

Background: Tiopronin is a small molecule that selectively kills multidrug-resistant cancer cells.
Results: Inhibition of glutathione peroxidase by tiopronin leads to elevated reactive oxygen species in MDR cells.
Conclusion: Tiopronin mediates the killing of MDR cells by inhibition of glutathione peroxidases.
Significance: Glutathione peroxidase inhibition may be a viable strategy for modulation of multidrug resistance in cancer.
Keywords: Cell Death, Chemoresistance, Enzyme Inhibitor, Oxygen Radicals, Reactive Oxygen Species (ROS), Glutathione Peroxidase
Abstract
Multidrug resistance (MDR) is a major obstacle to the successful chemotherapy of cancer. MDR is often the result of overexpression of ATP-binding cassette transporters following chemotherapy. A common ATP-binding cassette transporter that is overexpressed in MDR cancer cells is P-glycoprotein, which actively effluxes drugs against a concentration gradient, producing an MDR phenotype. Collateral sensitivity (CS), a phenomenon of drug hypersensitivity, is defined as the ability of certain compounds to selectively target MDR cells, but not the drug-sensitive parent cells from which they were derived. The drug tiopronin has been previously shown to elicit CS. However, unlike other CS agents, the mechanism of action was not dependent on the expression of P-glycoprotein in MDR cells. We have determined that the CS activity of tiopronin is mediated by the generation of reactive oxygen species (ROS) and that CS can be reversed by a variety of ROS-scavenging compounds. Specifically, selective toxicity of tiopronin toward MDR cells is achieved by inhibition of glutathione peroxidase (GPx), and the mode of inhibition of GPx1 by tiopronin is shown in this report. Why MDR cells are particularly sensitive to ROS is discussed, as is the difficulty in exploiting this hypersensitivity to tiopronin in the clinic.
Introduction
Chemotherapy remains the main treatment option available for combating hematologic and disseminated malignancies. However, the development of resistance to chemotherapeutics presents a significant challenge to current cancer treatment, drug design, and drug development strategies (1). Cellular alterations acquired during the development of multidrug resistance (MDR)2 include expression of the ATP-binding cassette family of efflux transporters including ABCB1 (also termed P-glycoprotein (P-gp)) and a range of alterations that arise as a result of adaptation to individual drugs in pathways involved in DNA repair, death signaling, drug detoxification, lysosomal changes, and acquisition of point mutations in drug targets (2). Given this broad range of alterations, there is an unmet need for therapeutic strategies that are active against drug-resistant malignancies (3).
One approach is to exploit resistance by identifying compounds that are selectively cytotoxic to MDR cells (4). Using a model tissue culture system, the selectivity and potency of compounds can be assessed by determining the cytotoxicity (IC50) of a compound against a parental, drug-sensitive cell line relative to its MDR subline (5). This activity profile, termed “collateral sensitivity” (or MDR selectivity) (6), has been demonstrated using compounds such as NSC73306 (7) and verapamil, which selectively kill P-gp-expressing cells and reduce P-gp expression at concentrations that are subtoxic to parental cells (8). The phenomenon is essentially a form of synthetic lethality wherein the acquisition of an MDR phenotype is accompanied by secondary exploitable genetic alterations (9). Expression of drug transporters is not the only mechanism that results in collateral sensitivity. For example, the natural product austocystin D is more toxic to MDR cell lines because up-regulation of cytochrome P450s in MDR cells leads to increased activation of the drug (10), and cisplatin-resistant cells with down-regulated topoisomerase I expression show hypersensitivity to topoisomerase II inhibitors (11). These observations suggest that a range of known molecular alterations in MDR cells can be selectively targeted. However, the mechanism of action of P-gp-specific collateral sensitivity remains to be determined (12).
We recently reported the finding that some MDR cell lines were collaterally sensitive to the simple thiol-substituted N-propanoyl form of glycine called tiopronin (N-(3-mercaptopropanoyl) glycine; Fig. 1) (13). Tiopronin is a Food and Drug Administration-registered orphan drug that has been used for over 30 years to treat a diverse range of pathophysiological conditions (14). Although tiopronin showed MDR-selective activity against some P-gp and multidrug resistance protein 1 (MRP1)-expressing cells, it did not kill all P-gp-expressing cell lines tested (both drug-selected or transfected). Also, inhibition of P-gp did not abrogate selective killing, indicating that tiopronin selectivity was not dependent on P-gp function. A range of other thiol compounds tested did not possess the same activity profile but structural analogs of tiopronin generated by varying the amino acid side chain retained activity. The basis of the MDR-selective activity of tiopronin remained to be identified (13).
FIGURE 1.
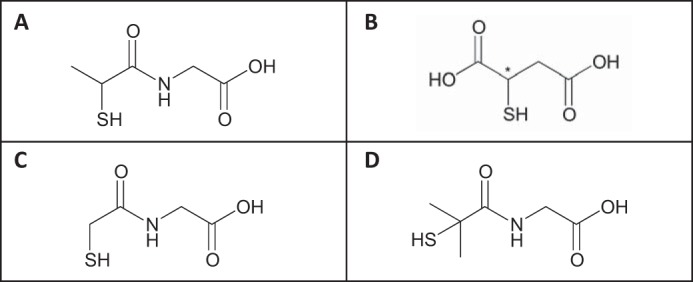
Chemical structures of principal compounds examined. A, tiopronin ((2-mercaptopropanoyl)aminoacetic acid). Tiopronin is the condensation product of glycine and thiolactic acid. B, mercaptosuccinic acid (thiomalic acid, 2-sulfanylbutanedioic acid). C, demethyl tiopronin ((2-mercaptoethanoyl)aminoacetic acid). D, dimethyl tiopronin ((2-mercapto-2-methylpropanoyl)aminoacetic acid). Tiopronin and mercaptosuccinic acid were tested as the racemate.
Tiopronin is believed to exert biological activity via a number of modalities including redox coupling (cystinurea treatment), reactive oxygen species (ROS) scavenging, and chelation, largely mediated by its thiol group (13). Tiopronin has also been reported by Chaudiere et al. (15) to inhibit members of the glutathione peroxidase (GPx) family. GPx 1–4 are a family of selenocysteine-containing enzymes that catalytically degrade H2O2 and organic hydroperoxides (16). Because tiopronin is a GPx inhibitor and given the view that ROS might play a role in the mechanism of action of other CS agents (4, 17), we hypothesized that some MDR cells may be collaterally sensitive to tiopronin via GPx inhibition, leading to ROS generation.
To examine this hypothesis, we assessed several tipronin analogs as GPx inhibitors and have shown selective toxicity toward MDR KB-V1 adenocarcinoma cells compared with the parent (drug-sensitive) KB-3-1 cell line. Mass spectrometry was used to identify the mode of binding to GPx of tiopronin and another inhibitor, mercaptosuccinate, to GPx. Knockdown of GPx isoforms using specific shRNA also reduced tiopronin toxicity against MDR cells. To confirm that CS was ROS-mediated, we examined the effect of a series of ROS scavengers (N-acetylcysteine, catalase, and pyruvate) on the CS activity of tiopronin to determine whether these reversed toxicity. The generation of ROS in cells in the presence of tiopronin was examined by confocal microscopy (CellROX) and a quantitative plate-based assay (dihydrofluorescein diacetate) using dyes that fluoresce upon oxidation.
EXPERIMENTAL PROCEDURES
Chemicals
Tiopronin, N-acetylcysteine (NAC), mercaptosuccinic acid, dimercaptosuccinic acid, catalase, glucose oxidase, H2O2, dicumyl peroxide, luperox, and dimethyl sulfoxide were purchased from Sigma-Aldrich; CellROX Deep Red reagent, Hoechst 33342, and dihydrofluorescein diacetate (DHFDA) were purchased from Invitrogen. All other reagents were purchased from Sigma-Aldrich unless otherwise stated.
