

Background: CRISPR/Cas9-directed cleavages may result in genomic deletion.
Results: CRISPR/Cas9-produced genomic deletion frequency is inversely related to deletion size, with large deletions and inversions practicable and biallelic deletions exceeding probabilistic expectation.
Conclusion: Biallelic, large genomic deletions are efficiently engineered in mammalian cells by CRISPR/Cas9.
Significance: CRISPR/Cas9-mediated genomic deletion represents a robust method for loss-of-function studies in mammalian cells.
Keywords: Gene Expression, Gene Knock-out, Gene Regulation, Genetics, Genomics, CRISPR, Cas9, Genome Engineering, Genomic Deletion
Abstract
The clustered regularly interspaced palindromic repeats (CRISPR)/CRISPR-associated (Cas) 9 nuclease system has provided a powerful tool for genome engineering. Double strand breaks may trigger nonhomologous end joining repair, leading to frameshift mutations, or homology-directed repair using an extrachromosomal template. Alternatively, genomic deletions may be produced by a pair of double strand breaks. The efficiency of CRISPR/Cas9-mediated genomic deletions has not been systematically explored. Here, we present a methodology for the production of deletions in mammalian cells, ranging from 1.3 kb to greater than 1 Mb. We observed a high frequency of intended genomic deletions. Nondeleted alleles are nonetheless often edited with inversions or small insertion/deletions produced at CRISPR recognition sites. Deleted alleles also typically include small insertion/deletions at predicted deletion junctions. We retrieved cells with biallelic deletion at a frequency exceeding that of probabilistic expectation. We demonstrate an inverse relationship between deletion frequency and deletion size. This work suggests that CRISPR/Cas9 is a robust system to produce a spectrum of genomic deletions to allow investigation of genes and genetic elements.
Introduction
Recent studies have revealed a prokaryotic adaptive immune system that may be repurposed to allow for genomic engineering of eukaryotic genomes (1–4). The Streptococcus pyogenes type II CRISPR4/Cas9 adaptive immune system relies on three genes as follows: two noncoding CRISPR RNAs (crRNAs) including a trans-activating crRNA (tracrRNA) and a precursor crRNA, as well as the CRISPR-associated Cas9 nuclease. The precursor crRNA is transcribed from an array that contains repetitive elements with interspersed unique sequences (spacers) derived from exogenous DNA. Once processed and after interaction with the tracrRNA, the mature crRNA guides Cas9 to direct cleavage of foreign DNA (1–3, 5). This system has been repurposed for mammalian genome engineering using Cas9 along with a fusion of the tracrRNA and mature crRNA to create a chimeric single guide RNA (sgRNA) (2, 3, 6). Site-specific cleavage is directed by complementarity of the sgRNA to a 20-bp genomic sequence (protospacer) immediately 5′ of a protospacer-adjacent motif (PAM), which is NGG for SpCas9. This recruits Cas9 to introduce site-specific double strand breaks (DSBs) repaired by either homology-directed repair or by insertion/deletion (indel)-forming nonhomologous end joining (NHEJ) (2, 3). Heterologous expression of the CRISPR system components has been shown to be a facile method of genome engineering as compared with previous systems, such as zinc finger nucleases or TAL effector nucleases, in part because of the ease with which individual sgRNAs may be designed and produced (7).
The CRISPR/Cas9 system has already demonstrated wide applicability for efficient genome editing in a variety of model systems (8–13), which has spawned an era of unprecedented control over the genome. This includes applications such as genome editing in clonal cell lines in a matter of weeks (6), CRISPR interference-mediated gene regulation with a catalytically inactive Cas9 (14), pooled sgRNA library screening for functional genomics (15–17), and potential CRISPR-based therapy highlighted by its recent use for gene correction in both murine and human stem cells (18, 19).
A strategy of using two DSBs to create a deletion of the intervening segment by NHEJ has previously been successfully applied using zinc finger nucleases, TAL effector nucleases, and CRISPR systems (6, 20–25). However, the efficiency, reliability, and genomic outcomes of using pairs of CRISPRs to introduce genomic deletions remain incompletely characterized. Here, we sought to test the capability and efficiency of creating deletions in mammalian cell lines. Our results indicate that the CRISPR/Cas9 system is a powerful tool for the robust and dependable generation of genomic deletions.
EXPERIMENTAL PROCEDURES
CRISPR Design and Creation
sgRNA-specifying oligo sequences were chosen to minimize the likelihood of off-target cleavage based on publicly available on-line tools (6). Each sgRNA specified sequences exonic, intronic, or intergenic (within 3.5 kb of a gene body) with respect to a RefSeq gene (Fig. 1, A and B). “CACC” was added to the 5′ end of the sgRNA-specifying oligo sequence and “AAAC” was added to the 5′ end of the reverse complement of the sgRNA-specifying oligo for cloning using the BbsI restriction enzyme. G was added immediately following CACC if the first nucleotide was A, T, or C (in these cases C was added at the 3′ end of the reverse complement oligo). The two oligos were phosphorylated and annealed using the following conditions: guide sequence oligo (10 μm); guide sequence reverse complement oligo (10 μm); T4 ligation buffer (1×) (New England Biolabs, Ipswich, MA); and T4 polynucleotide kinase (5 units) (New England Biolabs) with the following temperature conditions: 37 °C for 30 min; 95 °C for 5 min; and then ramp down to 25 °C at 5 °C/min. The annealed oligos were cloned into pSpCas9(BB) (pX330; Addgene plasmid ID 42230) using a Golden Gate Assembly strategy with the following conditions: 100 ng of circular pX330 vector; annealed oligos (0.2 μm); 2.1 buffer (1×) (New England Biolabs); BbsI restriction enzyme (20 units) (New England Biolabs); ATP (0.2 mm) (New England Biolabs); BSA (1×) (New England Biolabs); and T4 DNA ligase (750 units) (New England Biolabs) with the cycling conditions of 20 cycles of 37 °C for 5 min, 20 °C for 5 min, and 80 °C for 20 min. The sgRNAs were not pre-screened for editing efficiency prior to genomic deletion experiments presented herein.
FIGURE 1.

