

Background: The SUMO system is involved in double-strand break (DSB) repair.
Results: SUMO2/3 is required for the major NHEJ pathway; SUMO1 stimulates all DSB repair pathways, and a non-conjugatable form of SUMO1 stimulates DSB repair pathways involving DNA end resection.
Conclusion: SUMO1 and SUMO2/3 have distinct activities in DSB repair.
Significance: SUMO1 can stimulate DSB repair as a free protein.
Keywords: DNA Damage Response, Homologous Recombination, Small Ubiquitin-like Modifier (SUMO), SUMO-interacting Motif (SIM), Sumoylation, UBC9, Double-strand Break Repair, Non-homologous End Joining
Abstract
Small ubiquitin-like modifier (SUMO) proteins act in DNA double-strand break (DSB) repair, but the pathway specificity of the three major isoforms has not been defined. In experiments in which we depleted the endogenous SUMO protein by RNAi, we found that SUMO1 functioned in all subpathways of either homologous recombination (HR) or non-homologous end joining (NHEJ), whereas SUMO2/3 was required for the major NHEJ pathway, called conservative NHEJ, but dispensable in other DSB repair pathways. To our surprise, we found that depletion of UBC9, the unique SUMO E2 enzyme, had no effect in HR or alternative NHEJ (Alt-NHEJ) but was required for conservative NHEJ. Consistent with this result, both non-conjugatable mutant and wild-type SUMO1 proteins functioned similarly in HR and Alt-NHEJ. These results detail the functional roles of specific SUMO isoforms in DSB repair in mammalian cells and reveal that SUMO1 functions in HR or Alt-NHEJ as a free protein and not as a protein conjugate.
Introduction
DNA double-strand breaks (DSB)2 present a major problem in genome maintenance because the repair machinery must bridge a gap of indeterminate composition. Two mechanistically distinct pathways are present for DSB repair in mammalian cells: homologous recombination (HR) and non-homologous end joining (NHEJ), competing for the repair of DSBs (1–3). There are two major mechanisms present in HR: the error-free homology-directed repair (HDR) pathway and the error-prone single-strand annealing (SSA). HDR and SSA pathways utilize sequence homology and DNA end resection for repair of DSBs (3). Similarly, eukaryotic cells utilize two pathways of NHEJ, the major NHEJ, called conservative NHEJ (C-NHEJ), in which DSB ends are ligated without homology and which protects DSB ends with minimal processing (4, 5), and the alternative NHEJ (Alt-NHEJ), which depends on DNA end resection at the DSB to generate single strands that anneal via microhomology (5–7).
In vertebrates, there are three functional forms of SUMO family proteins: SUMO1, SUMO2, and SUMO3. SUMO2 and SUMO3 share about 95% sequence identity but are only 45% identical in sequence to SUMO1, thus forming a distinct subfamily as SUMO2/3 (8). The conjugation of SUMO isoforms onto target protein is designated as SUMOylation, an enzymatic cascade triggered by an E1 SUMO-activating enzyme (SAE1/SAE2), followed by a single E2-conjugating enzyme, UBC9, and an E3 SUMO ligase, resulting in a covalent isopeptide bond between the lysine of target protein and glycine-glycine dipeptide at the carboxyl terminus of the activated SUMO (9, 10).
The SUMO system has been shown to have strong ties to DSB repair. Abolition of activity of SUMO E3 enzymes in human cells impairs DSB repair (11–13). Mutation of the single SUMO E2 enzyme UBC9 in yeast or human cells results in defects in DNA repair, including recombination abnormality and impaired DSB repair (14–16). Furthermore, many DSB repair proteins are modified by SUMO (11, 12, 14, 17–22). Nonetheless, cells expressing individual SUMOylation-defective HR protein mutants often lack notable phenotypes (20, 23). SUMO modification of individual NHEJ proteins however, regulates their function in DSB repair (14, 21, 22).
SUMO1 has been discovered as a non-covalent binding partner via SUMO-interacting motifs (SIMs) for human HR proteins including RAD51, RAD52, and replication protein A (RPA) (24–28). The SIM-dependent non-covalent binding to SUMO1 is required for loading of the recombinase RAD51 onto resected DSB ends for HR-mediated repair (19, 29).
In this study, we identified the roles for SUMO isoforms in all four DSB repair subpathways. We found that SUMO1 stimulated all four pathways whereas SUMO2/3 was required only in the C-NHEJ pathway. Surprisingly, the single SUMO E2 enzyme UBC9 was dispensable for HR and Alt-NHEJ, and the conjugation-deficient SUMO1 mutant protein was competent for HR and Alt-NHEJ repair. In contrast, UBC9 was required for C-NHEJ and the SUMO1 mutant was defective in this pathway as compared with the wild type. We conclude that although C-NHEJ is SUMOylation-dependent, the HR and Alt-NHEJ pathways are stimulated by non-covalent SUMO1 interactions.
