

Background: Jak2 mediates cytokine-stimulated physiological events, but the mechanism of its activation is still unknown.
Results: IL-3 stimulated c-Abl kinase activity leading to Jak2 activation through direct interaction with c-Abl.
Conclusion: c-Abl activates Jak2 in response to IL-3 in normal hematopoietic cells.
Significance: Our findings reveal a novel role of c-Abl kinase in Jak2 activation.
Keywords: Cytokine Induction, Hematopoiesis, Janus Kinase (JAK), Leukemia, Oncogene
Abstract
Jak2 is involved in cytokine growth factor-stimulated signal transduction, but the mechanism of its activation is largely unknown. Here, we investigated Jak2 activation in a normal hematopoietic cell line, 32D mouse myeloid cells. The bimolecular fluorescence complementation studies showed that c-Abl formed a stable complex with Jak2 in live cells. Co-immunoprecipitation results showed that c-Abl bound to the βc chain of IL-3/IL-5/GM-CSF receptors. The kinase activities of both c-Abl and Jak2 were stimulated by IL-3 in 32D cells. Decreasing c-Abl protein expression in 32D cells by inducible shRNA decreased Jak2 activity and resulted in the failure of Jak2 activation in response to IL-3. Treatment of IL-3 and serum-starved 32D cells with 1 μm imatinib mysylate inhibited IL-3 stimulated kinase activities of both c-Abl and Jak2. In addition, the kinase-deficient Bcr-Abl mutant (p210K1172R) was defective for activation of Jak2 in 32D cells and impaired IL-3 independent growth, which was rescued by overexpression of c-Abl (+Abl). IL-3 efficiently inhibited apoptosis of 32Dp210K/R+Abl cells induced by imatinib mysylate but not Jak2 kinase inhibitor TG101209. In summary, our findings provide evidence that the kinase function of c-Abl and its C-terminal CT4 region is crucial for its interaction with Jak2 and its activation. c-Abl kinase activity induced by IL-3 is required for IL-3-stimulated Jak2 and Jak1 activation. Our findings reveal a novel regulatory role of c-Abl in Jak2 activation induced by IL-3 cytokine growth factor in 32D hematopoietic cells.
Introduction
Janus kinase 2 (Jak2) is a receptor-associated tyrosine kinase, which is widely expressed and involved in transducing signals for a variety of cytokines, interferons (IFNs), and growth factors through interaction with the receptors (1, 2). Jak2 affects hematopoiesis, body growth, lactation, and immunity in receptor-mediated signaling in different cell types (3–5). JAK2−/− mice die in mid-gestation and exhibit impaired erythropoiesis, indicating a critical role of Jak2 in cytokine receptor signaling in erythropoiesis (6–8).
Jak2 kinase activity is stimulated by cytokines through their receptors. In particular, IL-3 and GM-CSF ligands induce the formation of a dodecamer receptor structure that facilitates Jak2 trans-phosphorylation of tyrosine 1007 in the activation loop of Jak2, leading to its kinase activation (3, 9, 10). Because of its important physiological function, Jak2 activity is critically regulated through phosphorylation of residues within the seven domains of Jak2. The regulation on Jak2 activation also includes interaction with the cytokine signaling suppressor, LNK, tyrosine phosphatases, and the pseudokinase domain (JH2) of Jak2 (4, 11–13).
The JH2 domain of Jak2 kinase negatively regulates Jak2 activity by interacting with the activation loop of the kinase domain (14, 15). Spontaneous mutations in the JH2 domain such as V617F and E695K are defective in its inhibitory function on the Jak2 kinase, resulting in constitutive activation of Jak2 kinase and the activation of downstream events such as STAT-mediated transcription (4). Abnormal Jak2 activity and mutations in the JH2 domain have been identified in more than 50% of patients with Philadelphia chromosome-negative myeloproliferative neoplasms such as polycythemia vera, essential thrombosis, primary myelofibrosis, and chronic myelomonocytic leukemia (16–22). Additional JAK2-activating mutations, such as JAK2T875N in the kinase domain (23), JAK2ΔIREED (a deletion in the JH2 domain) (24), and JAK2 exon 12 mutations (proximal to the JH2 domain) (25) have been found in a small number of patients with myeloproliferative disease lacking JAK2V617F (26).
In Bcr-Abl-positive CML,2 Bcr-Abl expression induces strong activation of Jak2 that activates Lyn kinase through the SET-PP2A-Shp1 pathway (27). Jak2 also activates the Gab2/PI3K/Akt pathway and is involved in c-Myc expression induced by Bcr-Abl, which leads to increased cell growth in the absence of cytokines (e.g. IL-3) (28–30). In turn, Jak2 regulates stimulation of the Ras/Raf/PI3K pathways in Bcr-Abl-transformed cells by phosphorylating tyrosine 177 within the Bcr region of Bcr-Abl and maintains Bcr-Abl protein stability (31). Jak2 has also been reported to act at nuclear sites of cells by phosphorylating histone H3 (32). Persistent Jak2 activation can lead to oncogenic activation and genomic instability through phosphorylation of H3 Tyr-41, resulting in displacement of HP1α from heterochromatin. However, little is known about the signaling pathway that leads to Jak2 activation in normal hematopoietic cells.
The c-abl proto-oncogene is a nonreceptor tyrosine kinase that is a key element in intracellular signaling and is involved in diverse biological processes, including regulation of cytoskeletal reorganization, cell migration and morphogenesis, cell differentiation, proliferation, adhesion, cell death, stress responses, and gene expression (33–36). c-Abl is located in multiple cellular compartments, including the nucleus and cytoplasm, and its activity is modulated by various stimuli (37–39). The c-Abl kinase is activated in response to growth factors such as platelet-derived growth factor (PDGF) and epidermal growth factor (EGF) through Src family members (38). Abnormal c-Abl activity is involved in leukemia as well as in solid tumors (13, 29, 40–43, 57). In CML, Bcr fuses with the second exon of c-Abl that disrupts the self-inhibition of c-Abl and contributes to the constitutively kinase-activated Bcr-Abl kinase. In our previous studies, we found that the kinase domain and C-terminal region 4 (CT4) of c-Abl are involved in Jak2 binding (30).
In this study, we used the BiFC system (44) to confirm our earlier findings that c-Abl directly binds to Jak2 through its CT4 region and its kinase domain (30), and we further demonstrated that this direct interaction between c-Abl and Jak2 is required for Jak2 activation. We found that c-Abl associates with the common β chain (βc) of the interleukin 3/interleukin 5/granulocyte-macrophage colony-stimulating factor (IL-3/IL-5/GM-CSF) receptors, and its activity is efficiently stimulated by IL-3. Importantly, c-Abl is critically involved in IL-3-stimulated Jak2 activation in the 32D mouse myeloid cell line. c-Abl overexpression allows IL-3-independent growth and Jak2 activation in 32D cells expressing the kinase-deficient Bcr-Abl K1172R mutant. The novel finding of the requirement of c-Abl kinase in Jak2 activation stimulated by IL-3 leads to a further understanding of the mechanism of Jak2 activation in the normal hematopoietic cells.
