

Background: Lysine acetylation sites have been mapped, but information on stoichiometry is lagging.
Results: We developed and utilized the first direct, unbiased method for quantifying site-specific acetylation stoichiometry of a proteome without antibody enrichment.
Conclusion: High stoichiometry is associated with central metabolism, transcription, and translation. Loss of deacetylase CobB affects site-specific and global acetylation stoichiometry, altering acetyl-CoA metabolism.
Significance: Stoichiometry provides functional insight into protein acetylation.
Keywords: Acetyl-CoA Synthetase, Escherichia coli (E. coli), Mass Spectrometry (MS), Metabolism, Proteomics, Acetylation, Stoichiometry
Abstract
Acetylation of lysine ϵ-amino groups influences many cellular processes and has been mapped to thousands of sites across many organisms. Stoichiometric information of acetylation is essential to accurately interpret biological significance. Here, we developed and employed a novel method for directly quantifying stoichiometry of site-specific acetylation in the entire proteome of Escherichia coli. By coupling isotopic labeling and a novel pairing algorithm, our approach performs an in silico enrichment of acetyl peptides, circumventing the need for immunoenrichment. We investigated the function of the sole NAD+-dependent protein deacetylase, CobB, on both site-specific and global acetylation. We quantified 2206 peptides from 899 proteins and observed a wide distribution of acetyl stoichiometry, ranging from less than 1% up to 98%. Bioinformatic analysis revealed that metabolic enzymes, which either utilize or generate acetyl-CoA, and proteins involved in transcriptional and translational processes displayed the highest degree of acetylation. Loss of CobB led to increased global acetylation at low stoichiometry sites and induced site-specific changes at high stoichiometry sites, and biochemical analysis revealed altered acetyl-CoA metabolism. Thus, this study demonstrates that sirtuin deacetylase deficiency leads to both site-specific and global changes in protein acetylation stoichiometry, affecting central metabolism.
Introduction
Acetylation of the ϵ-amino group of lysine residues is now considered a major regulatory protein modification that influences many cellular processes, including protein-protein interactions, protein-DNA interactions, stability, cellular localization, and enzymatic activity (1, 2). Acetylation occurs by enzymatic and nonenzymatic mechanisms (3). Nonenzymatic acetylation occurs through nucleophilic attack of the lysine side chain on esters of the metabolic intermediates, acetyl-CoA and acetyl phosphate (4, 5). In prokaryotes, acetyl phosphate was reported to serve as the major acetyl donor (4). Removal of acetyl modifications requires protein deacetylases (6).
Mass spectrometry-based proteomic studies have mapped thousands of acetylated sites in a wide range of organisms (7–11). Recently, acetyl proteome measurements have utilized labeling strategies, such as tandem mass tags or stable isotope labeling by amino acids in cell culture, followed by immunoenrichment of acetylated peptides and mass spectrometry (8, 12–15). Although these approaches are valuable for comparing relative changes across conditions, they do not provide direct information on stoichiometry at individual sites (1, 14). Stoichiometric information is essential to accurately interpret the biological significance of these acetylation sites. For example, if only relative acetylation can be determined, as is the case with current acetylation methods, then a 1–5% and a 20–100% change would appear identical, i.e. a 5-fold change. The biological significance of these changes is entirely different, especially in cases in which acetylation is inhibitory.
Mass spectrometry has been used to examine phosphorylation stoichiometry in large scale studies (14, 16, 17). One central issue that is addressed by these approaches is that post-translationally modified peptides and their corresponding unmodified counterparts have varying ionization efficiencies, and thus, they cannot be directly compared. The issue of differing ionization efficiency can be addressed by making indirect measurements that incorporate correlated changes in phosphorylated and nonphosphorylated versions of peptides that are corrected for overall protein level changes across experiments with very different amounts of phosphorylation (18). Very recently, this indirect strategy was applied to estimate acetylation stoichiometry in log phase yeast compared with growth-arrested yeast (19). Yet, this strategy requires quantifying modified and unmodified versions of the same peptide, which are difficult to detect even with enrichment. It is biased in that it can only measure stoichiometry for peptides where PTM2 levels are dramatically different across conditions. Furthermore, it is unclear how the estimates behave when the changes in PTM status across conditions are small. In the context of phosphorylation, these challenges can be addressed by treating an identical stable isotope-labeled sample with a phosphatase and measuring and comparing the increased abundance of the unphosphorylated peptide (17). The percent increase provides a measurement of the stoichiometry of a phosphosite that circumvents the need for enrichment and the requirement for a change in the level of a PTM site. Yet no such approach exists for lysine acetylation.
Here, we developed and utilized the first unbiased and direct method for quantifying the stoichiometry of site-specific acetylation at the proteome-wide scale, without enrichment. Our approach uses a stable isotope chemical labeling step that acetylates all unmodified lysines resulting in a “heavy” and “light” acetyl-lysine pair across the entire proteome. This pair is then analyzed using high resolution, high accuracy mass spectrometry to yield proteome-wide, site-specific stoichiometry. Using this approach, we interrogated the acetyl proteome of Escherichia coli and investigated the functional consequences of genetically removing the sole NAD+-dependent protein deacetylase, CobB, on both site-specific and global acetylation. We quantified 2206 peptides from 899 proteins in the range of less than 1% up to 98% acetylation. Loss of CobB resulted in a slight increase of global acetylation at low stoichiometry sites, as well as site-specific changes at high stoichiometry sites. Proteins with the highest stoichiometry included those involved in central metabolism, transcription, and translation.
EXPERIMENTAL PROCEDURES
Cell Culture and Sample Preparation
E. coli BL21 (DE3) wild type and ΔCobB (with empty pQE80) were grown in 2× YT media under ampicillin selection overnight. Cells were harvested, flash-frozen, and stored in −80 °C. Frozen cells were resuspended in ∼10 volumes of 8 m urea solution (8 m urea, 5 mm DTT, 100 mm ammonium bicarbonate, pH 8) and lysed by sonication. After centrifugation, the protein concentration was determined using Bradford reagent (Bio-Rad).
AceCS2 Expression
AceCS2 pQE80 was transformed into E. coli BL21 (DE3), and protein expression was performed as described previously (20).