Cell Lines
The cell lines used were the human cervical epithelial adenocarcinoma cell line KB-3-1 (a HeLa derivative) and its P-gp-expressing MDR subline KB-V1. All cell lines were grown at 37 °C in 5% CO2. The cell lines were cultured in DMEM supplemented with 10% fetal bovine serum, 5 mm l-glutamine, 50 units/ml penicillin, and 50 μg/ml streptomycin, all obtained from Invitrogen. KB-V1 cells were additionally cultured in vinblastine (1 μg/ml) (18, 19).
Stable Knockdown Cells
To stably knock down expression of GPx1, a pLKO.1-puro plasmid-based shRNA targeting the sequence GCAAGGTACTACTTATCGAGA (clone ID TRCN0000046228; Sigma-Aldrich) was employed (GPx1-shRNA). Similarly, to stably knock down expression of GPx4, a pLKO.1-neo plasmid-based shRNA targeting the sequence GTGAGGCAAGACCGAAGTAAA (clone ID TRCN0000046249; Sigma-Aldrich) was employed (GPx4-shRNA). Additionally, a nontargeting shRNA vector (NT-shRNA) that targets no known human sequence was utilized as a control. A primer containing the target sequence (CTGGTTACGAAGCGAATCCTT) along with a stem loop followed by the reverse target sequence was annealed to a complementary primer and inserted into the EcoRI and AgeI sites of the pLKO.1-puro vector (Addgene number 10878). Lentiviral particles were produced via Lipofectamine 2000 (Invitrogen)-mediated triple transfection of 293T cells with either the GPx1-shRNA, GPx4-shRNA, or the NT-shRNA along with the lentiviral envelope plasmid (pMD2.G, Addgene number 12259) and the lentiviral packaging plasmid (psPAX2, Addgene number 12260). Target cells (KB-3-1 or KB-V1) were transduced with either NT-shRNA, GPx1-shRNA, and/or GPx4-shRNA containing lentiviral particles in the presence of 8 μg/ml Polybrene, and stable cells were selected using either puromycin or G418 antibiotics. For puromycin selection in KB-3-1 cells, 5 μg/ml puromycin was used, and for selection in KB-V1 cells, 10 μg/ml puromycin, along with 10 μm cyclosporine A (as puromycin is a P-gp substrate), was employed. For neomycin selection, 500 μg/ml G418 was used for both cell lines.
Cytotoxicity Assays
Cytotoxicity was measured with Cell TiterGlo luminescence-based viability assay (Promega). Cells (4,000 cells/well on opaque 96-well plates) were allowed to attach for 24 h. Stock solutions of compounds were prepared and then serially diluted in medium to give a range of final tissue culture concentrations. After 72 h, medium on cells was replaced with drug-free medium, and cell viability was examined as described by the manufacturer. Cytotoxicity (IC50) was defined as the drug concentration that reduced cell viability to 50% of the untreated control. Selectivity ratios (SRs) are also reported for each cell line pair, determined by dividing the IC50 of the parental cell line by that of the transporter-expressing cell line. An SR value of >1 indicates that the MDR cell population is collaterally sensitive to the tested drug, whereas an SR of <1 indicates that the MDR cells are resistant to the drug relative to the parental cell line (4). Results were confirmed using a colorimetric cell viability assay incorporating 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (Molecular Probes, Eugene, OR), as previously described (20). The data are expressed as means ± S.D. from at least nine observations for cytotoxicity assays.
Anoxic Cytotoxicity Assay
The cells were plated and allowed to attach for 24 h as described above. Stock solution of tiopronin was serially diluted in medium and added to the plates in a CO2 environment. The plates were then immediately placed in a modular incubator chamber (Billups-Rothenberg, San Diego, CA) in a CO2 atmosphere. The container was then incubated for 72 h, and the assay was carried out as described above.
Confocal Microscopy
To gain visual confirmation that tiopronin increases ROS in the MDR KB-V1 cell line more so than in the parental KB-3-1 cell line, CellROX Deep Red reagent, a dye that fluoresces when oxidized by ROS, was added to the cells and observed by confocal microscopy. The cells were seeded on an eight-chambered #1.0 borosilicate coverglass system (Nalge Nunc International, Penfield, NY) at 2.0 × 105 cells/well and incubated 24 h at 37 °C for attachment. Cells were treated with tiopronin (1 mm) for 1 or 24 h. As positive controls, cells were treated with H2O2 (200 μm) for 1 h. For negative controls, cells were left untreated (i.e. dye only). All cells received Hoechst 33342 (5 μg/ml) and CellROX Deep Red reagent (5 μm).
Plate-based ROS Assay
ROS were measured using DHFDA. DHFDA (100 mm in DMSO) was prepared fresh and then diluted 1:1,000 in Iscove's modified Dulbecco's medium. The cells were plated at a concentration of 1.0 × 105 cells/well on a black 96-well cell culture plate. The cells were incubated in tiopronin for either 24 h or 1 h then washed in PBS and incubated in DHFDA (100 μm) for 10 min (excitation = 485 nm; emission = 538 nm). To examine whether cells were able to consume ROS after incubation with tiopronin, cells were incubated with tiopronin for 24 h, and then H2O2 (1.5 mm) was added for 30 min and treated as described above.
Mass Spectrometry
The molecular weights of species were determined by MALDI-TOF MS on a MALDI micro MX (Waters, Milford, MA). Compounds were diluted 1:1 in α-cyano-4-hydroxycinnamic acid and analyzed using reflectron mode. To verify that GPx becomes bound to tiopronin and mercaptosuccinate, three samples were prepared for mass spectrometry. Purified GPx (77 nm) dissolved in Tris buffer, pH 8 (20 mm), was diluted 1:1 in Tris buffer, pH 8 (20 mm), alone or containing tiopronin (260 μm) or mercaptosuccinate (260 μm). GPx was digested in solution with trypsin at 37 °C for 16 h, and the resultant peptides were desalted by C18 ZipTip (Millipore). They were analyzed by MALDI-TOF MS and tandem MS on an LTQ-XL mass spectrometer (Thermo, Waltham, MA).
GPx Assay
The assay was performed as described in the manufacturer's instructions (glutathione peroxidase assay kit; Cayman Chemical Company, Ann Arbor, MI). Briefly, wells containing bovine erythrocyte GPx were incubated alone or with either cell lysate or in the presence of tiopronin, mercaptosuccinate, dimethyl tiopronin, or demethyl tiopronin. The assay was initiated by addition of the GPx substrate cumene hydroperoxide, and the loss of absorbance at 340 nm (corresponding to the oxidation NADPH to NADP+) was measured each minute for 10 min. The GPx rate was determined.
Synthesis of Tiopronin Analogs
All commercially available organic precursors and dry solvents were obtained from Sigma-Aldrich and used as received unless otherwise noted. The amino acid methyl ester hydrochlorides were obtained from Bachem (Torrance, CA) and used as received unless otherwise noted. Reactions were magnetically stirred under an argon atmosphere and monitored by TLC with 0.25 mm Sigma-Aldrich precoated aluminum-backed silica gel plates with fluorescent indicator. TLC visualization was achieved using 254- or 360-nm UV lamp detection and/or staining with cerium molybdate (Hannesian's stain), phosphomolybdic acid, or potassium permanganate. Flash column chromatography was performed on an Ana-Logix IntelliFlash 280 system, using Biotage® SNAP cartridges and SNAP sampler cartridges with KP-Silica 60 μm. Analytical HPLC analyses were performed on an Agilent 1200 Series instrument equipped with multiwavelength detectors using a Zorbax Stable Bond C-18 column (4.6 × 50 mm, 3.5 μm) with a flow rate of 0.5 ml/min or 1.0 ml/min. Solvent A was 0.05% TFA in H2O, solvent B was 0.05% TFA in acetonitrile, and a linear gradient of 5% B to 95% B over 10 min was used. Electrospray ionization or atmospheric pressure chemical ionization MS was performed on an LC/MSD TrapXCl Agilent Technologies instrument or on a 6130 Quadrupole LC/MS Agilent Technologies instrument equipped with a diode array detector. Microwave (μλ) irradiation was carried out in a CEM Discover Synthesizer with 150 watts max power. 1H and 13C NMR spectra were recorded on a Varian spectrometer operating at 400 and 100 MHz, respectively. Chemical shifts are reported relative to either chloroform (δ 7.26), dichloromethane (δ 5.32), or deuterium oxide (δ 4.79) for 1H NMR and chloroform (δ 77.0) or dichloromethane (δ 54.0) for 13C NMR. Synthetic schemes for demethyl tiopronin and dimethyl tiopronin are shown in Figs. 3 and 4.