Schema for CRISPR/Cas9-mediated genomic deletion. A, exonic, intronic, and intergenic sgRNA deletion strategies. The sgRNA sites are shown in relation to an idealized gene. The PAM sequence (purple) is shown on the top strand for simplicity, but PAMs on both the top (Watson) and bottom (Crick) strands were used in different combinations. The red line indicates the predicted Cas9 cleavage between positions 17 and 18. The blue arrows indicate the position of PCR primers for deletion band amplification, and the red arrows indicate the position of PCR primers for nondeletion band amplification. B, sgRNA localization (exonic/intronic/intergenic), deletion size, chromosome, genomic coordinates (mm10), and sequence for each sgRNA pair. Loci 1–4 used for further sequence analysis are indicated. C, CRISPR/Cas9-mediated genomic deletion strategy for MEL cells. 2 × 106 cells were electroporated with 5 μg of each sgRNA construct and 0.5 μg of a GFP expression construct. The top 3% of GFP+ cells were sorted 1–3 days post-electroporation and plated at limiting dilution. 7–10 days after plating, gDNA was extracted, and clones were screened for deletion by PCR. A representative screening agarose gel shows the detection of two nondeletion clones, two monoallelic deletion clones, and two biallelic deletion clones. The red ND refers to the nondeletion amplicon as schematized in A, and the blue D refers to the deletion amplicon as schematized in A. Upon inversion analysis, clones were further classified as nondeletion/noninversion, compound inversion/scar-complex, compound deletion/scar-complex, and compound deletion/inversion. The distinction between scar and complex was established by the presence or absence of PCR amplification flanking both sgRNA target recognition sites. Inv refers to inversion amplicons flanking left and right sgRNA recognition sites (L and R, respectively). D, gDNA was extracted from cells prior to (unsorted) and after sorting the top 3% of GFP+ cells (sorted). Deletion enrichment was calculated by RT-quantitative PCR, and data were normalized to the unsorted cells using the 2−ΔΔCt method. A biallelic deletion clone for each locus was used as a positive control and nonedited parental gDNA as a negative control. E, primers flanking the sgRNA recognition sites (shown in green and purple) were used to amplify 500–700-bp regions around each sgRNA site on nondeletion/noninversion alleles (primers 1/2 and 3/4, top panel). Inversion PCR utilized primer pairs (primers 1/3 and 2/4, respectively; bottom panel) in which both primers were in the same orientation, one inside and one outside the intended deletion.
Cell Culture, Transfection, and Screening Clones
Murine erythroleukemia (MEL) cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2% penicillin/streptomycin, and 1% l-glutamine at 37 °C with 5% CO2 (Invitrogen). Despite a complex karyotype, MEL cells exhibit karyotypic stability and are disomic for most chromosomes by karyotype reconstruction analysis (26, 27). Analysis of the MEL cells used in these experiments revealed a karyotype consistent with these previous reports. Two copies were present for each chromosome studied (data not shown). 2 × 106 cells were electroporated with 0.5 μg of pmaxGFP plasmid (Lonza, Allendale, NJ) and 5 μg each of pX330-sgRNA-A and pX330-sgRNA-B plasmids using the ECM 830 Square Wave Electroporation System (Harvard Apparatus, Holliston, MA) for a total of 10.5 μg of plasmid (6). Preliminary experiments showed this concentration of cells and plasmids to be effective for production of genomic deletions. Extensive analyses regarding optimal concentrations of cells and plasmids were not performed. Cells were resuspended in 100 μl of BTX solution and electroporated at 250 V, 5 ms, in 2-mm cuvettes (Harvard Apparatus). Cells were placed in 1 ml of culture media immediately following electroporation.
To enrich for deletion, the top 3% GFP+ cells were sorted via FACSAria cell sorter (BD Biosciences) 1–3 days post-electroporation (Fig. 1, C and D). Preliminary experiments showed these time points to be effective for production of genomic deletions. Extensive analyses regarding optimal time points for cell sorting were not performed. Cells were plated at 30 cells per 96-well plate to isolate single cell-derived clones. After 7–10 days for expansion, clones were screened for CRISPR-mediated deletion (Fig. 1C). Genomic DNA (gDNA) was extracted by resuspending cells in 50 μl of QuickExtract DNA extraction solution per well and incubating at the following conditions: 65 °C for 6 min; 98 °C for 2 min (Epicenter, Madison, WI). Polymerase chain reaction (PCR) was performed using two sets of primers (Fig. 1, A and C) as follows: one set to amplify a sequence within the segment to be deleted (“nondeletion band”) and one set that was only amplified in the presence of a deletion (“deletion band”) using the Qiagen HotStarTaq 2× master mix and the following cycling conditions: 95 °C for 15 min; 35 cycles of 95 °C for 15 s, 60 °C for 1 min, and 72 °C for 1 min; 72 °C for 10 min.
Monoallelic deletion clones were defined as having PCR amplification of both the nondeletion band and deletion band. Biallelic deletion clones were defined as having PCR amplification of the deletion band and absence of the nondeletion band. Clones with PCR amplification of the nondeletion band and absence of the deletion band were defined as nondeletion clones (Fig. 1C).
Nondeletion/noninversion alleles were analyzed using PCR primers flanking the sgRNA recognition sites to amplify 500–700-bp regions around each sgRNA site with one primer inside and one outside the intended deletion (Fig. 1E, top panel). Monoallelic and biallelic inversion clones were defined by amplification of inversion bands at each inversion junction, using primer pairs in which both primers were in the same orientation, one inside and one outside the intended deletion (Fig. 1E, bottom panel). Specifically, monoallelic inversion clones were defined as having PCR amplification using one or both inversion primer pairs. Biallelic inversion clones were defined as having PCR amplification of one or both inversion primer pairs in conjunction with neither deletion band PCR amplification nor amplification of sequences flanking the sgRNA recognition site.
Deletion, nondeletion, and inversion amplicons from nondeletion, monoallelic, and biallelic deletion clones were subjected to Sanger sequencing. The deletion amplicons from biallelic deletion clones were separately amplified by Phusion Hot Start PCR (New England Biolabs) with the following conditions: Phusion High Fidelity DNA polymerase (0.5 units), dNTPs (2 mm each), GC Buffer (1×), 3% dimethyl sulfoxide (DMSO), forward and reverse primers (0.1 μm each) with the cycling conditions of 95 °C for 15 min; 40 cycles of 95 °C for 15 s, 60 °C for 30 s, and 72 °C for 30 s; 72 °C for 5 min. Amplicons were either gel-purified and directly Sanger-sequenced or cloned with Zero Blunt PCR cloning kit (Invitrogen) if two bands could not be resolved by gel electrophoresis. At least four clones each were subjected to Sanger sequencing. Deletion frequency was calculated as shown in Equation 1.
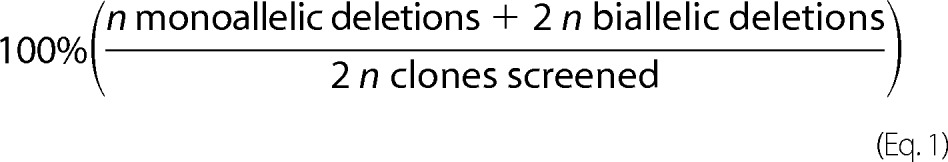 |
Inversion frequency was also calculated using Equation 1.
