a phosphohistidine intermediate (18). The phosphate group from the substrate is transferred to the catalytic histidine of the enzyme. A glutamate residue has been known to play the role of proton donor during this phosphorylation. The same residue accepts a proton during dephosphorylation of the histidine. Although MtbGpgP exhibits the RHG motif, its primary sequence shares only 31% identity with its closest known homologue, PhoE, a promiscuous phosphatase from Bacillus stearothermophilus. Therefore, we sought to obtain a structural view of the protein to find out how MtbGpgP differs structurally from its homologues. The structure was also likely to explain the molecular basis for conversion of GPG to GG by MtbGpgP, which is currently unknown.
Here, we describe the crystal structures of apo-, vanadate-bound, and phosphate-bound MtbGpgP representing the unliganded, reaction intermediate mimic, and product-bound views of the enzyme. Comparison of the structures reveals pivotal conformational changes in a loop region located in proximity of the active site, providing insights into maneuvers of structural elements during the course of catalysis. Structure-guided site-directed mutagenesis and results of activity assays of mutants have helped identify amino acids essential for catalysis. The structures together with mutagenesis and biochemical data provide a framework for understanding the dephosphorylation of GPG by MtbGpgP.
EXPERIMENTAL PROCEDURES
Cloning, Expression, and Purification
The open reading frame corresponding to Rv2419c (MtbGpgP) was amplified from the genomic DNA of H37Rv strain of Mycobacterium tuberculosis by PCR and inserted between the BamHI and XhoI restriction sites of pET28a vector (Novagen). This construct expressed protein with an N-terminal His6 tag. Point mutations (R10A, N17A, Q23A, R60A, E84Q, H159A, and L209E) were introduced into this construct using QuikChange site-directed mutagenesis kit (Invitrogen) following the manufacturer's instructions. All the constructs were verified by sequencing the entire gene prior to expression.
MtbGpgP was overexpressed in Escherichia coli BL21 (DE3) (TransGen Biotech). Cells were grown aerobically at 37 °C in Luria-Bertani medium supplemented with 100 μg/ml kanamycin. When the cell density (A600 nm) reached 0.6, the culture was first cooled for 2 h at 4 °C and then induced at 16 °C for 18 h with 0.5 mm isopropyl β-d-thiogalactoside. The cells were harvested by centrifugation at 8,000 × g for 15 min. The cell pellet was resuspended in McAc0 buffer (25 mm Tris-HCl, 500 mm NaCl, pH 8.0) and lysed by sonication. Unbroken cells and debris were removed by spinning the lysate at 12,000 × g for 40 min. Supernatant containing soluble target protein was then loaded onto a nickel-nitrilotriacetic acid column (GE Healthcare) previously equilibrated with McAc0 buffer. After thorough washing with buffer, protein bound to the column was eluted with McAc500 buffer supplemented with 500 mm imidazole. Imidazole was removed by size exclusion chromatography. A Superdex G200 column (GE Healthcare) equilibrated with 25 mm HEPES, 100 mm NaCl, pH 7.5, was used for size exclusion chromatography. The protein was purified further by an anion exchange chromatography step using a Resource Q column (GE Healthcare). Protein bound to the column was eluted using a linear gradient of 1 m NaCl. Fractions containing protein were analyzed by SDS-PAGE. Fractions exhibiting a single band on SDS-PAGE corresponding to the molecular weight of the protein were pooled, concentrated to 15–20 mg/ml, and stored at −20 °C until further use.
Mutants of MtbGpgP were expressed and purified using the same protocol as the wild type protein. The purity of the proteins was estimated to be >95% as judged by SDS-PAGE analysis.
Crystallization and Data Collection
Crystals of MtbGpgP were grown at 293 K using the sitting drop vapor diffusion technique. 1 μl of protein solution (20 mg/ml, 25 mm HEPES, 150 mm NaCl, pH 7.5) was mixed with 1 μl of reservoir solution and equilibrated over 100 μl of reservoir solution. Complexes of MtbGpgP were prepared by mixing the enzyme with ammonium metavanadate and p-nitrophenyl phosphate (pNPP), respectively. Crystals for apo-MtbGpgP, MtbGpgP-VO3, and MtbGpgP-PO4 were obtained under the following conditions: 0.1 m HEPES, pH 7.5, 10% 2-propanol, 28% (w/v) polyethylene glycol 4000; 1.26 m sodium phosphate monobasic monohydrate, 0.14 m potassium phosphate dibasic, pH 5.6; and 0.1 m HEPES, pH 7.5, 10% 2-propanol, 20% (w/v) polyethylene glycol 4000, respectively. Crystals were cryoprotected by adding 20% glycerol to the crystallization solution before being flash frozen in liquid nitrogen. X-ray diffraction data for MtbGpgP were collected at Beamline BL17U of Shanghai Synchrotron Radiation Facility. Data sets for the MtbGpgP-VO3 and MtbGpgP-PO4 complexes were collected on Beamline 5A of Photon Factory (Japan). All of the data were processed with the HKL2000 suite of programs (19) (see Table 1).
TABLE 1.