Protein Chemical Acetylation and Digestion
An equal amount of protein (100 μg) was diluted using 8 m urea solution (2 μg/μl) and incubated at 60 °C, 1000 rpm, for 30 min on the Thermomixer (Eppendorf). Cysteine alkylation with iodoacetamide was performed for 30 min in the dark. Chemical acetylation was performed as described previously, with slight modification (21). An equal volume of ammonium acetate solution (1 m NH4OAc, 8 m urea, 100 mm ammonium bicarbonate, pH 8) was added to each sample. ∼20 μmol of acetic anhydride (acetic anhydride, acetic anhydride-d6, or acetic anhydride-13C4,d6 (Sigma)) was added to each sample and incubated on a Thermomixer at 4 °C, 1000 rpm, for 20 min. After incubation, the pH was raised to ∼8 using ammonium hydroxide and checked using litmus paper. Chemical acetylation was repeated two more times, and buffer exchange occurred with 50 mm ammonium bicarbonate (pH 8), 10% methanol using 10K MWCO spin filters (Millipore). Trypsin was added at a 1:100 ratio and incubated overnight, 37 °C, 300 rpm on a Thermomixer.
LC-MS/MS
Peptides were separated with a Dionex Ultimate 3000 RSLCnano HPLC using a Waters Atlantis dC18 (100 μm × 150 mm) reverse phase column. The mobile phase consisted of (A) water with 0.1% formic acid and (B) acetonitrile with 0.1% formic acid. Peptides were eluted with a linear gradient of 2–40% B at a flow rate of 0.7 μl/min over 120 min and introduced into a hybrid quadrupole-Orbitrap mass spectrometer (Thermo Q Exactive) by nanoelectrospray ionization (Thermo Nanospray Flex). The MS survey scan was performed in positive ion mode with a resolution of 70,000, AGC of 1E6, maximum fill time of 100 ms, and scan range of 400 to 2000 m/z. Data-dependent MS/MS was performed with a resolution of 17,500, AGC of 1E5, maximum fill time of 64 ms, isolation window of 1.5 Da, underfill ratio of 0.1%, normalized collision energy of 28, dynamic exclusion of 20 s, and a loop count of 20. The source voltage was set at 2000 V and capillary temperature at 250 °C.
MS Data Analysis
Data were analyzed using a modified version of the December 25th, 2013, release of open-source, quantitative mass spectrometry analysis tool PVIEW (22, 23). We generated a peptide database with cleavage only at arginine, as trypsin did not cleave at acetylated lysines. Up to two missed cleavages were allowed. MS1 precursor tolerance was set to ±10 ppm, and MS2 fragment mass tolerance was set to ±15 ppm. Peptide identifications were obtained at a false discovery rate of 1%. Estimation of false discovery rate was carried out using only peptide spectrum matches from paired extracted ion chromatograms (XICs).
Acetylation Stoichiometry Calculations
Equation 1 was used to calculate stoichiometry of single lysine-containing peptides,
 |
and Equation 2 for double lysine-containing peptides,
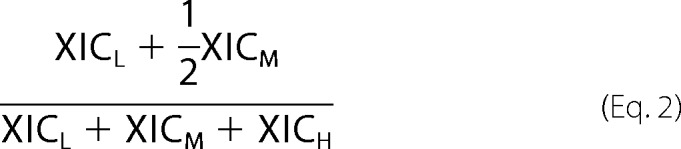 |
where XICL is the extracted ion chromatogram of the light acetyl peptide; XICH is the extracted ion chromatogram of the heavy acetyl peptide (acetic anhydride-d6 or acetic anhydride-13C4,d6), and XICM, when present, is the extracted ion chromatogram for the double lysine-containing peptide with a light and heavy acetyl-lysine.
Because of the isotopic purity of the commercially available acetic anhydride-13C4,d6 labeling reagent (97 atom % D, 99 atom % 13C), a global correction factor was applied to the stoichiometry of the Δ5-acetic anhydride-labeled sample.
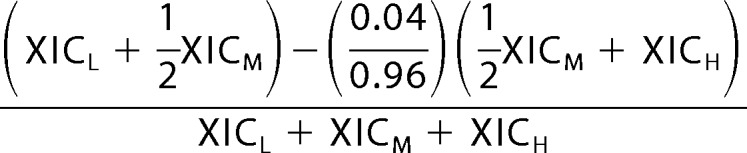 |
Determination of Chemical Labeling Efficiency
An equal amount of protein (BSA or E. coli lysate with expressed mouse AceCS2) was prepared as described above. An unlabeled sample was used as a control. Protein digestion, LC-MS/MS, and database search were performed as described above.
Unmodified tryptic peptides of the labeled and control sample were monitored and compared to determine labeling efficiency. Briefly, the peak area for a lysine-containing peptide in the control sample was measured. In the labeled sample, the same lysine site would be acetylated to some degree, generating a larger acetyl peptide and also the unmodified peptide proportionate to the degree of chemical labeling. As a result, the unmodified peptide in both the labeled and control samples can be compared to determine labeling efficiency. For normalization across samples, a peptide lacking lysine residues was used. Peak areas for each peptide were determined using Xcalibur 2.2 SP1 (Thermo Scientific).
Stoichiometry Curve Determination
An equal amount of BSA was chemically acetylated with acetic anhydride or acetic anhydride-13C4,d6 and digested as described above. Light and heavy BSA peptides were resuspended to equal concentrations and then mixed at varying ratios corresponding to 1, 5, 10, 20, 25, 40, 50, 60, 75, 80, 90, 95, and 99% acetylation. The BSA samples were then added to a trypsin-digested E. coli proteome at equal concentrations, analyzed by LC-MS/MS, and data processed as described above.
Bioinformatics
Gene ontology (GO) and pathway analysis was performed using DAVID version 6.7 (24, 25). For the enrichment analysis, an unlabeled, trypsin-digested, E. coli proteome was used as the background. Network analysis was performed using STRING version 9.1 with proteins having an acetylation stoichiometry greater than 7% as the input (26, 27). The network was visualized using Cytoscape version 3.1.0 (28).