FIGURE 3.
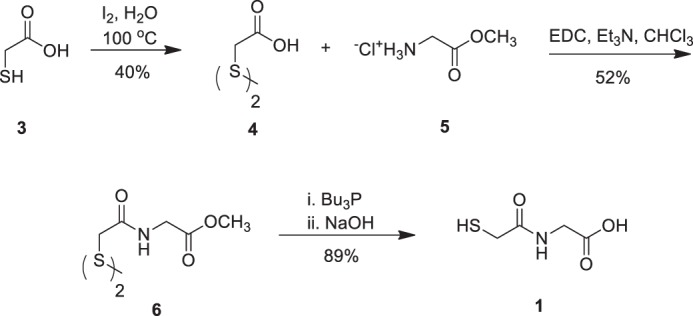
The synthesis of demethyl tiopronin analog 1 is displayed in this scheme. Commercially available thiolglycolic acid (3) was oxidized to disulfide (4) in 40% yield with iodine. The bisacid disulfide (4) was then coupled with glycine methyl ester hydrochloride (5) via N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC) activation of (4) to afford bisamide disulfide (6) in 52% yield. Disulfide reduction mediated by Bu3P and methyl ester saponification with 2 m NaOH of (6) was carried out in a one-pot operation to afford the target demethyl tiopronin analog (1) in 89% yield.
FIGURE 4.
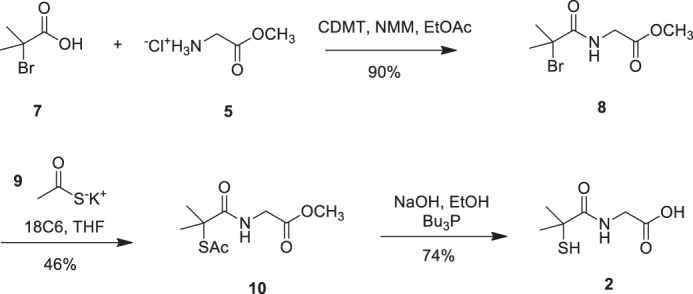
The route utilized to obtain dimethyl tiopronin analog 2 is shown. Commencing from commercially available 2-bromo-2-methylpropionic acid (7), 2-chloro-4,6-dimethoxy-1,3,5-triazine (CDMT)-mediated coupling with glycine methyl ester hydrochloride (5) afforded bromoamide 8 in 90% yield, which was then reacted with thioacetate salt (9) to give (10) in 46% yield. Compound (10) was subjected to saponification with 2 n NaOH in the presence of tributylphosphine to afford dimethyl tiopronin analog (2) in 74% yield.
Dithiodiethanoic Acid (4) (C4H6O4S2, mw = 182.22 gmol−1)
Iodine (4.00 g, 15.9 mmol) was added in portions to a solution of thioglycolic acid (3) (2.66 g, 28.9 mmol) in H2O (10 ml). The resulting reaction mixture was heated to 100 °C for 2 h. The reaction mixture was allowed to cool to room temperature and quenched by addition of a saturated aqueous solution of Na2S2O3. It was then filtered through celite, and the filtrate was extracted with ethyl acetate twice. The combined organic layers were washed with saturated Na2S2O3 (twice), dried over magnesium sulfate (MgSO4), and concentrated. The residue was triturated from toluene to afford (4) (1.04 g, 40% yield) as white crystals (1H NMR (400 MHz, CD3OD): δ 3.61 (s, 4H); mp 130–132 °C (literature value 132 °C) (21)).
Bisamide Disulfide (6) C10H16N2O6S2, mw = 324.37 gmol−1)
A solution of N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (1.05 g, 5.49 mmol) in CHCl3 (5 ml) was added slowly to a mixture of bisacid disulfide 4 (400 mg, 2.19 mmol), glycine derivative (5) (606 mg, 4.83 mmol), and triethylamine (489 mg, 4.83 mmol) in CHCl3 at 0 °C. The resulting reaction mixture was allowed to stir for 4 h, as the temperature rose to room temperature. TLC (EtOAc) showed reaction completion. The reaction mixture was filtered and concentrated, and the residue was purified by flash column chromatography using silica gel and EtOAc to afford 6 (402 mg, 52% yield) as a white solid (1H NMR (400 MHz, CDCl3): δ 7.40 (bs, 1H), 4.10 (d, J = 5.6 Hz, 2H), 3.76 (s, 3H), 3.57 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 170.8, 168.9, 52.5, 42.5, 41.5. mercaptosuccinate (m/z) = 325.0 (M+1)+).
Demethyl Tiopronin (1) (C4H7NO3S, mw = 149.17 gmol−1)
A solution of the bisamide disulfide (4) (400 mg, 1.23 mmol) in 20% H2O in tetrahydrofuran (v/v, 10 ml) was thoroughly degassed by bubbling argon through the solution for 5–10 min. Bu3P (748 mg, 3.70 mmol) was added slowly, and the resulting reaction mixture was allowed to stir at room temperature for 5 min and monitored by TLC (EtOAc). The reaction mixture was diluted with 4 ml EtOH, 2 m NaOH was added (3.1 ml), and stirring continued for 1 h. The reaction mixture was diluted with EtOH and concentrated under reduced pressure. The residue was taken up in H2O and extracted twice with EtOAc, and the organic layers were discarded. The aqueous layer was treated with Dowex 50W-X8 acid exchange resin for 5 min or until pH 4–5 was reached. The resin was filtered, and the aqueous filtrate was lyophilized to afford demethyl tiopronin analog 1 as a white waxy solid (327 mg, 89% yield; 1H NMR (400 MHz, D2O): δ 4.04 (s, 2H), 3.34 (s, 2H). 13C NMR (100 MHz, D2O): δ 174.1, 173.3, 41.4, 26.9. mercaptosuccinate (m/z) = 150.1 (M+1)+).
Bromoamide (8) (C7H12BrNO3, mw = 238.08 gmol−1)
A solution of N-methylmorpholine (1.06 g, 10.5 mmol) in EtOAc (5 ml), was added slowly to a 0 °C mixture of the 2-bromo-2-methylpropionic acid 7 (500 mg, 2.99 mmol), glycine methyl ester hydrochloride (5) (451 mg, 3.59 mmol), and 2-chloro-4,6-dimethoxy-1,3,5-triazine (631 mg, 3.59 mmol) in EtOAc (30 ml). The resulting reaction mixture was allowed to stir at 0 °C for 5 min at room temperature for 2 h. TLC (1:1 hexane:EtOAc) showed completion. The reaction mixture was partitioned between EtOAc and H2O, and the organic layer was separated and washed with 1 m HCl (twice), dried over MgSO4, and concentrated. Crude (8) (647 mg, 90% yield) was used without further purification (1H NMR (400 MHz, CDCl3): δ 7.18 (bs, 1H), 4.05 (d, J = 4.8 Hz, 2H), 3.78 (s, 3H), 1.97 (s, 6H). 13C NMR (100 MHz, CDCl3): δ 172.3, 169.9, 61.8, 52.4, 42.0, 32.4. mercaptosuccinate (m/z) = 239.7 (M+1)+).