EXPERIMENTAL PROCEDURES
Homologous Recombination and Non-homologous End-joining Assays
HDR and SSA assays were performed as described previously in HeLa cells (30, 31). The repair of double-strand break by Alt-NHEJ pathway was based on a vector kindly provided by Jeremy Stark (Beckman Research Institute of the City of Hope) (32) stably integrated into HeLa genome. On day 1, the appropriate cell line was seeded in 15.6-mm-diameter wells. The next day, cells, 50% confluent, were transfected with 30 pmol of each siRNA in the presence of 3 μl of Oligofectamine (Life Technologies). On day 3, cells were transferred to 35-mm-diameter wells. At 48 h after transfection, cells were retransfected with 50 pmol of the same siRNA in the presence of 5 μl of Lipofectamine 2000 (Life Technologies), plus 3 μg of an I-SceI endonuclease expression vector, which causes a DSB cut in the recombination substrate integrated in the genome. In each transfection, the total siRNA amount was adjusted to be the same in each well by adding siControl. On day 7, cells were trypsinized, and 10,000 cells from each well were counted using a FACSCalibur flow cytometer (BD Biosciences) in the Ohio State University Comprehensive Cancer Center Analytical Cytometry shared resource for the percentage of GFP-positive cells.
The C-NHEJ assay utilized quantitative real-time PCR and was carried out as described (33) with the following modifications in 293 cells. Two rounds of transfection procedure were done as above. The genomic DNA isolated 3 days after transfection of the I-SceI plasmid was digested with the restriction enzyme XhoI and purified by Qiagen PCR purification kit before real-time PCR was applied. RPS17 probe (Hs00734303_g1, Applied Biosystems) was used as an internal control, and the quantitative ΔΔCT method was used to analyze the data.
For plasmid add-back in the rescue assay for all four DSB repair pathways, the transfection procedure was the same except for the amount of reagents used; in the first transfection, 30 pmol of siSUMO1-3′ and 0.75 μg of SUMO1 expression plasmid were added to the cells in the presence of 1.5 μl of Lipofectamine 2000. At 48 h after transfection, cells were retransfected with 50 pmol of siSUMO1-3′ and 1.5 μg of SUMO1 plasmid plus 1.5 μg of I-SceI expression vector in 2.5 μl of Lipofectamine 2000.
RNA Interference and Plasmids
The following siRNAs were used were produced by Sigma: siControl targeting the luciferase gene, 5′-CGUACGCGGAAUACUUCGA-3′(30); siBRCA1, 5′-GCUCCUCUCACUCUUCAGU-3′(30); siLigase IV, 5′-AGGAAGUAUUCUCAGGAAUUA-3′(11); siSUMO1-1, 5′-CUGGGAAUGGAGGAAGAAG-3′(34); siSUMO2/3, 5′-GUCAAUGAGGCAGAUCAGA-3′(35); siUBC9, 5′-CAAAAAAUCCCGAUGGCAC-3′(34); siSUMO1-3′ starting at nucleotide 850, 5′-GGAAAUUGCACAUGGUACA-3′. I-SceI expression plasmid has been previously described (30) and was a kind gift from Maria Jasin (Memorial Sloan-Kettering Cancer Institute). Wild-type SUMO1 expression plasmid (pFLAG-CMV-2-SUMO1) (36) was a kind gift from Lirim Shemshedini (University of Toledo). SUMO1-ΔGG expression plasmid was constructed by PCR amplification from the wild-type SUMO1 plasmid using the following primers: forward 5′-CGGATCCATGTCTGACCAG-3′; reverse 5′-CCCGGGTCACGTTTGTTCCTG-3′. The PCR-amplified fragment was then ligated into pFLAG-CMV-2 vector by digestion using BamHI and SmaI.
Immunoblot Analysis
Whole cell lysates were harvested in lysis buffer (50 mm Tris, pH 7.9, 300 mm NaCl, 0.5% Nonidet-40, 1 mm EDTA, 5% glycerol, 1 mm phenylmethylsulfonyl fluoride, 1 mm dithiothreitol, 1× complete protease inhibitor mixture from Sigma) after 3 days following the second transfection step in HeLa cells and subjected to immunoblot analysis using the following primary antibodies: anti-SUMO1 (34), anti-SUMO2/3 (Abcam), anti-UBC9 (BD Transduction Laboratories), anti-β-actin (Cell Signaling), anti-GAPDH (Advanced ImmunoChemical Inc.), anti-MRE11 (Calbiochem), anti-Ku70 (Neomarkers, Ab-5), anti-Ku80 (Neomarkers, Ab-7), anti-RAD51 (Santa Cruz Biotechnology, H-92), anti-RAD52 (purified from rabbit serum), anti-RNA helicase A (RHA) (purified from rabbit serum), and anti-FLAG M2, affinity-purified (Sigma).
Statistical Analysis
Data were compared between different groups for each sample by unpaired and two-tailed Student's t test (*, **, and *** represent p < 0.05, p < 0.01, and p < 0.001, respectively).