Metabolite Analysis
To measure intracellular metabolite levels, cells were grown in liquid culture with 2× YT media to early stationary phase. Metabolites were extracted by filtering a 5-ml culture on nylon filters (Millipore) and quenching the cells in cold extraction solvent (40:40:20 methanol/acetonitrile/water). Cell extracts were centrifuged followed by one round of re-extraction. Supernatants from two rounds of extraction were combined, dried under N2, and resuspended in HPLC water for analysis. The metabolites were analyzed by LC-MS as described previously (29). Briefly, LC separation on a Synergy Fusion-RP column (100 mm × 2 mm, 2.5-μm particle size, Phenomenex, Torrance, CA) using a gradient of solvent A (97:3 H2O/MeOH with 10 mm tributylamine and 5 mm ammonium bicarbonate), and solvent B (100% MeOH) was coupled by negative mode electrospray ionization to a stand-alone Q Exactive Orbitrap mass spectrometer (Thermo Scientific).
RESULTS
Method Development and Validation
To measure acetylation stoichiometry on the proteome scale, we developed a robust workflow using chemical acetylation with isotopic acetic anhydride followed by trypsin digestion and high resolution mass spectrometry (Fig. 1). The general strategy involves preparing denatured protein extracts from cells and chemically acetylating free lysine residues with isotopic acetic anhydride, followed by trypsin cleavage and MS analysis. Using this process, every lysine bears an acetyl group, either the light version derived from endogenous acetylation or the heavy version originating from in vitro chemical acetylation. The trypsin cleavage step produces chemically identical peptides, which have matching retention times. The corresponding pairs of peptides that differ in mass are resolved using mass spectrometry, and direct stoichiometry is determined by dividing the light peak area over the sum of the light and heavy peak areas.
FIGURE 1.

Diagram of method for determining direct acetylation stoichiometry. Extracted protein was denatured, chemically acetylated using isotopic acetic anhydride followed by trypsin digestion. Peptides were analyzed by LC-MS/MS and quantified to determine site-specific stoichiometry.
To ensure accurate determination of stoichiometry, we first evaluated acetic anhydride labeling efficiency within a simple and complex protein sample. An E. coli whole-cell lysate was chemically modified with acetic anhydride and qualitatively assessed for the degree of acetylation. Western blot analysis revealed that acetic anhydride chemically acetylated E. coli proteins across the full range of protein sizes (Fig. 2A). To quantitatively assess the degree of chemical acetylation in a simple protein sample, equal amounts of labeled and unlabeled bovine serum albumin (BSA) were digested and analyzed by mass spectrometry. Unmodified BSA tryptic peptides in both samples were monitored and compared to determine the degree of acetylation. The labeling efficiency across eight out of nine BSA peptides was greater than 98% (Fig. 2B). To determine the labeling efficiency in a complex sample, the mouse AceCS2 protein was recombinantly expressed in BL21 (DE3) bacteria. The cells were lysed; protein was labeled with increasing amounts of acetic anhydride, and the labeling efficiency was calculated for peptides of AceCS2 protein. Across the eight sites in AceCS2, the labeling efficiency was >98% (Table 1). These results indicate that proteins in complex mixtures can be chemically acetylated with high efficiency.
FIGURE 2.

Acetic anhydride comprehensively modifies lysine residues on a wide range of protein sizes and to a high degree. Chemical acetylation by acetic anhydride was assessed qualitatively and quantitatively. A, Western blotting analysis with α-acetyl-lysine antibody with an unmodified and modified E. coli sample shows chemical labeling across a full range of proteins in a complex mixture. B, degree of acetylation was determined using bovine serum albumin. The indicated peptides were quantified in the unmodified and modified samples. The abundance of each peptide correlates to the degree of chemical acetylation by acetic anhydride.
TABLE 1.
Degree of chemical acetylation with increasing amount of acetic anhydride in a complex mixture
An E. coli lysate with expressed AceCS2 protein (Uniprot ID Q99NB1) was used to determine acetylation efficiency by acetic anhydride. Nonacetylated tryptic peptides of the labeled and control sample were monitored and compared to determine labeling efficiency. The XIC for a lysine-containing peptide in the control sample was quantified. In the labeled sample, the same lysine site would be acetylated to some degree, generating two peptides after trypsin cleavage as follows: 1) a larger acetyl peptide, and 2) the unmodified peptide, proportionate to the degree of chemical labeling. Therefore, the unmodified peptide from the labeled and control samples can be compared to determine labeling efficiency. For normalization across samples, a peptide lacking lysine residues is used.
| Peptide | Nanomoles of acetic anhydride/μg of protein |
||
|---|---|---|---|
| ∼200 | ∼300 | ∼400 | |
| K_AVITFNQGLR | 0.998 | 0.998 | 0.998 |
| K_GNVLEGGDVSGALCISQAWPGMAR | 0.924 | 0.931 | 0.967 |
| K_INQFYGAPTAVR | 0.980 | 0.989 | 0.990 |
| K_SCPTVQHVLVAHR | 0.991 | 0.999 | 0.997 |
| K_SPETIALIWER | 0.997 | 0.998 | 0.997 |
| R_GQDLGDTTTLEDPSVITEILSAFQK | 0.969 | 0.992 | 0.994 |
| R_LGTAEIEDAMADHPAVPETAVIGYPHDIK | 0.990 | 0.994 | 0.997 |
| R_TLGSVGEPINHEAWEWLHK | 0.972 | 0.989 | 0.992 |
We developed a computational method for stoichiometric determination of acetylation that avoids the need for immunoenrichment. Existing proteome-wide acetylation studies require an enrichment step to increase sensitivity in the PTM analysis (14). Sensitivity is achieved by the increased abundance and subsequent identification of acetylated peptides. In this study, we achieved high sensitivity by coupling a novel computational algorithm, implemented in a peptide quantification software tool called PVIEW, to the chemical acetylation strategy. This algorithm identifies acetyl peptides (heavy or light) using one of two databases (Fig. 3A) and alternates between iterations of heavy-to-light and light-to-heavy XIC pairing (Fig. 3, B–D), using the high abundant species as a proxy to identify the corresponding, lower abundant, peptide pair. In essence, the algorithm identifies the best acetyl peptide and based on charge and isotopic shift, the corresponding peptide pair is identified, matched, and quantified. In this manner, our algorithm performs an in silico enrichment of acetylated peptides.
