

Background: Rubisco fixes atmospheric CO2 to organic carbon and sustains life on earth.
Results: A form II Rubisco structure has been solved, and functional analysis was conducted with divergent residues.
Conclusion: The unique structure combined with functional analysis can help better understand and improve Rubisco catalysis.
Significance: This is the first high resolution structure of an activated transition-state analog (CABP)-bound form II Rubisco.
Keywords: Carbon Fixation; Enzyme Catalysis; Enzyme Structure; Ribulose-1,5-bisphosphate Carboxylase/Oxygenase (Rubisco); Site-directed Mutagenesis
Abstract
The first x-ray crystal structure has been solved for an activated transition-state analog-bound form II ribulose-1,5-bisphosphate carboxylase/oxygenase (Rubisco). This enzyme, from Rhodopseudomonas palustris, assembles as a unique hexamer with three pairs of catalytic large subunit homodimers around a central 3-fold symmetry axis. This oligomer arrangement is unique among all known Rubisco structures, including the form II homolog from Rhodospirillum rubrum. The presence of a transition-state analog in the active site locked the activated enzyme in a “closed” conformation and revealed the positions of critical active site residues during catalysis. Functional roles of two form II-specific residues (Ile165 and Met331) near the active site were examined via site-directed mutagenesis. Substitutions at these residues affect function but not the ability of the enzyme to assemble. Random mutagenesis and suppressor selection in a Rubisco deletion strain of Rhodobacter capsulatus identified a residue in the amino terminus of one subunit (Ala47) that compensated for a negative change near the active site of a neighboring subunit. In addition, substitution of the native carboxyl-terminal sequence with the last few dissimilar residues from the related R. rubrum homolog increased the enzyme's kcat for carboxylation. However, replacement of a longer carboxyl-terminal sequence with termini from either a form III or a form I enzyme, which varied both in length and sequence, resulted in complete loss of function. From these studies, it is evident that a number of subtle interactions near the active site and the carboxyl terminus account for functional differences between the different forms of Rubiscos found in nature.
Introduction
The rates of consumption and release of atmospheric CO2 largely determine the severity of ecological problems such as global warming, pollution, and food and energy shortages. Autotrophic organisms play a key role in maintaining this balance because of their ability to fix inorganic carbon dioxide from the environment into reduced organic carbon that is then utilized by all organisms. The enzyme ribulose-1,5-bisphosphate carboxylase/oxygenase (Rubisco)3 accounts for most of the biologically fixed CO2 on earth (1–3). It catalyzes the rate-limiting reaction of the Calvin-Benson-Bassham (CBB) reductive pentose phosphate pathway whereby CO2 attacks the enediol of the keto sugar ribulose 1,5-bisphosphate (RuBP) to form two molecules of 3-phosphoglycerate. There has been much interest in manipulating Rubisco because it is an inefficient biological catalyst, and the carboxylation efficiency of the enzyme is further diminished by its competition with O2. With increased potential to use Rubisco and other catalysts for enhancing carbon capture strategies and to engineer autotrophic organisms for biofuel production, it is clear that even small but incremental additions to knowledge about the catalytic mechanism of this enzyme may have a significant impact (4, 5).
There is natural variation in both structural and functional properties of Rubiscos from diverse sources (6–8). Although traditionally associated with its role in the CBB pathway for CO2 assimilation in plants and autotrophic bacteria, the involvement of archaeal Rubiscos in nucleic acid metabolism and that of the Rubisco-like proteins (RLPs) in sulfur metabolism exemplify the diverse physiological contexts in which this enzyme family may function (6). With an explosion in data obtained from “omics” approaches, it is becoming clear that several organisms utilize more than one form of Rubisco to cater to the diverse physiological needs arising from changing environmental conditions (6, 8). Because of its abundance and importance, Rubisco is one of the most highly sequenced genes with 14 unique structures and several variations also available in the protein database. Eleven of these structures are of homologous form I enzymes, all of which assemble as hexadecamers with eight large, catalytic subunits (∼475 amino acids) and eight small subunits (∼125 amino acids) to form an L8S8 structure. There is one form II structure (Rhodospirillum rubrum L2 dimer) and two archaeal form III structures (Pyrococcus horikoshii L8 octamer and Thermococcus kodakaraensis L10 decamer) (9). The average sequence identity between the catalytic subunits of different forms of Rubiscos is about 30%, and the in-group identity ranges from about 50 to 85% (8). The existing structures of R. rubrum Rubisco, the only form II enzyme studied in detail so far, have been useful for deducing the roles of several active site residues in the reaction mechanism (10–13). However, the identification of structural determinants of functional differences between the different forms of Rubisco has been somewhat limited by the absence of a form II structure bound to the transition-state analog 2-carboxyarabinitol 1,5-bisphosphate (CABP). CABP locks the enzyme into a “closed” conformation and positions active site residues in those conformations that likely resemble the arrangement in the enzyme bound to the carboxylation intermediate (14). By contrast, excellent transition-state analog-Rubisco structural complexes are available for form I (several) and form III (T. kodakaraensis) Rubiscos. In part, the current study was undertaken to provide the structure of a form II transition-state analog complex that might provide a further understanding as to how unique enzymatic properties attributed to form II enzymes might be resolved at the structural level.
Previous studies have shown that the genomes of Rhodobacter sphaeroides, Rhodobacter capsulatus (15), and more recently Rhodopseudomonas palustris (16) all encode for form II Rubiscos, which are homologs of the R. rubrum Rubisco, sharing about 75% sequence identity among each other. R. palustris is a metabolically versatile Gram-negative purple non-sulfur bacterium best known for its ability to switch between the different modes of metabolism that support life, thereby making this organism suitable for use in biotechnological applications like biodegradation and hydrogen production (16). The amino acid sequence of R. palustris form II Rubisco is 74% identical and 83% similar to its R. rubrum homolog. In addition to the form II Rubisco, the genome of R. palustris encodes for a form I enzyme as well as two RLPs (RLP1 and RLP2). Whereas the regulation and functions of Rubiscos (forms I and II) as part of the CBB pathway have been well studied (17, 18), the precise physiological role(s) of the RLPs is yet to be determined. The evolution of both structure and function of Rubiscos and RLPs appears to be linked (8). The form II enzyme from R. palustris was chosen for analysis as part of a broader investigation to understand the role of its Rubisco function in the physiological context. From a structural perspective, it will be useful to find out how these divergent active sites may account for functional differences between the different forms of Rubiscos and RLPs and whether Rubiscos may complement for RLP function (8).
Studies aimed at better understanding the reaction mechanism for engineering a more efficient Rubisco have relied on biological selection/directed enzyme evolution carried out with different Rubiscos in the metabolically diverse native/non-native host systems provided by Chlamydomonas, Escherichia coli, and R. capsulatus (19–21). With versatile growth capabilities, simple medium requirements, and the potential to use foreign Rubiscos for complementing autotrophic growth requirements, R. capsulatus has proven to be a valuable heterologous host for the selection and eventual identification of several residues and their functional significance (20, 22, 23). Thus, in the current study, structural analysis was combined with functional assessment using the Rubisco deletion strain of R. capsulatus complemented with wild-type and mutant enzymes of R. palustris form II Rubisco.
The x-ray crystal structure of the activated CABP-bound R. palustris form II Rubisco was solved in this study, making this the first instance of a form II-CABP enzyme-activated structural complex. Two residues near the active site were chosen for further analysis via site-directed mutagenesis based on previous studies with the R. rubrum (form II) (13, 24), Chlamydomonas (25), Synechococcus (26), and tobacco (27) form I enzymes. The carboxyl terminus, which varies both in length and composition among different Rubiscos, was also targeted for structure-function analysis by constructing chimeric enzymes. Using random mutagenesis, a second-site suppressor was isolated for a negative mutant using a previously described biological selection system (23). The structure-function analyses presented here help in better understanding the structural basis for catalysis in form II Rubisco, and these studies might prove useful for engineering a Rubisco with optimized properties for a variety of applications (4, 5).
EXPERIMENTAL PROCEDURES
Plasmids, Strains, DNA Manipulations, and Culture Conditions
Genomic DNA from R. palustris strain CGA009 was used as a template to amplify the cbbM gene, which encodes for the form II Rubisco. The amplified gene with its native ribosome binding site (15 bp upstream of the start codon) was cloned as a blunt end PCR product into the plasmid pCR Blunt II-TOPO using the Zero Blunt TOPO PCR cloning kit (Invitrogen) to generate pCR-RpcbbM. Site-directed mutagenesis was performed with the QuikChange kit (Agilent) using appropriate primers and pCR-RpcbbM as the template. Chimeric carboxyl-terminal sequences were introduced using inverse PCR with pCR-RpcbbM as the template. The PCR primers, which annealed to the coding sequence for the carboxyl terminus, were designed to introduce the substituted sequences in one or two successive rounds of PCRs depending on the length of the chimera. After verifying the constructs via DNA sequencing at the Plant-Microbe Genomics Facility at The Ohio State University, the exact coding regions were amplified from wild-type and mutant plasmids with primers that included an NdeI site overlapping the start codon and a BamHI site immediately 3′ of the stop codon. The 1.5-kb PCR products were digested with the restriction enzymes and cloned between NdeI and BamHI sites in plasmid pET11a or pET28a (Novagen) for recombinant protein expression in E. coli strain BL21(DE3). Use of pET28a resulted in the introduction of an amino-terminal hexahistidine tag to the various proteins, thereby facilitating protein purification via a single step nickel affinity column chromatography. All the aforementioned PCRs were carried out using either Phusion (New England Biolabs) or PrimeSTAR GXL (Clontech) high fidelity DNA polymerase.