Thioacetate (10) C9H15NO4S, mw = 233.28 gmol−1)
Potassium thioacetate (672 mg, 5.88 mmol) and 18-crown-6 (155 mg, 0.59 mmol) were added in one portion to a solution of bromoamide (8) (700 mg, 2.94 mmol) in tetrahydrofuran (20 ml) mixed with 4 Å molecular sieves (∼1 g). The resulting reaction mixture was heated to reflux for 2 h. TLC (40% EtOAc in hexanes) still showed the presence of (8). Another portion of potassium thioacetate was added (672 mg, 5.88 mmol), and the reaction was heated for a further 2 h. The reaction mixture was allowed to cool to room temperature and filtered through celite, and the filtrate was taken up in EtOAc, washed with 1 n HCl (twice), dried over MgSO4, and concentrated. The residue was purified by flash column chromatography with silica gel and 35% EtOAc in hexane to afford thioacetate (10) (493 mg, 46% yield) as a tan oil (1H NMR (400 MHz, CDCl3): δ 7.15 (bs, 1H), 4.03 (d, J = 4.8 Hz, 2H), 3.76 (s, 3H), 2.32 (s, 3H), 1.62 (s, 6H). 13C NMR (100 MHz, CDCl3): δ 195.6, 173.5, 170.3, 52.3, 52.1, 41.7, 30.6, 25.8. mercaptosuccinate (m/z) = 234.1 (M+1)+).
Dimethyl Tiopronin (2) C6H11NO3S, mw = 177.22 gmol−1)
A solution of thioacetate 10 (438 mg, 1.88 mmol) in EtOH (15 ml) was degassed by bubbling argon for 5 min. Bu3P (380 mg, 1.88 mmol) was added, followed by 2 m NaOH (4.7 ml), and the resulting reaction mixture was allowed to stir for 45 min. The reaction mixture was concentrated. The residue was taken up in H2O and extracted twice with EtOAc, and the organic layers were discarded. The aqueous layer was treated with Dowex acid exchange resin for 5 min or until pH 4–5 was reached. The resin was filtered, and the aqueous filtrate was lyophilized to afford dimethyl tiopronin analog (2) as an off-white solid (247 mg, 74% yield; 1H NMR (400 MHz, D2O): δ 3.97 (s, 2H), 1.61 (s, 6H). 13C NMR (100 MHz, D2O): δ 178.8, 173.9, 45.9, 42.0, 28.8. mercaptosuccinate (m/z) = 177.7 (M+1)+).
RESULTS
Glutathione Peroxidase Inhibitors Elicit CS
The cytotoxicity of tiopronin (Fig. 2A) was confirmed with parental KB-3-1 human adenocarcinoma cells and its MDR subline expressing P-gp, developed during long term selection in vinblastine (1 μg/ml) (19). Rather than demonstrating cross-resistance, tiopronin was found to selectively kill MDR KB-V1 cells, a mode of biological activity termed collateral sensitivity. The degree of collateral sensitivity is defined by a SR, which is calculated as the ratio of the IC50 of a compound for parental cells divided by its IC50 for MDR cells (in this case ((KB-3-1 IC50)/(KB-V1 IC50)). A SR value > 1 indicates that the compound kills MDR cells more effectively than parental cells (4). In the case of tiopronin, cytotoxicity against parental KB-3-1 cells (IC50 = 3.90 ± 1.1 mm) is lower than against KB-V1 cells (IC50 = 0.143 ± 0.007 mm) with a SR of 27 (Fig. 2A), similar to our previously reported values and with a slight biphasic response in the MDR cells (13). Although the inherent toxicity of tiopronin is low, we have previously demonstrated that acidity of the carboxylic acid functional group does not contribute to the selective toxicity (13).
FIGURE 2.

Collateral sensitivity of KB-V1 cells to inhibitors of GPx, assessed by cytotoxicity assay. A and B, dose-response curves demonstrating the effect of tiopronin (A) and mercaptosuccinate (B) on KB-3-1 and KB-V1 cells treated with tiopronin for 72 h. C, dose-response curves demonstrating the effect of (nontoxic) concentrations of N-acetylcysteine (2 and 5 mm) on the cytotoxicity of mercaptosuccinate toward KB-V1 and KB-3-1 cells. D–F, dose-response curves demonstrating the effects of dimercaptosuccinate (D), demethyl tiopronin (E), and dimethyl tiopronin (F) on KB-V1 and KB-3-1 cells. G, inhibition of bovine erythrocyte GPx activity by mercaptosuccinate, tiopronin, demethyl tiopronin, and dimethyl tiopronin (200 μm) compared with control (DMSO only) conditions.
Tiopronin has been reported by Chaudiere et al. (15) to inhibit members of the glutathione peroxidase family (GPx) at a concentration of 200 μm, resulting in 61% inhibition of purified GPx. Human GPx1–4 are selenocysteine-containing enzymes that catalytically degrade H2O2 and (unlike catalase) organic hydroperoxides such as tert-butyl hydroperoxide (16). This is achieved by utilizing glutathione as a co-factor and electron source to reduce H2O2 to water (and organoperoxides to alcohols).
To examine whether the CS activity of tiopronin was related to the inhibition of GPx, we examined mercaptosuccinic acid (thiomalic acid; Fig. 1B) and 2,3-dimercaptosuccinic acid, identified by Chaudiere et al. (15) as inhibitors of GPx. Mercaptosuccinate demonstrated selective killing of KB-V1 cells (IC50 = 0.14 ± 0.01 mm) compared with KB-3-1 cells (IC50 = 6.15 ± 0.52 mm), with a SR of 45 (Fig. 2B and Table 1). These values are similar to those of tiopronin (above); 2,3-dimercaptosuccinate (Fig. 2D) also demonstrated MDR-selective killing (KB-V1 IC50 = 0.049 ± 0.001 mm, KB-3-1 IC50 = 0.97 ± 0.10 mm, SR = 20).
TABLE 1.
Determination of cytotoxicity (IC50, mm) of tiopronin and collateral sensitivity (SR) of parental and MDR sublines (indented)
SR was calculated as IC50 parental/IC50 MDR cell line.