FIGURE 3.

Diagram of LC-MS/MS data analysis algorithm used for identifying isotopic pairs for stoichiometry determination. The pairing algorithm was carried out using XICs. The peaks here are for illustration purposes only. A, we created two separate MS2 peptide search databases as follows: 1) a “light” database with native acetylation as a fixed modification for each lysine; and 2) a “heavy” database where every lysine is modified by the isotopic acetic anhydride. The algorithm used two iterations as follows: light-to-heavy pairing when the native acetyl peptide was the most highly abundant species (B) and a heavy-to-light pairing when chemically labeled peptide was the most highly abundant species (C). In each respective iteration for an XIC with an associated MS2 spectrum, we determined the best matching peptide based on MS2 search score using the light or heavy database. We used the expected isotopic shift to find the corresponding MS1 peak. D, for low stoichiometry sites, the algorithm is able to perform in silico enrichment. The algorithm uses the MS2 spectrum and MS1 signal of the heavy acetyl peptide as a proxy for the native acetyl peptide. Using this proxy information, the algorithm identifies a low level signal from the native acetyl peptide.
We demonstrated the capability of measuring acetylation stoichiometry across a broad range. BSA protein was chemically acetylated with either acetic anhydride (light) or isotopic acetic anhydride (heavy) followed by trypsin digestion. Peptides were then mixed at distinct ratios corresponding to 1–99% acetylation and added to a trypsin-digested E. coli proteome. This experiment served to mimic the full range of endogenous acetylation that might be encountered from diverse biological conditions. Samples were analyzed by LC-MS/MS, and the stoichiometries of five BSA acetyl peptides were quantified using our novel quantification algorithm. As a representative, the MS/MS spectrum for the acetyl peptide, ALK(Ac)AWSVAR, is shown in Fig. 4A. MS1 spectra corresponding to 10, 50, and 90% stoichiometry are shown in Fig. 4, B–D, respectively. The stoichiometry curve encompassing 1–99% stoichiometry is shown in Fig. 4E. Linear regression analysis of the stoichiometry curves from all five acetyl-BSA peptides show a high correlation with R2 values ranging between 0.970 and 0.995 (Fig. 4F). These results establish the feasibility of accurately determining stoichiometry across a wide range of values in a complex mixture.
FIGURE 4.

Acetylation stoichiometry is detected and quantified across a full range of values. Light and Δ5-acetic anhydride-labeled BSA protein digests were mixed at different ratios corresponding to 1–99% acetylation followed by MS analysis and quantitation. A, MS/MS spectrum for the acetylated BSA peptide, ALK(Ac)AWSVAR, is shown. (Product ions shown are charge 1+.) B–D, MS1 spectrum showing light and heavy forms of the peptide in A (charge 2+) corresponding to 10, 50, 90% acetylation, respectively, with a mass shift of ∼2.5 m/z. E, scatterplot comparing percent volume input (light/total) to the corrected measured stoichiometry of the BSA peptide, ALK(Ac)AWSVAR. Stoichiometry values were corrected due to the isotopic purity of ∼96%. F, linear regression analysis of all the acetyl peptides quantified shows the best fit line and high R2 value.
Acetylation Stoichiometry Values from Proteomes of WT and ΔCobB Bacteria
Many bacteria, like E. coli, contain a single NAD+-dependent protein deacetylase that is the ortholog to CobB from Salmonella enterica. In S. enterica, CobB regulates metabolic pathways that permit growth on alternative carbon sources such as short chain fatty acids (SCFA) (30, 31) and citrate (32). Here, we applied our new method to investigate the role of CobB on both site-specific and global acetylation stoichiometries.
We determined protein acetylation of BL21 (DE3) wild type and ΔCobB strains, analyzing three biological replicates for each strain. Cells were grown in rich media, harvested during stationary phase, and processed as outlined in Fig. 1, using acetic anhydride-d6 (Δ3-acetic anhydride) or acetic anhydride-13C4,d6 (Δ5-acetic anhydride) for the isotopic chemical labeling. We identified a total of 4025 and 2659 acetyl-lysine sites corresponding to 852 and 576 proteins for the Δ3 and Δ5 acetic anhydride labeling, respectively (Table 2 and supplemental Tables S2 and S4). As a consequence of all lysines bearing an acetyl moiety, trypsin cleavage occurs only at arginine residues (33, 34). Using Δ3 and Δ5 acetic anhydride-labeling strategies, we quantified stoichiometry for peptides bearing one and two lysine residues, corresponding to 3123 and 2099 peptides, respectively (Table 2).
TABLE 2.
Acetylation stoichiometry values
The number of acetyl peptides was quantified across the experimental conditions and strains. This includes the total number of acetylation sites quantified, 1K and 2K peptides, and total proteins identified.
| Acetic anhydride |
ΔCobB | WT | Total | ||
|---|---|---|---|---|---|
| Δ3 | Δ5 | ||||
| Lysine sites detected | 4025 | 2659 | 2573 | 1945 | 2206 |
| Single lysine peptides with stoichiometry | 2221 | 1539 | 1451 | 1103 | 1602 |
| Double lysine peptides with stoichiometry | 902 | 560 | 561 | 421 | 603 |
| Proteins identified | 852 | 576 | 827 | 680 | 899 |
We observed a wide distribution of acetyl stoichiometry, ranging from less than 1% up to 98% (Fig. 5A). Among the peptides quantified, ∼2500 displayed stoichiometry between 0 and 10%, ∼500 were acetylated between 10 and 20%, ∼60 between 20 and 30%, and 70 displayed stoichiometry greater than 30%. Thus, the largest fraction of peptides exhibiting stoichiometry greater than 20% acetyl-lysine is 4% of the quantified proteome (Fig. 5B).
FIGURE 5.