EXPERIMENTAL PROCEDURES
Chemicals and Antibodies
IM was purchased from LC Laboratories. TG101029 was supplied under a Material Transfer Agreement from TargeGen Inc. Commercially available antibodies used were anti-phospho-Jak2Y1007 (Millipore, catalog no. 04-1098), phospho-SrcY416 (Cell Signaling, catalog no. 6943), phospho-LynY396 (Gene Tex, catalog no. GTX61275), Lyn (Cell Signaling, catalog no. 2732), Jak2 (Cell Signaling, catalog no. 3230), c-Abl (Cell Signaling, catalog no. 2862), phosphor-Abl (Tyr-412) (Millipore, catalog no. 07-788), phosphotyrosine (4G10) (Millipore, catalog no. 05-321), α-tubulin (B-7) (Santa Cruz Biotechnology, catalog no. sc-5286), β-actin (N-21) (Santa Cruz Biotechnology, catalog no. sc-130656), IL-3/IL-5/GM-CSF common β chain (clone K-17) (Santa Cruz Biotechnology, catalog no. sc-678). Sepharose bead-conjugated Jak2 antibody was purchased from Cell Signaling (catalog no. 4089). The recombinant mouse IL-3 was purchased from Roche Applied Science.
Constructs
MIGR1 Bcr-Ablp210 K1172R was cloned by digesting Bcr-AblK1172R mutant gene from pSG5 Bcr-Abl K1172R vector with EcoRI and cloned into the retroviral MIGR1. For BiFC constructs, human c-Abl and Abl mutants (Abl ΔCT4 and Abl KNΔCT4) were amplified by PCR using the following primers: 5′ cctccggaatggggcagcagcctgg 3′; 5′ cctctagactacctctgcactatgtc 3′; and 5′ cctctagattatggcagggccgaggatg 3′. Mouse Jak2 was amplified using the following primers: 5′ cctccggaatgggaatggcctgc 3′ and 5′ cgagggccctcacgcagctatactg 3′.
The PCR products were cloned into the BspEI and XbaI site (Abl) and BspEI and ApaI (Jak2) of BiFC Venus vectors kindly provided by Dr. Stephen W. Michnick (University of Montreal, Canada). Human c-Abl was cloned into MIGR-1 mCherry vector kindly provided by Dr. Mallampati (University of Texas, M.D. Anderson Cancer Center). TRIPZ-inducible lentiviral c-Abl shRNA (Clone ID V2THS_198745) and nontargeted shRNA control (Clone ID RHS4743) were purchased from Open Biosystems (Thermo Scientific). The sequences of all the constructs were confirmed by DNA sequencing. The cDNAs of interleukin 3 receptor α (IL-3Rα) and the common βc chain of IL-3/IL-5/GM-CSF receptor (βc) were kindly provided by Dr. James McCubrey (East Carolina Medical School, Greenville, NC).
Cell Culture
IL-3-dependent 32D cells and the Bcr-Abl+ 32D cells were grown in RPMI 1640 medium supplemented with 10% FBS, 2 mm glutamine, penicillin/streptomycin, and 10% WEHI media. 32Dp210, 32Dp210K/R+Abl, and Wehi3b cells were maintained in RPMI 1640 medium supplemented with 10% FBS, 2 mm glutamine, penicillin/streptomycin. 293T cells were grown in DMEM supplemented with 10% FBS, 2 mm l-glutamine, and penicillin/streptomycin. All cells were incubated with 5% CO2 at 37 °C.
BiFC
Human c-Abl constructs (WT, ΔCT4, and KDΔCT4) and mouse Jak2 cloned into the Venus vectors (44) were co-transfected into 293T cells. Live fluorescent images were obtained 48 h after transfection.
Lentivirus/Retrovirus Preparation and Stable Cell Line Establishment
Lentiviruses were prepared by co-transfection of pCMV_8.2, pMD.G and pTRIPZ containing c-Abl shRNA or nontargeted shRNA into 293T cells using FuGENE 6 reagent (Roche Applied Science) as described previously (45). Retroviral constructs MIGR1-Bcr-Ablp2101172R and MIGR1-mCherry-Abl were transfected into Phoenix packaging cells (kindly provided by Dr. Sue-Hwa Lin, University of Texas M.D. Anderson Cancer Center, Houston, TX) (31). 32D cells were infected with the desired viral supernatants mixed with an equal volume of RPMI 1640 medium plus 20% WEHI-conditioned medium containing 8 μg/ml Polybrene at 37 °C for 16 h, followed by removing the virus/media mixture and continuing cell culture with fresh RPMI 1640 medium with WEHI-conditioned medium for 48 h. Positive infected cells were obtained either by selection in the presence of 3 μg/ml puromycin for 10 days or by identification of green or mCherry fluorescent positive cells using flow cytometry. To enrich the population of cells expressing c-Abl shRNA or nontargeted shRNA, puromycin-resistant cells were cultured in the presence of 2 μg/ml doxycycline for 3 days followed by cell sorting to harvest cells with a high red fluorescent signal.
Immunoprecipitation and Kinase Assay
Cells were lysed in 1% Nonidet P-40 buffer containing a mixture of protease and phosphatase inhibitors (Thermo Scientific, catalog no. PI-78442) as described previously (46). For immunoprecipitation, 400 μg of cell lysates were incubated with a specific primary antibody overnight at 4 °C with rotation and then with 20 μl of protein A/G-Sepharose for 1 h at 4 °C. The agarose beads were collected by centrifugation at 2000 rpm for 2 min at 4 °C. The agarose beads were washed four times with cold PBS, followed by boiling in 70 μl of SDS sample buffer for 5 min. For Western blotting (WB), cell lysates or immunoprecipitates were subjected to SDS-PAGE as described (27). The blots were incubated with specific primary antibodies overnight at 4 °C. The immunoreactive bands were visualized by Amersham Biosciences ECL Western blotting detection reagents (GE Healthcare, catalog no. RPN2106). In vitro kinase assays for Jak2 (autophosphorylation) was carried as described (41).
Apoptotic Assays
The apoptosis analysis was performed by using FITC annexin V apoptosis detection kit (BD Biosciences, catalog no. 556547) or PE annexin V apoptosis detection kit (BD Biosciences, catalog no. 559763). Briefly, aliquots of 1 × 105 cells were washed twice with cold PBS, resuspended in 100 μl of binding buffer containing 5 μl of FITC-conjugated annexin V antibody plus 7 μl of propidium iodide (PI) reagents, or 5 μl of PE-conjugated annexin V antibody and 7 μl of 7-aminoactinomycin D reagents, at room temperature in the dark for 15 min. After washing with PBS twice, cell pellets were suspended by 400 μl of binding buffer and subjected to flow cytometry analysis.
Statistical Analysis
Results are shown as the mean ± S.E. of values obtained in independent experiments.