| Cytotoxicity (IC50) |
SR | ||
|---|---|---|---|
| KB-3-1 | KB-V1 | ||
| mm | |||
| Tiopronin | 3.9 ± 1.1 | 0.1 ± 0.01 | 28 |
| Tiopronin + N-acetylcysteine (1 mm) | 2.4 ± 0.8 | 0.4 ± 0.1 | 6.6 |
| Tiopronin + N-acetylcysteine (2 mm) | 2.6 ± 1.4 | 0.5 ± 0.05 | 5.1 |
| Tiopronin + N-acetylcysteine (5 mm) | 2.5 ± 1.1 | 1.0 ± 0.2 | 2.5 |
| Tiopronin + anoxia | 3.9 ± 1.3 | 2.3 ± 0.9 | 1.4 |
| Tiopronin + catalase (1,000 units/ml) | 16.5 ± 0.9 | 7.4 ± 0.8 | 2.2 |
| Tiopronin + catalase (inactivated) | 14.7 ± 4.3 | 0.3 ± 0.3 | 49 |
| Tiopronin + RBC lysate | 4.1 ± 0.1 | 3.2 ± 0.3 | 1.3 |
| Tiopronin + RBC lysate (inactivated) | 9.4 ± 3.7 | 0.5 ± 0.02 | 20.1 |
| Mercaptosuccinate | 6.2 ± 0.5 | 0.1 ± 0.01 | 45 |
| Mercaptosuccinate + N-acetylcysteine (2 mm) | 6.0 ± 0.9 | 5.3 ± 1.9 | 1.1 |
| Mercaptosuccinate + N-acetylcysteine (5 mm) | 3.5 ± 0.3 | 6.9 ± 0.1 | 2.0 |
| Dimercaptosuccinate | 1.0 ± 0.10 | 0.05 ± 0.001 | 19.8 |
| Desmethyl tiopronin | 18.4 ± 0.2 | 0.4 ± 0.1 | 43.7 |
| Dimethyl tiopronin | 15.1 ± 0.2 | 9.6 ± 0.7 | 1.6 |
| H2O2 | 0.10 ± 0.02 | 0.1 ± 0.01 | 0.9 |
| H2O2 + pyruvate | 0.46 ± 0.06 | 0.3 ± 0.06 | 1.4 |
| NSC73306 | 0.018 ± 0.001 | 0.003 ± 0.0004 | 6.6 |
| NSC73306 + N-acetylcysteine (4 mm) | 0.015 ± 0.007 | 0.003 ± 0.0003 | 5.8 |
| NSC73306 + N-acetylcysteine (10 mm) | 0.0117 ± 0.003 | 0.0020 ± 0.0004 | 5.9 |
| NSC73306 + anoxia | 0.0159 ± 0.006 | 0.0027 ± 0.0004 | 10.4 |
| NSC73306 + catalase (1,000 units/ml) | >0.050 | >0.050 | |
In humans, GPx1–4 proteins contain the unusual amino acid selenocysteine (Sec or U), and although there are no high affinity inhibitors of GPx and little mechanistic understanding of how existing inhibitors work, the prevailing view appears to be that mercaptocarboxylic acids interact via their thiol directly with the GPx selenol group (16), forming a thioselenate adduct in the active site. To explore the relationship between GPx inhibition and selective cytotoxicity toward KB-V1 cells, we generated two analogs of tiopronin modified at the carbon alpha to the thiol group (Fig. 1, C and D; synthetic schemes shown in Figs. 3 and 4 and characterization outlined under “Experimental Procedures”) (21–23). The first, a demethyl analog of tiopronin (termed demethyl tiopronin) was synthesized without the methyl substituent alpha to the thiol (Fig. 1C), removing steric bulk making the thiol more accessible. The second, a dimethyl analog of tiopronin (termed dimethyl tiopronin), was synthesized with a second methyl substituent alpha to the thiol (Fig. 1D), adding steric bulk making the thiol less accessible. To assess whether these structure-activity relationships affected GPx inhibitory activity, we tested mercaptosuccinate, tiopronin, demethyl tiopronin, and dimethyl tiopronin (all at 200 μm) in a bovine GPx activity assay (Cayman Chemical) (Fig. 2G). Mercaptosuccinate (71 ± 4%, compared with control activity of 100%) and tiopronin (60 ± 3%) both inhibited GPx when compared with control, as previously reported (15). As predicted from its structure, demethyl tiopronin (54 ± 1%) showed even stronger GPx inhibition. However, the nonselective dimethyl tiopronin analog showed no inhibition of GPx.
When these compounds were tested against KB-3-1 and KB-V1 cells, the demethyl tiopronin compound showed improved selectivity over tiopronin (Fig. 2E; KB-V1 IC50 = 0.42 ± 0.09 mm, KB-3-1 IC50 = 18.37 ± 0.22 mm, SR = 44). However, the dose-response curve for demethyl tiopronin against KB-V1 cells displayed a strong biphasic recovery of cell viability at concentrations >2 mm, much exaggerated compared with the slight biphasic recovery reported previously for tiopronin (13). Biphasic activity of thiol compounds has been observed previously and rationalized as the thiol group scavenging ROS at higher concentrations (24). In contrast to tiopronin and desmethyl tiopronin, dimethyl tiopronin (containing greater steric shielding of the thiol) did not selectively kill KB-V1 cells (Fig. 2F; KB-V1 IC50 = 7.80 ± 1.28 mm, KB-3-1 IC50 = 9.50 ± 1.15 mm, SR = 1.2) but had a similar level of baseline killing against KB-3-1 cells.
Tiopronin Inhibits GPx by Covalent Binding
No structural and little mechanistic insight appear to exist concerning the mode of inhibition of GPx by tiopronin and mercaptosuccinate (or any other known inhibitor). Protein mass spectrometry was employed to identify whether inhibitors form a covalent linkage to the enzyme (at the Sec site or elsewhere). Bovine GPx was incubated with tiopronin or mercaptosuccinate (or control), concentrated, and trypsinized for protein mass spectrometry (untrypsinized GPx1 could not be observed by mass spectrometry). A single trypsinized fragment of bovine GPx (M + H+ m/z = 1,138.52, peptide NDVSWNFEK) that did not contain the Sec residue showed mass increases of 148.59 and 161.69 that correspond to the molecular weights of mercaptosuccinic acid (H3A m/z = 150.00 g mol−1, HA2− m/z = 147.98) and tiopronin (H2A m/z = 163.03 mol−1, HA− m/z = 162.02), respectively (Fig. 5).
FIGURE 5.

Interaction of tiopronin and mercaptosuccinate with GPx. A, mass spectra of the tryptic digest of bovine GPx (top spectrum), GPx preincubated with mercaptosuccinate (middle spectrum) and tiopronin (bottom spectrum). Peaks that arose because of binding of mercaptosuccinate or tiopronin to the fragment at m/z = 1,139.50 and their subsequent oxidation are indicated by arrows, and their absence is highlighted in the control GPx spectrum. B, rendering of human GPx1 structure highlighting the Lys residue to which mercaptosuccinate and tiopronin bind and the site of the Sec residue at the active site. C, mass spectrum demonstrating the lack of reaction between Tio (middle spectrum) and mercaptosuccinate (bottom spectrum) with the Lys-containing peptide (control, top spectrum). D, mass spectra of the reaction of a Sec-containing peptide (control, top spectrum) with Tio (middle spectrum) and Tio followed by the reductant DTT (bottom spectrum). E, mass spectra of the reaction of the Sec- and Lys-containing peptide (control, top spectrum) with Tio (middle spectrum) and Tio followed by the reductant DTT (bottom spectrum).
Tandem mass spectrometry of the NDVSWNFEK tryptic peptide from GPx1 samples treated with mercaptosuccinate (Fig. 6A) and tiopronin (Fig. 6B) was examined to identify the amino acid to which these compounds were bound. In both cases, all fragments containing the Lys residue (y ion series) were of a greater molecular mass because of the covalent binding of the GPx inhibitors (the tandem mass spectrum of this fragment in control GPx is shown in Fig. 6C). Conversely, all fragments in which the Lys residue was missing (b ion series) had an unchanged molecular mass corresponding to the unmodified peptide ions. This indicated that both mercaptosuccinate and tiopronin were bound to the lysine of the NDVSWNFEK fragment in GPx1. The WXFXKL motif is conserved in all four human and four bovine glutathione peroxidases and more generally in eukaryotes, plants, insects, and virus (16, 25), suggesting that the mechanism of inhibition is not unique to the bovine GPx isoform we examined.
FIGURE 6.