Site-specific and global acetylation stoichiometry in BL21 (DE3) WT and ΔCobB strains. A, distribution of acetylation stoichiometry obtained in WT and ΔCobB strains and B, a pie chart showing the distribution of stoichiometry across the entire E. coli proteome. C, acetylated peptides of all biological conditions from the Δ5-acetic anhydride labeling were compared. Left of diagonal shows the scatterplot of acetyl peptides from each biological condition. Right of diagonal is the Spearman's correlation coefficient. Correlation within biological replicates is high (0.811–0.849 for WT and 0.813–0.845 for ΔCobB), whereas the correlation decreased across biological conditions (0.5632–0.6208). Number in parentheses represents the total number of peptides analyzed in scatterplot. D, histogram of log2 fold change between ΔCobB and WT acetyl peptides. The plot shows a normal distribution and slight shift toward the right.
To assess reproducibility and reveal global differences between strains, we compared peptide level acetylation stoichiometry across all Δ5-acetic anhydride-prepared biological samples. Stoichiometry values of acetylated peptides within the same biological condition show the highest correlation with a Spearman's correlation coefficient of 0.811–0.849 for wild type and 0.813–0.845 for ΔCobB (Fig. 5C). However, when stoichiometries are compared across biological conditions, a decrease in correlation was observed (0.563–0.621), suggesting that the ΔCobB strain differs substantially in the acetylation profile compared with the WT strain. To assess global alterations in acetylation, we plotted a histogram of peptide level ΔCobB/WT log2 fold change. The ΔCobB strain displayed a slight global increase in acetylation when compared with WT, suggesting that CobB alters the E. coli acetyl proteome under stationary phase growth (Fig. 5D).
We next determined site-specific acetyl stoichiometry changes upon CobB deletion by generating a stoichiometry line plot that compares the acetylation of WT and ΔCobB peptides and quantifies the magnitude change between the two conditions, revealing peptides that have higher stoichiometry in either ΔCobB (red lines) or WT (black lines) strains (Fig. 6 and supplemental Tables S1 and S3). This analysis also revealed three groups of peptide stoichiometry profiles as follows: 1) peptides with low acetylation stoichiometry and low magnitude change; 2) peptides with higher acetylation stoichiometry and low magnitude change; and 3) peptides with different stoichiometries and a high magnitude change between conditions. Group 1 represents the majority of peptides, which is reflected by the low average global acetylation stoichiometry (Fig. 6). Interestingly, group 2 peptides had a higher stoichiometry regardless of CobB status, which suggests these acetyl-lysine sites are not targets of CobB. Finally, group 3 peptides had the highest magnitude change between WT and ΔCobB and included acetyl peptides that increase in the CobB deletion. These include tryptophan-tRNA ligase (TrpS), 30 S ribosomal protein S16 (RpsP), peptide deformylase (Def), xanthine phosphoribosyltransferase (Gpt), and acetyl-CoA synthetase (Acs).
FIGURE 6.

Dynamics of acetylation stoichiometry in WT and ΔCobB strains. Stoichiometry line plot demonstrates peptide level stoichiometry in WT and ΔCobB and the magnitude change between strains. Peptides are shown that have a higher stoichiometry in WT (black lines) and ΔCobB (red lines).
Bioinformatic and Network Analysis of Acetylation Stoichiometry
To examine whether proteins that display substantial stoichiometry are enriched in specific cellular processes, we performed an enrichment analysis of our unique stoichiometry results using the database for annotation, visualization, and integrated discovery (DAVID) (24, 25). Certain metabolic pathways and ribosomal proteins were significantly enriched above background (Fig. 7A). Gene Ontology (GO) analysis also revealed that acetyl-CoA metabolic processes, transcription, translation, and tRNA aminoacylation were significantly enriched (Fig. 7B). A network analysis of the identified pathways was performed using the STRING Database (26, 27). Acetylation stoichiometries along with the number sites were superimposed across the protein interaction network and visualized using Cytoscape (28). Acetylation sites per protein varied across the proteome (Fig. 7C); therefore, we used the highest stoichiometry quantified for each protein in the network. The pentose phosphate pathway, glycolysis, pyruvate metabolism, and the TCA cycle revealed proteins with sites of higher stoichiometry (Fig. 7D). The proteins in these pathways were also enriched in total acetylation, as many proteins in these pathways had more than seven acetyl-lysine sites (Fig. 7, D and F). Proteins involved in transcription and translation displayed many sites of acetylation and high stoichiometries (Fig. 7, E and F). This unique network analysis revealed the range of stoichiometries in major cellular pathways and processes.
FIGURE 7.

Bioinformatic and network analysis of acetylation stoichiometry. KEGG pathway analysis (A) and gene ontology (B) of biological process for acetylated proteins in both WT and ΔCobB using an acetylation stoichiometry greater than 7% (average stoichiometry). Pathways and biological processes are significantly enriched (p < 0.05) above an unmodified, trypsin-digested E. coli proteome (background proteome used for DAVID analysis). C, pie chart of acetylation sites detected per protein. Network analysis of metabolic pathways (D) and transcriptional and translational biological processes (E). F, legend for D and E. Shapes represent the number of acetylation sites detected per protein. The size of the shape is proportional to the highest acetyl peptide stoichiometry of a protein in either WT or ΔCobB. Proteins are colored according to pathway or biological process.
Total acetylation site number and high stoichiometry enrichment in central metabolic pathways suggest that CobB deletion affects these pathways. To visualize these results, we plotted the TCA cycle and pyruvate metabolism pathways with acetyl-CoA at the crossroads of these two pathways (Fig. 8). As evident from this analysis, enzymes that either utilize or generate acetyl-CoA exhibited peptides with higher stoichiometry, such as acetyl-CoA carboxylase subunit α (AccA), biotin carboxylase (AccC), formate acetyltransferase (PflB), phosphate acetyltransferase (Pta), acetyl-CoA synthetase (Acs), citrate synthase (GltA), and aldehyde-alcohol dehydrogenase (AdhE) (Fig. 8). Also, proteins in these pathways displayed a higher number of quantifiable peptides with above average stoichiometry. For example, pyruvate kinase II (PykA), phosphoenolpyruvate synthase (PpsA), Pta, aconitate hydratase 1 and 2 (AcnA and AcnB), α-ketoglutarate dehydrogenase E1 component (SucA) and AdhE had at least six peptides with a stoichiometry higher than 7%.