RESULTS
CRISPR/Cas9 Is a Robust System for the Production of Genomic Deletions
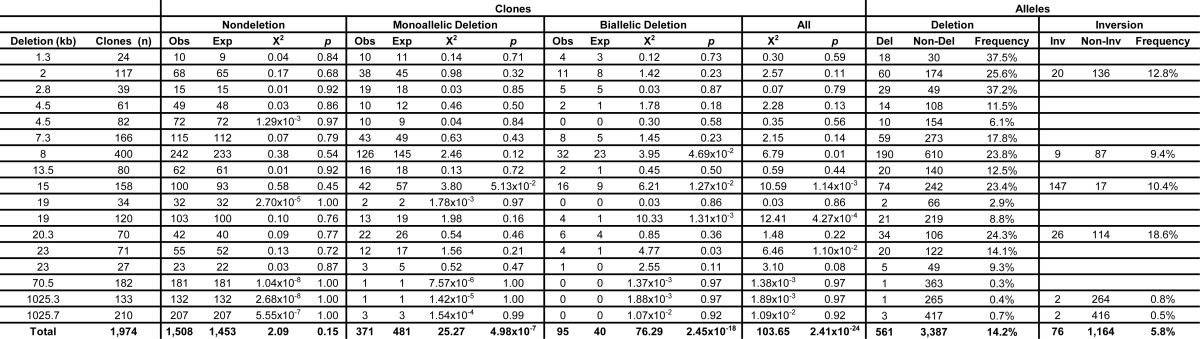
Seventeen sgRNA pairs at 12 genomic loci were assayed in MEL cells to determine their ability to engender genomic deletions and to determine the robustness of the approach. At each locus, a pair was composed of an sgRNA 5′ with respect to the top (Watson) strand, indicated as sgRNA-A, and another 3′ with respect to the Watson strand, sgRNA-B (Fig. 1, A and B). The ability to create interstitial deletion of the segment AB was tested by conventional PCR. These sgRNAs were either exonic, intronic, or intergenic (within 3.5 kb of a gene body) with respect to RefSeq genes (Fig. 1, A and B). None of the genes was known to be essential for cell viability. The sgRNA pairs were designed to create a spectrum of deletions, ranging in size from 1.3 to 1,026 kb (Fig. 1B). 1,974 clones across 17 sgRNA pairs were screened for deletions. Clones with deleted alleles were observed for all tested sgRNA pairs (Table 1).
TABLE 1.
Observed biallelic deletion frequency exceeds probabilistic expectation
The expected (Exp) number of clones in each category (nondeletion clones, monoallelic deletion clones, biallelic deletion clones, and all clones) was calculated based on the observed (Obs) deletion frequency using the quadratic Equation 2 (analogous to Hardy-Weinberg equilibrium). Monoallelic deletion clones included compound deletion/scar, compound deletion/complex, and compound deletion/inversion clones. sgRNA pairs were analyzed individually and collectively for agreement with expected number of nondeletion, monoallelic, and biallelic deletion clones obtained. χ2 statistics and p values were generated by Pearson's χ2 tests. Inversion frequency was evaluated for the four pairs studied in detail as well as for pairs 16 and 17.
Indels Are Often Formed at the Predicted Deletion Junctions
The deleted allele was examined in monoallelic and biallelic deletion clones at four loci, referred to as loci 1–4, spanning a range of intended deletion sizes (2.0, 8.0, 15.0, and 20.3 kb) (Fig. 1B). One primer from upstream of sgRNA-A and another primer from downstream of sgRNA-B were used to amplify and sequence across the deletion junction (Figs. 1, A and C, 2A, 3A, 4A, and 5A). Indel sizes were enumerated as the sum of the number of base pairs inserted (positive values) or deleted (negative values) relative to the predicted deletions, assuming cleavage between positions 17 and 18 as specified by each sgRNA (Fig. 1A) (2, 3, 6). Indels ranged from −176 to +538 bp in monoallelic deletion clones and from −286 to +449 bp in biallelic deletion clones. Both monoallelic and biallelic deletion clones showed the preponderance of indels clustering between −10 and 0 bp (Fig. 6A). Notably, the absence of indels occurred as the most frequent outcome at each sgRNA-predicted cleavage site in both monoallelic deletion clones (77/174 sites on 87 alleles from 87 clones, 44.3%) and biallelic deletion clones (17/80 sites on 40 alleles from 31 clones, 21.3%) (Fig. 6A). Furthermore, the precise predicted deletion (i.e. absence of indels at both sites on a deleted allele) occurred in 31.0% (27/87) of monoallelic deletion clones and 15.0% (6/40) of alleles from biallelic deletion clones. Positive indels (i.e. relative insertions to the predicted deletion) had homology to sequences flanking the predicted deletion site except in one case where a 538-bp insertion with sequence homology to a portion of the pX330 plasmid was identified. However, this accounting is likely an underestimate of the full spectrum of indels because the PCR-based screening method would not identify large deletions or insertions. In several instances, we identified large insertions and deletions around cleavage sites not detectable by the screening PCR strategy (data not shown).
FIGURE 2.

Indels at the nondeletion/noninversion allele and indels at the predicted deletion/inversion junction at locus 1. A, sequencing of the deletion allele in compound deletion/scar-complex-inversion clones and biallelic deletion clones at locus 1 (intended 2.0-kb deletion). Top row indicates sequence of unmodified allele. sgRNA sequences are shown in green and PAM sequences in red. Deletion events are shown by an equivalent number of dash marks and insertions are highlighted in blue. Vertical lines indicate predicted cleavage site, between positions 17 and 18 of the sgRNA. B, sequencing of the nondeletion/noninversion allele in compound deletion/scar-complex clones and compound inversion/scar-complex clones. Top row indicates sequence of the unmodified allele. sgRNA sequences are shown in green and PAM sequences in red. Deletion events are shown by an equivalent number of dash marks and insertions are highlighted in blue. Vertical lines indicate predicted cleavage site, between positions 17 and 18 of the sgRNA. C, sequencing of the inversion allele in compound deletion/inversion clones and compound inversion/scar-complex clones. Top row indicates perfect inversion of intervening segment between predicted cleavage sites. sgRNA sequences are shown in green, PAM sequences in red, and inverted sequence in purple. Deletion events are shown by an equivalent number of dash marks and insertions are highlighted in blue. Vertical lines indicate predicted cleavage site, between positions 17 and 18 of the sgRNA. MD indicates monoallelic deletion; MI indicates monoallelic inversion, and BD indicates biallelic deletion.
FIGURE 3.