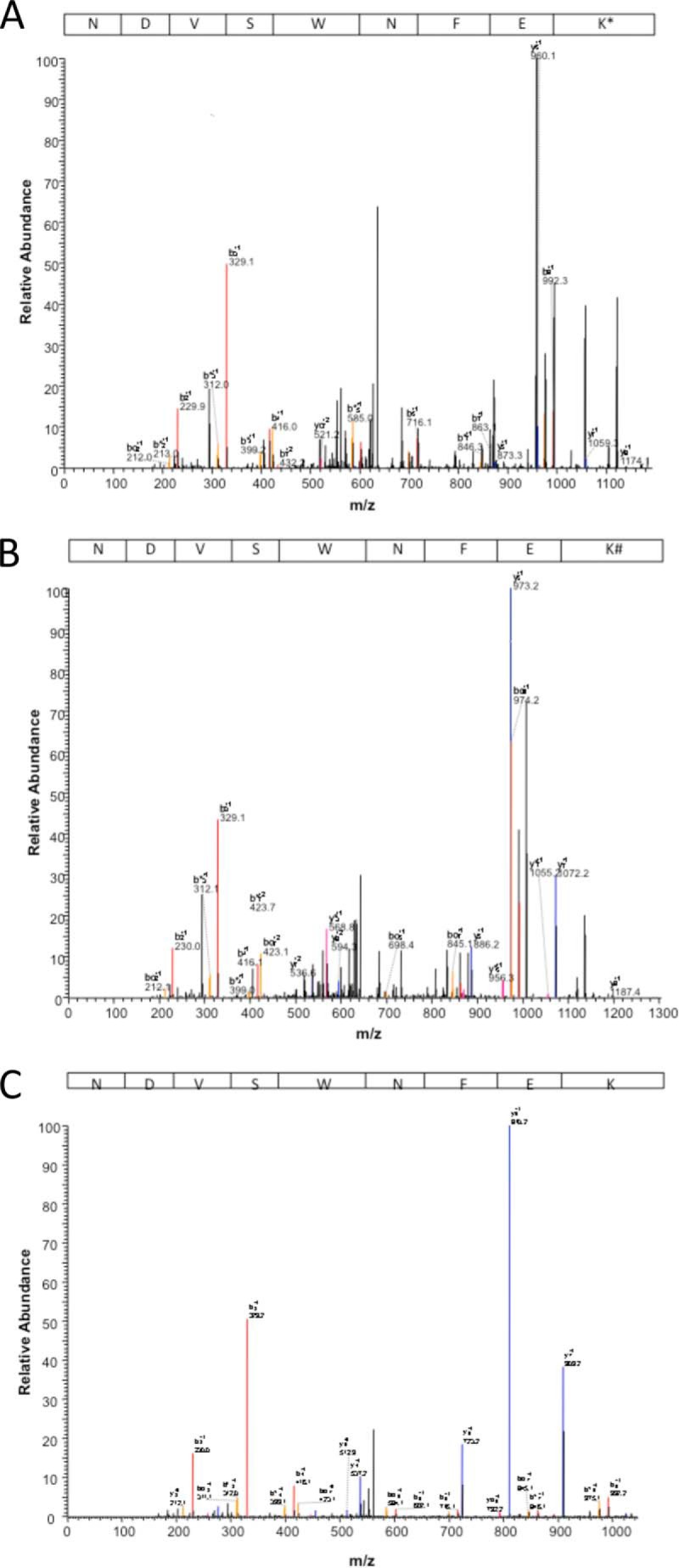
Representative MS/MS spectra of [M+2H]2+ peptides from bovine GPx1 treated with mercaptosuccinate (A) or tiopronin (B), compared with control (C), showing sites of modification (black peaks). The peptide sequence for each spectrum is shown, with the sites of mercaptosuccinate or tiopronin modification marked as an asterisk and pound symbol, respectively. The peptide fragments observed in the unmodified state for each treatment condition are shown as colored peaks (observed in the control spectrum). The peptide sequence for each spectrum is shown.
In the MALDI-TOF mass spectrum of the reaction of each compounds with GPx1, a peak 32.09 mass units higher than the compound fragment adduct was observed (Fig. 5A), which corresponds to the addition of two oxygen atoms (2O m/z = 31.99), consistent with oxidation of the sulfur in the adduct to a sulfonylamino group (Tio-S(O2)-N-Lys, or mercaptosuccinate-S(O2)-N-Lys). In the rendering of a monomer of crystallized human GPx1 (Fig. 5B, generated from the structure of the C47G mutant; Protein Data Bank 2F8A), the lysine residue (red) resides at the N-terminal end of a β-sheet domain with the side chain oriented toward the loop where the Sec residue resides (magenta).
Given that Tio and mercaptosuccinate may initially interact with the glutathione binding site of GPx1 by virtue of their chemical properties, we hypothesized that the molecules may first oxidatively react with the Sec residue, before reactive transfer to the Lys residue. To address this question, we incubated the two compounds with synthetic peptides representing the tryptic fragments containing Sec (VLLIENVASLUGTTVR, [M + H]+ m/z = 1,735.89) and Lys (NDVSWNFEK, [M + H]+ m/z = 1,138.52) residues. No direct reaction of the Lys-containing peptide with mercaptosuccinate or tiopronin could be observed; only the parent peak was evident by mass spectrometry (Fig. 5C). In contrast, both tiopronin and mercaptosuccinate reacted with the Sec-containing peptide, producing peaks of expected mass increase, in each case a loss of three protons being due to the thiol and selenol oxidative reaction and deprotonation of a carboxylate (Tio: m/z 1,896.3 = Sec peptide + Tio - 3H+, Fig. 5C; and mercaptosuccinate: 1,883.3 = Sec peptide + mercaptosuccinate - 3H+, Fig. 7). The reaction between the Sec-containing peptide and mercaptosuccinate or Tio could be reversed by subsequent addition of the reductant DTT (m/z = 154.01), evidenced by loss of their respective conjugate peaks in the mass spectrum, and a new peak in each spectrum that arose because of excess DTT reacting with the peptide (m/z 1,887.5 = Sec peptide + DTT - 3H+; Figs. 5D and 7). The reductive removal of Tio and mercaptosuccinate adducts from the Sec peptide support the hypothesis of the formation of an initial R-S-Se-R′ product (where R is tiopronin, for example, and R′ is the Sec residue). It is possible that when GPx1 protein was analyzed, the trypsin enzyme digest process facilitated a similar reductive removal of Tio and mercaptosuccinate, masking this addition.
FIGURE 7.

Interaction of mercaptosuccinate with GPx peptides. A, mass spectra of the reaction of a Sec-containing peptide (control, top spectrum) with mercaptosuccinate (middle spectrum), and mercaptosuccinate followed by the reductant DTT (bottom spectrum). B, mass spectrum of the reaction of the Sec- and Lys-containing peptide (control, top spectrum) and with mercaptosuccinate (middle spectrum) and mercaptosuccinate followed by the reductant DTT (bottom spectrum). C, scheme demonstrating the proposed reaction between thiol-containing Tio or mercaptosuccinate and GPx.
To assess whether Sec-mediated transfer of Tio and mercaptosuccinate to a Lys residue, we reacted the two compounds with a synthetic fusion peptide containing both Lys and Sec residues (ASLUNDVSWNFEK, [M + H]+ m/z = 1,560.62). In both cases, a Tio and mercaptosuccinate adduct with the Sec- and Lys-containing peptide was observed (Tio: m/z 1,721.1 = Sec- and Lys-containing peptide + Tio - 3H+, Fig. 5E; mercaptosuccinate: 1,708.0 = Sec- and Lys-containing peptide + mercaptosuccinate - 3H+, Fig. 7). Addition of the reductant DTT did not remove either the Tio or mercaptosuccinate adducts on the peptide. This suggests that this model peptide, in the case of Tio, facilitated the transfer of Tio to the Lys residue. The proposed reaction scheme between Tio or mercaptosuccinate and GPx is shown in Fig. 7C.