RESULTS
c-Abl Associates with Jak2 in Live Cells
Our previous cell-based pulldown experiments showed the binding of c-Abl and Jak2 is facilitated by the C-terminal domain (CT4) and the kinase domain of c-Abl (30). To further investigate this interaction in live cells, we applied the BiFC assay (44) by constructing vectors that fused the N-terminal half of the fluorescent protein (YFP) to the N terminus of c-Abl (YFP(N)-Abl) and the C-terminal half of YFP to the N terminus of Jak2 (YFP(C)-Jak2). 293T cells co-transfected with YFP(N)-Abl and YFP(C)-Jak2 showed strong fluorescent signals (Fig. 1A), although neither vector alone gave any fluorescent signals (data not shown). Interestingly, we observed an intense localized YFP signal in the cytoplasm of cells expressing wild-type YFP(N)-Abl and YFP(C)-Jak2 (Fig. 1A, white arrowheads). This signal was decreased in cells expressing Abl lacking CT4 (Abl ΔCT4) and Abl kinase-dead mutant lacking CT4 (Abl KN ΔCT4). Next, we examined the Jak2 activity shown by co-expressing either wild-type c-Abl or Abl mutants. We found that Jak2 is activated by co-expressing wild-type c-Abl, as indicated by the increased phosphorylation on Tyr-1007/1008 of Jak2 (Fig. 1B, lanes 1 and 4). The absence of direct interaction between c-Abl and Jak2 down-regulates the Jak2 kinase activity, as shown by the reduction of Jak2 activation when co-expressing the KN, ΔCT4 Abl mutants (Fig. 1B, lanes 2 and 3), indicating the critical roles of these two regions of c-Abl in regulating Jak2 activity.
FIGURE 1.
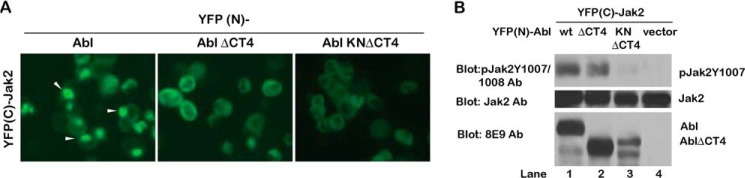
Direct interaction of Jak2 and c-Abl in living cells. A, YFP signal formation in 293T cells co-transfected with YFP(C)-Jak2 and various forms of YFP (N)-Abl (WT, ΔCT4 and KNΔCT4). The intense localized YFP signal is indicated by the white arrowheads. The images were captures 2 days after transfection under the same exposure. B, regulation of Jak2 activity by c-Abl. Jak2 activity in the cell lysate from the 293T cells co-transfected with the Jak2 and Abl BiFC constructs was examined using the Jak2 Tyr(P)-1007/1008 antibodies (Ab).
c-Abl Associates with the IL-3Rβ Chain in Normal Hematopoietic Cells
Our previous studies (30) and current BiFC results (Fig. 1) document the interaction between c-Abl and Jak2. Jak2 associates with the membrane-proximal region of erythropoietin receptors and the common βc of IL-3/IL-5/GM-CSF receptors (47). Jak2 kinase is activated in response to erythropoietin (48), IL-3 (49), growth hormone (50), GM-CSF (47), and prolactin (51). Thus, we wanted to know whether c-Abl associates with IL-3 receptor β (IL-3Rβ) chain. To detect this, IP experiments were performed in 32D cells, a clone of mouse myeloid cells requiring IL-3 for survival. We observed c-Abl protein in the IL-3Rβ immune complex, suggesting a direct interaction between c-Abl and IL-3Rβ (Fig. 2A). This was confirmed in 293T cells co-expressing c-Abl and the common β chain of the IL-3/IL-5/GM-CSF receptors (βc chain) (Fig. 2A). IP results showed Jak2 protein interacted with IL-3Rβ in 32D and 293T cells co-expressing the βc chain and HA-tagged Jak2 (Fig. 2B), which is consistent with previous reports (1, 47). We also observed that IL-3 stimulation had no effect on c-Abl or Jak2 interaction with IL-3Rβ chains in 293T cells co-expressing c-Abl and both the IL-3 receptor α (IL-3Rα) and βc chains (Fig. 2C), These results indicate that c-Abl and Jak2 both can interact with the IL-3Rβ chain, and this association is independent of IL-3.
FIGURE 2.

c-Abl binds to IL-3 receptor β chain and is involved in IL-3 stimulation of Jak2 activation. A, cell lysates from 32D and 293T cells co-transfected with c-Abl and IL-3Rβ were immunoprecipitated with IL-3Rβ antibody (Ab) or control rabbit IgG, followed by WB with c-Abl antibody. B, left panel, cell lysates from 32D cells were immunoprecipitated with anti IL-3Rβ antibody or control rabbit IgG, followed by WB with Jak2 antibody. Right panel, cell lysate from 293T cells co-transfected with HA-tagged Jak2 and IL-3Rβ were immunoprecipitated with HA tag antibody followed by WB with IL-3Rβ antibody. C, 293T cells were co-transfected with c-Abl and IL-3 receptor (IL-3Rα and IL-3Rβ). Transfected 293T cells were stimulated with IL-3 for 10 min. Cell lysates were immunoprecipitated with IL-3Rβ antibody or rabbit IgG and followed by WB with the indicated antibodies. D, 32D cells were starved of IL-3 and FBS for 6 h followed by 3 ng/ml IL-3 stimulation or IM (1 and 3 μm) plus IL-3 treatment for 1 and 2 h. Cell lysates were analyzed by WB using the indicated antibodies. E, 293T cells co-transfected with c-Abl and IL-3 receptor (α and β subunits) were treated with or without IL-3 (3 ng/ml) for 10 min. Cell lysates were analyzed by WB with the indicated antibodies. F, 32D cells expressing nontarget shRNA (32D shNT) or Abl shRNA (32D shAbl) were treated with doxycycline for 4 days and were starved of IL-3 and FBS for 5 h followed by stimulation with 3 ng/ml IL-3 for 2 and 4 h. Cell lysates were immunoblotted with the indicated antibodies. C, control. G, in vitro kinase assay of Jak2 activity in 32D NT shRNA and 32D KD-Abl under the same treatment as the description in F. Cell lysates were immunoprecipitated with Jak2 antibody, and the immunoprecipitate was incubated with cold ATP in the kinase mix. After WB, the blots were processed with anti-Jak2 Tyr(P)-1007/1008 antibodies. H, c-Abl is involved in kinase activities of Jak1 stimulated by IL-3. 32D cells were starved of IL-3 and FBS for 6 h followed by 3 ng/ml IL-3 stimulation or imatinib (1 and 3 μm) plus 3 ng/ml IL-3 treatments for 1 and 2 h. Cell lysates were Western-blotted with pJak1Y1022/1023 and Jak1 antibodies.
Both c-Abl and Jak2 Kinases Are Activated in Response to IL-3
Next, we investigated whether c-Abl is activated in response to IL-3 stimulation in hematopoietic cells. To test this, 32D cells were starved of IL-3 and fetal bovine serum (FBS) for 6 h followed by IL-3 (3 ng/ml) stimulation for 1 and 2 h. c-Abl kinase activity as measured by Western blotting with antibody against c-Abl phosphorylation at tyrosine residue 412 was significantly increased after 1 and 2 h of IL-3 stimulation (Fig. 2D). Jak2 activity detected by the level of Jak2 phosphorylation on tyrosine residue 1007 was elevated in response to IL-3, as was the case for c-Abl kinase (presence of Abl Tyr(P)-412). To further examine the effects of increased Jak2 activity following IL-3 stimulation, the tyrosine phosphorylation of STAT3, which is a downstream target of Jak2, was examined. We found STAT3 activity also markedly increased upon 1 and 2 h of IL-3 stimulation, which paralleled the increased level of Jak2 activity (Fig. 2D). IL-3-induced activation of both c-Abl and Jak2 kinase was also observed in 293T cells co-expressing c-Abl and both IL-3Rα and βc chains. After IL-3 stimulation for 10 min, the kinase activities of both Jak2 and c-Abl increased (Fig. 2E), suggesting that c-Abl may be involved in Jak2 activation in response to IL-3.
c-Abl Is Required for Jak2 Activation Induced by IL-3
To examine whether c-Abl regulates IL-3-stimulated Jak2 activity, we examined kinase activities of both c-Abl and Jak2 in 32D cells treated with IM, a c-Abl kinase inhibitor. Followed by starvation of IL-3 and FBS for 6 h, 32D cells were incubated with either IL-3 alone or IL-3 with different doses of IM (1 or 3 μm) for 1 and 2 h. Western blotting showed that c-Abl kinase activity as well as Jak2 and STAT3 activities slightly decreased under IL-3 and IM treatment for 1 h but dramatically decreased after 2 h of treatment (Fig. 2D). These results demonstrate that inhibition on c-Abl kinase activity by IM down-regulates Jak2 and STAT3 kinase activities, suggesting that c-Abl mediates IL-3-induced Jak2 activation in normal hematopoietic cells.