Random mutagenesis was carried out with negative mutant pCR-RpcbbM plasmids as templates. This was accomplished using error-prone PCR amplification in the presence of 0.1 mm MnCl2 and the enzyme Taq DNA polymerase (Invitrogen). The forward and reverse primers contained the recognition sites for the restriction enzymes KpnI and SacI, respectively. The KpnI site was designed such that the PCR product would include the native ribosome binding site for the cbbM gene. Wild-type and mutant genes were cloned into the broad host range vector pRPS-MCS3 (23) using T4 DNA Ligase (New England Biolabs). These constructs were transformed into E. coli strain S17–1 (ATCC47055) and mobilized into the Rubisco deletion strain of R. capsulatus (SB I/II−) via diparental matings to aid heterologous protein expression and functional analysis (23). The plasmid-complemented R. capsulatus colonies were selected either on rich peptone yeast extract medium (chemoheterotrophy) or on Ormerod's minimal medium plates with no organic carbon (photoautotrophic growth) as described previously (20). Liquid photoautotrophic growth curves were generated by inoculating 20 ml of Ormerod's minimal medium with a 1:100 inoculum and bubbling illuminated cultures with a mixture of either 1.5 or 5% CO2 balanced with H2. The cells for the inoculum were obtained by growing single colony isolates in liquid chemoheterotrophic medium for 2 days; subsequently, the preculture cells were harvested and washed thoroughly with minimal medium. Growth was monitored by measuring the optical density of the cultures at 660 nm using a Genesys 20 visible spectrophotometer (Thermo Scientific). The cultures for protein purification were scaled up to 3 liters in transparent culture bottles with home-made fittings that enabled constant gas bubbling.
All E. coli strains were grown in Luria-Bertani medium at 37 °C using 12.5 μg/ml tetracycline or 36 μg/ml kanamycin to maintain the plasmids. For growth of R. capsulatus strains, the plates and cultures were maintained in the dark (chemoautotrophic growth) or in light (photoautotrophic growth) at 30 °C. When appropriate, the antibiotics kanamycin and spectinomycin were used at 25 μg/ml and tetracycline was used at 2 μg/ml, respectively. The plates for photoautotrophic growth selection were packed in a tightly sealed jar that contained a palladium catalyst (for removing oxygen) and CO2/H2-generating kits, resulting in CO2 concentrations in the 5–10% range (Oxoid). Mixotrophic growth was assessed on rich peptone yeast extract medium with the plates incubated in tightly sealed jars containing air and a CO2/H2-generating kit (Oxoid).
Expression and Purification of Recombinant Proteins
For proteins expressed in E. coli strain BL21(DE3) with either pET11a or pET28a constructs of wild-type and mutant R. palustris cbbM genes, cells were routinely cultured in 500–1000 ml of LB medium. Once the cells reached an optical density of 0.4–0.6 (at 600 nm), protein expression was induced by adding isopropyl 1-thio-β-d-galactopyranoside to 1 mm final concentration. After 5–7 h, the cells were harvested, and the pellets were stored at −20 °C prior to protein purification. Prior to purification, cells were rinsed and resuspended in either a 50 mm phosphate buffer, pH 8.0 with 300 mm NaCl and 15 mm imidazole (for nickel-nitrilotriacetic acid affinity purification) or a 50 mm Tris-HCl buffer, pH 8.0 with 1 mm DTT, 10 mm MgCl2, and 10 mm NaHCO3 and lysed using a French pressure cell at 1000 p.s.i. (two passages). The lysates were clarified using high speed centrifugation at 4 °C.
Hexahistidine-tagged proteins were purified using one-step nickel affinity gravity flow chromatography (nickel-nitrilotriacetic acid-agarose, Qiagen). The recommended procedure (Qiagen) was followed but with a few modifications. The lysis and wash buffers were prepared with 15 and 30 mm imidazole, respectively, to reduce nonspecific binding of other cellular proteins. All the buffers also contained 20% glycerol. The imidazole in the eluted proteins was removed by dialysis against a buffer containing 20 mm Tris-HCl, pH 8.0, 10 mm MgCl2, 300 mm NaCl, 1 mm DTT, 10 mm NaHCO3, and 20% glycerol.
For purification of non-tagged proteins expressed in either E. coli or R. capsulatus, a two-step procedure was optimized. The first purification step utilized a Green Separopore resin (bioWORLD, Dublin, OH) in which Rubisco bound to the reactive green dye was eluted using a 0–1 m NaCl gradient in a buffer that contained 50 mm Tris-HCl, 10 mm MgCl2, 1 mm DTT, and 10 mm NaHCO3, pH 8.0. Fractions with Rubisco were identified via RuBP carboxylase activities as determined via radiometric assays in which the 14C from NaH14CO3 was incorporated into acid-stable 3-phosphoglycerate (28). The Rubisco fractions were pooled, dialyzed against a no-salt buffer, and subjected to ion exchange chromatography using a 12-ml UnoQ column (Bio-Rad). Again the Rubisco fractions were identified via radiometric assays, pooled, dialyzed against no-salt buffer, concentrated, and mixed with 20% glycerol. Dialysis and protein concentration were carried out over Amicon filters (50- or 100-kDa molecular mass cutoff; Millipore), and the protein concentrations were determined using the Bradford assay (29). Aliquots of each protein were snap frozen in liquid nitrogen and stored at −80 °C for further biochemical analysis.
Enzyme Assays and Kinetic Measurements
The kinetic constants kcat (for carboxylation), Kc (Km for CO2), and Ko (Km for O2) were determined from simultaneous radiometric assays performed in vials flushed with either 100% N2 or 100% O2 as outlined previously (20). The values for KRuBP (Km for RuBP) were obtained from similar assays performed in sealed vials by varying the RuBP concentration. The substrate specificity (Ω) values were measured with purified enzymes (25–250 μg) in 400-μl assays performed in 1.5-ml vials under saturating (1.23 mm) O2 concentrations (23). [1-3H]RuBP used in these assays was synthesized and purified as described elsewhere (30). Rates of Rubisco activation were measured with unactivated Rubisco enzymes using a modified procedure adapted from elsewhere (31). Purified enzymes (500–2500 μg) were unactivated by four to five buffer exchanges against a 100 mm HEPES-NaOH buffer, pH 8.0 using 0.5-ml Amicon Ultracel filters (50-kDa molecular mass cutoff; Millipore). The unactivated enzymes (in 500 μl) were flushed with N2 in sealed vials and incubated with 0.5 mm RuBP. After incubation at room temperature for 1–1.5 h, a 100-μl aliquot of the enzyme-RuBP complex was treated with 400 μl of “activation” buffer comprising 100 mm HEPES-NaOH, pH 7.7, 12.5 mm MgCl2, 25 mm NaHCO3, 1.25 mm DTT, and 0.5 mm RuBP. The time course of activation was followed by periodically removing 20-μl aliquots of this mixture and measuring RuBP carboxylase activities at 30 °C in 0.5-min reactions, following the stoichiometric incorporation of 14C from NaHCO3 into acid-stable 3-phosphoglycerate. The assay reaction mixtures contained 20 mm NaHCO3 (containing 8 μCi of NaH14CO3), 10 mm MgCl2, and 0.8 mm RuBP in 100 mm HEPES-NaOH, pH 8.0.
Crystallization, Data Collection, Structure Determination, and Refinement
Purified Rubisco was buffer-exchanged into 20 mm Tris-Cl, pH 8.0, 0.3 m NaCl, 10% glycerol, 10 mm MgCl2, and 20 mm NaHCO3 using a 5-kDa Ultra-15 concentrator (Amicon), and the protein concentration was determined to be 14 mg/ml (46 nm). CABP, which had been synthesized using previously published procedures (32), was added to a final concentration of 4 mm. Seven 96-well crystal screens were set up for crystallization using the sitting drop vapor diffusion method. Drops were prepared by mixing the Rubisco-CABP solution with an equal amount of crystallization reservoir solution (varying composition). The trays were incubated at 20 °C, and crystals were observed after 2 days. The best diffracting apo crystals were grown using a crystallization reservoir solution comprising 170 mm lithium sulfate, 85 mm Tris-HCl, pH 8.5, 24% (w/w) PEG 4000, and 14% (w/v) glycerol. Similarly, good quality CABP-bound crystals could be obtained in a reservoir solution with 60 mm Tris-Cl, pH 8.5, 20% (w/w) PEG 4000, and 10% (v/v) glycerol. The crystals were harvested and swiftly passed through a cryoprotectant solution before freezing at −170 °C for data collection. The cryoprotectant solution was prepared by mixing 15 parts of 100% glycerol with 85 parts of the mother liquor. X-ray diffraction data sets were collected at beam line 8.2.1 of the Advanced Light Source at Lawrence Berkeley National Laboratory that was equipped with an ADSC Quantum 315 charge-coupled device detector. Data were collected using a wavelength of 1.000 Å at −170 °C and processed using DENZO and SCALEPACK (33). The quality of anisotropic data for the sulfate-bound crystal was improved by imposing an ellipsoidal resolution boundary using the UCLA MBI Diffraction Anisotropy Server.
The sulfate-bound structure (Protein Data Bank code 4LF2), which had resulted from using high sulfate concentrations in the initial crystallization conditions, was solved by molecular replacement using Phaser (34) with the structure of R. rubrum Rubisco form II (Protein Data Bank code 5RUB) as a search model. The CABP-bound structure (Protein Data Bank code 4LF1) obtained subsequently with the use of sulfate-free crystallization buffer was also solved by molecular replacement using the sulfate-bound structure as the search model. Electron densities were observed for the CABP molecule and a coordinating Mg2+ ion in all six oligomers in the hexamer. Model building was performed manually with Coot (35), and the models were refined with REFMAC (36) in which anisotropic displacement parameters were modeled with translation/libration/screw group definitions and non-crystallographic symmetry restraints. Structure validation was done using the UCLA MBI SAVES and the Protein Data Bank ADIT Servers. Subunit-subunit interface areas were calculated from accessible surfaces on each subunit that were buried in either the interface with the paired subunit of the dimer or the interface with another subunit from the neighboring dimer. The calculations were done using the program AreaIMol (37, 38). Surface electrostatic potentials were calculated using the program Ezprot (39). All structure images were prepared using The PyMOL Molecular Graphics System (Schrödinger, LLC).