Knockdown of GPx1 and GPx4 Reverses Collateral Sensitivity to Tiopronin
Given the observation that tiopronin inhibits GPx1, we used shRNA to stably knock down GPx1, GPx4, and GPx1 and GPx4 together in KB-3-1 and KB-V1 cells (quantitative RT-PCR results showed that GPx2 and GPx3 are not expressed in these cells; data not shown). Knockdown of the corresponding GPx1 and GPx4 proteins was determined by Western blotting (Fig. 8A). Control cells transfected with nontargeting (NT) shRNA showed elevated levels of GPx1 and GPx 4 in the MDR KB-V1 cells compared with KB-3-1 cells. In KB-3-1 cells, silencing of GPx4 resulted in elevated GPx1 expression and silencing of GPx1 resulted in elevated expression of GPx4, suggesting some compensatory capacity and functional redundancy. The phenotypic effect of GPx1 and GPx4 silencing was assessed by the cytotoxicity of luperox (tert-butyl hydroperoxide), an organic peroxide. We hypothesized that silencing of GPx protein would sensitize cells to an organic peroxide because the capacity to detoxify it would be disrupted, and this indeed was found to be the case (Fig. 8B and Table 2). For example, KB-3-1 NT cells (luperox IC50 = 119.0 ± 10.5 μm) were less sensitive than KB-3-1 GPx1 (luperox IC50 = 39.4 ± 2.8 μm) or KB-3-1 GPx4 (luperox IC50 = 59.7 ± 21.4 μm), and KB-3-1 GPx1 + 4 cells were the most sensitive (luperox IC50 = 21.3 ± 6.9 μm). The KB-V1 GPx1 + 4 cells (luperox IC50 = 37.9 ± 0.7 μm) were also more sensitive than the KV-V1 NT cells (luperox IC50 = 104.2 ± 5.4 μm). Knockdown of GPx1, GPx4, or both reversed the hypersensitivity of KB-V1 cells to tiopronin (SR = ∼1 for all three cell line pairs), compared with the NT cell lines (SR = 29) (Table 2). This reversal of selectivity was largely due to the KB-V1 cell lines showing lower sensitivity. For example, KB-V1 GPx1 + 4 cells (tiopronin IC50 = 4.9 ± 0.07 mm) were 10-fold less sensitive than KB-V1 NT cells (tiopronin IC50 = 0.6 ± 0.02 mm), although all KB-3-1 GPx knockdown cells were ∼2–3-fold more sensitive to tiopronin (KB-3-1 NT tiopronin IC50 = 4.9 ± 0.07 mm; KB-3-1 GPx 1 + 4 tiopronin IC50 = 17.2 ± 0.4 mm).
FIGURE 8.

Knockdown of GPx1 and/or GPx4 reverses selective killing by tiopronin. A, Western blot of GPx1 and GPx4 in KB-3-1 and KB-V1 cells transfected with shRNA that is NT or targeted against GPx1, GPx4, or GPx1 and GPx4 (GPx1 + 4). B and C, dose-response curves of KB-V1 and KB-3-1 GPx1 + 4 and NT cells treated with luperox (B) or tiopronin (C) for 72 h.
TABLE 2.
Determination of cytotoxicity (IC50) of tiopronin and luperox (tert-butyl hydroperoxide) against GPX knockdown cell lines
SR was calculated as IC50 parental/IC50 MDR cell line.
| Cell line | Cytotoxicity (IC50) |
||
|---|---|---|---|
| Tiopronin | SR | Luperox | |
| mm | μm | ||
| KB-3-1 NT | 17 ± 0.4 | 119 ± 11 | |
| KB-V1 NT | 0.6 ± 0.02 | 29.0 | 104 ± 5.4 |
| KB-3-1 GPX1 | 9.9 ± 0.7 | 39 ± 2.8 | |
| KB-V1 GPX1 | 8.2 ± 0.4 | 1.2 | 83 ± 23 |
| KB-3-1 GPX4 | 9.9 ± 3.9 | 60 ± 21 | |
| KB-V1 GPX4 | 8.4 ± 0.9 | 1.2 | 266 ± 163 |
| KB-3-1 GPX1 + 4 | 6.4 ± 3.2 | 21 ± 6.9 | |
| KB-V1 GPX1 + 4 | 4.9 ± 0.7 | 1.3 | 38 ± 0.7 |
Tiopronin Elicits ROS Production in Cells
To determine whether inhibition of GPx in cells caused ROS formation, we first used confocal microscopy to examine the oxidation of a fluorescent ROS probe (CellROX, Molecular Probes; excitation = 644 nm, emission = 665 nm). The CellROX probe itself is not fluorescent (structure not disclosed by the vendor). However, oxidation of the molecule by ROS yields a fluorescent dye (26). Under baseline conditions, cells treated with CellROX probe (5 μm) show little or no fluorescence after a 1-h incubation (Fig. 9). Cells were co-treated with Hoechst 33342 dye to stain the nuclei of cells. The ROS probe can indicate the presence of ROS in cells but does not allow for quantification, partly because we found that the CellROX probe (or its oxidized product) is a P-gp substrate (data not shown), meaning that the ROS probe signal is diminished. To overcome this problem, we co-incubated KB-V1 cells with the P-gp inhibitor tariquidar (100 nm) to prevent efflux of the CellROX probe (tariquidar does not affect the MDR selectivity of tiopronin (13)). For each experiment, cells were incubated with a ROS-inducing compound for a given period of time, washed, and then incubated with CellROX probe to assess intracellular ROS status. Both KB-3-1 and KB-V1 cells treated with 200 μm H2O2 (Fig. 9A) or 200 μm menadione (2-methyl-1,4-napthaquinone, a ROS generator (27)) resulted in extranuclear fluorescence of CellROX probe. Both KB-3-1 and KB-V1 cells treated with 1 mm tiopronin for 1 h also elicited a fluorescent signal. Strikingly, treatment of cells with 1 mm tiopronin for 24 h followed by addition of CellROX probe resulted in fluorescence, indicating strong ROS formation in KB-V1, but not KB-3-1 cells. These experiments suggest that tiopronin may produce an initial burst of ROS in cells (observed at 1 h), but prolonged ROS was only observed in KB-V1 cells.
FIGURE 9.

Assessment of ROS levels in cells treated with tiopronin. A, CellROX Deep Red reagent, a dye that fluoresces when oxidized by ROS, was added to the cells and observed by confocal microscopy. KB-3-1 and KB-V1 cells were treated with tiopronin (1 mm) for 1 or 24 h. As positive controls, cells were treated with H2O2 (200 μm) for 1 h. For negative controls, cells were left untreated (i.e. dye only). Prior to imaging, all cells were incubated with Hoechst 33342 (5 μg/ml) and CellROX Deep Red reagent (5 μm). B, ROS levels generated in cells were measured by preincubating cells in 96-well plates with tiopronin for 24 h, followed by washing and incubation of cells with DHFDA (reduced form) for 30 min. The cells were then washed, and fluorescence arising from oxidation of DHFDA to fluorescein diacetate was measured. C, measurement of ROS as described above, but after the addition of dye, 1,500 μm H2O2 was applied to cells to observe the ROS levels in cells in the presence of an oxidative insult.
Because the confocal microscopy experiments are nonquantitative, we employed a lower sensitivity but quantitative plate-based assay using DHFDA (28). DHFDA was selected as has been demonstrated by Hempel et al. (29) to be more useful for in-cell experiments than other reduced dyes. As with the CellROX probe, DHFDA is not fluorescent until oxidized and is not specifically oxidized by a single ROS species (i.e. ROS generation can be measured, but the ROS species involved cannot be inferred). A further advantage of employing a plate-based assay is that if DHFDA or the oxidized product (fluorescein diacetate) is a P-gp substrate and extruded from cells, it does not interfere with the assay outcome because total fluorescence in the well (i.e. cells and medium) is measured. Cells were incubated with tiopronin for 24 h, washed, and then incubated with DHFDA for 30 min while measuring fluorescence. Under these conditions, fluorescence could only be observed at the highest concentration of tiopronin used (2 mm) (Fig. 9B). This is probably partly due to the detection limit of plate-based fluorimeters (compared with confocal microscopy), but also because ROS generated in cells over time (such as the 24-h tiopronin window employed here) is not accumulated but instead rapidly degraded.
The ROS experiments with CellRox probe demonstrated that tiopronin can induce an enduring ROS effect in KB-V1 cells. We investigated whether the ROS accumulation mediating tiopronin selectivity was due to the prevention of endogenous ROS from being degraded. This would be consistent with the effect expected by inhibition of GPx.
To test whether tiopronin differentially inhibits the depletion of ROS in KB-3-1 and KB-V1 cells, the cells were incubated with 1 mm tiopronin for 24 h as described above. The cells were then loaded with DHFDA dye for 30 min, washed, and exposed to H2O2 (1.5 mm) for 10 min, and fluorescence was measured. Although KB-3-1 cells did not show increased fluorescence (similar to Fig. 9B), KB-V1 cells showed an increase in fluorescence (5 mm) compared with control cells, indicating that the H2O2 added to KB-V1 cells was not being depleted by the cells (Fig. 9C). KB-3-1 and KB-V1 cells incubated with H2O2 alone showed identical ROS probe fluorescence (data not shown), confirming that tiopronin was necessary for altered H2O2 consumption.