Indels at the nondeletion/noninversion allele and indels at the predicted deletion/inversion junction at locus 2. A, sequencing of the deletion allele in compound deletion/scar-complex-inversion clones and biallelic deletion clones at locus 2 (intended 8.0-kb deletion). Top row indicates sequence of unmodified allele. sgRNA sequences are shown in green and PAM sequences in red. Deletion events are shown by an equivalent number of dash marks, and insertions are highlighted in blue. Vertical lines indicate predicted cleavage site, between positions 17 and 18 of the sgRNA. B, sequencing of the nondeletion/noninversion allele in compound deletion/scar-complex clones and compound inversion/scar-complex clones. Top row indicates sequence of unmodified allele. sgRNA sequences are shown in green and PAM sequences in red. Deletion events are shown by an equivalent number of dash marks and insertions are highlighted in blue. Vertical lines indicate predicted cleavage site, between positions 17 and 18 of the sgRNA. C, sequencing of the inversion allele in compound deletion/inversion clones and compound inversion/scar-complex clones. Top row indicates perfect inversion of intervening segment between predicted cleavage sites. sgRNA sequences are shown in green, PAM sequences in red, and inverted sequence in purple. Deletion events are shown by an equivalent number of dash marks, and insertions are highlighted in blue. Vertical lines indicate predicted cleavage site, between positions 17 and 18 of the sgRNA. MD indicates monoallelic deletion; MI indicates monoallelic inversion, and BD indicates biallelic deletion. Clone MD10 exhibited amplification using PCR primers flanking sgRNA-site-B (see Fig. 1E, top panel) and amplification using inversion PCR primers flanking sgRNA-site-A (see Fig. 1E, bottom panel), suggesting a complex inversion. Clone MD2 exhibited amplification using PCR primers flanking sgRNA-site-A (see Fig. 1E, top panel) and amplification using inversion PCR primers flanking both sgRNA-site-A and sgRNA-site-B (see Fig. 1E, bottom panel), suggesting at least three alleles at the tested locus, which could be consistent with rare tetraploidies observed in MEL cells by karyotype (data not shown) or with a mixed clone. This was the only clone out of the 278 clones examined in detail across the four loci to exhibit apparent allele number greater than 2.
FIGURE 4.

Indels at the nondeletion/noninversion allele and indels at the predicted deletion/inversion junction at locus 3. A, sequencing of the deletion allele in compound deletion/scar-complex-inversion clones and biallelic deletion clones at locus 3 (intended 15.0-kb deletion). Top row indicates sequence of unmodified allele. sgRNA sequences are shown in green and PAM sequences in red. Deletion events are shown by an equivalent number of dash marks and insertions are highlighted in blue. Vertical lines indicate predicted cleavage site, between positions 17 and 18 of the sgRNA. B, sequencing of the nondeletion/noninversion allele in compound deletion/scar-complex clones and compound inversion/scar-complex clones. Top row indicates sequence of unmodified allele. sgRNA sequences are shown in green and PAM sequences in red. Deletion events are shown by an equivalent number of dash marks and insertions are highlighted in blue. Vertical lines indicate predicted cleavage site, between positions 17 and 18 of the sgRNA. C, sequencing of the inversion allele in compound deletion/inversion clones, compound inversion/scar-complex clones, and biallelic inversion clones. Top row indicates perfect inversion of the intervening segment between predicted cleavage sites. sgRNA sequences are shown in green, PAM sequences in red, and inverted sequence in purple. Deletion events are shown by an equivalent number of dash marks and insertions are highlighted in blue. Vertical lines indicate predicted cleavage site, between positions 17 and 18 of the sgRNA. MD indicates monoallelic deletion; MI indicates monoallelic inversion; BD indicates biallelic deletion, and BI indicates biallelic inversion. Biallelic inversion alleles are labeled A–D (rather than merely A and B) because phase of inversion junctions within a clone was not resolved. Clone MD16 did not exhibit amplification using PCR primers flanking either sgRNA-site-A or sgRNA-site-B (see Fig. 1E, top panel) but did exhibit amplification using inversion PCR primers flanking sgRNA-site-B only (see Fig. 1E, bottom panel), suggesting a complex inversion.
FIGURE 5.

Indels at the nondeletion/noninversion allele and indels at the predicted deletion/inversion junction at locus 4. A, sequencing of the deletion allele in compound deletion/scar-complex-inversion clones and biallelic deletion clones at locus 4 (intended 20.3-kb deletion). Top row indicates sequence of unmodified allele. sgRNA sequences are shown in green and PAM sequences in red. Deletion events are shown by an equivalent number of dash marks, and insertions are highlighted in blue. Vertical lines indicate predicted cleavage site, between positions 17 and 18 of the sgRNA. B, sequencing of the nondeletion/noninversion allele in compound deletion/scar-complex clones and compound inversion/scar-complex clones. Top row indicates sequence of unmodified allele. sgRNA sequences are shown in green and PAM sequences in red. Deletion events are shown by an equivalent number of dash marks and insertions are highlighted in blue. Vertical lines indicate predicted cleavage site, between positions 17 and 18 of the sgRNA. C, sequencing of the inversion allele in compound deletion/inversion clones, compound inversion/scar-complex clones, and biallelic inversion. Top row indicates perfect inversion of intervening segment between predicted cleavage sites. sgRNA sequences are shown in green, PAM sequences in red, and inverted sequence in purple. Deletion events are shown by an equivalent number of dash marks and insertions are highlighted in blue. Vertical lines indicate predicted cleavage site, between positions 17 and 18 of the sgRNA. MD indicates monoallelic deletion; MI indicates monoallelic inversion; BD indicates biallelic deletion, and BI indicates biallelic inversion. Biallelic inversion alleles are labeled A–D (rather than merely A and B) because phase of inversion junctions within a clone was not resolved. Clones MD1, MD5, MD8, and MD25 did not exhibit amplification using PCR primers flanking either sgRNA-site-A or sgRNA-site-B (see Fig. 1E, top panel) but did exhibit amplification using inversion PCR primers flanking sgRNA-site-A only (see Fig. 1E, bottom panel), suggesting complex inversions. Clone MD17 exhibited amplification using PCR primers flanking sgRNA-site-B (see Fig. 1E, top panel) and amplification using inversion PCR primers flanking sgRNA-site-A (see Fig. 1E, bottom panel), suggesting a complex inversion.
FIGURE 6.

Indels at deleted, scarred, and inverted alleles. A, left panel, frequency distribution of indel formation at the predicted deletion junctions from the deleted alleles of monoallelic and biallelic deletion clones across four loci examined in detail. Indels ranged from −176 to +538 bp in monoallelic deletion clones and from −286 to +449 bp in biallelic deletion clones with the majority found at −10 to 0 bp. A, right panel, distribution of negative indels on the deletion allele produced by sgRNA-A and sgRNA-B from both monoallelic and biallelic deletion clones. Positive indels could not be reliably mapped to individual sites, so were excluded from analysis. B, left panel, frequency distribution of indel formation (scarring) on the nondeleted allele from compound deletion/scar-complex clones and on the noninverted allele from compound inversion/scar-complex clones across eight sgRNA sites from four loci examined in detail. Indels ranged from −83 to +1 bp for the nondeletion alleles and from −134 to +2 bp for the noninversion alleles. B, right panel, distribution of indels on the nondeleted allele in compound deletion/scar clones and the noninverted allele in compound inversion/scar clones in which sequences surrounding both sgRNA-A and sgRNA-B sites were amplified. C, left panel, frequency distribution of indel formation at the predicted inversion junctions from the inversion alleles of compound inversion/scar-complex-deletion clones across all four loci examined in detail. Indels ranged from −115 to +138 bp with the majority found at −10 to 0 bp. C, right panel, distribution of indels on the inverted allele produced by sgRNA-A and sgRNA-B from compound inversion/scar-complex-deletion clones.