Data collection and refinement statistics
| apo-MtbGpgP | MtbGpgP -VO3 | MtbGpgP -PO4 | |
|---|---|---|---|
| Data collection | |||
| Wavelength (Å) | 0.97930 | 1.03935 | 1.00000 |
| Resolutiona | 50.00–1.95 (1.98–1.95) | 50.00–2.30 (2.34–2.30) | 50.00–1.77 (1.80–1.77) |
| Space group | P212121 | P212121 | P212121 |
| Unit cell dimensions | |||
| a, b, c (Å) | 46.3, 77.0, 126.5 | 46.3, 82.8, 131.3 | 55.4, 76.9, 106.0 |
| α, β, γ (°) | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 |
| Rmerge (%)a,b | 6.2 (41.1) | 5.5 (27.0) | 6.7 (32.8) |
| I/σIa | 31.2 (5.3) | 46.4 (8.9) | 29.5 (5.6) |
| Completeness (%)a | 97.1 (100) | 96.5 (100) | 99.3 (89.5) |
| Redundancya | 6.7 (7.1) | 7.0 (7.0) | 6.7 (4.8) |
| Total reflections | 223,942 | 156,449 | 295,755 |
| Unique reflections | 33,241 | 22,409 | 44,306 |
| Refinement statistics | |||
| Resolution (Å) | 48.90–1.95 | 35.01–2.30 | 36.14–1.78 |
| No. atoms | |||
| Protein | 3,227 | 3,151 | 3,250 |
| Water | 302 | 117 | 386 |
| Ligand | 0 | 8 | 10 |
| B-factor | |||
| Protein | 29.25 | 46.41 | 23.73 |
| Water | 32.87 | 38.73 | 32.47 |
| Ligand | N/A | 35.42 | 17.58 |
| Rwork/Rfreec | 20.3/23.9 | 19.20/24.48 | 18.2/21.9 |
| RMSDs | |||
| Bond lengths (Å) | 0.007 | 0.012 | 0.007 |
| Bond angles (°) | 1.158 | 1.232 | 1.172 |
| Observed residues | |||
| Chain A | 2–215 | 3–215 | 3–219 |
| Chain B | 3–216 | 3–215 | 2–218 |
| Ramachandran plot (%)d | |||
| Favored | 98.05 | 96.25 | 99.04 |
| Allowed | 1.95 | 3.00 | 0.24 |
| Outliers | 0 | 0.50 | 0.72 |
| MOLPROBITY scored | 1.75 | 2.12 | 1.66 |
a The numbers in parentheses are for the highest resolution shell.
b Rmerge = Σ|I − <I>|/Σ<I>, where I is the observed intensity, and <I> is the average intensity of multiple observations of symmetry related reflections.
c Rwork/Rfree = Σ||Fo| − |Fc||/Σ|Fo|, where Fo and Fc are the observed and calculated structure factors, respectively.
d The Ramachandran plot and MOLPROBITY score were calculated with MOLPROBITY.
Phasing, Model Building, and Refinement
The crystal structure of MtbGpgP was solved by molecular replacement with BALBES software suite (20) using the structure of PhoE (Protein Data Bank code 1H2E) as the search template. Automated model rebuilding by ARP/wARP was then performed with the structure solution obtained with BALBES (21). The atomic model was refined to an Rfree value of 0.247 by iterative cycles of refinement involving manual model adjustment with Coot (22) and Phenix.refine (23). For the complex of MtbGpgP-VO3 and MtbGpgP-PO4, the native structure was used as a search template for molecular replacement with Phaser (24). Ligand fitting into the corresponding difference electron density maps calculated from the precalculated map coefficients after Phenix.refine runs were carried out by Phenix.ligandfit (25). Both complex structures were finalized by several rounds of manual building in Coot and refinement using Phenix.refine. Electron density for AA 197–198 of chain A and AA 17–19 and 197–199 of chain B of apo-MtbGpgP; AA 197–200 of chain A and AA 145–148 and 196–203 of chain B of MtbGpgP-VO3; and AA 16–20 and 197–201 of chain A of MtbGpgP-PO4 was missing. All structures were judged to have good stereochemistry according to the Ramachandran plot calculated by MolProbity (26). A summary of the data collection, phasing, and structure refinement statistics is listed in Table 1.
Enzyme Assays
Phosphatase activity of wild type and mutant MtbGpgP was estimated using pNPP as substrate. The reaction mixture consisted of 20 mm Bis-Tris HCl, 2.5 mm MgCl2, 3 mm pNPP, pH 7.0, and pure MtbGpgP or its mutant (1 mg/ml). Incubation was carried out at 37 °C for 15 min. The amount of p-nitrophenol released was measured by reading the absorbance at 405 nm as described previously (27).
Multiangle Light Scattering Analysis
Multiangle light scattering (MALS) analyses were performed at room temperature and coupled up with size exclusion chromatography using an 18-angle DAWN HELLOS II instrument equipped with an OptilabrEX refractive index detector (28). All samples were diluted to 3 mg/ml and injected into a Superdex 200 10/300 GL column (GE Healthcare) equilibrated with 25 mm HEPES, 150 mm NaCl, pH 7.5. Calibration of the light scattering detector was performed with albumin monomer standards before conducting the assays. The data were analyzed using the ASTRA software (28).
RESULTS
Overall Structure of MtbGpgP
Recombinant MtbGpgP was expressed in E. coli with an N-terminal His6 tag. The enzyme was purified to homogeneity using a number of chromatography steps like nickel affinity, ion exchange, and gel filtration. The enzyme was active when tested for phosphatase activity. This enzyme was used for structural studies. We solved the crystal structures of the apo-form of GpgP (apo-MtbGpgP), the complex of GpgP with a transition state mimic, vanadate (MtbGpgP-VO3), and the complex of GpgP with one of the reaction products, orthophosphate (MtbGpgP-PO4). These structures were refined to 1.95, 2.30, and 1.77 Å resolution, respectively. The structure of the apo-enzyme was solved by molecular replacement using the structure of a phosphatase from B. stearothermophilus (29) (PhoE; Protein Data Bank code 1H2E) as the search template. PhoE shares 31% sequence identity with MtbGpgP. Subsequent structures of the binary complexes were solved by molecular replacement using the structure of the apo-enzyme as the search template. The final model of all three structures consists of residues 3–215 and exhibits good stereochemistry. Intriguingly, electron density for amino acids 197–200 was missing for all the chains, except one. These amino acids are part of a loop connecting strand β4 with β5. Notably, this region is protruding out of the protein but is located far away from the bound ligands. There are two protomers in the asymmetric unit, suggesting the possibility of a dimer being the minimal functional unit of MtbGpgP. The overall structures of apo-MtbGpgP, MtbGpgP-VO3, and MtbGpgP-PO4 are similar, with a root mean square deviation (RMSD) of less than 0.75 Å between the Cα atoms. Data collection and refinement statistics for the structures are listed in Table 1.