RESULTS
SUMO1 and SUMO2/3 Function Differently in DSB Repair Pathways
SUMO proteins have been shown to be involved in DSB repair (11, 12, 14, 19, 21). We used HeLa- or 293-derived cell lines with the specific recombination substrate DNA integrated in the genome to test the specificity of SUMO isoforms in each DSB repair pathway (Fig. 1, A–D, right). siRNA-dependent depletion of each isoform in the appropriate cell line specifically probes the two HR pathways, HDR and SSA, and the two NHEJ pathways, Alt-NHEJ and C-NHEJ (30–33). BRCA1 and Ligase IV, which are known to regulate DSB repair (30, 31, 37, 38), served as positive controls in three functional DSB repair assays: BRCA1 in HDR and SSA (Fig. 1, A and B) and Ligase IV in C-NHEJ (Fig. 1D). Depletion of SUMO1 (Fig. 1E) reduced repair in all four subpathways tested to about 62% HDR, 31% SSA, 41% Alt-NHEJ, and 39% C-NHEJ, respectively, relative to the control siRNA (Fig. 1, A–D), suggesting that the SUMO1 isoform is stimulatory in all mechanisms of DSB repair. On the other hand, depletion of SUMO2/3 had as significant a deficit in the C-NHEJ pathway as did depletion of Ligase IV (Fig. 1D). In the assays for the other three DSB pathways, depletion of SUMO2/3 increased the levels of HDR, SSA and Alt-NHEJ, although these increases were not statistically significant (Fig. 1, A–C). This result reveals that, in contrast to SUMO1, the SUMO2/3 isoforms are required for C-NHEJ, and either these isoforms do not participate in homologous recombination and Alt-NHEJ or these isoforms have a modest inhibitory activity.
FIGURE 1.

SUMO isoforms function differently in DSB repair pathways. A–C, the recombination substrates are diagrammed on the right with details described previously (30–32). iGFP indicates inactive GFP gene. HeLa-derived cell lines for HDR (A), SSA (B), and Alt-NHEJ (C) were subjected to two rounds of siRNA transfection, as indicated, followed by transfection of the I-SceI expression plasmid to induce DSB. After 3 days, the percentages of GFP-positive cells were determined by flow cytometry. In each experiment, the percentage of GFP-positive cells from control siRNA transfections was set equal to 1, and the fraction of GFP-positive cells was determined relative to the control siRNA (Con) to measure HDR, SSA, and Alt-NHEJ, respectively. Results (mean ± S.E.) are from three independent experimental replicates. NT indicates no transfection of the I-SceI-expressing plasmid. D, the C-NHEJ repair substrate in the genome of 293 cells is diagrammed on the right as described previously (33). C-NHEJ assay was done by transfecting cells with the indicated siRNAs as in panel A. After 3 days, the repair efficiency was measured by quantitative real-time PCR on extracted genomic DNA, represented by the percentage on the y axis. In each experiment, the yield of repaired DNA was normalized relative to the value of the result from the control siRNA transfection. E, immunoblots show the depletion of indicated protein by RNAi interference in HeLa cells. Upon siSUMO2/3 transfection, the bottom band (∼15 kDa) of the doublet was depleted. GAPDH and β-actin were used as loading controls. The positions of the molecular mass markers in kDa are indicated at the left. F, results (mean ± S.E.) from each functional DSB repair assay were summarized for the indicated siRNA transfection.
Studies have shown that SUMO1 and SUMO2/3 serve distinct functions in mammalian cells as they are conjugated to different target proteins in vivo (8, 39, 40), which is consistent with our result that SUMO1 and SUMO2/3 function differently in the DSB repair pathways. Co-depletion of SUMO1 and SUMO2/3 in C-NHEJ resulted in a similar level of repair as single depletion of SUMO2/3, indicating that the SUMO isoforms were participating in the same pathway (Fig. 1D). Co-depletion of SUMO1 and SUMO2/3 reduced HDR to a similar level as SUMO1 depletion alone, and single depletion of SUMO2/3 suggested that these isoforms do not function in HDR (Fig. 1A). By contrast, co-depletion of SUMO2/3 partially rescued the defect caused by SUMO1 depletion in SSA and Alt-NHEJ, consistent with the concept that SUMO2/3 has a modest repressive effect on these pathways of DSB repair (Fig. 1, B and C).
Depletions of protein by siRNAs were confirmed by immunoblot (examples shown in Fig. 1E). The above results from functional DSB repair assays were summarized in Fig. 1F. Depletion of SUMO2/3 minimally impacts homologous recombination or Alt-NHEJ, but these isoforms are required in C-NHEJ. By contrast, SUMO1 is stimulatory in all DSB repair subpathways.
Because SUMO isoforms have a major influence on DSB repair pathway choice, it is necessary to rule out the possibility that depletion of SUMO proteins affects cell cycle progression in mammalian cells. We depleted endogenous SUMO1, SUMO2/3, or UBC9, respectively, by siRNA transfection in HeLa cells and assessed whether the population of cells was blocked at a certain cell cycle stage by flow cytometry measurement at time points of 48, 72, and 96 h after transfection (Fig. 2A). Depletion of either protein had no effect on passage through the normal cell cycle, indicating that the DSB repair deficits caused by loss of the endogenous SUMO isoforms or UBC9 protein are not due to cell cycle blockage. Next we tested whether a secondary effect on DSB repair protein stability is present upon depleting SUMO isoforms because SUMOylation occurs commonly as a post-translational modification that might regulate target protein degradation. Following depletion of SUMO isoforms or UBC9 by siRNA transfection in HeLa cells, we assessed protein abundance of SUMOylation targets in DSB repair: RAD52, MRE11, and Ku70/80 complex, as well as RAD51, which is not a known SUMOylation target. We found that the depletions had no effect on protein abundance of these repair factors as compared with control siRNA (Fig. 2B).