Sanger sequencing of biallelic deletion clones revealed 9/31 (29.0%) to be compound heterozygotes based on differing indels at the predicted deletion sites on each allele. The remaining 22/31 (71.0%) had only a single deletion junction identified by both amplicon sequencing as well as sequencing of multiple clones of the PCR product. This finding may suggest that in these cells both alleles were independently repaired in an identical manner or that one allele served as a template for homology-directed repair of the other allele. Other possibilities include uniparental disomy, monosomy, or a large insertion/deletion at one allele. Each of the biallelic deletion clones was confirmed to result in loss of expression of the deleted gene by RT-quantitative PCR (data not shown).
Editing (“Scarring”) Occurs in the Absence of Deletion
Deletion is only one possible outcome following two DSBs at a locus. Another outcome is local NHEJ repair of each DSB without deletion of the intervening segment. We examined the nondeleted allele using Sanger sequencing of the monoallelic deletion clones from the same four sgRNA pairs targeting different loci spanning a range of intended deletion sizes (2.0, 8.0, 15.0, and 20.3 kb) (Figs. 2B, 3B, 4B, and 5B). Primers flanking the sgRNA recognition sites were used to amplify 500–700-bp regions around each sgRNA site (Fig. 1E, top panel). Three amplification patterns were observed for each sgRNA pair as follows: amplification at both sgRNA-site-A and sgRNA-site-B (50/87, 57.5%); amplification at either sgRNA-site-A or sgRNA-site-B (11/87, 12.6%), or amplification at neither sgRNA-site-A nor sgRNA-site-B (26/87, 29.9%). For each locus, both sgRNA sites amplified from parental gDNA. The sites lacking amplification could represent large indels, inversions, or structural aberrancy and were excluded from this analysis. Clones with both sites amplifying were classified as “compound deletion/scar” clones, whereas clones with one site or both sites not amplifying were classified as “compound deletion/complex” clones. Sanger sequencing of the amplifying sgRNA sites revealed indels at 99% (92/93) of eight sgRNA predicted cleavage sites across the four loci. Only one site exhibited either noncleavage or cleavage with perfect repair (Figs. 2B, 3B, 4B, and 5B). No monoallelic deletion clones were obtained that exhibited a precise predicted deletion in combination with an absence of scarring of the nondeleted allele. The high frequency of indel formation across all four loci suggested that the frequency of indel formation was independent of the intended deletion size. Indels ranged from −83 to +1 bp with most of the indels clustering between −10 and −1 bp (Fig. 6B). This high frequency of indel formation at the sgRNA sites on the nondeleted allele of monoallelic deletion clones indicates that the sgRNA recognition and/or PAM sequence is often obliterated (Figs. 2B, 3B, 4B, 5B, and 6B). Therefore, re-targeting the nondeleted allele in monoallelic deletion clones would likely require independent sgRNAs.
From the nondeletion clones, 131/161 were chosen for analysis using the same primers flanking the sgRNA recognition sites to amplify 500–700-bp regions around each sgRNA site (Fig. 1E, top panel). The same three amplification patterns were observed for each sgRNA pair as follows: amplification at both sgRNA-site-A and sgRNA-site-B (110/131, 84.0%); amplification at either sgRNA-site-A or sgRNA-site-B (10/131, 7.6%), or amplification at neither sgRNA-site-A nor sgRNA-site-B (11/131, 8.4%). Of the 120 clones with at least one PCR amplification band, 108 clones were selected and subjected to Sanger sequencing. 24.1% (26/108) of these clones were subsequently determined to be monoallelic or biallelic inversion clones. Analysis of the remaining 82 sequenced nondeletion clones, classified as “nondeletion/noninversion” clones, revealed indel formation of at least one allele in 75.6% (62/82) of instances with no apparent relationship between editing frequency and intended deletion size (data not shown). 24.4% (20/82) of the nondeletion/noninversion clones exhibited wild-type sequencing, which could result from insufficient or absent sgRNA/Cas9 expression, perfect repair, or the presence of a large indel on one allele and an unmodified (or perfectly repaired) other allele. Based on the high frequency of indel formation, it is apparent that a large fraction of nondeletion clones was exposed to Cas9, sgRNA-A, and sgRNA-B. Therefore, the induction of deletion is not simply limited by delivery of both CRISPR plasmids. However, the reduced rate of indel formation in nondeletion clones as compared with monoallelic deletion clones could indicate reduced delivery of one or both CRISPR plasmids to the cells, suggesting that deletion may be sensitive to sgRNA/Cas9 dose.
Inversion Is a Frequent Outcome
The alleles from monoallelic deletion clones exhibiting amplification at neither sgRNA-site-A nor sgRNA-site-B (26/87, 29.9%) on the nondeletion allele (Fig. 1E, top panel) were screened for inversions by PCR (Fig. 1E, bottom panel). Notably, 100% of these clones (26/26) demonstrated inversions. Each of the nondeletion clones previously analyzed (n = 131) was evaluated for inversions. This analysis revealed 29.8% (39/131) of nondeletion clones to be monoallelic inversion clones and 1.5% (2/131) biallelic inversion clones. Notably, for the 8.4% (11/131) of nondeletion clones with PCR amplification at neither sgRNA-site-A nor sgRNA-site-B (11/131, 8.4%) (Fig. 1E, top panel), 9/11 were determined to be monoallelic inversion clones and 2/11 were determined to be biallelic inversion clones by inversion PCR (Fig. 1E, bottom panel). Sanger sequencing of the inversion amplicons revealed indels ranging from −115 to +138 bp for compound inversion/scar-complex-deletion clones at the predicted inversion junction (Figs. 2C, 3C, 4C, 5C, and 6C). This included a +35-bp insertion with homology to Cas9 and a +138-bp insertion with homology to a different chromosomal locus. Monoallelic inversion clones showed the preponderance of indels clustering between −10 and 0 bp (Fig. 6C). Two biallelic inversion clones were identified and also exhibited small indel formation at the inversion junction (Figs. 4C and 5C).