Interestingly, we found Jak1 kinase activity behaved similarly as Jak2 in response to IL-3 stimulation. Western blotting results showed that Jak1 kinase activity evaluated by pJak1Y1022/1023 level was significantly increased in IL-3- and FBS-starved 32D cells after 2 h of IL-3 stimulation (Fig. 2H). However, this increased Jak1 activity was slightly reduced under IL-3 and IM treatment for 1 h but markedly decreased after 2 h of treatment (Fig. 2H). These data indicate that Jak1 kinase activity is induced by IL-3, and inhibition on c-Abl kinase activity by IM caused a decrease of Jak1 kinase activity, suggesting c-Abl is also involved in IL-3-induced Jak1 activation in normal hematopoietic cells.
To test whether c-Abl is required for Jak2 activity in normal hematopoietic cells, c-Abl was knocked down by lentiviral transduction of doxycycline (Doxy)-inducible c-Abl shRNA in 32D cells, which we termed 32D shAbl cells. 32D cells overexpressing nontargeted shRNA (32D shNT) was used as the control. Western blotting results showed that c-Abl protein level was reduced after Doxy induction for 3 days and recovered after Doxy removal for 4 days (Fig. 2F, lanes 5, 9, and 13). Importantly, Jak2 activity was dramatically decreased in 32D shAbl cells after 3 days of Doxy induction. In contrast, Jak2 activity remained the same in Doxy-treated 32D shNT cells and non-Doxy-treated 32D shAbl cells (Fig. 2F, lanes 1, 5, and 9). After 4 days of Doxy withdrawal, Jak2 activity was restored in parallel with the recovery of c-Abl protein levels (Fig. 2F, lane 13). Xie et al. (30) applied in vitro kinase assay by incubating c-Abl with either wild-type or Tyr to Phe mutant Jak2 synthetic peptides, and they found c-Abl kinase directly phosphorylates Jak2 at Tyr-1007. These results indicate that c-Abl kinase activity and protein expression are required for Jak2 activation in 32D cells.
Next we wanted to determine whether c-Abl is required for IL-3-stimulated Jak2 activation in 32D cells. To investigate this, Jak2 activity was examined in IL-3- and FBS-starved 32D cells expressing the desired shRNA upon IL-3 stimulation. After starvation, Jak2 activity was greatly reduced in both Doxy-treated 32D shNT and non-Doxy-treated 32D shAbl (Fig. 2F, lanes 2 and 6). Upon IL-3 stimulation, Jak2 activity gradually increased in these two cells (Fig. 2F, lanes 3, 4, 7, and 8). This increase of Jak2 activity was not observed in Doxy-treated 32D shAbl cells under the same treatment (Fig. 2F, lanes 10–12). To further confirm the above results, we used Jak2 IP kinase assay to confirm that Jak2 activity responded to IL-3 stimulation only in 32D shNT cells but not in Doxy-treated 32D shAbl cells (Fig. 2G). Our results show that the inhibition of c-Abl kinase by IM and c-Abl knockdown impaired IL-3-stimulated Jak2 activation, indicating c-Abl was required for stimulation of Jak2 activity by IL-3.
Kinase-deficient BCR-ABL Impairs Jak2 Activation and IL-3 Independence in 32D Cells
We compared wild-type Bcr-Abl (b3a2-p210) and kinase-deficient Bcr-Abl (p210K1172R) for their abilities to provide cytokine independence and Jak2 activation. First, we tested the requirement of IL-3 for the survival of 32D cells expressing wild-type Bcr-Abl (32Dp210) (see Fig. 4A) and 32D cells expressing kinase-deficient Bcr-Abl (32Dp210K/R). Flow cytometry data showed that 32D cells exhibited 68 and 99% apoptosis upon IL-3 withdrawal for 48 and 72 h, respectively (Fig. 3A). Similarly, 65 and 92% of apoptotic cells were observed in 32Dp210K/R cells under the same conditions (Fig. 3A), indicating that Bcr-Abl kinase activity is required for IL-3-independent cell growth. As expected, Bcr-Abl+ 32 D cells remained viable in the absence of IL-3 (Fig. 4A).
FIGURE 4.

Overexpression of c-Abl restored Jak2 activity and IL-3 independence in 32D cells expressing kinase-deficient Bcr-Abl mutant. A, apoptosis of 32D, 32Dp210, 32D+Abl, 32Dp210K/R, and 32Dp210K/R+Abl cells. After washing four times with cold PBS, cells were cultured in the absence of IL-3 for 72 h. Apoptotic cells were analyzed by annexin V-FITC/PI or annexin V-PE/7-aminoactinomycin D staining. B, total tyrosine phosphorylation of proteins in 32D, 32Dp210, 32Dp210K/R, and 32Dp210K/R+Abl cells. Cell lysates were analyzed by WB with anti-phosphotyrosine antibody (Ab) 4G10. * identifies the band c-Abl. C, activity of Bcr-Abl, Jak2, and Lyn in 32D, 32Dp210, 32Dp210K/R, and 32Dp210K/R+Abl cells. Cell lysates were analyzed by WB with the indicated antibodies. D, cell lysates of 32Dp210K/R+Abl cells treated with various doses of IM (0.2, 0.3, and 0.4 μm) for 16 h were analyzed by WB with the indicated antibodies.
FIGURE 3.

Kinase-deficient Bcr-Abl is deficient in Jak2 activity and IL-3 independent growth in 32D cells but did not prevent IL-3 stimulation of Jak2 activation. A, apoptosis of 32D and 32Dp210K/R cells cultured without IL-3 for 48 and 72 h. Cells were washed with cold PBS four times to completely remove IL-3 and resuspended in IL-3 free medium for 48 and 72 h. Apoptosis was analyzed by staining with annexin V-FITC/PI (for 32D cells) or annexin V-PE/7-aminoactinomycin D (for 32Dp210K/R cells). B, Bcr-Abl protein level and Jak2 activity in 32D, 32Dp210 and 32Dp210K/R cells. Cell lysates were analyzed by WB with the indicated antibodies. C, kinase-deficient Bcr-Abl does not impair Jak2 activation stimulated by IL-3. 32Dp210K/R cells were starved of IL-3 and FBS for 6 h followed by stimulation with 3 ng/ml IL-3 for 1 and 2 h. Cell lysates were Western blotted with pJak2Y1007/1008 and Jak2 antibodies.