FIGURE 2.

SUMO isoform depletion has no effect on cell cycle progression and DSB repair protein stability. A, endogenous SUMO1, SUMO2/3, or UBC9 was depleted by two rounds of siRNA transfection in HeLa cells. Cell cycle analysis by flow cytometry was carried out at 48, 72, and 96 h after the second transfection. DNA content of the HeLa cells, as determined by staining with propidium iodide, was measured by FACS analysis. B, depletion of SUMO isoforms or UBC9 by two rounds of siRNA transfection was done in HeLa cells as in A. Con, control; S1, SUMO1; S2/3, SUMO2/3. Analysis of immunoblots was applied to measure protein abundance using specific antibody against the DSB repair protein. RNA helicase A (RHA) was used as a loading control, and the positions of the molecular mass markers in kDa are indicated at the left.
UBC9 Is Dispensable for HR or Alt-NHEJ
UBC9 is the only SUMO E2-conjugating enzyme, and it has been implicated in the DNA damage response in animal models and human cells (11, 13, 16, 22, 41, 42). Depletion of UBC9 was included in the experiments in Fig. 1 to test simultaneously for all three SUMO isoforms, and we were surprised to note that UBC9 depletion did not affect three of the four subpathways tested (Fig. 1 and summarized in Fig. 1F). UBC9 was effectively depleted (Fig. 1E), and there was a phenotype due to UBC9 depletion in the C-NHEJ assay, indicating that the observed level of depletion was sufficient to cause a repair defect. UBC9 depletion reduced the C-NHEJ repair efficiency to about 19% as compared with the control siRNA, similar in magnitude to depletion of Ligase IV, suggesting that UBC9 was important for this pathway, consistent with the result that SUMO2/3 was required for C-NHEJ (Fig. 1D).
Because UBC9 was dispensable in three pathways and required in C-NHEJ, we hypothesized that SUMO1 function in HDR, SSA, and Alt-NHEJ was independent of conjugation to another protein. There have been other examples in the published literature of SUMO proteins functioning independent of covalent binding to a target protein. As an example, UBC9 has been shown to have no effect on SUMO1-mediated repression of BRCA1-induced transcriptional activity stimulated by DNA damage (43). Together, our results suggest that the stimulatory effect on homologous recombination or Alt-NHEJ by SUMO1 is not mediated by SUMO conjugation.
Free, Non-conjugated SUMO1 Stimulates HR and Alt-NHEJ
Conjugation of SUMO onto substrates requires the covalent interaction between the carboxyl terminus of SUMO and lysine acceptors on target proteins via an isopeptide bond (9, 10, 44, 45). Deletion of the carboxyl-terminal two glycines from the SUMO1 protein renders it incompetent for covalent conjugation to another protein (25, 36, 46). To test whether SUMO1 functions in homologous recombination and Alt-NHEJ without covalent modification, we transfected into cells an siRNA targeting the SUMO1 3′-UTR and a plasmid expressing the SUMOylation-incompetent SUMO1-ΔGG protein, which truncates the critical two carboxyl-terminal glycine residues and which is resistant to the SUMO1-targeting siRNA. Depletion of endogenous SUMO1 and expression of a FLAG-tagged wild-type SUMO1 rescued the DSB repair defect in all four DSB repair assays (Fig. 3, A–D). The exogenous SUMO1 was expressed at slightly lower levels than the endogenous protein (Fig. 3E, upper panel, lanes 1, 3, and 4), and in immunoblots probing for the FLAG epitope, we could detect the major SUMOylation modification of RanGAP1 migrating at a position consistent with a mass of 90 kDa (8, 46) (Fig. 3E, middle panel, lane 3). The SUMO1-ΔGG protein was expressed at similar levels as the wild-type protein and did not result in the SUMO1 conjugation product (Fig. 3E, middle panel, lane 4). Using this protocol to replace the endogenous SUMO1 with either wild-type or conjugation-defective SUMO1, we assayed for the specific DSB repair assays as in Fig. 1. Just as observed in Fig. 1, transfection of this 3′-UTR-specific siRNA (SUMO1-3′) depleted endogenous SUMO1 protein (Fig. 3E, upper panel, lane 2) and reproducibly yielded inhibition of all four DSB repair pathways, as did the siRNA targeting the SUMO1 coding region (Fig. 3, A–D, lane 3). When the 3′-UTR specific siRNA was co-transfected with a plasmid expressing wild-type SUMO1 resistant to this siRNA, repair efficiency in HDR was restored back to 87% of activity relative to the control siRNA (Fig. 3A, lane 4). Transfection of the non-conjugatable SUMO1-ΔGG plasmid rescued DNA repair in the HDR, SSA, and Alt-NHEJ assays to a similar amount as had the wild-type SUMO1 (Fig. 3, A–C, lane 5). These results unambiguously demonstrate that the stimulatory function of SUMO1 in the HDR, SSA, and Alt-NHEJ pathways was independent of conjugation to a target protein. By contrast, in the C-NHEJ assay, transfection of the wild-type SUMO1-expressing plasmid partially rescued repair, but transfection of the SUMO1-ΔGG-expressing plasmid did not (Fig. 3D). These results together with the data of UBC9 effect on DSB repair pathways (Fig. 1F) further support a SUMOylation-independent mechanism for the action of SUMO1 on homologous recombination and alternative NHEJ, as well as a conjugation-dependent role of SUMO protein, SUMO1, and also SUMO2/3, in conservative-NHEJ.