The noninversion alleles from the monoallelic inversion clones were analyzed for editing in the absence of inversion (Fig. 1E, top panel). Sanger sequencing of the amplifying sgRNA sites revealed indels at 100% (45/45) of sgRNA cleavage sites analyzed, which ranged from −134 to +2 bp (Figs. 2B, 3B, 4B, 5B, and 6B). No clones were obtained that exhibited the predicted inversion in combination with an absence of scarring on the noninversion allele. Of the monoallelic inversion clones, 59.0% (23/39) were “compound inversion/scar” clones and 41.0% (16/39) “compound inversion/complex” clones.
Pairs 16 and 17 each possess greater than 1 Mb between sgRNA-A and sgRNA-B (Fig. 1B). Inversions were identified in 1.5% (2/133) and 1.0% (2/210) of clones, respectively (Table 1). These data indicate that both large scale deletions and inversions of at least 1 Mb can be produced using two sgRNAs.
Deletion Occurs More Frequently Than Inversion
The four loci analyzed in detail include a total of 278 clones (558 alleles). Deletion and inversion frequencies were calculated on a per allele basis, which revealed a deletion frequency of 26.8% (149/558) and an inversion frequency of 12.9% (72/558) (Fig. 7A). Alleles characterized by nonediting, scarring, and complex indels remained the most common outcome (60.3%, 335/556). Clones were classified into eight categories based on combination of deletion, inversion, scar/nonedited, and complex alleles. The assignment of scarring required PCR amplification of both the sgRNA-A and sgRNA-B target sites from the nondeletion or noninversion allele. If one or both sites failed to amplify, alleles were classified as complex. Nondeletion/noninversion clones were the most frequent outcome (42.8%, 119/278). Monoallelic deletion with scarring on the nondeletion allele was second most common (14.7%, 41/278) (Fig. 7B). Although biallelic deletion was the third most common (11.2%, 31/278), biallelic inversion was the least common (0.7%, 2/278). This distribution may reflect the complicated repair processes cells undergo after a pair of DSBs initiated by CRISPR/Cas9.
FIGURE 7.

Deletion/inversion frequency and characterization of outcomes when using two sgRNAs. A, deletion and inversion frequency were calculated on a per allele basis for all 278 clones (556 alleles) across the four loci examined in detail. B, 278 clones from across the four loci examined in detail were classified into eight categories based on the presence of deletion, inversion, scar, and complex alleles. C, deletion frequency inversely related to deletion size. Genomic deletion sizes ranged from 1.3 to 1,026 kb, which demonstrate a decrease in the frequency of deletion events as deletion size increases. The size of the circles corresponds to the number of clones screened for the corresponding sgRNA pair. The best fit relationship was determined by a weighted (by number of clones screened) nonlinear regression of the form function(deletion size) = k1 + k2(deletion size in kb)−k3, where k1, k2, and k3 represent constants: function(deletion size in kb) = −2.84 + 41.41(deletion size in kb)−0.36; R2 = 0.62. The weighted nonlinear regression was computed using the fitnlm function available in MATLAB R2013b software (MathWorks, Natick, MA).
Observed Frequency of Monoallelic and Biallelic Deletion Clones Deviates from Probabilistic Expectation
The quadratic Equation 2 (analogous to Hardy-Weinberg equilibrium) was used to calculate the expected number of monoallelic and biallelic deletion clones for each deletion based on the observed deletion frequency,
where f represents allele frequency. A Pearson χ2 test demonstrated that the observed frequency of nondeletion, monoallelic, and biallelic deletion clones did not match the expectation (p = 2.41 × 10−24). The deviation from probabilistic expectation occurred due to an increased frequency of biallelic deletion clones at the expense of the observed number of monoallelic deletion clones (Table 1). This suggests that the deletion of one allele may increase the likelihood of deletion of the other allele. This finding could be consistent with the hypothesis that high expression of components of the CRISPR/Cas9 system favor production of genomic deletions. In any event, this observation is favorable for the creation of biallelic deletions for the study of genes or regulatory elements.
Deletion Frequency Varies Inversely to Deletion Size
Genomic deletion demonstrated an inverse relationship between genomic deletion size and frequency of deletion over the range of 1.3 to 1026 kb (Fig. 7C). The best fit relationship was determined by a weighted (by number of clones screened) nonlinear regression of the form function(deletion size in kb) = k1 + k2(deletion size in kb)−k3, where k1, k2, and k3 represent constants (R2 = 0.62). These data appear consistent with a competitive relationship between exposed cleaved ends at sgRNA sites-A and -B for local A-A indel repair and long range A-B deletion repair (as well as inversion repair). Practically, these results suggest that more clones need to be screened as intended deletion size increases to reliably retrieve biallelic deletion clones.
DISCUSSION
Efficient genomic deletion may be useful for creation of specific and reproducible gene knockouts as compared with frameshift mutations produced by a single sgRNA-mediated cleavage followed by indel-forming NHEJ. First, only 2/3 indels would result in a frameshift. Second, many frameshift mutations escape nonsense-mediated decay (28). Third, alternative splicing may result in isoforms that may compensate or change gene function.
Given the high observed frequency of scarring of the nondeleted allele and the inverse relationship between deletion size and frequency, we suggest that two exonic sgRNAs designed to create a disruptive interstitial deletion of modest size (see Fig. 1A, left panel) could be an efficient strategy to produce gene knock-out clones. Rapid screening via PCR would identify clones with an appropriate deletion. Even in the event that adequate biallelic deletion clones could not be easily obtained, a monoallelic deletion would enrich for compound heterozygous loss-of-function alleles, which could be confirmed by measurement of RNA and protein levels.
Off-target cleavages from CRISPR/Cas9 have been reported, particularly at sites of sequence match in the PAM-proximal seed region (29–31). The full extent of CRISPR/Cas9-mediated off-target events remains incompletely characterized. An unbiased evaluation of off-target events is a challenge for the field and may require deep whole genome sequencing. The introduction of two sgRNAs as described in our deletion schema theoretically carries twice the frequency of off-target mutations as compared with a single sgRNA. Off-target effects may be minimized using a double nickase strategy for DSBs (6, 22), truncated sgRNAs (32), or dimeric Cas9-FokI fusions (33, 34), but it remains to be determined how these approaches might affect deletion frequency. Complementation of the deletion phenotype by reintroduction of the deleted sequence would verify the association of the deletion to the phenotype but may be laborious to achieve. Alternatively, we suggest that multiple sgRNA pairs with nonoverlapping sequences be utilized as a practical measure to control for off-target effects. Consistent phenotypes associated with deletions produced by unique reagents would seem unlikely due to off-target mutations.
These results identify an inverse relationship between deletion size and frequency. Of course a chromosome is not organized as a purely one-dimensional structure but rather as three-dimensional chromatin. The relationship between genome editing and chromatin context remains poorly understood and is an important topic for future investigation. The loci analyzed in this study mainly represent euchromatin. Given the relationship between deletion size and efficiency, an estimate may be made of a minimal number of clones needed to be screened to obtain biallelic deletion clones for a given deletion size. If biallelic deletion frequency was found below probabilistic expectation, it could suggest that full loss-of-function is cell-lethal. Alternatively, lower than expected apparent biallelic deletion frequency in conjunction with the presence of apparent monoallelic deletion clones could indicate a copy number greater than two, particularly in cancer cell lines.