MtbGpgP consists of a single domain with a central twisted β-sheet that is flanked by α-helices on either side (Fig. 2A). The β-sheet is made up of five β-strands. Strands β1, β2, β3, and β5 run parallel, whereas strand β4 runs anti-parallel. Strand β5 is positioned at the dimer interface. Each protomer of MtbGpgP contains seven α-helixes. The arrangement of the structural elements is reminiscent of the classical α/β/α sandwich architecture of the canonical cofactor-dependent phosphoglycerate mutase (dPGM) fold (29, 30). Although the last five amino acids are missing in all the structures of MtbGpgP, the C-terminal tail is unlikely to extend into the active site as observed for PhoE and other dPGMs (29, 31, 32). A search for structural homologues using the Dali server (33) revealed that the overall structure of MtbGpgP is similar to the PhoE phosphatase (Table 2) (Protein Data Bank code 1EBB; Z score 24.5). The Cα atoms of the two structures could be superimposed with an RMSD of 2.1 Å for 197 matching residues (Fig. 2B). Other significant structural matches included phosphoserine phosphatase 1 (34) (PsP1; Protein Data Bank code 4IJ5) and phosphoglycerate mutase (35) (PGM; Protein Data Bank code 3EZN). Although Psp1 shares only 29% sequence identity with MtbGpgP, the overall structure of both of these enzymes is similar (Fig. 2B). In addition, the mode of dimerization observed in the crystal structures of the two enzymes is also similar. However, unlike MtbGpgP, the exact substrate of this phosphatase is currently not known (14). In contrast to PsP1, the substrate specificities of PGMs are known. They catalyze the conversion of 3-phosphoglycerate to 2-phosphoglycerate with high specificity (31). This sets them apart from MtbGpgP, which shows greater dephosphorylation activity against GPG when compared with its PGM activity. Despite the differences in the reactions catalyzed, these two enzymes share a common fold, which is evident from a high Z score of 22.4 and a low RMSD of 2.2 Å for 195 matching Cα atoms when the two structures were superimposed (Fig. 2B).
FIGURE 2.
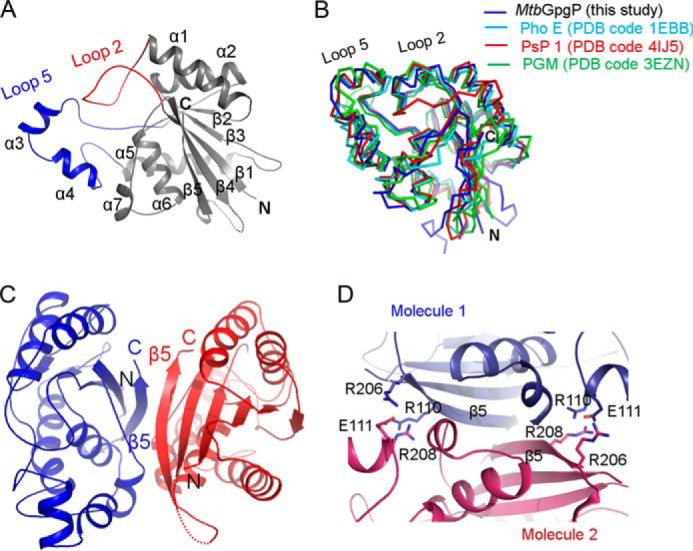
Structure of apo-MtbGpgP. A, cartoon representation of the structure of MtbGpgP. Loop 2 (red) connecting β1 with α1 plays an important role in catalysis. Loop 2 together with loop 5 (blue) covers the active site partially. B, superimposition of the structures of MtbGpgP homologues. The structures are shown as ribbons. C, cartoon representation of a dimer of MtbGpgP. The dimer interface is extensive and involves β5. D, homotypic, ionic interactions at the dimer interface. Interacting residues are shown as sticks. The N and C termini are marked as N and C, respectively. PDB, Protein Data Bank.
TABLE 2.
Structural homologues of MtbGpgP
Dali analysis retrieved >200 hits with a Z score above 6. Surprisingly, sequence conservation amongst the homologues is low. The top 10 hits are listed in the table.
| No. | Protein Data Bank Chain | Z-score | RMSD | Identity | Name of protein |
|---|---|---|---|---|---|
| % | |||||
| 1 | 4IJ6-B | 26.9 | 1.7 | 29 | Phosphoserine phosphatase of H. thermophilus |
| 2 | 1EBB-A | 24.5 | 2.1 | 31 | Phosphatase PhoE of B. stearothermophilus |
| 3 | 3GP3-A | 22.4 | 2.2 | 26 | Phosphoglycerate mutase of B. pseudomallei |
| 4 | 3D8H-B | 22.3 | 2.3 | 23 | Phosphatase of P. falciparum |
| 5 | 1FZT-A | 22.1 | 2.4 | 24 | Phosphoglycerate mutase of S. pombe |
| 6 | 2A9J-A | 22.0 | 2.2 | 24 | Bisphosphoglycerate mutase of Homo sapiens |
| 7 | 1RII-D | 22.0 | 2.3 | 29 | Phosphoglycerate mutase of M. tuberculosis |
| 8 | 3KKK-C | 22.0 | 2.4 | 24 | Phosphoglycerate mutase of P. falciparum |
| 9 | 3DCY-A | 21.9 | 2.6 | 27 | TP53-induced regulator of H. sapiens |
| 10 | 4EMB-A | 21.8 | 2.4 | 26 | Phosphoglycerate mutase of B. burgdorferi |
In all the structures of MtbGpgP, two protomers present in the asymmetric unit are observed forming a dimer that involves the participation of the terminal β5 strand of each protomer (Fig. 2C). The strands lie adjacent to each other, giving the appearance of a contiguous 10-stranded β-sheet traversing the dimer interface. The two protomers interact extensively via homotypic interactions. Most of these interactions involve amino acids from strand β5 of each protomer (Fig. 2D). Although the interactions are primarily hydrophobic, Arg110, Glu111, Arg206, and Arg208 located at the dimer interface contribute ionic interactions. Dimerization results in burial of a total of 1,600 Å2 of solvent-accessible surface area per subunit. MALS analysis of MtbGpgP suggested that the protein exists as a dimer in solution. Thus, MtbGpgP forms a homotypic dimer with the active sites of the monomers located in diagonally opposite clefts (Fig. 2C).