Molecular Dynamics Simulations and in Silico Analysis
The wild-type protein model was generated in silico by removing non-standard molecules from the CABP-bound R. palustris form II Rubisco structure that was solved as part of this study. Side chains of residues Ala47 and Met331 were mutated in silico to Val and Ala, respectively, in statistically more favorable conformations to obtain the protein model of the A47V/M331A double mutant enzymes. Molecular dynamics simulations were performed on these two protein models using the program Crystallography and NMR System (CNS; version 1.1) and hydrogen bond restraints described elsewhere (40, 41). Using a simulated annealing protocol, the structures were heated to 1500 K followed by stepwise cooling with increments of 25 K until 0 K. Shape complementarity and Protein Interfaces, Surfaces and Assemblies (PISA) analyses were carried out as described elsewhere (42, 43).
RESULTS
Structure Determination and Features of R. palustris Form II Rubisco
X-ray crystal structures of both the activated apo- and CABP-bound proteins were determined. The apo crystal structure was solved first and revealed two sulfate ions bound at each active site coordinated by the same residues that would interact with two phosphate groups of the CABP molecule. Attempts to obtain CABP-bound protein crystals using these crystallization conditions were unsuccessful, and in retrospect, it is obvious that the high sulfate concentration allowed sulfate ions to outcompete CABP molecules for active site occupancy. The CABP-bound crystal structure was subsequently solved from crystals obtained under phosphate- and sulfate-free conditions. Both structures show a novel hexameric arrangement in the same P1 space group with similar unit cell dimensions and an overall root mean square deviation of only 0.554 Å between the α-carbon positions of the two enzymes (Table 1). The hexameric assembly is consistent with native molecular weight determinations from size exclusion chromatography and static light scattering experiments yielding an approximate molecular mass of 300 kDa.4 It is thus likely that this quaternary structure arrangement is biologically relevant. The average root mean square deviation between α-carbon chains of all the dimers from both structures is 0.318 Å with a range from 0.11 to 0.45 Å. The mean value for individual subunits is 0.249 Å with a range from 0.05 to 0.45 Å. The overall arrangement of all secondary structural elements is identical in both sulfate-bound and CABP-bound structures of the activated R. palustris enzyme. Structural features around the active site region of our sulfate-bound structure are quite different from those in the previously solved sulfate-bound structure of the unactivated form I Rubisco from Galdieria partita (44). As the CABP-bound structure of the activated enzyme is comparable with the reaction intermediate-bound Rubisco during catalysis, further analysis and discussions presented below will focus on this structure. The hexamer is a trimer of homodimers with the three dimers arranged along the 3-fold symmetry axis forming a toroid tertiary structure (Fig. 1A). The two subunits within each dimer are positioned in a “head-to-tail” fashion forming two active sites per dimer with both subunits contributing to each active site, similar to what is found in other Rubisco structures (45). However, unlike the arrangement in form I (for example, Synechococcus sp. strain PCC6301 Rubisco) or form III (for example, T. kodakaraensis Rubisco), the inclination of the dimer interface relative to the cylindrical axis passing through the solvent channel is 39° to the right in the R. palustris form II enzyme (Fig. 1B). Furthermore, tertiary structure alignment of a subunit from this structure with an equivalent large subunit from other CABP-Rubisco structures shows the unique relative arrangement of amino and carboxyl termini with the termini positioned farther away from each other and pointing in different directions (Fig. 2). The closed conformation of catalytic loop 6 in the R. palustris structure is comparable with its position in all other analogous CABP structures. This is quite different, however, from the “open” arrangement of loop 6 in the activated, RuBP-bound structure of the homologous R. rubrum enzyme (Fig. 2A). Structure alignments with other representative Rubisco sequences highlight the presence of form II-specific α helices at both the amino (αA) and carboxyl (αI and αJ) termini of the R. palustris enzyme, similar to what was observed with R. rubrum form II Rubisco (Figs. 2 and 3) (45). As with the T. kodakaraensis and Galdieria enzymes, there is an α helix (α-1) preceding βC (44, 46) (Fig. 2B). However, unlike the equivalent residues from the form I and form III enzymes, residues in this region of the R. palustris enzyme are not involved in interdimer (L2-L2) interactions. Yet another distinguishing feature is the presence of a 310 helix in the amino-terminal domain (Helix 0) between αC and βE. The active site residue Asn112 is found in this structure (Fig. 3).
TABLE 1.
X-ray data collection and refinement statistics
rmsd, root mean square deviation.
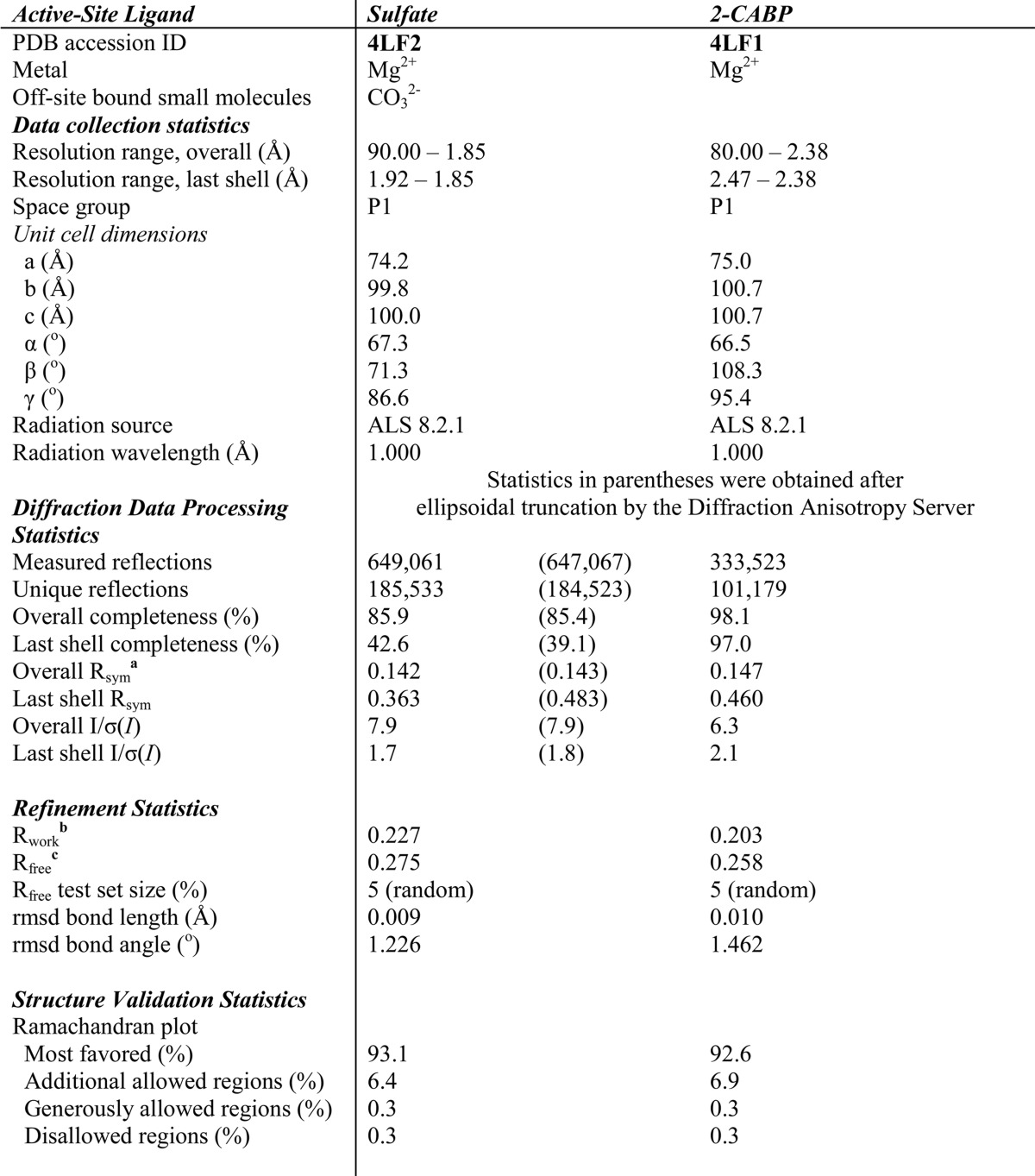
a Rsym(I) = Σhkl((Σi|Ihkl,i − <Ihkl>|)/ΣiIhkl,i).
b Rwork = Σhkl|Fobs − Fcalc|/ΣhklFobs.
c Rfree = Σhkl|Fobs − Fcalc|/ΣhklFobs where all reflections belong to a test set of 5% of randomly selected data.
FIGURE 1.

Structure of the hexameric R. palustris form II Rubisco with three head-to-toe dimers in cyclic arrangement. A, top view down the 3-fold symmetry axis of the three dimers (composed of subunits A and B, C and D, and E and F) and the 2-fold symmetry axes of the dimers radiating from the center out into the toroid plane and side view of the hexamer rotated 90° around the horizontal axis with the approximate dimensions of the hexamer specified. B, top view comparison of the hexameric R. palustris form II Rubisco with other selected form I and form III holoenzymes. Deviations in the dimer unit inclinations along the toroid plane were calculated using the Synechococcus form I Rubisco structure (Protein Data Bank code 1RBL) as reference. The inclination of dimer units in the R. palustris (Rp) Rubisco is to the right of the vertical axis passing through the central solvent channel, whereas all others are inclined to the left at various angles. For the sake of clarity, the non-catalytic small subunits have been omitted from the form I structures. Se, Synechococcus elongatus; So, Spinacia oleracea; Ph, P. horikoshii; Tk, T. kodakaraensis.
FIGURE 2.

A, superimposed large subunit monomers from R. palustris (chain A; red) and R. rubrum form II Rubiscos (Protein Data Bank code 9RUB; chain A; green). The CABP (red) and Mg2+ ion (cyan) shown in the figure are from the R. palustris structure, and the RuBP (green) is from the R. rubrum structure. Amino (Nt) and carboxyl termini (Ct) are indicated. B, stereoview of the secondary structure elements (α helices, red; β strands, yellow; loops and termini, gray) in the structure of R. palustris Rubisco (chain A) with CABP and the Mg2+ ion (cyan).