FIGURE 3.

Non-conjugated SUMO1 stimulates HR and Alt-NHEJ. A–D, the appropriate cell line was transfected with siSUMO1-3′ targeting the 3′-UTR of the SUMO1 mRNA plus a wild-type SUMO1 or SUMO1-ΔGG expression plasmid or an empty vector and assayed in DSB repair as in Fig. 1. The fraction of GFP-positive HeLa cells was determined by flow cytometry as a measure of repair efficiency of HDR, SSA, and Alt-NHEJ, respectively (A–C), or genomic DNA was extracted from 293 cells for quantitative real-time PCR analysis (D). NT indicates no transfection of the I-SceI-expressing plasmid. Results are mean ± S.E. E, whole cell lysates were extracted from HeLa cells 3 days after the second transfection and subjected to immunoblot analysis. SUMO1 specific antibody detected both endogenous SUMO1 and expressed FLAG-SUMO1 protein. FLAG specific antibody detected FLAG-SUMO1-conjugated protein. The asterisk indicates a nonspecific band. The β-actin protein was a loading control. The positions of the molecular mass markers in kDa are indicated at the left. Vec, vector; Con, control siRNA.
DISCUSSION
Together, our findings identify that SUMO isoforms act differently in double-strand break repair pathways. SUMO1 stimulates all subpathways; SUMO2/3, on the other hand, is required for the C-NHEJ pathway but dispensable for the other pathways. Strikingly, our data reveal a novel role of SUMO1 as a free protein, not a protein conjugate in homologous recombination and alternative NHEJ.
A previous observation had shown that overexpression of either wild type or SUMO1-ΔGG mutant inhibited homologous recombination in mammalian cells (25). The present study has different results, perhaps because we did not utilize overexpression, but rather depletion, and clearly indicate that SUMO1 stimulates homologous recombination. Two recent studies have found that non-covalent interaction between yeast SUMO and RAD51 via its SIM has an important role in RAD51 accumulation at DNA damage sites, a crucial step in HR-mediated DSB repair (19, 29). It is then not totally surprising to now discover that SUMO conjugation is dispensable in HR and Alt-NHEJ.
We propose a model in which SUMO1 acts via different mechanisms in response to DSB damage in mammalian cells (Fig. 4). Following DSB, either end resection or end protection occurs depending on the specific DSB repair mechanism utilized to repair the damage. HDR and SSA require more extensive end resection than Alt-NHEJ because microhomology can be revealed by minimal resection near DNA ends. In the HDR pathway, SUMO1 binds non-covalently to an HR factor, such as RAD51, via its SIM (19, 26, 29, 47). Although RAD51 is known not to be SUMOylated, this interaction is crucial for the loading of RAD51 onto the resected DNA ends (19, 29) (Fig. 4A). Similarly, when the DSB is repaired via the SSA or Alt-NHEJ pathway, an interaction between SUMO1 and the SIM of a given repair factor at the resected ends stimulates the efficient repair independent of covalent SUMOylation (Fig. 4, B and C). By contrast, the C-NHEJ pathway, in which DSB ends are ligated without homology and resection, requires conjugation of SUMO1 and SUMO2/3 to the repair protein substrates for DSB repair (Fig. 4D). Therefore, end resection upon DSB might be a common step stimulated by non-conjugatable SUMO1.
FIGURE 4.
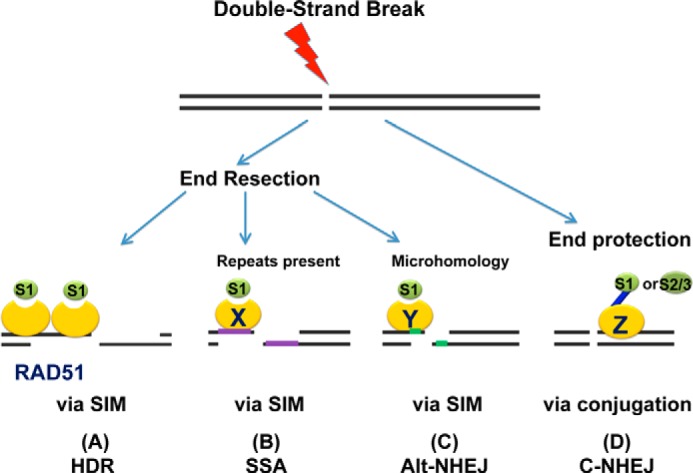
Model for SUMO proteins function in DSB repair. Following DSB, end resection is required for HR or Alt-NHEJ repair; otherwise, DNA ends are protected from minimal processing and C-NHEJ is employed to repair the damage. A, in the HDR pathway, SUMO1 (S1) stimulates the repair process by a non-covalent binding to SIM of RAD51 or other repair proteins. We model that the SUMO1-SIM interaction regulates RAD51 accretion on resected DSB ends. B and C, when SSA or Alt-NHEJ is utilized in response to a DSB, SUMO1 interacts non-covalently with an unknown repair factor (X or Y) via the SIM of the protein at sites of DSB to promote the subsequent repair events. D, in C-NHEJ, in which DNA ends are protected, SUMO1 and SUMO2/3 (S2/3) conjugation to an unknown target repair mediator (Z) is essential for efficient repair.