Structural View of the Phosphohistidine Transition State Mimic
Vanadate compounds have been used previously to study catalytic mechanism of phosphatases (29). Therefore, to gain insights into the mechanism of the MtbGpgP catalyzed reaction, we cocrystallized MtbGpgP with ammonium metavanadate. Vanadate was expected to form a covalent bond with the catalytic histidine mimicking the characteristic phosphohistidine reaction intermediate of histidine phosphatases. As observed for other phosphatases previously (29), the structure of the binary complex of MtbGpgP with vanadate shows vanadate covalently linked to His11 (Fig. 3A). Vanadate is entrenched in a cavity above the central β-sheet (Fig. 3B). Notably, the cavity is shielded by two long loops. Loop 2, connecting β1 with α1, towers over vanadate, whereas loop 5 connecting β3 with α4 is located adjacent to loop 2 (Fig. 3B). Together, these two loops are observed partially covering the active site and demarcating a large boundary of the active site pocket. One molecule of vanadate could be modeled into the electron density. Interestingly, the density merged with that of the aromatic ring of His11 (Fig. 3A). The vanadate bond to His11 mimicked the phosphohistidine reaction intermediate during catalysis. Similar covalent tethering of vanadate by a histidine has been observed before for PhoE (29). Other residues interacting with the vanadate moiety include Arg10, Asn17, Gln23, Arg60, Glu84, and His159 (Fig. 3C and Table 3). Notably, side chains of Gln23 and Asn17 move inwards to make contact with vanadate. As a result, the position of loop 2 is different during the transition state than that assumed by the enzyme in absence of substrate. Mutating either of these residues to alanine reduced the enzyme activity dramatically, indicating an important role for this maneuver of loop 2 during catalysis (Fig. 3D). Further, mutating Arg10, Thr14, Arg60, or His159 to alanine almost completely abolished the enzymatic activity, revealing the essentiality of these amino acids in dephosphorylation of the substrate (Fig. 3D). The structure of vanadate-bound MtbGpgP provides crucial mechanistic insights and helps identify key residues for dephosphorylation of GPG.
FIGURE 3.

Structure of the reaction intermediate mimic of MtbGpgP. A, 2Fo − Fc electron density for the vanadate covalently linked to His11 contoured at 1σ. B, vanadate covalently linked to His11 (shown as sticks) maps the location of the active site. Loop 2 (red) and loop 5 (green) cover the active site partially. The N and C termini of the protein are marked as N and C, respectively. C, amino acids interacting with the reaction intermediate mimic vanadate (V, shown as sticks) are shown as magenta sticks. Hydrogen bonds are depicted as dashed lines. D, relative activity of mutants of MtbGpgP. The error bars represent S.E. for three independent assays conducted under identical conditions. The significance of difference in phosphatase activity of wild type and mutants was analyzed by Student's t test. *, p < 0.05 (n = 3); **, p < 0.01 (n = 3).
TABLE 3.
Hydrogen bonds formed between MtbGpgP and VO3 group
| Molecule/residue name/atom name | Distance | |
|---|---|---|
| Å | ||
| VO3/O1 | MtbGpgP/Arg10/Nη2 | 2.8 |
| VO3/O1 | MtbGpgP/His11/Nϵ2 | 2.8 |
| VO3/O1 | MtbGpgP/Asn17/Nδ2 | 2.9 |
| VO3/O1 | MtbGpgP/Gln23/Nϵ2 | 2.8 |
| VO3/O2 | MtbGpgP/Arg10/Nϵ | 2.6 |
| VO3/O2 | MtbGpgP/His11/Nϵ2 | 3.0 |
| VO3/O3 | MtbGpgP/His11/Nϵ2 | 2.9 |
| VO3/O3 | MtbGpgP/His159/Nδ1 | 2.8 |
| VO3/O3 | MtbGpgP/Arg60/Nη1 | 2.5 |
Structure of Inorganic Phosphate-bound MtbGpgP
MtbGpgP shows low activity against pNPP (14). To gain further insights into the mechanism of catalysis, we cocrystallized pNPP with MtbGpgP. The substrate seems to have been dephosphorylated by MtbGpgP during the course of the crystallization. Electron density for only orthophosphate noncovalently bound to MtbGpgP, depicting the post catalytic state of the enzyme, was observed (Fig. 4A). The position of the phosphate overlaps with the position of vanadate. However, the aromatic ring of His11 has stepped back by 1.5 Å after the dephosphorylation and is now observed forming a hydrogen bond with the oxygen atom of the phosphate (Fig. 4B). Interestingly, the side chain of Glu84 retains its previous position of the transition state and is observed forming a hydrogen bond with the oxygen atom of the phosphate group. In fact, with the exception of Asn17 and Gln23 of loop 2, all other residues interacting with phosphate retain their positions of the transition state. The side chain of Gln23 that formed a hydrogen bond with the vanadate, is now 4.6 Å away from the phosphate group. More importantly, Asn17 has moved out (away from the active site) by 10.5 Å. As a result of the movement of Asn17 and Gln23, the position of loop 2 in the MtbGpgP-PO4 structure is different from that observed in other structures of MtbGpgP (Fig. 4B). The loop has moved away from the active site. Such an open conformation of the active site observed in the structure of MtbGpgP-PO4 denotes completion of catalysis. MtbGpgP probably assumes such a conformation during product release. Details of residues interacting with the phosphate and their distances are listed in Table 4.
FIGURE 4.

Conformational changes of MtbGpgP during product release. A, 2Fo − Fc electron density for the orthophosphate contoured at 1σ. B, superimposition of the vanadate bound structure of MtbGpgP (blue) over the structure of the phosphate-bound MtbGpgP (red) reveals conformational changes in loop 2 of the enzyme. Notably, Asn17 has moved >10 Å away from the active site (inset). C, location of Leu209 with respect to the dimer interface and the active site. Leu209 and phosphate are shown as sticks. D, on-site MALS during size exclusion chromatography of WT and L209E mutant of MtbGpgP. Comparison of the elution time with those of standards revealed that WT enzyme elutes as a dimer (blue curve), whereas the mutant elutes as a monomer (red curve).