FIGURE 3.

Structure-based sequence alignment of catalytic large subunits from R. palustris (Rp) form II, R. rubrum (Rr) form II (Protein Data Bank code 9RUB), Synechococcus (S. elongatus (Se)) form I (Protein Data Bank code 1RBL), spinach (S. oleracea (So)) form I (Protein Data Bank code 8RUC), P. horikoshii (Ph) form III (Protein Data Bank code 2D69), and T. kodakaraensis (Tk) form III (Protein Data Bank code 3A12) enzymes. The secondary structural assignments for the R. palustris enzyme are displayed above the sequences using a schematic that is consistent with that of Fig. 2B. The asterisks below the sequences designate strictly conserved residues among known Rubiscos, and the inverted triangles above the sequences indicate the positions of the active site residues. Residues involved in interactions with the dimer-forming neighboring subunit are colored green; the highlighted residues (in salmon color) are involved in interactions with neighboring subunit(s) other than the dimer partner. The dotted boxes indicate contiguous regions in different Rubiscos that appear to be involved in interactions with a neighboring subunit, excluding its dimer partner, as also highlighted in the space-filling surface representations (Fig. 4A).
It could be concluded from subunit interface analyses that the region involved in interactions with a neighboring subunit within the same dimer of the R. palustris form II Rubisco (Fig. 4A) is identical to what has been described before for other Rubiscos (9) (supplemental Movie S1). However, different forms appear to utilize different sets of regions for interactions with other neighboring subunits, including small subunits in the case of form I enzymes. The R. palustris form II enzyme uses a central core region (supplemental Movie S1), whereas the form III enzymes seem to use two flanking regions for these interactions (Fig. 4A). The form I Rubiscos utilize all these regions and a few more residues from elsewhere for the interdimer and large subunit-small subunit interactions. The interdimer interfaces of R. palustris and R. rubrum form II enzymes both have a higher concentration of positively charged residues, although the latter exists only as a free dimer. The equivalent surfaces in the form I and form III enzymes analyzed here are predominantly negatively charged (Fig. 4B).
FIGURE 4.

A, a space-filling surface comparison of the large subunit dimers in different Rubiscos at two different angles 90° apart relative to the vertical axis. In each structure, one of the chains forming a dimer is shown in gray with reduced transparency. Residues in the other subunit forming the dimer that are involved in interactions with the dimer partner are colored green. Salmon-colored residues are involved in interactions with neighboring subunit(s) other than its dimer partner. White-colored surfaces are solvent-exposed. Areas circled with dotted lines indicate the common regions utilized by different Rubiscos for interactions with neighboring subunits other than its dimer partner. The color scheme and dotted lines are consistent with what is shown in Fig. 3. B, electrostatic surface representation of large subunit monomers at view angle 2 showing the differences in the interdimer interaction surfaces among the different Rubiscos. For R. rubrum Rubisco, which forms only a dimer, the analogous region is shown for comparison. The dotted yellow circle highlights the analogous regions in the different enzymes with the most variable electrostatic surfaces.
Mutagenesis, Screening for Function, and Suppressor Selection
Comparison of all residues within 4 Å of CABP in the active sites of previously solved Rubisco structures highlights the conservation and predominantly polar nature of the active site (Figs. 5 and 6). Despite the involvement of a common set of active site residues in the reaction mechanism, there are subtle differences in both the spatial arrangement of the active site and identities of two vicinal residues between the structures of the R. palustris form II Rubisco and other homologs (Fig. 5). The active sites of sulfate-bound and CABP-bound R. palustris form II Rubisco structures are virtually superimposable with the exception of the position of residue Glu49, which appears to be closer to all other active site residues in the sulfate-bound structure (Fig. 5A). A representative comparison of the CABP structure of R. palustris enzyme with its form I homolog from Synechococcus (Fig. 5A) and phylogenetic comparisons of residue identities in this region (Fig. 5B) elicit other differences. These differences could potentially explain the functional distinctness of the different enzyme forms. Although not ascribed a direct role in catalysis, Thr173 (Synechococcus Rubisco numbering) is two residues away from the well characterized active site Lys175, and its identity is conserved as a Thr residue in all form I enzymes. However, its equivalent is an Ile in form II (Ile165 in R. palustris enzyme) and a Val in form III sequences (Fig. 5B). Situated on the opposite side of the active site is loop 6, which undergoes substantial conformational changes during catalysis (9). Lys334 (Synechococcus Rubisco numbering) from loop 6 has long been known to play a role in coordinating with the incoming CO2 molecule during catalysis (27). The side chain of the neighboring residue (Leu335 in tobacco) is hydrophobic in nature and is either a Met (Met331 in R. palustris form II Rubisco) or a Leu in other Rubisco sequences (Fig. 5). Based on phylogenetic comparisons, residue Ile165 was changed to either Thr (as found in form I) or Val (as found in form III), and Met331 was changed to a Leu (as in forms I and III). A neutral substitution was also introduced in the form of an Ala at these positions. A double mutant (I165T/M331L) was also created to mimic a form I-like sequence. When identical constructs in the broad host range vector were mobilized into R. capsulatus Rubisco deletion strain SBI/II−, only the wild type and mutant I165V supported autotrophic CO2-dependent growth (Fig. 7), indicating that the rest of the mutations led to structural and/or functional defects. However, it was evident from multiple growth experiments that the mutant I165V enzyme supported significantly poorer growth relative to the wild-type enzyme.
FIGURE 5.

Comparison of residues within 4 Å of CABP in several Rubisco structures. A, active site arrangements in the x-ray crystal structures of sulfate-bound (Protein Data Bank code 4LF2) and CABP-bound (Protein Data Bank code 4LF1) R. palustris form II Rubisco and CABP-bound Synechococcus form I Rubisco (Protein Data Bank code 1RBL). Electron density maps contoured between 1.0 and 2.0 σ for optimal visualization are displayed for the active site ligands and amino acid side chains with dissimilar conformations or identities. B, phylogenetic comparison of amino acid identities in this region. Sequences shown are representatives from the three forms of Rubisco. Residues appearing at different positions in the amino acid sequence are segregated into different columns, and the R. palustris form II Rubisco sequence numbers are indicated above the alignment along with a dot, which is aligned with the corresponding residue labels. Multiple residues within a column denote residues that are continuous in the amino acid sequence, and the sequence numbers correspond to those of the first residue within a column. Divergent residues chosen for analysis in this study and the analogous residues from other Rubisco sequences are underlined.
FIGURE 6.

Comparison of the active site regions in R. palustris form II (A) and Synechococcus form I (B) Rubiscos with contributing residues from two subunits (chains A and B) in each structure. The carboxyl-terminal domains of chain A (in red, yellow, green, and gray) and selected secondary structure elements in chain B (in blue and magenta) comprise key regions that affect catalysis. The active site residues, CABP, and other residues of interest are shown in stick representation. The carboxyl terminus with dissimilar conformation among the two structures that was also targeted for analysis via the construction of chimeric enzymes is colored green in both. In the Synechococcus form I structure, Lys128 (magenta) is interlocked between the carboxyl terminus (Ct) and loop 6 via multiple van der Waals interactions; the equivalent residue is Gly117 in the R. palustris form II structure. The relative positions of the R. palustris Ile165, Met330, and Ala47 and the analogous residues in the Synechococcus structure are all shown in the context of other active site residues.
FIGURE 7.

Photoautotrophic growth phenotypes conferred by the wild-type, mutant, and chimeric enzymes of R. palustris form II Rubisco complemented in a Rubisco deletion strain of R. capsulatus. Growth with CO2 as the sole carbon source was monitored in liquid minimal medium by flushing the cultures with a mixture of 5% CO2 and 95% H2. Data are representative of three independent growth experiments with similar results. RrCt, SyCt, and TkCt correspond to chimeric R. palustris Rubiscos with carboxyl-terminal sequences from R. rubrum, Synechococcus, or T. kodakaraensis, respectively.
Several studies have established the carboxyl terminus as a hot spot for affecting Rubisco catalysis (47–49). Structure comparisons and unique placement of the carboxyl terminus in the activated CABP-bound R. palustris Rubisco (Fig. 6) prompted further analysis of phylogenetic differences in this region. Chimeric cbbM constructs were prepared by replacing the native R. palustris carboxyl-terminal sequence with equivalent divergent sequences from R. rubrum (form II), Synechococcus (form I), or T. kodakaraensis (form III) (Fig. 8). Functional complementation analysis in R. capsulatus showed that only the chimeric RrCt with a form II-like carboxyl terminus could support CO2-dependent growth and at a rate comparable with the R. palustris wild-type enzyme (Fig. 7). After expression in either R. capsulatus or E. coli, equal amounts of all mutant and chimeric proteins could be purified, indicating that the substitutions did not destabilize the holoenzyme structure. Furthermore, size exclusion chromatography confirmed that the sizes of all mutant proteins were identical to that of wild-type protein (data not shown).
FIGURE 8.

Alignment of carboxyl-terminal sequences from representative Rubiscos. The variable sequences, which were swapped at different positions for the R. palustris sequence, are all underlined. The starting positions for swapping out the R. palustris sequence for construction of RrCt (Lys457), SyCt, or TkCt (Pro442) chimeras are marked with an asterisk above the sequences.
Random mutagenesis was carried out in vitro, and suppressor mutations were selected with the R. capsulatus strain SB I/II− system as described previously (20). Whereas only true revertants (i.e. T165I) and pseudo revertants (i.e. A165V) could be obtained from Ile165 mutants, a second-site suppressor was isolated for the negative mutant M331A. This resulted from a mutation in the codon for Ala47 that substitutes it with a Val. To identify the mechanism of suppression, the mutation resulting in an A47V substitution was introduced into an otherwise wild-type gene. Unlike the double mutant A47V/M331A, which could barely support CO2-dependent growth of the Rubisco deletion strain of R. capsulatus, the suppressor-alone mutation could complement for near wild-type levels of photoautotrophic growth (Fig. 7). After several rounds of mutagenesis experiments, no suppressors could be isolated for mutants M331L, I165T/M331L, SyCt, or TkCt. It is thus likely that mutations introduced via the random mutagenesis technique described here were insufficient to sample the sequence space for those mutations that may be required to suppress the defects of some of the negative mutants analyzed in this study. However, these single base mutations were sufficient to result in true reversion, pseudo reversion, or second-site suppression of mutants I165T, I165A, and M331A, respectively.