The results of this study apply to the cell in which a single DNA break is induced. However, it is possible that covalent conjugation of SUMO1 may be important under other circumstances not tested in this study. For example, conjugation of SUMO1 has been found to be important for the HR-mediated repair of replication forks, elimination of protein conjugates from DNA ends, and removal of interstrand DNA crosslinks, or when the DNA damage load is extensive, such as after exposure of cells to a high radiation dose or a DNA-alkylating agent (19, 47–50).
Notably, cells expressing individual SUMOylation-defective HR protein mutants often exhibit mild phenotypes (20, 23). Consistent with these data, our model suggests that non-covalent SUMO1 interaction mediated by SIM of the substrate may represent a mechanism that could stimulate both homologous recombination and alternative NHEJ in DSB repair.
Acknowledgments
We are grateful to Jeremy Stark (Beckman Research Institute of the City of Hope) and to Lirim Shemshedini (University of Toledo, Department of Biological Sciences) for the kind gift of plasmid reagents used in this study. We are grateful to Fen Xia (Ohio State University) for the kind gift of the C-NHEJ cell line and various DNA repair antibodies.
This work was supported, in whole or in part, by National Institutes of Health Grant CA141090 (to J. D. P.).
- DSB
- double-strand break
- HR
- homologous recombination
- HDR
- homology-directed repair
- SSA
- single-strand annealing
- NHEJ
- non-homologous end joining
- C-NHEJ
- conservative NHEJ
- Alt-NHEJ
- alternative NHEJ
- SIM
- SUMO-interacting motif
- si
- siRNA.
REFERENCES
- 1. Bunting S. F., Callén E., Wong N., Chen H. T., Polato F., Gunn A., Bothmer A., Feldhahn N., Fernandez-Capetillo O., Cao L., Xu X., Deng C. X., Finkel T., Nussenzweig M., Stark J. M., Nussenzweig A. (2010) 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 141, 243–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Daley J. M., Palmbos P. L., Wu D., Wilson T. E. (2005) Nonhomologous end joining in yeast. Annu. Rev. Genet. 39, 431–451 [DOI] [PubMed] [Google Scholar]
- 3. Pâques F., Haber J. E. (1999) Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 63, 349–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Burma S., Chen B. P., Chen D. J. (2006) Role of non-homologous end joining (NHEJ) in maintaining genomic integrity. DNA Repair 5, 1042–1048 [DOI] [PubMed] [Google Scholar]
- 5. Guirouilh-Barbat J., Huck S., Bertrand P., Pirzio L., Desmaze C., Sabatier L., Lopez B. S. (2004) Impact of the KU80 pathway on NHEJ-induced genome rearrangements in mammalian cells. Mol. Cell 14, 611–623 [DOI] [PubMed] [Google Scholar]
- 6. Corneo B., Wendland R. L., Deriano L., Cui X., Klein I. A., Wong S. Y., Arnal S., Holub A. J., Weller G. R., Pancake B. A., Shah S., Brandt V. L., Meek K., Roth D. B. (2007) Rag mutations reveal robust alternative end joining. Nature 449, 483–486 [DOI] [PubMed] [Google Scholar]
- 7. Haber J. E. (2008) Alternative endings. Proc. Natl. Acad. Sci. U.S.A. 105, 405–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Saitoh H., Hinchey J. (2000) Functional heterogeneity of small ubiquitin-related protein modifiers SUMO-1 versus SUMO-2/3. J. Biol. Chem. 275, 6252–6258 [DOI] [PubMed] [Google Scholar]
- 9. Bergink S., Jentsch S. (2009) Principles of ubiquitin and SUMO modifications in DNA repair. Nature 458, 461–467 [DOI] [PubMed] [Google Scholar]
- 10. Hay R. T. (2005) SUMO: a history of modification. Mol. Cell 18, 1–12 [DOI] [PubMed] [Google Scholar]
- 11. Galanty Y., Belotserkovskaya R., Coates J., Polo S., Miller K. M., Jackson S. P. (2009) Mammalian SUMO E3-ligases PIAS1 and PIAS4 promote responses to DNA double-strand breaks. Nature 462, 935–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Morris J. R., Boutell C., Keppler M., Densham R., Weekes D., Alamshah A., Butler L., Galanty Y., Pangon L., Kiuchi T., Ng T., Solomon E. (2009) The SUMO modification pathway is involved in the BRCA1 response to genotoxic stress. Nature 462, 886–890 [DOI] [PubMed] [Google Scholar]