We assumed the inability of IL-3-independent growth of 32Dp210K/R cells may due to the lack of Jak2 activity. Thus, we examined Jak2 activity under the regulation of either wild-type Bcr-Abl or the kinase-deficient Bcr-Abl mutant. Jak2 kinase activity was significantly increased in 32Dp210 cells (Fig. 3B). This increased Jak2 activity was not observed in 32Dp210K/R cells having equal protein expression of kinase-deficient Bcr-Abl compared with wild-type Bcr-Abl as detected by 8E9 anti-Abl Western blotting (Fig. 3B), suggesting that the down-regulation of Jak2 activity resulted from the deficiency of Bcr-Abl kinase activity but not from the difference in the protein expression level of wild-type or mutated Bcr-Abl. Importantly, kinase-deficient Bcr-Abl mutant did not impair IL-3 stimulation of Jak2 activation (Fig. 3C), suggesting the IL-3-mediated signal pathway is not blocked by deficient Bcr-Abl.
Overexpression of c-Abl Rescues IL-3 Independent Growth of 32D Cells Expressing Kinase-deficient Bcr-Abl
Fig. 2, F and G, showed that c-Abl is required for IL-3-stimulated Jak2 activation in 32D cells. It was of interest to explore whether kinase-active c-Abl could activate Jak2 in 32D cells expressing kinase-deficient Bcr-Abl. To address this, c-Abl was overexpressed in 32D and 32Dp210K/R cells to generate 32D+Abl and 32Dp210K/R overexpressing c-Abl, respectively. First, we examined the IL-3 dependence of these cells. After IL-3 withdrawal for 72 h, more than 90% apoptosis was detected in 32D and 32Dp210K/R cells (Fig. 4A). Interestingly, less than 10% of total cell population of 32Dp210K/R cells transduced with c-Abl was able to survive in the absence of IL-3 (viable cells were termed 32Dp210K/R+Abl cells). However, overexpression of c-Abl alone in 32D cells did not allow any detectable cell survival without IL-3. These results suggest that additional factors and functions of kinase-deficient Bcr-Abl such as Bcr sequences, including tyrosine residue 177, might be needed for c-Abl to transform 32D to IL-3-independent growth, as Jak2 phosphorylates Tyr-177 of Bcr-Abl (28).
We found the total tyrosine phosphorylation pattern detected by Western blotting with 4G10 antibody is higher in 32Dp210 and 32Dp210K/R+Abl cells compared with 32D, 32D+Abl, and 32Dp210K/R cells (Fig. 4B). Moreover, 32Dp210K/R+Abl cells showed stronger and additional tyrosine phosphorylation bands (Fig. 4B). One of these bands around 150 kDa in 32Dp210K/R+Abl cells is kinase-activated c-Abl, which was confirmed by Western blotting with Abl Tyr(P)-412 antibody (Fig. 4D). IM inhibited kinase activities of both c-Abl and Jak2 in 32Dp210K/R+Abl cells, as 0.3 μm IM significantly inhibited c-Abl and Jak2 kinase activities but not total protein levels of both c-Abl and Jak2 in 32Dp210K/R+Abl cells (Fig. 4D). These results suggest overexpression of c-Abl coupled with kinase-deficient Bcr-Abl restored the extensive protein tyrosine phosphorylation pattern seen in 32D cells expressing Bcr-Abl.
Based on our previous results of c-Abl and Jak2 interaction, we reasoned that Jak2 and its regulated proteins may be activated in 32Dp210K/R cells upon c-Abl overexpression. As Fig. 4C shows, c-Abl overexpression activated Jak2 in 32Dp210K/R+Abl cells to the same level as observed in 32Dp210 cells. This c-Abl-induced Jak2 activation was significantly abolished by various doses of IM (Fig. 4D). This activated Jak2 would contribute to phosphorylation of Bcr-Abl at Tyr-177 in 32Dp210K/R+Abl cells, which is consistent with our previous publication indicating that Bcr-Abl Tyr(P)-177 is phosphorylated by Jak2 (28). As a downstream target of Jak2 (e.g. Lyn kinase (43)), Lyn kinase was strongly activated in 32Dp210K/R+Abl cells compared with 32Dp210 cells (Fig. 4C). These data suggest that only in the presence of kinase-deficient Bcr-Abl could kinase-active c-Abl rescue IL-3 independence and restore Jak2/Lyn signaling pathways.
Jak2 May Play a Role in the Survival of 32Dp210K/R+Abl Cells
Next, we wanted to know whether the c-Abl kinase function is required for IL-3 independent growth of 32Dp210K/R+Abl cells. Cells were treated with different doses of IM (1 and 5 μm) in the presence or absence of IL-3 for 48 h. We found 53 and 97% of apoptotic cells in 32Dp210K/R+Abl cells treated with 1 and 5 μm IM, respectively, in the absence of IL-3 (Fig. 5A, left). Interestingly, the apoptotic rate is reduced to 12% in the presence of IL-3 (Fig. 5A, right). However, the rescue by IL-3 was not observed in 32Dp210K/R+Abl cells treated with TG101209, a selective Jak2 inhibitor (Fig. 5B). A similar percentage of apoptosis was induced by 5 and 10 μm TG101209 no matter whether IL-3 was present or not (Fig. 5B). Although these high levels of TG101209 inhibitor may have off-target effects, these results suggest that Jak2 is required for IL-3 independent growth in 32Dp210K/R+Abl cells, implying an important role of Jak2 in myeloid cells involving IL-3. Preliminary results obtained by Jak2 knockdown (results not shown) are consistent with a role of the Jak2 kinase in restoration of IL-3 independent growth in 32Dp210K/R mutant cells.
FIGURE 5.

IL-3 rescued apoptosis induced by IM but not TG101209 in 32Dp210K/R+Abl cells. Apoptosis of 32Dp210K/R+Abl cells treated with 1 and 5 μm IM (A) or 5 and 10 μm TG101209 (B) cultured with or without IL-3 for 48 h. 1% WEHI medium was added into IM- or TG101209-treated cells as a supplement of IL-3.
DISCUSSION
We showed that Jak2 activity is activated by c-Abl kinase in response to IL-3 in normal hematopoietic cells through their direct interaction (Figs. 1A and 2, A–D). IL-3 is able to stimulate both c-Abl and Jak2 kinase activities in IL-3 and serum-starved 32D cells (Fig. 2D). Decreasing c-Abl protein expression by inducible c-Abl shRNA in 32D cells reduced Jak2 activity and abolished its activation in response to IL-3 (Fig. 2F). Our studies showed that compared with the wild-type Bcr-Abl, kinase-defective Bcr-Abl (p210K1172R) had impaired IL-3 independence and Jak2 activity in 32D cells (Figs. 3A and 4A), which could be overcome by atopic expression of c-Abl (Fig. 4, A and C). IL-3 efficiently rescued apoptosis induced by IM but not by TG101209 (Fig. 5, A and B), indicating Jak2 activity is required for IL-3-independent growth.