Purification and Biochemical Characterization of Wild-type, Mutant, and Chimeric Rubiscos
Following previously published procedures for the expression and purification of common bacterial Rubisco enzymes, the wild-type and mutant proteins were initially expressed as recombinant proteins in E. coli (20, 22, 50). The use of an amino-terminal hexahistidine fusion tag facilitated purification of all proteins using single step nickel affinity chromatography. However, upon determination of enzymatic properties from these histidine-tagged proteins, the Km value for CO2 (Kc) determined for the recombinant A47V/M331A double mutant enzyme was substantially high (∼1500 mm) and failed to explain why it was recovered as a suppressor (Fig. 7). A reciprocal experiment was conducted in which the R. capsulatus Rubisco deletion strain was complemented with the histidine-tagged A47V/M331A double mutant gene. However, this construct failed to support CO2-dependent autotrophic growth, further confirming that the histidine tag was possibly interfering with the function of the enzyme. This prompted reanalysis of all enzymatic properties with untagged proteins that had been found to complement R. capsulatus strain SB I/II− for autotrophic growth. A two-step protein purification starting with large batches of autotrophically grown R. capsulatus yielded proteins that were sufficiently pure (about 75%) to facilitate further analysis. The autotrophic growth requirement for Rubisco expression precluded the purification of negative mutant enzymes from R. capsulatus. However, they were purified as recombinant proteins from E. coli after overexpression from respective pET11 (non His-tagged) constructs following isopropyl 1-thio-β-d-galactopyranoside induction. The properties of the wild-type enzyme, which was used as a control for E. coli expression, as well as the other enzymes that had been purified from R. capsulatus were identical to those of the respective recombinant proteins purified from E. coli. Whereas the specific activities of R. rubrum and R. palustris wild-type and the chimeric RrCt enzymes were all in the range of 2–3 μmol of CO2 fixed/min/mg, mutant I165T/M331L and chimeric SyCt and TkCt enzymes had insignificant levels of activity, precluding them from further analysis. The specific activities of I165V (about 30% of wild type) and all other mutant enzymes (10–15%) were sufficient for the determination of enzymatic properties.
The Ω values determined from HPLC fractionation of tritium-labeled carboxylation and oxygenation products were identical for the wild-type form II enzymes (R. palustris and R. rubrum), chimeric RrCt enzyme, and mutant A47V enzyme (Table 2). The kcat value (for carboxylation) measured for the chimeric RrCt enzyme was 37% higher relative to the value for R. palustris wild type and 55% higher than what was measured for the R. rubrum wild-type enzyme. Mutant enzymes I165V, A47V, and A47V/M331A all have lower kcat values, ranging from 27 to 5% of the wild-type value (Table 2). With all other properties comparable with the wild type, the improved kcat of the chimeric RrCt enzyme does not appear to confer a discernible growth advantage to the R. capsulatus host strain under the conditions reported here (Fig. 7 and Table 2). Comparable values for Ω and Km (for CO2 and RuBP) and an 82% lower kcat possibly explain the moderate effect in growth complementation observed for the mutant A47V. Substantially lower values of Ω and higher values of Km (for CO2 and RuBP) likely explain the slower growth conferred by mutants I165V and A47V/M331A (Fig. 7). The higher Km values for O2 (Ko) suggest that these mutant enzymes are less inhibited by O2. With only 4% of the wild-type value of kcat, it is not surprising that the A47V/M331A suppressor mutant confers the longest lag and slowest growth among all the mutants that can complement for Rubisco function. With such poor enzymatic properties, it seems likely that this suppressor may not have been identified if selection had been carried out under ambient CO2 concentrations. In fact, when autotrophic growth of the mutant A47V/M331A was monitored in liquid medium bubbled with either 5 or 1.5% CO2, the measured lag time increased from ∼100 to ∼150 h. There was very little difference in the lag times measured for the wild type and other mutants (data not shown).
TABLE 2.
Kinetic properties of recombinant R. palustris Rubisco enzymes purified from either autotrophically grown R. capsulatus or E. coli
RrcbbM WT refers to R. rubrum form II wild-type enzyme.
| Enzyme | Ωa (VcKo/VoKc) | kcata | Kca | Koa | Ko/Kcb | KRuBPa |
|---|---|---|---|---|---|---|
| s−1 | μm | μm | μm | |||
| Wild type | 12 ± 1 | 4.9 ± 0.2 | 104 ± 22 | 277 ± 15 | 2.7 | 10 ± 2 |
| A47V | 11 ± 1 | 0.9 ± 0.2 | 99 ± 34 | 82 ± 31 | 0.8 | 6 ± 2 |
| A47V/M331A | 6 ± 1 | 0.2 ± 0.1 | 433 ± 61 | 697 ± 77 | 1.6 | 20 ± 6 |
| I165V | 4 ± 1 | 1.3 ± 0.2 | 178 ± 47 | 391 ± 45 | 2.2 | 35 ± 9 |
| RrCt | 13 ± 1 | 6.7 ± 0.7 | 111 ± 28 | 270 ± 71 | 2.4 | 8 ± 2 |
| RrcbbM WT | 13 ± 1 | 4.3 ± 0.5 | 125 ± 12 | 143 ± 3 | 1.1 | 5 ± 2 |
a Values are the means ± S.D. (n − 1) of at least three separate enzyme preparations.
b Calculated values.
The mutant enzymes, which failed to complement R. capsulatus for CO2-dependent autotrophic growth, all had varying levels of detrimental changes to the catalytic properties. Barring the mutant I165A enzyme, which still retained 16% of the wild-type level of kcat, the other mutant enzymes retained only 4–6% of the wild-type kcat. Although the Ω values of mutant enzymes I165T and M331L were comparable with the wild-type value, the Kc (I165T, 218 μm; M331L, 322 μm) and KRuBP (I165T, 58 μm; M331L, 320 μm) values for these enzymes were both substantially higher. Mutant enzymes I165A and M331A both had Ω values of 8. The lower Ω value combined with a kcat value of only 0.2/s and high substrate Km values (Kc, 345 μm; KRuBP, 115 μm) possibly explains the negative phenotype attributable to the mutant M331A. However, with superior values for kcat (0.8/s), Kc (151 μm), and KRuBP (13 μm) relative to those measured for the suppressor mutant A47V/M331A enzyme (0.2/s, 433 μm/ and 20 μm, respectively), it was surprising that the mutant I165A confers a “negative” phenotype in R. capsulatus (Fig. 7 and Table 2).
Time courses for activation were measured with the intent to explain the possible differences in the phenotypes conferred by mutants I165A and A47V/M331A. Sugar phosphates, including RuBP, are known to bind the unactivated form of Rubisco (i.e. in the absence of CO2 and Mg2+), thereby forming an inactive complex in vivo (51). Reactivation of the enzyme involves release of the inhibitor from the active site followed by the carbamylation of an active site Lys residue and coordination with Mg2+. The tightness of inhibitor binding influences the rate of enzyme activation in vivo and is a well known mechanism to regulate Rubisco activity in various organisms (51). Upon inhibition of the unactivated form of the enzyme by RuBP, the rates of reactivation were measured for wild-type and mutant I165A and A47V/M331A enzymes. The mutant I165A enzyme was indeed the slowest to be reactivated when compared with the wild-type and mutant A47V/M331A enzymes (Fig. 9). Initial slopes from these activation profiles were used to calculate levels of activation per unit time for each enzyme (Fig. 9). The calculated values were 63%/min for the wild type, 14%/min for the double mutant A47V/M331A, and 5%/min for the mutant I165A. The rapid reactivation of the wild-type enzyme indicates weak inactivation by RuBP binding. Unlike eukaryotic and cyanobacterial Rubisco enzymes, which are known to interact with accessory proteins to relieve sugar phosphate inhibition in vivo (31, 51), no such regulatory proteins have been identified in R. palustris or R. capsulatus. It might be tentatively concluded that inactivation of the wild-type enzyme by RuBP (and potentially other sugar phosphates) is probably too weak to have a potential regulatory role in vivo. However, detrimental changes to the catalytic properties combined with tight binding inhibition caused by various sugar phosphates could account for the negative phenotype conferred by the mutant I165A enzyme and the poor but positive complementation conferred by suppressor mutant A47V/M331A (Fig. 7). To summarize, although the residues Ile165 and Met331 have not been ascribed direct roles in catalysis, even subtle alterations to the side chains of these residues appear to cause significant changes in the functional properties of the enzyme.
FIGURE 9.

Time courses for Rubisco activation measured from purified recombinant wild-type and mutant I165A and A47V/M331A enzymes. Percentages of maximal specific activity (measured with respective enzymes that had been fully activated) were plotted against the time allowed for activation after being inactivated by the formation of the enzyme-inhibitor complex. The maximal specific activity values measured were 2.2 μmol/min/mg for the wild type, 0.24 μmol/min/mg for mutant I165A, and 0.05 μmol/min/mg for mutant A47V/M331A. Data are representative of two identical experiments carried out with two separate enzyme preparations, which gave similar results.