TABLE 4.
Hydrogen bonds formed between MtbGpgP and PO4 group
| Molecule/atom name | Molecule/residue name/atom name | Distance |
|---|---|---|
| Å | ||
| PO4/O1 | MtbGpgP/Arg10/Nη2 | 2.9 |
| PO4/O2 | MtbGpgP/Arg60/Nϵ | 2.8 |
| PO4/O2 | MtbGpgP/Arg60/Nη2 | 3.3 |
| PO4/O2 | MtbGpgP/His159/Nδ1 | 2.8 |
| PO4/O2 | MtbGpgP/His11/Nϵ2 | 2.6 |
| PO4/O3 | MtbGpgP/Arg10/Nϵ | 2.8 |
| PO4/O4 | MtbGpgP/Arg60/Nη2 | 3.2 |
Thus, the structure of the binary complex of MtbGpgP with phosphate validates inferences about the location of the active site and the identity of residues involved in catalysis. In addition, it reveals that loop 2 undergoes a pivotal conformational change, possibly to assist product release or to bind substrate.
A Dimer Is the Minimal Functional Unit of MtbGpgP
Previously, MtbGpgP was shown to exist as dimers in solution (14). Our crystal structures of MtbGpgP provide information about the location and nature of the dimer interface. Based on these structures, amino acids of the dimer interface were selected and mutated to break the dimer interface. Estimation of the activity of the resulting monomeric MtbGpgP was expected to shed light on the essentiality of dimerization of MtbGpgP for dephosphorylation of its substrate. Among the point mutations tested, L209E mutation disrupted dimerization of the enzyme (Fig. 4C). MALS analysis of the mutant and the wild type enzyme under identical conditions revealed that the L209E mutant of MtbGpgP existed as a monomer in solution (Fig. 4D). Leu209 is located in the middle of strand β5 that mediates dimerization. However, this residue is located far away from the active site, and hence the L209E mutation is unlikely to have any direct effect on the integrity of the active site. Estimation of the enzymatic activity indicated that the monomeric MtbGpgP had lost its ability to perform dephosphorylation (Fig. 3D). Taken together, results of our structural and mutagenesis studies clearly show that dimerization of MtbGpgP is essential for its dephosphorylation activity. A dimer constitutes the minimal functional unit of MtbGpgP.
DISCUSSION
Monomerization of MtbGpgP abolished enzymatic activity. This is in stark contrast to the structural homologue closest to MtbGpgP: PhoE that functions as a monomer (29, 30). Although intriguing, it is not completely surprising for a phosphatase to be catalytically competent only when it is in its dimeric state. For example, the human prostatic acidic phosphatase (EC 3.1.3.2; Protein Data Bank code 1CVI) functions as a dimer. The structures of MtbGpgP described here reveal that the dimer interface is located far away from the bound ligands, and therefore it may not contribute residues directly for catalysis. To find out why monomeric MtbGpgP could not catalyze dephosphorylation, we examined the region around the dimer interface. A close inspection revealed intermolecular ionic interactions between amino acids from β5 of one monomer and loop 5 of another monomer. Incidentally, strand β5 is part of the dimer interface, whereas loop 5 is a long loop that partially covers the active site. Two intermolecular interactions—the salt bridge between Glu111 and Arg206 and stacking of guanidium groups of Arg110 and Arg208 against each other—probably play a role in localizing loop 5 such that it permits nonintrusive entry of the substrate into the active site (Fig. 5A). In addition, Arg208 from one monomer might assist in docking the substrate into the active site of another monomer (Fig. 5A). Although these observations help partially explain the need of dimerization for enzymatic activity, a structural view of MtbGpgP in complex with GPG is likely to further clarify the requirement of dimerization for enzymatic activity.
FIGURE 5.

Mechanism of catalysis. A, intermolecular ionic interactions help stabilize conformation of loop 5 during catalysis. Within a dimer of MtbGpgP, one monomer is colored blue, whereas the other is colored gray. Loop 2 and loop 5 of monomer 1 are colored in green and magenta, respectively. Residues involved in interactions and orthophosphate are shown as sticks. The hydrogen bond is shown as a dashed line. B, location of conserved residues (left panel) based on ConSurf analysis of primary sequence of 151 homologues of MtbGpgP (38). The surface of MtbGpgP is colored according to conservation depicted in scale. Residues around phosphate (green sticks) are highly conserved. The right panel shows a stereo view of close up of some of the conserved residues (sticks) around the phosphate (green sticks). C, diagrammatic representation of conformational changes of MtbGpgP during catalysis. Key conformational changes of loop 2 (red) during the conversion of GPG to GG have been captured in the crystal structures of MtbGpgP. D, catalysis proceeds in two steps. In step 1 (left panel), His11 (magenta sticks) mounts a nucleophilic attack on P center of phosphate (sticks). Glu84 (magenta sticks) donates a proton to the substrate. The substrate (GPG) is depicted as phosphate (sticks) linked to GG by a covalent bond shown in blue. In step 2 (right panel), a water molecule (cyan sphere) activated by Glu84 mounts an attack on the P center of the reaction intermediate. Amino acids stabilizing reaction intermediates are shown as green sticks. For clarity, Asn17, Gln23, and His159 participating in the stabilization are not shown.