These data suggest that both genomic deletions and inversions are a common outcome of a pair of DSBs initiated by CRISPR/Cas9. Despite relatively high deletion and inversion frequency, the paucity of biallelic inversion as compared with biallelic deletion clones may reflect a more complicated mechanism of inversion resolution as compared with deletion production. It is possible that some of the excess of biallelic deletion clones indicates circumstances in which the first deletion allele served as template for homology-directed repair of the second allele. For pairs of sgRNAs separated by greater than 1 Mb, we observed a similar frequency of deletions and inversions (0.58% or 4/686 for both inversions and deletions; Fig. 1B and Table 1). These results suggest that this technique may be suitable for the production of even relatively large intrachromosomal inversions and deletions.
The high scarring frequency observed related to indel production at the sgRNA recognition sites on nondeleted/noninverted alleles provides insight into the efficiency of the CRISPR/Cas9 system. Even transient transfection of components of this system can induce extensive on-target editing in the form of deletions, inversions, and scarring (i.e. small indels) in selected subpopulations. One practical consequence is that retargeting monoallelic deletion clones with the same sgRNAs may be difficult because in many cases the recognition sites would be altered by indels. Although small indels at deletion and inversion junctions are common, we also observed alleles with the precise predicted deletion or inversion without additional indels.
It has been previously observed that individual sgRNAs in the presence of Cas9 may show differences in editing efficiency (32). We observed relatively substantial editing efficiencies for all tested sgRNA pairs (Table 1). This finding could indicate that enrichment via marker-based sorting can minimize differences in sgRNA efficiency, although the efficiency of these same sgRNA pairs to produce deletion without marker-based sorting was not characterized in detail.
The CRISPR-mediated deletion strategy appears particularly suited for the study of noncoding regulatory DNA where frameshift mutations do not pertain (21). The importance of efficient strategies for assessment of regulatory DNA function is emphasized by recent reports that have suggested the majority of common variants associated with human disease resides in noncoding regulatory DNA (35). Combinatorial deletion of regulatory elements and/or genes may be a powerful method for studying pathways, the hierarchy of regulatory elements, and gene-regulatory element interactions. This study demonstrates that the CRISPR/Cas9 system is a robust tool to produce biallelic genomic deletions for prospective functional genetics.
Acknowledgments
We thank Matthew Porteus for suggesting genomic deletions by genome editing as a strategy to produce loss-of-function alleles; Jason Wright for suggesting the Golden Gate Assembly cloning strategy; Dana-Farber Cancer Institute FACS facility for cell sorting; Lisa Livanos (from KaryoLogic, Research Triangle Park, NC) for karyotype analysis; and members of the Orkin laboratory, particularly Jian Xu, Guoji Guo, Elenoe C. Smith, and Partha Das, for helpful discussions and sharing results.
This work was supported, in whole or in part, by National Institutes of Health Grants R01HL032259 and P30DK049216 (Center of Excellence in Molecular Hematology to S. H. O.), Grant K08DK093705 from NIDDK (to D. E. B.), Grant R01AI084905 from NIAID (to T. M.), Grants T32HL007574 and F32DK098866 (to Y. Y. Y.), and Grants R01DK070838 and P01HL032262 (to B. H. P.).
This article was selected as a Paper of the Week.
- CRISPR
- clustered regularly interspaced palindromic repeat
- DSB
- double strand break
- NHEJ
- nonhomologous end joining repair
- sgRNA
- single guide RNA
- gDNA
- genomic DNA
- PAM
- protospacer-adjacent motif
- MEL
- murine erythroleukemia
- crRNA
- CRISPR RNA
- tracrRNA
- trans-activating crRNA
- indel
- insertion/deletion
- oligo
- oligonucleotide.
REFERENCES
- 1. Barrangou R., Fremaux C., Deveau H., Richards M., Boyaval P., Moineau S., Romero D. A., Horvath P. (2007) CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712 [DOI] [PubMed] [Google Scholar]
- 2. Cong L., Ran F. A., Cox D., Lin S., Barretto R., Habib N., Hsu P. D., Wu X., Jiang W., Marraffini L. A., Zhang F. (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mali P., Yang L., Esvelt K. M., Aach J., Guell M., DiCarlo J. E., Norville J. E., Church G. M. (2013) RNA-guided human genome engineering via Cas9. Science 339, 823–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J. A., Charpentier E. (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sampson T. R., Saroj S. D., Llewellyn A. C., Tzeng Y. L., Weiss D. S. (2013) A CRISPR/Cas system mediates bacterial innate immune evasion and virulence. Nature 497, 254–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ran F. A., Hsu P. D., Wright J., Agarwala V., Scott D. A., Zhang F. (2013) Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ding Q., Regan S. N., Xia Y., Oostrom L. A., Cowan C. A., Musunuru K. (2013) Enhanced efficiency of human pluripotent stem cell genome editing through replacing TALENs with CRISPRs. Cell Stem Cell 12, 393–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Xie K., Yang Y. (2013) RNA-guided genome editing in plants using a CRISPR-Cas system. Mol. Plant 6, 1975–1983 [DOI] [PubMed] [Google Scholar]
- 9. Bassett A. R., Liu J. L. (2014) CRISPR/Cas9 and genome editing in Drosophila. J. Genet. Genomics 41, 7–19 [DOI] [PubMed] [Google Scholar]
- 10. Waaijers S., Portegijs V., Kerver J., Lemmens B. B., Tijsterman M., van den Heuvel S., Boxem M. (2013) CRISPR/Cas9-targeted mutagenesis in Caenorhabditis elegans. Genetics 195, 1187–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yang H., Wang H., Shivalila C. S., Cheng A. W., Shi L., Jaenisch R. (2013) One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell 154, 1370–1379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Guo X., Zhang T., Hu Z., Zhang Y., Shi Z., Wang Q., Cui Y., Wang F., Zhao H., Chen Y. (2014) Efficient RNA/Cas9-mediated genome editing in Xenopus tropicalis. Development 141, 707–714 [DOI] [PubMed] [Google Scholar]