DISCUSSION
Distinguishing Features of the CABP-bound R. palustris Form II Rubisco Structure
The form II Rubisco structure solved as part of this study allows for a direct comparison with previously described CABP structural complexes of form I and form III Rubiscos. Besides the unique placement of the carboxyl terminus in the R. palustris form II enzyme, which could not be deduced from the previously solved structures of R. rubrum Rubisco, the surface areas involved in the interdimer interactions and the nature of the constituent surface residues are quite different among the three forms of Rubisco (Figs. 3 and 4 and Table 3). Structural alignments show that the residues involved in the interdimer interactions in the R. palustris form II Rubisco are indeed similar to those in the R. rubrum form II Rubisco (Figs. 2 and 3). However, subtle differences in residue identities and the consequential interactions involving at least two surface regions potentially explain the higher order hexameric association that is observed only with the R. palustris enzyme. In the R. palustris structure, the residue Arg149 in αE is stabilized by van der Waals interactions with Asp146 from the same chain and carbonyl oxygen of Leu359 from the adjacent dimer (Fig. 10). The R. rubrum enzyme has a more flexible Lys148 in place of Arg149 of the R. palustris enzyme; likewise Ala145 is found in place of the Asp146 residue of the R. palustris enzyme. Although the carbonyl oxygen of the R. rubrum Gly358 could serve the same function as the equivalent oxygen of Leu359 in R. palustris, any potential interaction to form a higher order structure would be sterically precluded by the side chain of Glu154 in the R. rubrum enzyme (Fig. 10C). Furthermore, the R. palustris structure has an intricate network of hydrogen bonds between adjacent chains involving three residues from the loop between α4 and β5. The side chain of Gln283 is stabilized by two carbonyl oxygen atoms from Ala256 and Ile259 of the same chain and by hydrogen bond interactions with the guanidyl group of Arg99 from the adjacent dimer. The carbonyl oxygen of the adjacent Lys282 is also within hydrogen bond distance from the guanidyl group of Arg99. This network is further stabilized by a water-mediated interaction of the side-chain amino group of Lys282 with the same Arg99 (Fig 10C). Such an intricate network seems unlikely with the R. rubrum structure where the equivalent of R. palustris Gln283 is a similar but shorter Asn282. Likewise, the R. rubrum equivalents of Lys282 and Arg99 are Asp281 and Lys98, respectively (Fig. 10C). In place of the four hydrogen bonds in R. palustris, only a single hydrogen bond would be formed between the side chains of Asp281 and Lys98 if the R. rubrum subunits assembled into a higher order oligomer. This is further substantiated by additional surface analysis. Subunits A and F from the CABP-bound R. palustris structure that contribute to one of the three interdimer interfaces were used as the reference (4LF1_A-F). Subunit A from the R. rubrum dimer structure (Protein Data Bank code 9RUB) was aligned with 4LF1_A and then combined with another 9RUB subunit, either chain A again or chain B, which was aligned with 4LF1_F. This generated two models (9RUB_A-A and 9RUB_A-B) that resemble the interdimer interface in 4LF1_A-F. Shape complementarity analysis gave a value of 0.538 for the 4LF1_A-F interface and much lower values of 0.231 and 0.299 for 9RUB_A-A and 9RUB_A-B interfaces, respectively. Higher values are indicative of higher interface-shape complementarity with most highly complementary protein interfaces receiving on average a value of 0.53 (42). Protein Interfaces, Surfaces and Assemblies (PISA) analyses estimated an interface solvation free energy (ΔiG) of −4.7 kcal/mol for the model 4LF1_A-F and 0.88 and 0.78 kcal/mol for models 9RUB_A-A and 9RUB_A-B, respectively. Higher interface affinities typically result in more negative ΔiG values. Positive values obtained for the R. rubrum models indicate that the interactions between the constituent subunit surfaces are too weak to allow for stable interface formation.
TABLE 3.
Comparison of buried intradimer (highlighted in green color) and interdimer (highlighted in salmon color) interaction surface areas calculated from available structures of various Rubisco enzymes in the Protein Data Bank
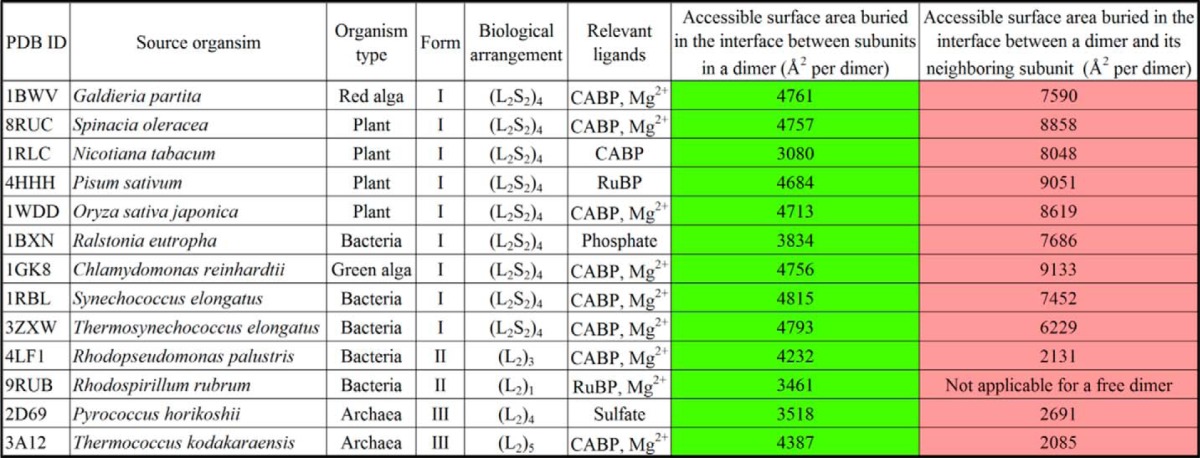
FIGURE 10.

Dimer-dimer interaction regions in R. palustris form II Rubisco (red and green) and the analogous region from R. rubrum form II (Protein Data Bank code 9RUB; dark and light gray) Rubisco. A, location of two groups of contact residues (dotted circles), which facilitate the association of dimer units via hydrogen bonds, as shown in one of the subunits (Rp Chain A; red ribbon) from the R. palustris enzyme. B, the association with neighboring subunit (Rp Chain F; green ribbon) is shown with an imaginary axis passing through the interface. Identical hydrogen bond contacts are present on the lower side of the dimer-dimer interface as well but are not shown in the figure. C, a close-up view of the two regions of contact aligned with the analogous region from R. rubrum Rubisco. Chain A from R. rubrum (Rr-A; dark gray) and R. palustris (Rp-A; red) enzymes were aligned, and chain A from another molecule of R. rubrum enzyme (Rr-A2F; light gray) was aligned to chain F of the R. palustris Rubisco (Rp-F; green). The hydrogen bond interactions present in the R. palustris structure are shown, and it is perceivable that similar interactions cannot be formed with analogous residues from the superimposed R. rubrum subunits. Ct, carboxyl terminus; Nt, amino terminus.
R. palustris Rubisco as a Model Enzyme for Carrying Out Structure-Function Analyses
The form II Rubisco from R. rubrum has been the preferred model enzyme for discerning the role of several active site residues in Rubisco catalysis (10–13). The activated CABP-bound structure of the R. palustris form II Rubisco now allows for some of these studies to be carried out with this enzyme. Moreover, the gene encoding the R. palustris Rubisco is conveniently expressed in the R. capsulatus strain SB I/II− host, allowing one to combine biological selection methodologies with protein structure-function studies. We report here one such analysis in which divergent residues near the active site and the carboxyl termini have both been targeted. Most substitutions at Ile165 and Met331 of the R. palustris Rubisco result in detrimental changes to both the enzymatic properties (Table 2) and the ability to complement for Rubisco function (e.g. CO2-dependent growth) in R. capsulatus strain SB I/II− (Fig. 7). Although these two residues have not been ascribed direct roles in the reaction mechanism (52), the results presented here underline their importance for catalysis in the context of the form II active site. This has been partially demonstrated in previous studies in which the R. rubrum equivalent of Ile165 was changed to Thr, Asn, or Asp. All substitutions led to substantial decreases in kcat and increases in KRuBP, but there was no convenient way to assess the functionality of these mutants in vivo (13). The reduced catalytic efficiency of R. palustris mutant I165T enzyme described in this study is consistent with what was measured for the R. rubrum mutant I164T enzyme. However, unlike the R. rubrum equivalent, there is no change in the specificity factor of the mutant I165T relative to the wild type (Table 2). In another study, mutant screening in Chlamydomonas identified a missense mutation at the equivalent position (residue 173) resulting in a Thr to Ile change. This led to complete loss of carboxylase activity (25). Because reciprocal substitutions at this residue in form I (Chlamydomonas; from Thr to Ile) and form II (R. palustris; Ile to Thr) led to loss of function, it must be concluded that other divergent residues complement for different requirements in the different enzyme forms. It appears that in form II enzymes an Ile at this position helps create a hydrophobic environment, which allows for seamless electron transfers mediated by the neighboring active site residues (Fig. 6). A Val substitution is thus expected to retain a similar steric and hydrophobic environment, leading to a milder impact on the catalytic activity as seen by the fact that this mutant enzyme retained functional competency. Nevertheless, the negative changes to the enzymatic properties measured for the mutant I165V enzyme suggest that the side-chain length in Ile may be optimal for this enzyme. Conversely, substitution with an Ala would create a void that may be large enough to allow an extra water molecule, thereby disrupting the balanced electrostatic environment. It is thus not surprising that the mutant I165A enzyme lacks functional competency.