GpgPs are grouped under halo acid dehalogenase-like hydrolase superfamily because they harbor a conserved characteristic DDDD sequence (36, 37). However, GpgP from Mtb is an exception to this grouping because it carries the RHG motif, a hallmark of histidine phosphatases (14). Because PGMs also exhibit RHG motifs, Rv2419c (MtbGpgP) was erroneously annotated as a PGM earlier. Interestingly, Rv2419c does exhibit low PGM activity. However, the phosphatase activity against GPG is much higher comparatively. In this context, MtbGpgP shows some promiscuity (14). Low dephosphorylation activities of MtbGpgP have been reported against mannosyl-3-phosphoglycerate, mannosylglucosyl-3-phosphoglycerate, and pNPP (14). The specific activities for these substrates are at least 10-fold less than those for GPG. Examination of the active sites of MtbGpgP structures reveals that the pocket can be extended to accommodate larger substrates. Especially displacement of Loop 2 could make room for larger substrates like mannosyl-3-phosphoglycerate and mannosylglucosyl-3-phosphoglycerate to dock into the active site. In contrast to MtbGpgP, PhoE is a highly promiscuous phosphatase (29, 30). Intriguingly, the apo- and ligand-bound crystal structures of PhoE reveal no movement of the loop region equivalent to the Loop 2 of MtbGpgP. Dynamics simulations analysis, however, did suggest the presence of flexible regions around the active site that could explain the substrate promiscuity of PhoE (29, 30).
Key catalytic residues of MtbGpgP like His11 and Glu84 involved in proton transfers during dephosphorylation are strictly conserved in PhoE. Other essential residues of MtbGpgP like Arg10, Arg60, and His159 are also strictly conserved in PhoE. Analysis of conservation based on alignment of primary sequences of members belonging to PGM family coupled with structural view of PhoE helped identify Gln22 as a signpost for phosphatase activity (29, 30). In agreement with the analysis, GpgP has Gln23 located at a structurally identical position. However, differences are observed in the composition of amino acids around Gln23 (Fig. 5B). This is not surprising because Gln23 is part of loop 2 (Arg10–Ser31), which plays a role in recognition of substrates, and both of these enzymes have different substrate specificities. In particular, Asp15, Gly19, and Ser20 of GpgP are replaced by lysine, glutamate, and arginine in PhoE, respectively, imparting different substrate specificities. ConSurf analysis of MtbGpgP (38) for structural conservation revealed that the region encompassing the active site is highly conserved, highlighting an evolutionarily conserved mechanism of catalysis (Fig. 5B). In contrast, the region in vicinity of the active site shows less conservation, indicating a role for this region in imparting substrate specificity.
The crystal structures of MtbGpgP described here unveil subtle structural maneuvers of the enzyme during catalysis. In particular, the movement of loop 2 is conspicuous in all the three structures. In its apo-enzyme form, the loop is partially covering the active site, conceivably to occlude nonsubstrate ligands. Once there is recognition, possibly by side chains of Asn17 and Gln23, the loop permits entry of the substrate into the active site. Amino acids like Arg10, Met22, Glu84, Trp90, His95, Trp109, Arg123, and Asn186 lining the active site probably assist in orienting and positioning the substrate optimally for catalysis. During catalysis, loop 2 covers the active site. This is clearly observed in the structure of the enzyme covalently bound with vanadate mimicking the transition state intermediate. Here, part of the loop assumes a one-turn helical conformation, resulting in the side chain of Asn17 moving inwards by 8.0 Å and rotating by 110° to make contact with the ligand. Once the catalysis is over, loop 2 moves away from the active site, permitting product release. This movement of loop 2 can be visualized by comparing the structures of vanadate-bound and PO4-bound MtbGpgP, depicting the transition state and product release states of the enzyme during catalysis, respectively. The side chain of Asn17 has moved more than 13.5 Å away and rotated by 180° from its position observed in the vanadate-bound structure (Fig. 4B). Thus, pivotal maneuvers of loop 2, as depicted in a model shown in Fig. 5C, probably assist in achieving substrate specificity and catalysis.
The structures of MtbGpgP reported here are the first for a GpgP belonging to the histidine phosphatase family. Because the RHG motif is highly conserved, the overall mechanism of catalysis of MtbGpgP is likely to be similar to other histidine phosphatases (31, 39–41). As proposed earlier for the structural homologue of MtbGpgP, PhoE (29), catalysis proceeds in two steps. In the first step, the phosphate group is transferred from glucosyl-3-phosphoglycerate to His11 of MtbGpgP. The NE1 nitrogen atom of His11 mounts a nucleophilic attack on the phosphorous atom of the phosphate moiety. A proton is shuttled from the carboxyl oxygen of Glu84 to the leaving group (Fig. 5D). This results in transfer of the phosphate group to His11 of MtbGpgP and release of the glucosylglycerate from the enzyme. Vanadate covalently linked to His11 in the MtbGpgP-VO3 structure mimics this phosphohistidine reaction intermediate. The excess charge on the phosphohistidine is probably stabilized by interactions with side chains of Arg10, Asn17, Gln23, Arg60, and His159 and the backbone amide nitrogen of Gly160. In the second half of the reaction, a water molecule activated by Glu84 mounts a nucleophilic attack on the phosphorous atom of the phosphohistidine (Fig. 5D). Here, the proton is returned back to Glu84, which can now serve as a proton donor again in the next cycle of catalysis. Numerous solvent molecules are observed around the vanadate and phosphate moieties in the structures of MtbGpgP, supporting such a role for water in catalysis. The nucleophilic attack by water results in release of orthophosphate and completion of the reaction (Fig. 5D). The GG thus formed is the substrate for di-glucosyl glycerate synthase that catalyzes the third step of the pathway leading to the biosynthesis of MGLPs.
The importance of MGLPs in the physiology of Mtb is highlighted by the fact that transposon-mediated mutagenesis has identified at least six essential genes: Rv3030, Rv3032, Rv1208, Rv0127, Rv1326c, and Rv1327c, that are likely to participate in the biosynthesis of MGLPs (42–47). GpgP (Rv2419c) catalyzes the second step of the pathway leading to the biosynthesis of MGLPs. Therefore, inhibitors targeting this enzyme could potentially aid in the elimination of the pathogen. In this context, the structures of MtbGpgP reported here could be invaluable for designing inhibitors of the enzyme with high specificity and potency.
Acknowledgments
We thank Z. Wang and L. Wang for help with MALS. We are grateful to the staff at Beamline BL17U of Shanghai Synchrotron Radiation Facility (China) and Beamline 5A of Photon Factory (Japan) for assistance in data collection.
This work was supported by Grants 2013CB733904 and 2014CB542800 from the Ministry of Science and Technology of China Project 973 and by National Science Foundation of China Grant 813300237.