FIGURE 8.

Acetylation of enzymes in metabolic pathways and metabolite analysis. A, enzymes of pyruvate metabolism and the TCA cycle are shown. Filled circles represent a peptide identified with an acetyl stoichiometry greater than 7% (including single and double lysine-containing peptides). The color of each circle represents the degree of acetylation according to heat map legend. Enzymes with the highest degree of acetylation include PykA, Pta, AccA, AccC, Acs, GltA, AdhE, and Msa. Enzymes with the highest number of acetyl-lysine peptides include PpsA, Pta, AdhE, AcnA, AcnB, and SucA. B, acetyl phosphate is significantly increased in the ΔCobB strain (p = 0.01).
Acetyl-CoA is at the junction of central metabolism. It is striking that most of the metabolic enzymes one reaction away from generating or utilizing acetyl-CoA have a higher acetylation stoichiometry in the ΔCobB strain during stationary phase, suggesting that CobB is involved in many facets of acetyl-CoA utilization.
Metabolite Analysis Reveals Increased Levels of Acetyl Phosphate in the CobB Mutant
NAD+-dependent protein deacetylases (sirtuins) regulate many metabolic enzymes in prokaryotes and eukaryotes (35). In prokaryotes, CobB regulates acetyl-CoA synthetase, which is required for cell growth on acetate (30, 31). Activation of Acs by reversible acetylation is thought to be essential for “the acetate switch” (36). This transition occurs when bacterial cells have depleted carbon sources such as glucose and begin to utilize environmental acetate for growth. If flux through Acs is inhibited, for example by acetylation, then acetate can be diverted through the acetate kinase, Pta pathway to generate acetyl-CoA (Fig. 8). The higher stoichiometry as well as multiple acetylation sites on Pta suggest an inhibitory effect on protein function. Because acetate is rerouted through the acetate kinase-Pta pathway due to Acs inhibition, we speculated that there would be a buildup of acetyl phosphate if Pta function were inhibited by acetylation. To test this, we measured acetyl phosphate in the WT and ΔCobB strains. Indeed, there was a significant increase of acetyl phosphate in the CobB mutant (Fig. 8B). This suggests that strains lacking CobB have an inability to efficiently utilize acetyl phosphate in the ΔCobB strain, consistent with inhibition of Pta activity.
DISCUSSION
In this work, we developed a novel method to quantify site-specific acetylation stoichiometry across an entire proteome. This method utilizes isotopic acetic anhydride to chemically acetylate all unmodified lysines in a protein sample. Coupling this protein chemistry to high resolution mass spectrometry and novel bioinformatic data processing, acetylation stoichiometry was quantified. Applying this method to bacterial cells, we describe the effects of the bacterial sirtuin member, CobB, on site-specific and global acetylation.
Site-specific Acetylation Stoichiometry
One role of the NAD+-dependent sirtuins is the regulation of metabolism by deacetylation of key transcription factors and metabolic enzymes themselves (6). CobB deacetylates acetyl-CoA synthetase at a conserved lysine residue, thereby activating Acs (30, 31). Acs forms acetyl-CoA from acetate and ATP, thus allowing bacterial cells to utilize alternative carbon sources for energy when glucose is depleted. The active site lysine was not identified in this study, likely due to the small size of the peptide (<350 m/z) upon trypsin cleavage. We did, however, identify and quantify another acetyl site on Acs, Lys-348, which displayed 32.5% acetylation in ΔCobB and 8.3% in WT. Further investigation is needed to determine whether this site is critical for Acs activity as it lies at the dimeric interface.
Metabolic enzymes of the TCA cycle, pyruvate metabolism, and glycolysis were also highly enriched in acetylation, most notably in the number of acetyl-lysine sites. Of these enriched metabolic pathways, enzymes one reaction away from acetyl-CoA displayed higher stoichiometry, including Pta, AccA, AccC, GltA, AdhE, and Msa (Fig. 8). It is noteworthy that the enzymes, which utilize or generate acetyl-CoA, could also be regulated by reversible acetylation. This observation coupled with the known inhibition of Acs, led us to examine and demonstrate increased levels of acetyl phosphate in the CobB-deficient strain, suggesting an inhibitory effect of acetylation on Pta activity.
Of the group 3 peptides (those showing substantially increased acetylation in the ΔCobB), RpsP, TrpS, Def, and Gpt had acetylation sites with the largest magnitude change in the CobB mutant. RpsP had a magnitude difference of 47% acetylation between the WT and ΔCobB strains. As a component of the 30 S ribosome, RpsP interacts with the RNA component of the ribosome. The ϵ-amino group of Lys-46 appears to make a critical contact with the 2′-OH of a guanine nucleotide of the RNA (37). Upon acetylation, this contact could destabilize the interaction between RpsP and RNA. TrpS generates tryptophanyl-tRNA that is used for protein synthesis, and Lys-312 has a magnitude difference of 64% acetylation between WT and ΔCobB. There is not a solved crystal structure for the E. coli form; however, modeling with the Yersinia pestis variant shows that the analogous Lys site, Lys-324, is 2.7 Å away from the backbone oxygen of Pro-86, which likely forms H-bonding contact between the two residues (38, 39). Deformylase removes the formyl group from the N-terminal methionine, and the peptide containing Lys-158/161 has a magnitude change of 39%. These sites are on the surface of the protein, and it is not known whether they are critical for protein-protein interaction. Xanthine phosphoribosyltransferase is involved in purine metabolism. The acetyl site, Lys-30, has a magnitude change of 27% between WT and ΔCobB strains. The crystal structure reveals this site is also surface-exposed, and it is unknown whether it plays a role in interactions (40).
Global Acetylation Stoichiometry
The ability to measure acetyl-lysine stoichiometry allowed us to estimate the amount of total acetate attached to protein. The average global acetyl stoichiometry was ∼7%, and we determined stoichiometry for ∼30% of the quantifiable lysines in the E. coli proteome. Using these values and the known calculated values of E. coli for cell volume (BNID 100004), protein concentration (BNID 108263), and the frequency of lysines present in E. coli (BNID 100636) (13), we estimated the amount of acetate on protein to be between 1.7 and 2.8 mm. This is striking because the concentration of acetyl-CoA is estimated to be less than 500 μm in E. coli (15). This analysis reveals the high amount of acetate stored on proteins, although future experiments will be needed to assess the turnover rate with respect to acetyl-CoA and acetyl phosphate consumption.