It was surmised that Met331, another divergent residue positioned on the other side of CABP (Fig. 6), could compensate for a change at Ile165. However, neither the M331L single mutant nor the I165T/M331L double mutant enzyme was able to complement for function in vivo. Clearly, there must be other subtle differences in the form I active site that would allow Thr173 (equivalent to Ile165) and Leu335 (equivalent to Met331) to function optimally at their respective positions. The substitution analogous to M331L in the R. rubrum enzyme (M330L) also resulted in similar changes to the kinetic constants, i.e. no change in Ω, reduced kcat, and higher Km values for RuBP, CO2, and O2 (24). Lys330 and Met331 are located in loop 6 (residues Gly324–Gly333), which closes or “plugs” the active site in conjunction with residues from a neighboring subunit of the homodimer (26) (supplemental Movie S2). This is also evident from various Rubisco structures in which the entire loop is disordered in the absence of an active site ligand. However, these two residues are part of the most conformationally stable region of this flexible loop in the closed conformation as indicated by the temperature factors in the crystal structures. Lys330 plays a direct role in catalysis, and hence no substitutions are tolerated at this position (26). Conformational stabilization of Met331 is achieved via van der Waals interactions with residues Gly115, Met116, Gly117, and the active site Asn112 from the neighboring subunit (Fig. 6). The form I equivalent, Leu335, in the Synechococcus 6301 Rubisco is likewise stabilized by interactions with Gly126, Phe127, and Lys128. Lys128, which is conserved in both forms I and III, is believed to stabilize the closed conformation by hydrogen bonding to the main chain carbonyl groups of residues in both loop 6 and the carboxyl terminus (Fig. 6). Substitutions at this residue in the Synechococcus 6301 Rubisco caused decreases in the affinity for RuBP and kcat and resulted in increased synthesis of a side reaction product (9). It has been shown before that activation and ligand binding in form I Rubisco result in significant movements around this region (53). The entire α helix B from the amino terminus shifts by about 2 Å closer to the active site accompanied by closure of loop 6 and the carboxyl terminus and insertion of Lys128 between them. It is thus conceivable that a negative change caused by an M331A substitution in the R. palustris form II enzyme is suppressed by the A47V substitution in α helix B (Fig. 6). Because the increase in KRuBP caused by an Ala substitution at position 331 was not as drastic as that caused by a change to Leu, it was possibly easier to obtain a suppressor for M331A and not M331L. The α-carbon of Ala47 is ∼15 Å away from Met331 in the crystal structure. Its side chain is in a hydrophobic environment within 4 Å of Ala70 and Val72. Substitution with a bulkier Val would be expected to push αB (with A47V) toward the active site. Glu49 of αB functions to stabilize Lys330 and assists in loop 6 closure during catalysis, and Thr54 in the adjoining loop is involved in binding the P1 phosphate of the substrate (53). Hence, the movement of αB likely nudges these catalytically important residues along with Asn112 closer to the active site (Fig. 6). This could also restore steric shielding around the active site that was lost when Met331 was substituted with a smaller Ala (supplemental Movie S2). Together, these structural changes likely account for the increased affinity for RuBP as well as the lower Kc value caused by an A47V substitution, which resulted in suppressing the negative effect of M331A in vivo (Table 2). This was further confirmed by in silico molecular dynamics simulations with a modeled structure of the A47V/M331A double mutant, which showed an increased distance between Ala70 and Val47 and decreased distances between the active site residues Thr54 and Asn112 from the side chain of Lys330 (data not shown).
Analysis of Carboxyl-terminal Chimera Accentuates the Importance of Carboxyl Terminus as a Potential Target for Enhancing Rubisco Catalysis
The most distinctive feature of the R. palustris form II-CABP structure is the position of its carboxyl terminus, i.e. pointed away from the active site region and with no contacts to the loop 6 residues (Fig. 6). When CABP is bound to the active site, the carboxyl terminus is folded over loop 6 in all other Rubisco structures available in the database. In form I Rubisco, the loop 6/carboxyl terminus arrangement has been linked to the regulation of active site opening and closure and is thus believed to be critical for optimal rates of catalysis (53). However, disruption of these interactions via substitutions at a conserved carboxyl-terminal residue could still be tolerated for the function of the enzyme in vivo in Chlamydomonas (49). When the active site is unliganded or occupied by loosely bound substrates, the carboxyl terminus is either disordered or pointed away from loop 6 (3, 7). The presence of αI in both R. palustris and R. rubrum form II enzymes and αJ in the former likely constrains this region from taking up an extended conformation on top of loop 6 (Figs. 3B and 6). The ability of the RrCt chimera to support wild-type levels of growth likely reflects the similarities and common structural requirements in R. palustris and R. rubrum form II enzymes. However, a deeper truncation of the native terminus and replacement with shorter sequences from either Synechococcus 6301 (SyCt) or T. kodakaraensis (TkCt) Rubiscos both resulted in negative phenotypes in vivo correlated with a complete loss of in vitro activity. This suggests that the terminal α helix I may play a critical role in the form II enzyme (Fig. 2B), which is also consistent with results from previous studies in which deeper truncations to the R. rubrum carboxyl terminus caused severe perturbations to both structure and function (54, 55). Furthermore, it is likely that the terminal residues introduced by substitutions in the SyCt and TkCt enzymes may not have steric compatibility with residues from loop 6 and the neighboring large subunit (Fig. 6) when these proteins adopt a form I- or form III-like closed conformation. The significant increase in the kcat of the chimeric RrCt enzyme indicates that a longer form II carboxyl terminus might be beneficial for the R. palustris enzyme (Table 2). Systematic future analyses will be needed to test this hypothesis, and such studies bode well for enhancing Rubisco efficiency.
Conclusions
The structure-function analyses presented here with the unique hexameric form II enzyme from R. palustris has identified a region in the amino terminus that has been proposed to undergo major conformational changes accompanying the reaction of the enzyme with its substrate RuBP. Tertiary structure alignments show this region to be highly variable among different forms of Rubisco, making it a promising target for future genetic engineering. Two substitutions created in the vicinity of the active site of R. palustris form II enzyme to mimic the arrangement in form I enzymes resulted in a negative phenotype. It should thus be concluded that other divergent residues elsewhere in the structure must contribute differently to catalysis in form II and form I enzymes. One such region could be the carboxyl terminus where chimeric replacements resulted in opposing changes to the enzymatic properties. The high resolution structure of the R. palustris form II Rubisco complexed with the transition-state analog CABP now allows for comparisons with similar form I and form III CABP structures, potentially leading to rational design of future experimental approaches to understand functional differences between these structurally distinct enzyme forms.
Supplementary Material
Acknowledgments
We thank Prof. David Eisenberg for invaluable and generous support for the project and Dr. Michael Sawaya for helpful advice regarding various aspects of x-ray crystallography and structural analyses. Dr. Mark Arbing and Dan E. McNamara helped with the determination of protein molecular weights using static light scattering and provided critical feedback on the manuscript. X-ray data collection was facilitated by the UCLA-DOE X-ray Crystallography Core Facility and Lawrence Berkeley National Laboratory Advanced Light Source. We also acknowledge Richa Patel and Katherine Huening for help with cloning and initial mutagenesis experiments. Comments from Drs. Duilio Cascio, Michael Sawaya, and Lorena Saelices were of great help in improving the quality of the manuscript, and we are grateful for that.
This work was supported, in whole or in part, by National Institutes of Health Grant GM095742. This work was also supported by Department of Energy Grant DE-FC02-02ER63421 (to the Protein Expression Technology Center Core Facility at UCLA).

This article contains supplemental Movies S1 and S2.
The atomic coordinates and structure factors (codes 4LF1 and 4LF2) have been deposited in the Protein Data Bank (http://wwpdb.org/).
S. Satagopan and M. Arbing, unpublished results.
- Rubisco
- ribulose-1,5-bisphosphate carboxylase/oxygenase
- RLP
- Rubisco-like protein
- RuBP
- ribulose 1,5-bisphosphate
- CBB
- Calvin-Benson-Bassham
- Ω
- substrate specificity
- Kc
- Michaelis constant for CO2
- Ko
- Michaelis constant for O2
- KRuBP
- Michaelis constant for RuBP
- CABP
- 2-carboxyarabinitol 1,5-bisphosphate.