- 13. Potts P. R., Yu H. (2005) Human MMS21/NSE2 is a SUMO ligase required for DNA repair. Mol. Cell. Biol. 25, 7021–7032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cremona C. A., Sarangi P., Yang Y., Hang L. E., Rahman S., Zhao X. (2012) Extensive DNA damage-induced sumoylation contributes to replication and repair and acts in addition to the Mec1 checkpoint. Mol. Cell 45, 422–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Maeda D., Seki M., Onoda F., Branzei D., Kawabe Y., Enomoto T. (2004) Ubc9 is required for damage-tolerance and damage-induced interchromosomal homologous recombination in S. cerevisiae. DNA Repair 3, 335–341 [DOI] [PubMed] [Google Scholar]
- 16. Mo Y. Y., Yu Y., Ee P. L., Beck W. T. (2004) Overexpression of a dominant-negative mutant Ubc9 is associated with increased sensitivity to anticancer drugs. Cancer Res. 64, 2793–2798 [DOI] [PubMed] [Google Scholar]
- 17. Altmannova V., Eckert-Boulet N., Arneric M., Kolesar P., Chaloupkova R., Damborsky J., Sung P., Zhao X., Lisby M., Krejci L. (2010) Rad52 SUMOylation affects the efficiency of the DNA repair. Nucleic Acids Res. 38, 4708–4721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dou H., Huang C., Singh M., Carpenter P. B., Yeh E. T. (2010) Regulation of DNA repair through deSUMOylation and SUMOylation of replication protein A complex. Mol. Cell 39, 333–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Psakhye I., Jentsch S. (2012) Protein group modification and synergy in the SUMO pathway as exemplified in DNA repair. Cell 151, 807–820 [DOI] [PubMed] [Google Scholar]
- 20. Sacher M., Pfander B., Hoege C., Jentsch S. (2006) Control of Rad52 recombination activity by double-strand break-induced SUMO modification. Nature Cell Biol. 8, 1284–1290 [DOI] [PubMed] [Google Scholar]
- 21. Yurchenko V., Xue Z., Sadofsky M. J. (2006) SUMO modification of human XRCC4 regulates its localization and function in DNA double-strand break repair. Mol. Cell. Biol. 26, 1786–1794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhao X., Blobel G. (2005) A SUMO ligase is part of a nuclear multiprotein complex that affects DNA repair and chromosomal organization. Proc. Natl. Acad. Sci. U.S.A. 102, 4777–4782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ohuchi T., Seki M., Branzei D., Maeda D., Ui A., Ogiwara H., Tada S., Enomoto T. (2008) Rad52 sumoylation and its involvement in the efficient induction of homologous recombination. DNA Repair 7, 879–889 [DOI] [PubMed] [Google Scholar]
- 24. Kerscher O. (2007) SUMO junction-what's your function? New insights through SUMO-interacting motifs. EMBO Rep. 8, 550–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li W., Hesabi B., Babbo A., Pacione C., Liu J., Chen D. J., Nickoloff J. A., Shen Z. (2000) Regulation of double-strand break-induced mammalian homologous recombination by UBL1, a RAD51-interacting protein. Nucleic Acids Res. 28, 1145–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shen Z., Pardington-Purtymun P. E., Comeaux J. C., Moyzis R. K., Chen D. J. (1996) UBL1, a human ubiquitin-like protein associating with human RAD51/RAD52 proteins. Genomics 36, 271–279 [DOI] [PubMed] [Google Scholar]
- 27. Song J., Durrin L. K., Wilkinson T. A., Krontiris T. G., Chen Y. (2004) Identification of a SUMO-binding motif that recognizes SUMO-modified proteins. Proc. Natl. Acad. Sci. U.S.A. 101, 14373–14378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Takahashi H., Hatakeyama S., Saitoh H., Nakayama K. I. (2005) Noncovalent SUMO-1 binding activity of thymine DNA glycosylase (TDG) is required for its SUMO-1 modification and colocalization with the promyelocytic leukemia protein. J. Biol. Chem. 280, 5611–5621 [DOI] [PubMed] [Google Scholar]
- 29. Shima H., Suzuki H., Sun J., Kono K., Shi L., Kinomura A., Horikoshi Y., Ikura T., Ikura M., Kanaar R., Igarashi K., Saitoh H., Kurumizaka H., Tashiro S. (2013) Activation of the SUMO modification system is required for the accumulation of RAD51 at sites of DNA damage. J. Cell Sci. 126, 5284–5292 [DOI] [PubMed] [Google Scholar]