It is known that IL-3/IL-5/GM-CSF receptor α and β chains form a dodecamer structure when a cytokine such as IL-3 interacts with the receptor. This leads to Jak2 association with common β chain and subsequently cross-phosphorylation of Jak2 on adjacent β chains, which results in Jak2 activation (13). Here, we show that c-Abl directly interacts with Jak2 (Fig. 1A), and c-Abl forms a stable complex with IL-3Rβ in an IL-3-independent way (Fig. 2, A–C). Based on these results, we hypothesize that c-Abl would form a complex with Jak2 on the IL-3Rβ chain, which structurally facilitates Jak2 activation by c-Abl as a result of IL-3 stimulation. We further propose that c-Abl may phosphorylate Jak2 on crucial tyrosine residues leading to Jak2 activation as part of the IL-3 dodecamer structure.
Our findings show that kinase-active c-Abl is critical for activation of Jak2 in 32D cells. The inhibition of c-Abl by IM significantly decreases Jak2 activity in 32D cells (Fig. 2D), which was also observed in IM-treated 32Dp210K/R+Abl (Fig. 4D) and 32D cells expressing wild-type Bcr-Abl (30). These latter results suggest that constitutively activated Bcr-Abl kinase is the major driving force for Jak2 activation in CML cells. Interestingly, the inhibition of Jak2 activity by 1 μm IM treatment can be overcome by the addition of IL-3 in 32D cells (Fig. 2D) as well as in 32Dp210K/R+Abl cells (data not shown) and 32Dp210 cells (49). These results suggest that Jak2 is required for the survival of both normal hematopoietic cells and CML cells. Further studies are underway to determine whether c-Abl kinase is still needed for Jak2 activation in CML cells.
Jak1 and Jak3 share a similar molecular structure as Jak2. Therefore, it was of interest to determine whether c-Abl is also involved in cytokine-mediated Jak1 and Jak3 activation. We found that Jak1 activity stimulated by IL-3 in 32D cells is efficiently inhibited by IM (Fig. 2H), indicating that c-Abl may also be involved in IL-3 stimulation of Jak1 activation. In support of this conclusion, Huang et al. (52) reported that Jak1 plays a role in Jak2 activation. Thus, c-Abl may regulate both Jak1 and Jak2 activation upon cytokine stimulation. Although Moresco (53) proposed that Abl is involved in regulating both Jak1 and Jak3 in response to IL-7-driven lymphoid development, we do not have clear data on whether c-Abl is involved in Jak3 activation in myeloid cells.
However, we do not know the mechanism of c-Abl kinase activation by IL-3 in normal hematopoietic cells. It is reported that c-Abl kinase activity is stimulated by PDGF in a manner dependent on Src family kinase and phospholipase C-γ1 (PLC-γ1) (37, 54). Based on this, we hypothesize that other kinases (e.g. Src and Fyn) may be involved in c-Abl activation in response to IL-3, and this c-Abl activation pathway may not be efficiently blocked by IM. We observed that overexpression of c-Abl alone in 32D cells was unable to activate Jak2 and render cells independent of IL-3 (Fig. 4A). However, co-expression of c-Abl and kinase-deficient Bcr-Abl in 32D cells was able to allow IL-3 independent growth (Fig. 4). These results suggest that additional factors, such as Bcr sequences, in particular Tyr-177 in Bcr-Abl as described above (31), may contribute to Jak2 activation in CML cells.
In CML, c-Abl sequences fused with part of Bcr to generate the Bcr-Abl fusion protein exhibit constitutive Abl kinase activity to continuously drive Jak2/Lyn and Stat5 signal pathways. Certain myeloproliferative neoplasms have mutations on Jak2 tyrosine kinase such as V617F resulting in uncontrolled Jak2 activity (17–19). Lu et al. (55) and Pradhan et al. (56) report that co-expression of cognate homodimeric cytokine receptor (e.g. erythropoietin receptor) or heterodimeric cytokine receptor (e.g. IL-27 receptor α) in BaF3 cells is necessary for the continuously activated Jak2V617F mutant and cytokine-independent growth. In addition, expression of either of the two subunits of the IL-3 receptor (IL-3Rα or IL-3Rβ) further enhanced JAK2V617F kinase activity mediated by the homodimeric receptor and its ability to transform hematopoietic cells (56). Our data showed that c-Abl directly binds to Jak2 (Fig. 1A) and that c-Abl binds to the βc of IL-3/Il-5/GM-CSF receptors in an IL-3-independent manner (Fig. 2C). c-Abl regulates Jak2 activity under IL-3 stimulation in normal hematopoietic cells through the direct interaction (Figs. 1A and 2, A–D). Based on the reports cited above (56) and our results, we propose that c-Abl may also play a role in regulating kinase activity of Jak2 mutants (e.g. Jak2V617F) in myeloproliferative neoplasms.
In conclusion, we provide new evidence that c-Abl activates Jak2 in normal hematopoietic cells. Our studies imply a possible role of c-Abl in Jak2 activation in various myeloid malignancies lacking the Philadelphia chromosome.
Acknowledgment
We thank Vicken Frankian for technical assistance.
This work was supported, in whole or in part, by National Institutes of Health Grant CA P01 49639, Project 5, from NCI.
- CML
- chronic myeloid leukemia
- BiFC
- bimolecular fluorescence complementation
- IM
- imatinib mysylate
- Doxy
- doxycycline
- IP
- immunoprecipitation
- WB
- Western blot
- βc
- β chain
- PI
- propidium iodide
- PE
- phycoerythrin.