Stoichiometry Versus Relative Quantitation
Knowledge of stoichiometry is a key piece of information to accurately interpret the functional roles of acetylation. Current acetyl proteomic methods rely on relative changes of peptides across conditions. In most cases, researchers have used an arbitrary 2-fold change cutoff as meaningful (4, 11), which can greatly skew interpretation. For example, in our study, the acetyl peptide from glutathione reductase (Gor) containing Lys-105/Lys-112 has a ΔCobB/WT fold change of 9.3. When the stoichiometry is considered, this peptide is 0.5% acetylated in WT and 4.3% in ΔCobB. This results in a magnitude change of 3.8% acetylation between the two conditions, which might only reflect biological variation. For the converse example, the acetyl peptide from 3-phosphoshikimate 1-carboxyvinyltransferase (AroA) containing Lys-354 has a ΔCobB/WT fold change of 0.6. However, the stoichiometry of this peptide is 38.8 and 69.5% for ΔCobB and WT, respectively (Fig. 6). This is a magnitude difference of 30.7% acetylation and is likely to have more of an impact on overall protein function than the former example.
Unique Features of Methodology
Major advantages/improvements of the presented method include the following. 1) Quantitation is independent of protein level because stoichiometry depends on the total protein in a given sample. This eliminates the requirement to normalize to total protein amount. 2) Stoichiometry as well as relative changes can be determined using this method. 3) There is no need for immunoenrichment of acetyl peptides. This avoids any issues toward antibody enrichment bias introduced in the analysis. This issue is circumvented with the pairing algorithm of PVIEW by performing an in silico enrichment of acetyl peptides. 4) Low amount of starting material is used. Because of the minimal sample processing, less protein can be used, facilitating the analysis of limited and difficult to obtain samples. 5) This method has broad applicability, from an isolated purified protein to cell culture or tissue sample.
Supplementary Material
Acknowledgments
We thank Jorge Escalante for providing the BL21 (DE3) ΔCobB E. coli strain and the Sushmita Roy laboratory for helpful discussions on performing the network analysis.
This work was supported, in whole or in part, by National Institutes of Health Grant GM065386 (to J. M. D.). This work was also supported by National Science Foundation Grant GRF DGE-1256259 (to J. B.).
This article was selected as a Paper of the Week.

This article contains supplemental Tables S1 to S4.
- PTM
- post-translational modification
- Pta
- phosphate acetyltransferase
- Acs
- acetyl-CoA synthetase
- XIC
- extracted ion chromatogram.
REFERENCES
- 1. Xiong Y., Guan K. L. (2012) Mechanistic insights into the regulation of metabolic enzymes by acetylation. J. Cell Biol. 198, 155–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yang X. J., Seto E. (2008) Lysine acetylation: codified crosstalk with other posttranslational modifications. Mol. ;cell 31, 449–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tanner K. G., Trievel R. C., Kuo M. H., Howard R. M., Berger S. L., Allis C. D., Marmorstein R., Denu J. M. (1999) Catalytic mechanism and function of invariant glutamic acid 173 from the histone acetyltransferase GCN5 transcriptional coactivator. J. Biol. Chem. 274, 18157–18160 [DOI] [PubMed] [Google Scholar]
- 4. Weinert B. T., Iesmantavicius V., Wagner S. A., Schölz C., Gummesson B., Beli P., Nyström T., Choudhary C. (2013) Acetyl phosphate is a critical determinant of lysine acetylation in E. coli. Mol. Cell 51, 265–272 [DOI] [PubMed] [Google Scholar]
- 5. Wagner G. R., Payne R. M. (2013) Widespread and enzyme-independent Nϵ-acetylation and Nϵ-succinylation of proteins in the chemical conditions of the mitochondrial matrix. J. Biol. Chem. 288, 29036–29045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Feldman J. L., Dittenhafer-Reed K. E., Denu J. M. (2012) Sirtuin catalysis and regulation. J. Biol. Chem. 287, 42419–42427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kim S. C., Sprung R., Chen Y., Xu Y., Ball H., Pei J., Cheng T., Kho Y., Xiao H., Xiao L., Grishin N. V., White M., Yang X. J., Zhao Y. (2006) Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol. Cell 23, 607–618 [DOI] [PubMed] [Google Scholar]
- 8. Choudhary C., Kumar C., Gnad F., Nielsen M. L., Rehman M., Walther T. C., Olsen J. V., Mann M. (2009) Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840 [DOI] [PubMed] [Google Scholar]
- 9. Hebert A. S., Dittenhafer-Reed K. E., Yu W., Bailey D. J., Selen E. S., Boersma M. D., Carson J. J., Tonelli M., Balloon A. J., Higbee A. J., Westphall M. S., Pagliarini D. J., Prolla T. A., Assadi-Porter F., Roy S., Denu J. M., Coon J. J. (2013) Calorie restriction and SIRT3 trigger global reprogramming of the mitochondrial protein acetylome. Mol. Cell 49, 186–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lundby A., Lage K., Weinert B. T., Bekker-Jensen D. B., Secher A., Skovgaard T., Kelstrup C. D., Dmytriyev A., Choudhary C., Lundby C., Olsen J. V. (2012) Proteomic analysis of lysine acetylation sites in rat tissues reveals organ specificity and subcellular patterns. Cell Reports 2, 419–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rardin M. J., Newman J. C., Held J. M., Cusack M. P., Sorensen D. J., Li B., Schilling B., Mooney S. D., Kahn C. R., Verdin E., Gibson B. W. (2013) Label-free quantitative proteomics of the lysine acetylome in mitochondria identifies substrates of SIRT3 in metabolic pathways. Proc. Natl. Acad. Sci. U.S.A. 110, 6601–6606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ong S. E., Blagoev B., Kratchmarova I., Kristensen D. B., Steen H., Pandey A., Mann M. (2002) Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 1, 376–386 [DOI] [PubMed] [Google Scholar]