- 30. Ransburgh D. J., Chiba N., Ishioka C., Toland A. E., Parvin J. D. (2010) Identification of breast tumor mutations in BRCA1 that abolish its function in homologous DNA recombination. Cancer Res. 70, 988–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Towler W. I., Zhang J., Ransburgh D. J., Toland A. E., Ishioka C., Chiba N., Parvin J. D. (2013) Analysis of BRCA1 variants in double-strand break repair by homologous recombination and single-strand annealing. Hum. Mutat. 34, 439–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bennardo N., Cheng A., Huang N., Stark J. M. (2008) Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. 4, e1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhuang J., Jiang G., Willers H., Xia F. (2009) Exonuclease function of human Mre11 promotes deletional nonhomologous end joining. J. Biol. Chem. 284, 30565–30573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu H. W., Zhang J., Heine G. F., Arora M., Gulcin Ozer H., Onti-Srinivasan R., Huang K., Parvin J. D. (2012) Chromatin modification by SUMO-1 stimulates the promoters of translation machinery genes. Nucleic Acids Res. 40, 10172–10186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Guo W. Z., Sugaya S., Satoh M., Tomonaga T., Nomura F., Hiwasa T., Takiguchi M., Kita K., Suzuki N. (2009) Nm23-H1 is responsible for SUMO-2-involved DNA synthesis induction after X-ray irradiation in human cells. Arch. Biochem. Biophys. 486, 81–87 [DOI] [PubMed] [Google Scholar]
- 36. Zheng Z., Cai C., Omwancha J., Chen S. Y., Baslan T., Shemshedini L. (2006) SUMO-3 enhances androgen receptor transcriptional activity through a sumoylation-independent mechanism in prostate cancer cells. J. Biol. Chem. 281, 4002–4012 [DOI] [PubMed] [Google Scholar]
- 37. Critchlow S. E., Jackson S. P. (1998) DNA end-joining: from yeast to man. Trends Biochem. Sci. 23, 394–398 [DOI] [PubMed] [Google Scholar]
- 38. Frank K. M., Sekiguchi J. M., Seidl K. J., Swat W., Rathbun G. A., Cheng H. L., Davidson L., Kangaloo L., Alt F. W. (1998) Late embryonic lethality and impaired V(D)J recombination in mice lacking DNA ligase IV. Nature 396, 173–177 [DOI] [PubMed] [Google Scholar]
- 39. Rosas-Acosta G., Russell W. K., Deyrieux A., Russell D. H., Wilson V. G. (2005) A universal strategy for proteomic studies of SUMO and other ubiquitin-like modifiers. Mol. Cell. Proteomics 4, 56–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vertegaal A. C., Andersen J. S., Ogg S. C., Hay R. T., Mann M., Lamond A. I. (2006) Distinct and overlapping sets of SUMO-1 and SUMO-2 target proteins revealed by quantitative proteomics. Mol. Cell. Proteomics 5, 2298–2310 [DOI] [PubMed] [Google Scholar]
- 41. Ishiai M., Kimura M., Namikoshi K., Yamazoe M., Yamamoto K., Arakawa H., Agematsu K., Matsushita N., Takeda S., Buerstedde J. M., Takata M. (2004) DNA cross-link repair protein SNM1A interacts with PIAS1 in nuclear focus formation. Mol. Cell. Biol. 24, 10733–10741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mabb A. M., Wuerzberger-Davis S. M., Miyamoto S. (2006) PIASy mediates NEMO sumoylation and NF-κB activation in response to genotoxic stress. Nat. Cell Biol. 8, 986–993 [DOI] [PubMed] [Google Scholar]
- 43. Park M. A., Seok Y. J., Jeong G., Lee J. S. (2008) SUMO1 negatively regulates BRCA1-mediated transcription, via modulation of promoter occupancy. Nucleic Acids Res. 36, 263–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Desterro J. M., Rodriguez M. S., Hay R. T. (1998) SUMO-1 modification of IκBα inhibits NF-κB activation. Mol. Cell 2, 233–239 [DOI] [PubMed] [Google Scholar]
- 45. Mahajan R., Gerace L., Melchior F. (1998) Molecular characterization of the SUMO-1 modification of RanGAP1 and its role in nuclear envelope association. J. Cell Biol. 140, 259–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yamada K., Muramatsu M., Saito D., Sato-Oka M., Saito M., Moriyama T., Saitoh H. (2012) Characterization of the C-terminal diglycine motif of SUMO-1/3. Biosci. Biotechnol. Biochem. 76, 1035–1037 [DOI] [PubMed] [Google Scholar]
- 47. Ouyang K. J., Woo L. L., Zhu J., Huo D., Matunis M. J., Ellis N. A. (2009) SUMO modification regulates BLM and RAD51 interaction at damaged replication forks. PLoS Biol. 7, e1000252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fontebasso Y., Etheridge T. J., Oliver A. W., Murray J. M., Carr A. M. (2013) The conserved Fanconi anemia nuclease Fan1 and the SUMO E3 ligase Pli1 act in two novel Pso2-independent pathways of DNA interstrand crosslink repair in yeast. DNA Repair 12, 1011–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Galanty Y., Belotserkovskaya R., Coates J., Jackson S. P. (2012) RNF4, a SUMO-targeted ubiquitin E3 ligase, promotes DNA double-strand break repair. Genes Dev. 26, 1179–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Macris M. A., Sung P. (2005) Multifaceted role of the Saccharomyces cerevisiae Srs2 helicase in homologous recombination regulation. Biochem. Soc. Trans. 33, 1447–1450 [DOI] [PubMed] [Google Scholar]