REFERENCES
- 1. Sandberg E. M., Wallace T. A., Godeny M. D., VonDerLinden D., Sayeski P. P. (2004) Jak2 tyrosine kinase: a true jak of all trades? Cell Biochem. Biophys. 41, 207–232 [DOI] [PubMed] [Google Scholar]
- 2. Reddy E. P., Korapati A., Chaturvedi P., Rane S. (2000) IL-3 signaling and the role of Src kinases, JAKs and STATs: a covert liaison unveiled. Oncogene 19, 2532–2547 [DOI] [PubMed] [Google Scholar]
- 3. Shuai K., Liu B. (2003) Regulation of JAK-STAT signalling in the immune system. Nat. Rev. Immunol. 3, 900–911 [DOI] [PubMed] [Google Scholar]
- 4. Vainchenker W., Constantinescu S. N. (2013) JAK/STAT signaling in hematological malignancies. Oncogene 32, 2601–2613 [DOI] [PubMed] [Google Scholar]
- 5. Herrington J., Carter-Su C. (2001) Signaling pathways activated by the growth hormone receptor. Trends Endocrinol. Metab. 12, 252–257 [DOI] [PubMed] [Google Scholar]
- 6. Neubauer H., Cumano A., Müller M., Wu H., Huffstadt U., Pfeffer K. (1998) Jak2 deficiency defines an essential developmental checkpoint in definitive hematopoiesis. Cell 93, 397–409 [DOI] [PubMed] [Google Scholar]
- 7. Parganas E., Wang D., Stravopodis D., Topham D. J., Marine J. C., Teglund S., Vanin E. F., Bodner S., Colamonici O. R., van Deursen J. M., Grosveld G., Ihle J. N. (1998) Jak2 is essential for signaling through a variety of cytokine receptors. Cell 93, 385–395 [DOI] [PubMed] [Google Scholar]
- 8. Krempler A., Qi Y., Triplett A. A., Zhu J., Rui H., Wagner K. U. (2004) Generation of a conditional knockout allele for the Janus kinase 2 (Jak2) gene in mice. Genesis 40, 52–57 [DOI] [PubMed] [Google Scholar]
- 9. Silvennoinen O., Ungureanu D., Niranjan Y., Hammaren H., Bandaranayake R., Hubbard S. R. (2013) New insights into the structure and function of the pseudokinase domain in JAK2. Biochem. Soc. Trans. 41, 1002–1007 [DOI] [PubMed] [Google Scholar]
- 10. Yoshimura A., Naka T., Kubo M. (2007) SOCS proteins, cytokine signalling and immune regulation. Nat. Rev. Immunol. 7, 454–465 [DOI] [PubMed] [Google Scholar]
- 11. Hansen G., Hercus T. R., McClure B. J., Stomski F. C., Dottore M., Powell J., Ramshaw H., Woodcock J. M., Xu Y., Guthridge M., McKinstry W. J., Lopez A. F., Parker M. W. (2008) The structure of the GM-CSF receptor complex reveals a distinct mode of cytokine receptor activation. Cell 134, 496–507 [DOI] [PubMed] [Google Scholar]
- 12. Dey R., Ji K., Liu Z., Chen L. (2009) A cytokine-cytokine interaction in the assembly of higher-order structure and activation of the interleukine-3:receptor complex. PLoS One 4, e5188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hercus T. R., Thomas D., Guthridge M. A., Ekert P. G., King-Scott J., Parker M. W., Lopez A. F. (2009) The granulocyte-macrophage colony-stimulating factor receptor: linking its structure to cell signaling and its role in disease. Blood 114, 1289–1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lindauer K., Loerting T., Liedl K. R., Kroemer R. T. (2001) Prediction of the structure of human Janus kinase 2 (JAK2) comprising the two carboxy-terminal domains reveals a mechanism for autoregulation. Protein Eng. 14, 27–37 [DOI] [PubMed] [Google Scholar]
- 15. Saharinen P., Vihinen M., Silvennoinen O. (2003) Autoinhibition of Jak2 tyrosine kinase is dependent on specific regions in its pseudokinase domain. Mol. Biol. Cell 14, 1448–1459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Baxter E. J., Scott L. M., Campbell P. J., East C., Fourouclas N., Swanton S., Vassiliou G. S., Bench A. J., Boyd E. M., Curtin N., Scott M. A., Erber W. N., Green A. R. (2005) Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 365, 1054–1061 [DOI] [PubMed] [Google Scholar]
- 17. James C., Ugo V., Le Couédic J. P., Staerk J., Delhommeau F., Lacout C., Garçon L., Raslova H., Berger R., Bennaceur-Griscelli A., Villeval J. L., Constantinescu S. N., Casadevall N., Vainchenker W. (2005) A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 434, 1144–1148 [DOI] [PubMed] [Google Scholar]
- 18. Kralovics R., Passamonti F., Buser A. S., Teo S. S., Tiedt R., Passweg J. R., Tichelli A., Cazzola M., Skoda R. C. (2005) A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 352, 1779–1790 [DOI] [PubMed] [Google Scholar]
- 19. Levine R. L., Wadleigh M., Cools J., Ebert B. L., Wernig G., Huntly B. J., Boggon T. J., Wlodarska I., Clark J. J., Moore S., Adelsperger J., Koo S., Lee J. C., Gabriel S., Mercher T., D'Andrea A., Fröhling S., Döhner K., Marynen P., Vandenberghe P., Mesa R. A., Tefferi A., Griffin J. D., Eck M. J., Sellers W. R., Meyerson M., Golub T. R., Lee S. J., Gilliland D. G. (2005) Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 7, 387–397 [DOI] [PubMed] [Google Scholar]
- 20. Kirk R. (2012) Haematological cancer: Hit the lymphoma, JAK. Nat. Rev. Clin. Oncol. 9, 608. [DOI] [PubMed] [Google Scholar]
- 21. Levine R. L., Pardanani A., Tefferi A., Gilliland D. G. (2007) Role of JAK2 in the pathogenesis and therapy of myeloproliferative disorders. Nat. Rev. Cancer 7, 673–683 [DOI] [PubMed] [Google Scholar]
- 22. Levine R. L., Loriaux M., Huntly B. J., Loh M. L., Beran M., Stoffregen E., Berger R., Clark J. J., Willis S. G., Nguyen K. T., Flores N. J., Estey E., Gattermann N., Armstrong S., Look A. T., Griffin J. D., Bernard O. A., Heinrich M. C., Gilliland D. G., Druker B., Deininger M. W. (2005) The JAK2V617F activating mutation occurs in chronic myelomonocytic leukemia and acute myeloid leukemia, but not in acute lymphoblastic leukemia or chronic lymphocytic leukemia. Blood 106, 3377–3379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mercher T., Wernig G., Moore S. A., Levine R. L., Gu T. L., Fröhling S., Cullen D., Polakiewicz R. D., Bernard O. A., Boggon T. J., Lee B. H., Gilliland D. G. (2006) JAK2T875N is a novel activating mutation that results in myeloproliferative disease with features of megakaryoblastic leukemia in a murine bone marrow transplantation model. Blood 108, 2770–2779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Malinge S., Ben-Abdelali R., Settegrana C., Radford-Weiss I., Debre M., Beldjord K., Macintyre E. A., Villeval J. L., Vainchenker W., Berger R., Bernard O. A., Delabesse E., Penard-Lacronique V. (2007) Novel activating JAK2 mutation in a patient with Down syndrome and B-cell precursor acute lymphoblastic leukemia. Blood 109, 2202–2204 [DOI] [PubMed] [Google Scholar]
- 25. Scott L. M., Tong W., Levine R. L., Scott M. A., Beer P. A., Stratton M. R., Futreal P. A., Erber W. N., McMullin M. F., Harrison C. N., Warren A. J., Gilliland D. G., Lodish H. F., Green A. R. (2007) JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N. Engl. J. Med. 356, 459–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Levine R. L., Gilliland D. G. (2008) Myeloproliferative disorders. Blood 112, 2190–2198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Samanta A. K., Chakraborty S. N., Wang Y., Kantarjian H., Sun X., Hood J., Perrotti D., Arlinghaus R. B. (2009) Jak2 inhibition deactivates Lyn kinase through the SET-PP2A-SHP1 pathway, causing apoptosis in drug-resistant cells from chronic myelogenous leukemia patients. Oncogene 28, 1669–1681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Samanta A. K., Lin H., Sun T., Kantarjian H., Arlinghaus R. B. (2006) Janus kinase 2: a critical target in chronic myelogenous leukemia. Cancer Res. 66, 6468–6472 [DOI] [PubMed] [Google Scholar]