The atomic coordinates and structure factors (codes 4PZ9, 4QIH, and 4PZA) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- Mtb
- Mycobacterium tuberculosis
- MGLPs
- methylglucose lipopolysaccharides
- GG
- glucosylglycerate
- GPG
- glucosyl-3-phosphoglycerate
- GpgP
- glucosyl-3-phosphoglycerate phosphatase
- PGM
- phosphoglycerate mutase
- MALS
- multiangle light scattering
- pNPP
- p-nitrophenyl phosphate
- RMSD
- root mean square deviation
- AA
- amino acid(s)
- Bis-Tris
- 2-(bis(2-hydroxyethyl)amino)-2-(hydroxymethyl)propane-1,3-diol.
REFERENCES
- 1. Daffé M., Draper P. (1998) The envelope layers of mycobacteria with reference to their pathogenicity. Adv. Microb. Physiol. 39, 131–203 [DOI] [PubMed] [Google Scholar]
- 2. Hett E. C., Rubin E. J. (2008) Bacterial growth and cell division: a mycobacterial perspective. Microbiol. Mol. Biol. Rev. 72, 126–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Brennan P. J. (2003) Structure, function, and biogenesis of the cell wall of Mycobacterium tuberculosis. Tuberculosis (Edinb.) 83, 91–97 [DOI] [PubMed] [Google Scholar]
- 4. Riley L. W. (2006) Of mice, men, and elephants: Mycobacterium tuberculosis cell envelope lipids and pathogenesis. J. Clin. Invest. 116, 1475–1478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ilton M., Jevans A. W., McCarthy E. D., Vance D., White H. B., 3rd, Bloch K. (1971) Fatty acid synthetase activity in Mycobacterium phlei: regulation by polysaccharides. Proc. Natl. Acad. Sci. U.S.A. 68, 87–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sassetti C. M., Boyd D. H., Rubin E. J. (2003) Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 48, 77–84 [DOI] [PubMed] [Google Scholar]
- 7. Tuffal G., Albigot R., Rivière M., Puzo G. (1998) Newly found 2-N-acetyl-2,6-dideoxy-β-glucopyranose containing methyl glucose polysaccharides in M. bovis BCG: revised structure of the mycobacterial methyl glucose lipopolysaccharides. Glycobiology 8, 675–684 [DOI] [PubMed] [Google Scholar]
- 8. Tuffal G., Ponthus C., Picard C., Rivière M., Puzo G. (1995) Structural elucidation of novel methylglucose-containing polysaccharides from Mycobacterium xenopi. Eur. J. Biochem. 233, 377–383 [DOI] [PubMed] [Google Scholar]
- 9. Kamisango K., Dell A., Ballou C. E. (1987) Biosynthesis of the mycobacterial O-methylglucose lipopolysaccharide. Characterization of putative intermediates in the initiation, elongation, and termination reactions. J. Biol. Chem. 262, 4580–4586 [PubMed] [Google Scholar]
- 10. Saier M. H., Jr., Ballou C. E. (1968) The 6-O-methylglucose-containig lipopolysaccharide of Mycobacterium phlei: complete structure of the polysaccharide. J. Biol. Chem. 243, 4332–4341 [PubMed] [Google Scholar]
- 11. Costa J., Empadinhas N., Gonçalves L., Lamosa P., Santos H., da Costa M. S. (2006) Characterization of the biosynthetic pathway of glucosylglycerate in the archaeon Methanococcoides burtonii. J. Bacteriol. 188, 1022–1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Machida Y., Bloch K. (1973) Complex formation between mycobacterial polysaccharides and fatty acyl-CoA derivatives. Proc. Natl. Acad. Sci. U.S.A. 70, 1146–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bergeron R., Machida Y., Bloch K. (1975) Complex formation between mycobacterial polysaccharides or cyclodextrins and palmitoyl coenzyme A. J. Biol. Chem. 250, 1223–1230 [PubMed] [Google Scholar]
- 14. Mendes V., Maranha A., Alarico S., da Costa M. S., Empadinhas N. (2011) Mycobacterium tuberculosis Rv2419c, the missing glucosyl-3-phosphoglycerate phosphatase for the second step in methylglucose lipopolysaccharide biosynthesis. Sci. Rep. 1, 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Empadinhas N., Marugg J. D., Borges N., Santos H., da Costa M. S. (2001) Pathway for the synthesis of mannosylglycerate in the hyperthermophilic archaeon Pyrococcus horikoshii. Biochemical and genetic characterization of key enzymes. J. Biol. Chem. 276, 43580–43588 [DOI] [PubMed] [Google Scholar]
- 16. Costa J., Empadinhas N., da Costa M. S. (2007) Glucosylglycerate biosynthesis in the deepest lineage of the bacteria: characterization of the thermophilic proteins GpgS and GpgP from Persephonella marina. J. Bacteriol. 189, 1648–1654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Empadinhas N., Albuquerque L., Henne A., Santos H., da Costa M. S. (2003) The bacterium Thermus thermophilus, like hyperthermophilic archaea, uses a two-step pathway for the synthesis of mannosylglycerate. Appl. Environ. Microbiol. 69, 3272–3279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rigden D. J. (2008) The histidine phosphatase superfamily: structure and function. Biochem. J. 409, 333–348 [DOI] [PubMed] [Google Scholar]