REFERENCES
- 1. Tabita F. R., Hanson T. E., Satagopan S., Witte B. H., Kreel N. E. (2008) Phylogenetic and evolutionary relationships of RubisCO and the RubisCO-like proteins and the functional lessons provided by diverse molecular forms. Philos. Trans. R. Soc. Lond. B Biol. Sci. 363, 2629–2640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Peterhansel C., Offermann S. (2012) Re-engineering of carbon fixation in plants—challenges for plant biotechnology to improve yields in a high-CO2 world. Curr. Opin. Biotechnol. 23, 204–208 [DOI] [PubMed] [Google Scholar]
- 3. Andersson I. (2008) Catalysis and regulation in Rubisco. J. Exp. Bot. 59, 1555–1568 [DOI] [PubMed] [Google Scholar]
- 4. Parry M. A., Andralojc P. J., Scales J. C., Salvucci M. E., Carmo-Silva A. E., Alonso H., Whitney S. M. (2013) Rubisco activity and regulation as targets for crop improvement. J. Exp. Bot. 64, 717–730 [DOI] [PubMed] [Google Scholar]
- 5. Guadalupe-Medina V., Wisselink H. W., Luttik M. A., de Hulster E., Daran J. M., Pronk J. T., van Maris A. J. (2013) Carbon dioxide fixation by Calvin-Cycle enzymes improves ethanol yield in yeast. Biotechnol. Biofuels 6, 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tabita F. R., Satagopan S., Hanson T. E., Kreel N. E., Scott S. S. (2008) Distinct form I, II, III, and IV Rubisco proteins from the three kingdoms of life provide clues about Rubisco evolution and structure/function relationships. J. Exp. Bot. 59, 1515–1524 [DOI] [PubMed] [Google Scholar]
- 7. Andersson I., Backlund A. (2008) Structure and function of Rubisco. Plant Physiol. Biochem. 46, 275–291 [DOI] [PubMed] [Google Scholar]
- 8. Tabita F. R., Hanson T. E., Li H., Satagopan S., Singh J., Chan S. (2007) Function, structure and evolution of the RubisCO-like proteins and their RubisCO homologs. Microbiol. Mol. Biol. Rev. 71, 576–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. van Lun M., van der Spoel D., Andersson I. (2011) Subunit interface dynamics in hexadecameric rubisco. J. Mol. Biol. 411, 1083–1098 [DOI] [PubMed] [Google Scholar]
- 10. Nakano T., Ashida H., Mizohata E., Matsumura H., Yokota A. (2010) An evolutionally conserved Lys122 is essential for function in Rhodospirillum rubrum bona fide RuBisCO and Bacillus subtilis RuBisCO-like protein. Biochem. Biophys. Res. Commun. 392, 212–216 [DOI] [PubMed] [Google Scholar]
- 11. Liggins J. R., Gready J. E. (2009) Putative functional role for the invariant aspartate 263 residue of Rhodospirillum rubrum Rubisco. Biochemistry 48, 2226–2236 [DOI] [PubMed] [Google Scholar]
- 12. Harpel M. R., Larimer F. W., Hartman F. C. (1998) Multiple catalytic roles of His 287 of Rhodospirillum rubrum ribulose 1,5-bisphosphate carboxylase/oxygenase. Protein Sci. 7, 730–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chène P., Day A. G., Fersht A. R. (1997) Role of isoleucine-164 at the active site of rubisco from Rhodospirillum rubrum. Biochem. Biophys. Res. Commun. 232, 482–486 [DOI] [PubMed] [Google Scholar]
- 14. Andersson I. (1996) Large structures at high resolution: the 1.6 Å crystal structure of spinach ribulose-1,5-bisphosphate carboxylase/oxygenase complexed with 2-carboxyarabinitol bisphosphate. J. Mol. Biol. 259, 160–174 [DOI] [PubMed] [Google Scholar]
- 15. Paoli G. C., Tabita F. R. (1998) Aerobic chemolithoautotrophic growth and RubisCO function in Rhodobacter capsulatus and a spontaneous gain of function mutant of Rhodobacter sphaeroides. Arch. Microbiol. 170, 8–17 [DOI] [PubMed] [Google Scholar]
- 16. Larimer F. W., Chain P., Hauser L., Lamerdin J., Malfatti S., Do L., Land M. L., Pelletier D. A., Beatty J. T., Lang A. S., Tabita F. R., Gibson J. L., Hanson T. E., Bobst C., Torres J. L., Peres C., Harrison F. H., Gibson J., Harwood C. S. (2004) Complete genome sequence of the metabolically versatile photosynthetic bacterium Rhodopseudomonas palustris. Nat. Biotechnol. 22, 55–61 [DOI] [PubMed] [Google Scholar]
- 17. Romagnoli S., Tabita F. R. (2006) A novel three-protein two-component system provides a regulatory twist on an established circuit to modulate expression of the cbbI region of Rhodopseudomonas palustris CGA010. J. Bacteriol. 188, 2780–2791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Joshi G. S., Zianni M., Bobst C. E., Tabita F. R. (2013) Regulatory twist and synergistic role of metabolic coinducer- and response regulator-mediated CbbR-cbbI interactions in Rhodopseudomonas palustris CGA010. J. Bacteriol. 195, 1381–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Spreitzer R. J., Salvucci M. E. (2002) Rubisco: structure, regulatory interactions, and possibilities for a better enzyme. Annu. Rev. Plant. Biol. 53, 449–475 [DOI] [PubMed] [Google Scholar]
- 20. Satagopan S., Scott S. S., Smith T. G., Tabita F. R. (2009) A Rubisco mutant that confers growth under a normally “inhibitory” oxygen concentration. Biochemistry 48, 9076–9083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mueller-Cajar O., Whitney S. M. (2008) Directing the evolution of Rubisco and Rubisco activase: first impressions of a new tool for photosynthesis research. Photosynth. Res. 98, 667–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Smith S. A., Tabita F. R. (2004) Glycine 176 affects catalytic properties and stability of the Synechococcus sp. strain PCC6301 ribulose-1,5-bisphosphate carboxylase/oxygenase. J. Biol. Chem. 279, 25632–25637 [DOI] [PubMed] [Google Scholar]
- 23. Smith S. A., Tabita F. R. (2003) Positive and negative selection of mutant forms of prokaryotic (cyanobacterial) ribulose-1,5-bisphosphate carboxylase/oxygenase. J. Mol. Biol. 331, 557–569 [DOI] [PubMed] [Google Scholar]
- 24. Terzaghi B. E., Laing W. A., Christeller J. T., Petersen G. B., Hill D. F. (1986) Ribulose 1,5-bisphosphate carboxylase. Effect on the catalytic properties of changing methionine-330 to leucine in the Rhodospirillum rubrum enzyme. Biochem. J. 235, 839–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Spreitzer R. J., Brown T., Chen Z., Zhang D., Al-Abed S. R. (1988) Missense mutation in the Chlamydomonas chloroplast gene that encodes the Rubisco large subunit. Plant Physiol. 86, 987–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lee G. J., McDonald K. A., McFadden B. A. (1993) Leucine 332 influences the CO2/O2 specificity factor of ribulose-1,5-bisphosphate carboxylase/oxygenase from Anacystis nidulans. Protein Sci. 2, 1147–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Whitney S. M., von Caemmerer S., Hudson G. S., Andrews T. J. (1999) Directed mutation of the Rubisco large subunit of tobacco influences photorespiration and growth. Plant Physiol. 121, 579–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Whitman W., Tabita F. R. (1976) Inhibition of D-ribulose 1,5-bisphosphate carboxylase by pyridoxal 5′-phosphate. Biochem. Biophys. Res. Commun. 71, 1034–1039 [DOI] [PubMed] [Google Scholar]
- 29. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 30. Kuehn G. D., Hsu T. C. (1978) Preparative-scale enzymic synthesis of D-[14C]ribulose 1,5-bisphosphate. Biochem. J. 175, 909–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li L. A., Tabita F. R. (1997) Maximum activity of recombinant ribulose 1,5-bisphosphate carboxylase/oxygenase of Anabaena sp. strain CA requires the product of the rbcX gene. J. Bacteriol. 179, 3793–3796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhu G., Jensen R. G. (1990) Status of the substrate binding sites of ribulose bisphosphate carboxylase as determined with 2-C-carboxyarabinitol 1,5-bisphosphate. Plant Physiol. 93, 244–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Otwinowski Z., Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 34. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Winn M. D., Isupov M. N., Murshudov G. N. (2001) Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr. D Biol. Crystallogr. 57, 122–133 [DOI] [PubMed] [Google Scholar]
- 37. Lee B., Richards F. M. (1971) The interpretation of protein structures: estimation of static accessibility. J. Mol. Biol. 55, 379–400 [DOI] [PubMed] [Google Scholar]
- 38. Saff E. B., Kuijlaars A. B. (1997) Distributing many points on a sphere. Math. Intell. 19, 5–11 [Google Scholar]
- 39. Yeates T. O. (1995) Algorithms for evaluating the long-range accessibility of protein surfaces. J. Mol. Biol. 249, 804–815 [DOI] [PubMed] [Google Scholar]
- 40. Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., Warren G. L. (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 41. Fabiola F., Bertram R., Korostelev A., Chapman M. S. (2002) An improved hydrogen bond potential: impact on medium resolution protein structures. Protein Sci. 11, 1415–1423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lawrence M. C., Colman P. M. (1993) Shape complementarity at protein/protein interfaces. J. Mol. Biol. 234, 946–950 [DOI] [PubMed] [Google Scholar]
- 43. Krissinel E., Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 44. Sugawara H., Yamamoto H., Shibata N., Inoue T., Okada S., Miyake C., Yokota A., Kai Y. (1999) Crystal structure of carboxylase reaction-oriented ribulose 1, 5-bisphosphate carboxylase/oxygenase from a thermophilic red alga, Galdieria partita. J. Biol. Chem. 274, 15655–15661 [DOI] [PubMed] [Google Scholar]
- 45. Schneider G., Lindqvist Y., Lundqvist T. (1990) Crystallographic refinement and structure of ribulose-1,5-bisphosphate carboxylase from Rhodospirillum rubrum at 1.7 Å resolution. J. Mol. Biol. 211, 989–1008 [DOI] [PubMed] [Google Scholar]
- 46. Kitano K., Maeda N., Fukui T., Atomi H., Imanaka T., Miki K. (2001) Crystal structure of a novel-type archaeal rubisco with pentagonal symmetry. Structure. 9, 473–481 [DOI] [PubMed] [Google Scholar]
- 47. Satagopan S., Spreitzer R. J. (2008) Plant-like substitutions in the large-subunit carboxy terminus of Chlamydomonas Rubisco increase CO2/O2 specificity. BMC Plant. Biol. 8, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Burisch C., Wildner G. F., Schlitter J. (2007) Bioinformatic tools uncover the C-terminal strand of Rubisco's large subunit as hot-spot for specificity-enhancing mutations. FEBS Lett. 581, 741–748 [DOI] [PubMed] [Google Scholar]
- 49. Satagopan S., Spreitzer R. J. (2004) Substitutions at the Asp-473 latch residue of Chlamydomonas ribulosebisphosphate carboxylase/oxygenase cause decreases in carboxylation efficiency and CO2/O2 specificity. J. Biol. Chem. 279, 14240–14244 [DOI] [PubMed] [Google Scholar]
- 50. Horken K. M., Tabita F. R. (1999) The “green” form I ribulose 1,5-bisphosphate carboxylase/oxygenase from the nonsulfur purple bacterium Rhodobacter capsulatus. J. Bacteriol. 181, 3935–3941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Parry M. A., Keys A. J., Madgwick P. J., Carmo-Silva A. E., Andralojc P. J. (2008) Rubisco regulation: a role for inhibitors. J. Exp. Bot. 59, 1569–1580 [DOI] [PubMed] [Google Scholar]
- 52. Cleland W. W., Andrews T. J., Gutteridge S., Hartman F. C., Lorimer G. H. (1998) Mechanism of rubisco: the carbamate as general base. Chem. Rev. 98, 549–562 [DOI] [PubMed] [Google Scholar]
- 53. Taylor T. C., Andersson I. (1996) Structural transitions during activation and ligand binding in hexadecameric Rubisco inferred from the crystal structure of the activated unliganded spinach enzyme. Nat. Struct. Biol. 3, 95–101 [DOI] [PubMed] [Google Scholar]
- 54. Morell M. K., Kane H. J., Andrews T. J. (1990) Carboxylterminal deletion mutants of ribulosebisphosphate carboxylase from Rhodospirillum rubrum. FEBS Lett. 265, 41–45 [DOI] [PubMed] [Google Scholar]
- 55. Ranty B., Lundqvist T., Schneider G., Madden M., Howard R., Lorimer G. (1990) Truncation of ribulose-1,5-bisphosphate carboxylase/oxygenase (Rubisco) from Rhodospirillum rubrum affects the holoenzyme assembly and activity. EMBO J. 9, 1365–1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.