- 29. Xie S., Lin H., Sun T., Arlinghaus R. B. (2002) Jak2 is involved in c-Myc induction by Bcr-Abl. Oncogene 21, 7137–7146 [DOI] [PubMed] [Google Scholar]
- 30. Xie S., Wang Y., Liu J., Sun T., Wilson M. B., Smithgall T. E., Arlinghaus R. B. (2001) Involvement of Jak2 tyrosine phosphorylation in Bcr-Abl transformation. Oncogene 20, 6188–6195 [DOI] [PubMed] [Google Scholar]
- 31. Samanta A., Perazzona B., Chakraborty S., Sun X., Modi H., Bhatia R., Priebe W., Arlinghaus R. (2011) Janus kinase 2 regulates Bcr-Abl signaling in chronic myeloid leukemia. Leukemia 25, 463–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dawson M. A., Bannister A. J., Göttgens B., Foster S. D., Bartke T., Green A. R., Kouzarides T. (2009) JAK2 phosphorylates histone H3Y41 and excludes HP1α from chromatin. Nature 461, 819–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Van Etten R. A. (1999) Cycling, stressed-out and nervous: cellular functions of c-Abl. Trends Cell Biol. 9, 179–186 [DOI] [PubMed] [Google Scholar]
- 34. Lanier L. M., Gertler F. B. (2000) From Abl to actin: Abl tyrosine kinase and associated proteins in growth cone motility. Curr. Opin. Neurobiol. 10, 80–87 [DOI] [PubMed] [Google Scholar]
- 35. Pendergast A. M. (2002) The Abl family kinases: mechanisms of regulation and signaling. Adv. Cancer Res. 85, 51–100 [DOI] [PubMed] [Google Scholar]
- 36. Glover R. T., Angiolieri M., Kelly S., Monaghan D. T., Wang J. Y., Smithgall T. E., Buller A. L. (2000) Interaction of the N-methyl-d-aspartic acid receptor NR2D subunit with the c-Abl tyrosine kinase. J. Biol. Chem. 275, 12725–12729 [DOI] [PubMed] [Google Scholar]
- 37. Lewis J. M., Baskaran R., Taagepera S., Schwartz M. A., Wang J. Y. (1996) Integrin regulation of c-Abl tyrosine kinase activity and cytoplasmic-nuclear transport. Proc. Natl. Acad. Sci. U.S.A. 93, 15174–15179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Plattner R., Kadlec L., DeMali K. A., Kazlauskas A., Pendergast A. M. (1999) c-Abl is activated by growth factors and Src family kinases and has a role in the cellular response to PDGF. Genes Dev. 13, 2400–2411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zipfel P. A., Grove M., Blackburn K., Fujimoto M., Tedder T. F., Pendergast A. M. (2000) The c-Abl tyrosine kinase is regulated downstream of the B cell antigen receptor and interacts with CD19. J. Immunol. 165, 6872–6879 [DOI] [PubMed] [Google Scholar]
- 40. Greuber E. K., Smith-Pearson P., Wang J., Pendergast A. M. (2013) Role of ABL family kinases in cancer: from leukaemia to solid tumours. Nat. Rev. Cancer 13, 559–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pardanani A., Hood J., Lasho T., Levine R. L., Martin M. B., Noronha G., Finke C., Mak C. C., Mesa R., Zhu H., Soll R., Gilliland D. G., Tefferi A. (2007) TG101209, a small molecule JAK2-selective kinase inhibitor potently inhibits myeloproliferative disorder-associated JAK2V617F and MPLW515L/K mutations. Leukemia 21, 1658–1668 [DOI] [PubMed] [Google Scholar]
- 42. Zhao H., Chen M. S., Lo Y. H., Waltz S. E., Wang J., Ho P. C., Vasiliauskas J., Plattner R., Wang Y. L., Wang S. C. (2013) The Ron receptor tyrosine kinase activates c-Abl to promote cell proliferation through tyrosine phosphorylation of PCNA in breast cancer. Oncogene 33, 1429–1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chakraborty S., Lin Y. H., Leng X., Miranda R. N., Medeiros L. J., Shpall E., Arlinghaus R. B. (2013) Activation of Jak2 in patients with blast crisis chronic myelogenous leukemia: inhibition of Jak2 inactivates Lyn kinase. Blood Cancer J. 3, e142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nyfeler B., Michnick S. W., Hauri H. P. (2005) Capturing protein interactions in the secretory pathway of living cells. Proc. Natl. Acad. Sci. U.S.A. 102, 6350–6355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ling X., Ma G., Sun T., Liu J., Arlinghaus R. B. (2003) Bcr and Abl interaction: oncogenic activation of c-Abl by sequestering Bcr. Cancer Res. 63, 298–303 [PubMed] [Google Scholar]
- 46. Tao W. J., Lin H., Sun T., Samanta A. K., Arlinghaus R. (2008) BCR-ABL oncogenic transformation of NIH 3T3 fibroblasts requires the IL-3 receptor. Oncogene 27, 3194–3200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Quelle F. W., Sato N., Witthuhn B. A., Inhorn R. C., Eder M., Miyajima A., Griffin J. D., Ihle J. N. (1994) JAK2 associates with the β c chain of the receptor for granulocyte-macrophage colony-stimulating factor, and its activation requires the membrane-proximal region. Mol. Cell. Biol. 14, 4335–4341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Witthuhn B. A., Quelle F. W., Silvennoinen O., Yi T., Tang B., Miura O., Ihle J. N. (1993) JAK2 associates with the erythropoietin receptor and is tyrosine phosphorylated and activated following stimulation with erythropoietin. Cell 74, 227–236 [DOI] [PubMed] [Google Scholar]
- 49. Silvennoinen O., Witthuhn B. A., Quelle F. W., Cleveland J. L., Yi T., Ihle J. N. (1993) Structure of the murine Jak2 protein-tyrosine kinase and its role in interleukin 3 signal transduction. Proc. Natl. Acad. Sci. U.S.A. 90, 8429–8433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Argetsinger L. S., Campbell G. S., Yang X., Witthuhn B. A., Silvennoinen O., Ihle J. N., Carter-Su C. (1993) Identification of JAK2 as a growth hormone receptor-associated tyrosine kinase. Cell 74, 237–244 [DOI] [PubMed] [Google Scholar]
- 51. Campbell G. S., Argetsinger L. S., Ihle J. N., Kelly P. A., Rillema J. A., Carter-Su C. (1994) Activation of JAK2 tyrosine kinase by prolactin receptors in Nb2 cells and mouse mammary gland explants. Proc. Natl. Acad. Sci. U.S.A. 91, 5232–5236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Huang H. M., Lin Y. L., Chen C. H., Chang T. W. (2005) Simultaneous activation of JAK1 and JAK2 confers IL-3 independent growth on Ba/F3 pro-B cells. J. Cell Biochem. 96, 361–375 [DOI] [PubMed] [Google Scholar]
- 53. Moresco E. M. (2006) in Abl Family Kinases in Development and Disease (Koleske A. J., ed) pp. 1–120, Landes Bioscience, Austin, TX [Google Scholar]
- 54. Plattner R., Koleske A. J., Kazlauskas A., Pendergast A. M. (2004) Bidirectional signaling links the Abelson kinases to the platelet-derived growth factor receptor. Mol. Cell. Biol. 24, 2573–2583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lu X., Huang L. J., Lodish H. F. (2008) Dimerization by a cytokine receptor is necessary for constitutive activation of JAK2V617F. J. Biol. Chem. 283, 5258–5266 [DOI] [PubMed] [Google Scholar]
- 56. Pradhan A., Lambert Q. T., Griner L. N., Reuther G. W. (2010) Activation of JAK2-V617F by components of heterodimeric cytokine receptors. J. Biol. Chem. 285, 16651–16663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lin J., Sun T., Ji L., Deng W., Roth J., Minna J., Arlinghaus R. (2007) Oncogenic activation of c-Abl in non-small cell lung cancer cells lacking FUS1 expression: inhibition of c-Abl by the tumor suppressor gene product Fus1. Oncogene 26, 6989–6996 [DOI] [PMC free article] [PubMed] [Google Scholar]