- 13. Milo R., Jorgensen P., Moran U., Weber G., Springer M. (2010) BioNumbers–the database of key numbers in molecular and cell biology. Nucleic Acids Res. 38, D750–D753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Olsen J. V., Mann M. (2013) Status of large-scale analysis of post-translational modifications by mass spectrometry. Mol. Cell. Proteomics 12, 3444–3452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Danchin A., Dondon L., Daniel J. (1984) Metabolic alterations mediated by 2-ketobutyrate in Escherichia coli K12. Mol. Gen. Genet. 193, 473–478 [DOI] [PubMed] [Google Scholar]
- 16. Olsen J. V., Vermeulen M., Santamaria A., Kumar C., Miller M. L., Jensen L. J., Gnad F., Cox J., Jensen T. S., Nigg E. A., Brunak S., Mann M. (2010) Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal. 3, ra3. [DOI] [PubMed] [Google Scholar]
- 17. Wu R., Haas W., Dephoure N., Huttlin E. L., Zhai B., Sowa M. E., Gygi S. P. (2011) A large-scale method to measure absolute protein phosphorylation stoichiometries. Nat. Methods 8, 677–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Steen H., Jebanathirajah J. A., Springer M., Kirschner M. W. (2005) Stable isotope-free relative and absolute quantitation of protein phosphorylation stoichiometry by MS. Proc. Natl. Acad. Sci. U.S.A. 102, 3948–3953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Weinert B. T., Iesmantavicius V., Moustafa T., Schölz C., Wagner S. A., Magnes C., Zechner R., Choudhary C. (2014) Acetylation dynamics and stoichiometry in Saccharomyces cerevisiae. Mol. Syst. Biol. 10, 716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hallows W. C., Lee S., Denu J. M. (2006) Sirtuins deacetylate and activate mammalian acetyl-CoA synthetases. Proc. Natl. Acad. Sci. U.S.A. 103, 10230–10235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Riordan J. F., Vallee B. L. (1972) [41] acetylation. Methods Enzymol. 25, 494–499 [DOI] [PubMed] [Google Scholar]
- 22. Khan Z., Amini S., Bloom J. S., Ruse C., Caudy A. A., Kruglyak L., Singh M., Perlman D. H., Tavazoie S. (2011) Accurate proteome-wide protein quantification from high resolution 15N mass spectra. Genome Biol. 12, R122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Khan Z., Bloom J. S., Garcia B. A., Singh M., Kruglyak L. (2009) Protein quantification across hundreds of experimental conditions. Proc. Natl. Acad. Sci. U.S.A. 106, 15544–15548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Huang da W., Sherman B. T., Lempicki R. A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 [DOI] [PubMed] [Google Scholar]
- 25. Huang da W., Sherman B. T., Lempicki R. A. (2009) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Franceschini A., Szklarczyk D., Frankild S., Kuhn M., Simonovic M., Roth A., Lin J., Minguez P., Bork P., von Mering C., Jensen L. J. (2013) STRING version 9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 41, D808–D815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Snel B., Lehmann G., Bork P., Huynen M. A. (2000) STRING: a web-server to retrieve and display the repeatedly occurring neighbourhood of a gene. Nucleic Acids Res. 28, 3442–3444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Saito R., Smoot M. E., Ono K., Ruscheinski J., Wang P. L., Lotia S., Pico A. R., Bader G. D., Ideker T. (2012) A travel guide to Cytoscape plugins. Nat. Methods 9, 1069–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lu W., Clasquin M. F., Melamud E., Amador-Noguez D., Caudy A. A., Rabinowitz J. D. (2010) Metabolomic analysis via reversed-phase ion-pairing liquid chromatography coupled to a stand alone Orbitrap mass spectrometer. Anal. Chem. 82, 3212–3221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Starai V. J., Celic I., Cole R. N., Boeke J. D., Escalante-Semerena J. C. (2002) Sir2-dependent activation of acetyl-CoA synthetase by deacetylation of active lysine. Science 298, 2390–2392 [DOI] [PubMed] [Google Scholar]
- 31. Starai V. J., Takahashi H., Boeke J. D., Escalante-Semerena J. C. (2003) Short-chain fatty acid activation by acyl-coenzyme A synthetases requires SIR2 protein function in Salmonella enterica and Saccharomyces cerevisiae. Genetics 163, 545–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang Q., Zhang Y., Yang C., Xiong H., Lin Y., Yao J., Li H., Xie L., Zhao W., Yao Y., Ning Z. B., Zeng R., Xiong Y., Guan K. L., Zhao S., Zhao G. P. (2010) Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science 327, 1004–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Olsen J. V., Ong S. E., Mann M. (2004) Trypsin cleaves exclusively C-terminal to arginine and lysine residues. Mol. Cell. Proteomics 3, 608–614 [DOI] [PubMed] [Google Scholar]
- 34. Beck H. C., Nielsen E. C., Matthiesen R., Jensen L. H., Sehested M., Finn P., Grauslund M., Hansen A. M., Jensen O. N. (2006) Quantitative proteomic analysis of post-translational modifications of human histones. Mol. Cell. Proteomics 5, 1314–1325 [DOI] [PubMed] [Google Scholar]
- 35. Schwer B., Verdin E. (2008) Conserved metabolic regulatory functions of sirtuins. Cell Metab. 7, 104–112 [DOI] [PubMed] [Google Scholar]
- 36. Wolfe A. J. (2005) The acetate switch. Microbiol. Mol. Biol. Rev. 69, 12–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pulk A., Cate J. H. (2013) Control of ribosomal subunit rotation by elongation factor G. Science 340, 1235970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kiefer F., Arnold K., Künzli M., Bordoli L., Schwede T. (2009) The SWISS-MODEL repository and associated resources. Nucleic Acids Res. 37, D387–D392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kopp J., Schwede T. (2004) The SWISS-MODEL repository of annotated three-dimensional protein structure homology models. Nucleic Acids Res. 32, D230–D234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vos S., Parry R. J., Burns M. R., de Jersey J., Martin J. L. (1998) Structures of free and complexed forms of Escherichia coli xanthine-guanine phosphoribosyltransferase. J. Mol. Biol. 282, 875–889 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.