- 19. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 20. Long F., Vagin A. A., Young P., Murshudov G. N. (2008) BALBES: a molecular-replacement pipeline. Acta Crystallogr. D Biol. Crystallogr. 64, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Langer G., Cohen S. X., Lamzin V. S., Perrakis A. (2008) Automated macromolecular model building for x-ray crystallography using ARP/wARP version 7. Nat. Protoc. 3, 1171–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 23. Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., Adams P. D. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Terwilliger T. C., Klei H., Adams P. D., Moriarty N. W., Cohn J. D. (2006) Automated ligand fitting by core-fragment fitting and extension into density. Acta Crystallogr. D Biol. Crystallogr. 62, 915–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Takai A., Mieskes G. (1991) Inhibitory effect of okadaic acid on the p-nitrophenyl phosphate phosphatase activity of protein phosphatases. Biochem. J. 275, 233–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Some D. (2013) Light-scattering-based analysis of biomolecular interactions. Biophys. Rev. 5, 147–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rigden D. J., Littlejohn J. E., Henderson K., Jedrzejas M. J. (2003) Structures of phosphate and trivanadate complexes of Bacillus stearothermophilus phosphatase PhoE: structural and functional analysis in the cofactor-dependent phosphoglycerate mutase superfamily. J. Mol. Biol. 325, 411–420 [DOI] [PubMed] [Google Scholar]
- 30. Rigden D. J., Mello L. V., Setlow P., Jedrzejas M. J. (2002) Structure and mechanism of action of a cofactor-dependent phosphoglycerate mutase homolog from Bacillus stearothermophilus with broad specificity phosphatase activity. J. Mol. Biol. 315, 1129–1143 [DOI] [PubMed] [Google Scholar]
- 31. Bond C. S., White M. F., Hunter W. N. (2001) High resolution structure of the phosphohistidine-activated form of Escherichia coli cofactor-dependent phosphoglycerate mutase. J. Biol. Chem. 276, 3247–3253 [DOI] [PubMed] [Google Scholar]
- 32. Fothergill-Gilmore L. A., Watson H. C. (1989) The phosphoglycerate mutases. Adv. Enzymol. Relat. Areas Mol. Biol. 62, 227–313 [DOI] [PubMed] [Google Scholar]
- 33. Holm L., Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chiba Y., Horita S., Ohtsuka J., Arai H., Nagata K., Igarashi Y., Tanokura M., Ishii M. (2013) Structural units important for activity of a novel-type phosphoserine phosphatase from Hydrogenobacter thermophilus TK-6 revealed by crystal structure analysis. J. Biol. Chem. 288, 11448–11458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Davies D. R., Staker B. L., Abendroth J. A., Edwards T. E., Hartley R., Leonard J., Kim H., Rychel A. L., Hewitt S. N., Myler P. J., Stewart L. J. (2011) An ensemble of structures of Burkholderia pseudomallei 2,3-bisphosphoglycerate-dependent phosphoglycerate mutase. Acta Crystallogr. Sect. F. Struct. Biol. Cryst. Commun. 67, 1044–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Koonin E. V., Tatusov R. L. (1994) Computer analysis of bacterial haloacid dehalogenases defines a large superfamily of hydrolases with diverse specificity. Application of an iterative approach to database search. J. Mol. Biol. 244, 125–132 [DOI] [PubMed] [Google Scholar]
- 37. Thaller M. C., Schippa S., Rossolini G. M. (1998) Conserved sequence motifs among bacterial, eukaryotic, and archaeal phosphatases that define a new phosphohydrolase superfamily. Protein Sci. 7, 1647–1652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ashkenazy H., Erez E., Martz E., Pupko T., Ben-Tal N. (2010). ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 38, W529–W533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lee Y. H., Ogata C., Pflugrath J. W., Levitt D. G., Sarma R., Banaszak L. J., Pilkis S. J. (1996) Crystal structure of the rat liver fructose-2,6-bisphosphatase based on selenomethionine multiwavelength anomalous dispersion phases. Biochemistry 35, 6010–6019 [DOI] [PubMed] [Google Scholar]
- 40. Yuen M. H., Mizuguchi H., Lee Y. H., Cook P. F., Uyeda K., Hasemann C. A. (1999) Crystal structure of the H256A mutant of rat testis fructose-6-phosphate,2-kinase/fructose-2,6-bisphosphatase. Fructose 6-phosphate in the active site leads to mechanisms for both mutant and wild type bisphosphatase activities. J. Biol. Chem. 274, 2176–2184 [DOI] [PubMed] [Google Scholar]
- 41. Bond C. S., White M. F., Hunter W. N. (2002) Mechanistic implications for Escherichia coli cofactor-dependent phosphoglycerate mutase based on the high-resolution crystal structure of a vanadate complex. J. Mol. Biol. 316, 1071–1081 [DOI] [PubMed] [Google Scholar]
- 42. Stadthagen G., Sambou T., Guerin M., Barilone N., Boudou F., Korduláková J., Charles P., Alzari P. M., Lemassu A., Daffé M., Puzo G., Gicquel B., Rivière M., Jackson M. (2007) Genetic basis for the biosynthesis of methylglucose lipopolysaccharides in Mycobacterium tuberculosis. J. Biol. Chem. 282, 27270–27276 [DOI] [PubMed] [Google Scholar]
- 43. Empadinhas N., Albuquerque L., Mendes V., Macedo-Ribeiro S., da Costa M. S. (2008) Identification of the mycobacterial glucosyl-3-phosphoglycerate synthase. FEMS Microbiol. Lett. 280, 195–202 [DOI] [PubMed] [Google Scholar]
- 44. Mendes V., Maranha A., Alarico S., Empadinhas N. (2012) Biosynthesis of mycobacterial methylglucose lipopolysaccharides. Nat. Prod Rep. 29, 834–844 [DOI] [PubMed] [Google Scholar]
- 45. Mendes V., Maranha A., Lamosa P., da Costa M. S., Empadinhas N. (2010) Biochemical characterization of the maltokinase from Mycobacterium bovis BCG. BMC Biochem. 11, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Garg S. K., Alam M. S., Kishan K. V., Agrawal P. (2007) Expression and characterization of α-(1,4)-glucan branching enzyme Rv1326c of Mycobacterium tuberculosis H37Rv. Protein Expr. Purif. 51, 198–208 [DOI] [PubMed] [Google Scholar]
- 47. Syson K., Stevenson C. E., Rejzek M., Fairhurst S. A., Nair A., Bruton C. J., Field R. A., Chater K. F., Lawson D. M., Bornemann S. (2011) Structure of Streptomyces maltosyltransferase GlgE, a homologue of a genetically validated anti-tuberculosis target. J. Biol. Chem. 286, 38298–38310 [DOI] [PMC free article] [PubMed] [Google Scholar]