- 13. Niu Y., Shen B., Cui Y., Chen Y., Wang J., Wang L., Kang Y., Zhao X., Si W., Li W., Xiang A. P., Zhou J., Guo X., Bi Y., Si C., Hu B., Dong G., Wang H., Zhou Z., Li T., Tan T., Pu X., Wang F., Ji S., Zhou Q., Huang X., Ji W., Sha J. (2014) Generation of gene-modified cynomolgus monkey via Cas9/RNA-mediated gene targeting in one-cell embryos. Cell 156, 836–843 [DOI] [PubMed] [Google Scholar]
- 14. Larson M. H., Gilbert L. A., Wang X., Lim W. A., Weissman J. S., Qi L. S. (2013) CRISPR interference (CRISPRi) for sequence-specific control of gene expression. Nat. Protoc. 8, 2180–2196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang T., Wei J. J., Sabatini D. M., Lander E. S. (2014) Genetic screens in human cells using the CRISPR-Cas9 system. Science 343, 80–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shalem O., Sanjana N. E., Hartenian E., Shi X., Scott D. A., Mikkelsen T. S., Heckl D., Ebert B. L., Root D. E., Doench J. G., Zhang F. (2014) Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhou Y., Zhu S., Cai C., Yuan P., Li C., Huang Y., Wei W. (2014) High-throughput screening of a CRISPR/Cas9 library for functional genomics in human cells. Nature 509, 487–491 [DOI] [PubMed] [Google Scholar]
- 18. Schwank G., Koo B.-K., Sasselli V., Dekkers J. F., Heo I., Demircan T., Sasaki N., Boymans S., Cuppen E., van der Ent C. K., Nieuwenhuis E. E., Beekman J. M., Clevers H. (2013) Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell 13, 653–658 [DOI] [PubMed] [Google Scholar]
- 19. Wu Y., Liang D., Wang Y., Bai M., Tang W., Bao S., Yan Z., Li D., Li J. (2013) Correction of a genetic disease in mouse via use of CRISPR-Cas9. Cell Stem Cell 13, 659–662 [DOI] [PubMed] [Google Scholar]
- 20. Lee H. J., Kim E., Kim J. S. (2010) Targeted chromosomal deletions in human cells using zinc finger nucleases. Genome Res. 20, 81–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bauer D. E., Kamran S. C., Lessard S., Xu J., Fujiwara Y., Lin C., Shao Z., Canver M. C., Smith E. C., Pinello L., Sabo P. J., Vierstra J., Voit R. A., Yuan G. C., Porteus M. H., Stamatoyannopoulos J. A., Lettre G., Orkin S. H. (2013) An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science 342, 253–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ran F. A., Hsu P. D., Lin C. Y., Gootenberg J. S., Konermann S., Trevino A. E., Scott D. A., Inoue A., Matoba S., Zhang Y., Zhang F. (2013) Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154, 1380–1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhou J., Wang J., Shen B., Chen L., Su Y., Yang J., Zhang W., Tian X., Huang X. (2014) Dual sgRNAs facilitate CRISPR/Cas9-mediated mouse genome targeting. FEBS J. 281, 1717–1725 [DOI] [PubMed] [Google Scholar]
- 24. Xiao A., Wang Z., Hu Y., Wu Y., Luo Z., Yang Z., Zu Y., Li W., Huang P., Tong X., Zhu Z., Lin S., Zhang B. (2013) Chromosomal deletions and inversions mediated by TALENs and CRISPR/Cas in zebrafish. Nucleic Acids Res. 41, e141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gupta A., Hall V. L., Kok F. O., Shin M., McNulty J. C., Lawson N. D., Wolfe S. A. (2013) Targeted chromosomal deletions and inversions in zebrafish. Genome Res. 23, 1008–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mamaeva S. E., Tsvileneva N. N. (1985) A study of chromosome content of Friend virus-induced mouse erythroleukemia cells (clone M2) via karyotype reconstruction. Cancer Genet. Cytogenet. 16, 199–205 [DOI] [PubMed] [Google Scholar]
- 27. Miller D. A., Tantravahi R., Newman B., Dev V. G., Miller O. J. (1979) Karyotype of Friend virus-induced mouse erythroleukemia cells. Cancer Genet. Cytogenet. 1, 103–113 [Google Scholar]
- 28. Lappalainen T., Sammeth M., Friedländer M. R., 't Hoen P. A., Monlong J., Rivas M. A., Gonzàlez-Porta M., Kurbatova N., Griebel T., Ferreira P. G., Barann M., Wieland T., Greger L., van Iterson M., Almlöf J., Ribeca P., Pulyakhina I., Esser D., Giger T., Tikhonov A., Sultan M., Bertier G., MacArthur D. G., Lek M., Lizano E., Buermans H. P., Padioleau I., Schwarzmayr T., Karlberg O., Ongen H., Kilpinen H., Beltran S., Gut M., Kahlem K., Amstislavskiy V., Stegle O., Pirinen M., Montgomery S. B., Donnelly P., McCarthy M. I., Flicek P., Strom T. M., Geuvadis Consortium, Lehrach H., Schreiber S., Sudbrak R., Carracedo A., Antonarakis S. E., Häsler R., Syvänen A.-C., van Ommen G. J., Brazma A., Meitinger T., Rosenstiel P., Guigó R., Gut I. G., Estivill X., Dermitzakis E. T. (2013) Transcriptome and genome sequencing uncovers functional variation in humans. Nature 501, 506–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fu Y., Foden J. A., Khayter C., Maeder M. L., Reyon D., Joung J. K., Sander J. D. (2013) High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 31, 822–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hsu P. D., Scott D. A., Weinstein J. A., Ran F. A., Konermann S., Agarwala V., Li Y., Fine E. J., Wu X., Shalem O., Cradick T. J., Marraffini L. A., Bao G., Zhang F. (2013) DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 31, 827–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wu X., Scott D. A., Kriz A. J., Chiu A. C., Hsu P. D., Dadon D. B., Cheng A. W., Trevino A. E., Konermann S., Chen S., Jaenisch R., Zhang F., Sharp P. A. (2014) Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat. Biotechnol. 32, 670–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fu Y., Sander J. D., Reyon D., Cascio V. M., Joung J. K. (2014) Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat. Biotechnol. 32, 279–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tsai S. Q., Wyvekens N., Khayter C., Foden J. A., Thapar V., Reyon D., Goodwin M. J., Aryee M. J., Joung J. K. (2014) Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat. Biotechnol. 32, 569–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Guilinger J. P., Thompson D. B., Liu D. R. (2014) Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification. Nat. Biotechnol. 32, 577–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Maurano M. T., Humbert R., Rynes E., Thurman R. E., Haugen E., Wang H., Reynolds A. P., Sandstrom R., Qu H., Brody J., Shafer A., Neri F., Lee K., Kutyavin T., Stehling-Sun S., Johnson A. K., Canfield T. K., Giste E., Diegel M., Bates D., Hansen R. S., Neph S., Sabo P. J., Heimfeld S., Raubitschek A., Ziegler S., Cotsapas C., Sotoodehnia N., Glass I., Sunyaev S. R., Kaul R., Stamatoyannopoulos J. A. (2012) Systematic localization of common disease-associated variation in regulatory DNA. Science 337, 1190–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]