The CS Activity of Tiopronin Is Reversed by ROS Scavengers
Following the observation that the CS activity of tiopronin may be driven by GPx inhibition and ROS production, we assessed whether a series of compounds that scavenge ROS could reverse the selectivity of tiopronin. We first assessed the cytotoxicity of tiopronin in the presence of 1, 2, and 5 mm NAC (all concentrations are nontoxic), a cell-permeable precursor of cysteine that increases glutathione levels, directly scavenges ROS and reverses ROS-mediated cell killing (30). Tiopronin cytotoxicity toward KB-V1 cells (0.14 ± 0.007 mm) was lowered in the presence of 1 (IC50 = 0.36 ± 0.101 mm), 2 (IC50 = 0.51 ± 0.050 mm) and 5 mm NAC (IC50 = 0.99 ± 0.22 mm), altering the dose-response curve toward that of the parental KB-3-1 cells (Fig. 10A). In contrast, cytotoxicity toward KB-3-1 cells was unaffected (Table 1). To ensure that this desensitization by NAC was not the result of a direct oxidative interaction between NAC and tiopronin, MALDI-TOF mass spectrometry was employed to demonstrate that auto-oxidation of tiopronin in the presence of NAC or oxidation between tiopronin, and NAC was not observed (data not shown). The selective cytotoxicity of mercaptosuccinate toward KB-V1 cells was also reversed by NAC to levels approximating those of KB-3-1 cells (Fig. 2C). The addition of 2 mm NAC abrogated the SR of mercaptosuccinate from 45 to almost parity (SR = 1.1). The selective killing of 2,3-dimercaptosuccinate was also reversed by NAC (data not shown).
FIGURE 10.

Reversal of collateral sensitivity of KB-V1 cells to tiopronin by ROS scavengers, assessed by cytotoxicity assay. A, dose-response curves of the P-gp-expressing subline KB-V1 and the parental KB-3-1 human adenocarcinoma cell line treated with tiopronin for 72 h, showing the effect of increasing (nontoxic) concentrations of N-acetylcysteine (1, 2, and 5 mm) on the cytotoxicity of tiopronin toward KB-V1 and KB-3-1 cells. B, effect of anoxic conditions on the cytotoxicity of tiopronin toward KB-V1 and KB-3-1 cells. C, effect of catalase and inactivated catalase (generated by heating lysate at 65 °C for 4 h) added to growth media on the cytotoxicity of tiopronin toward KB-V1 and KB-3-1 cells. D, effect of erythrocyte (RBC) lysate and inactivated RBC lysate (generated by heating lysate at 65 °C for 4 h), added to growth media on the cytotoxicity of tiopronin toward KB-V1 and KB-3-1 cells.
NAC is a nonspecific ROS reversing agent. We sought to determine the specific ROS species being scavenged by NAC, thereby reversing activity. Several strategies were employed to gain this insight. First, O2 is necessary for the formation of ROS, so we hypothesized that cells incubated with tiopronin in an anoxic environment would not show collateral sensitivity. This proved to be the case (Fig. 10B), with KB-V1 cells in anoxic conditions (IC50 = 2.3 ± 0.94 mm) showing similar sensitivity to KB-3-1 cells under the same conditions (IC50 = 3.9 ± 1.3 mm) or regular culture conditions (IC50 = 3.9 ± 1.1 mm).
The enzyme catalase specifically converts H2O2 to H2O and O2 with high efficiency (31). Although catalase cannot enter cells, it can indirectly reduce H2O2 concentrations in cells when added to culture medium (culture medium contains no detectable catalase activity, probably because the enzyme in fetal bovine serum is sensitive to freeze-thawing), reversing H2O2-mediated toxicity. This is possible because H2O2 is relatively stable and cell-permeable, probably via the aquaporin proteins (32), and rapid consumption of H2O2 outside cells creates a concentration gradient that draws H2O2 from cells, resulting in a lower cellular concentration (26). When cells were dosed with tiopronin in the presence of nontoxic amounts of catalase (1,000 units/ml), collateral sensitivity of KB-V1 cells to tiopronin was reduced (KB-V1 IC50 = 7.4 ± 0.8 mm, KB-3-1 IC50 = 17 ± 0.9 mm), similar to NAC and anoxic conditions (Fig. 10C). To confirm that the reversal effect was not mediated by nonspecific effects of catalase, deactivated enzyme (achieved by multiple freeze-thaw cycles or heating at 60 °C for 2 h, confirmed by an absence of oxygen bubbles upon addition of H2O2) did not reverse collateral sensitivity (Fig. 10C). Erythrocyte cytoplasm should be expected to have a similar effect on the activity of tiopronin as purified catalase enzyme (33). Cell lysate prepared from ruptured erythrocytes that retained catalase activity (visually confirmed by adding H2O2) reversed tiopronin selectivity (Fig. 10D). As a control, deactivated lysate prepared by heating the erythrocyte lysate at 57 °C for 3 h (confirmed as above) had no effect on the selectivity of tiopronin. In contrast to these results supporting the involvement of H2O2 in the toxicity of tiopronin, the cell-permeable small molecule superoxide dismutase mimic manganese(III) tetrakis(4-benzoic acid)porphyrin chloride had no effect on the activity of tiopronin (data not shown).
The CS Activity of NSC73306 Is Not Directly Mediated by ROS
For comparison, we also examined the effect of reversing ROS on the MDR-selective compound NSC73306 (1-isatin-4-(4′-methoxyphenyl)-3-thiosemicarbazone) (7). The MDR selectivity of NSC73306 has been shown against all P-gp-expressing cells tested (34). Against KB-V1 cells, selectivity has been shown to be reversed by inhibition or silencing of the P-gp efflux transporter (7, 34). Unlike tiopronin, the selective killing of KB-V1 cells by NSC73306 was unaffected by NAC or anoxia. Both normal (1,000 units/ml) and inactivated catalase inhibited cytotoxicity against both KB-3-1 and KB-V1 cells, suggesting a protein binding effect (Fig. 11). These data demonstrate that ROS-mediated killing is not necessarily a general feature of CS agents.
FIGURE 11.

Collateral sensitivity of KB-V1 cells to NSC73306, assessed by cytotoxicity assays. A, dose-response curves of NSC73306 against the P-gp-expressing sublines KB-V1 and the parental KB-3-1 human adenocarcinoma cell line treated with NSC73306 for 72 h. B, effect of increasing (nontoxic) concentrations of N-acetylcysteine on the cytotoxicity of NSC73306 toward KB-V1 and KB-3-1 cells. C, effect of anoxic conditions on the cytotoxicity of NSC73306 toward KB-V1 and KB-3-1 cells. D, false-positive effect of inactivated catalase (generated by freeze-thaw cycles) on the cytotoxicity of NSC73306 toward both KB-V1 and KB-3-1 cells.
H2O2 Cannot Directly Mediate CS
Given the evidence that H2O2 may play a role in the toxicity of tiopronin, we assessed the direct cytotoxicity of H2O2 on KB-3-1 and KB-V1 cells (Fig. 12). H2O2 showed equal toxicity toward both cell lines (similar results were found with tert-butyl hydroperoxide and cumene hydroperoxide; data not shown), and subtoxic H2O2 did not increase the selectivity of tiopronin. ROS-generating molecules such as rotenone also showed equal toxicity toward both cell lines (Fig. 12). Using a colorimetric peroxide detection assay (35, 36), equivalent numbers of KB-3-1 and KB-V1 cells demonstrated equivalent consumption of H2O2 over time, with a half-life of ∼5 min (Fig. 12). This is consistent with other studies examining the depletion of peroxide in other cell types such as cultured astrocytes (half-life of ∼5 min) (37).
FIGURE 12.

Sensitivity of KB-3-1 and KB-V1 cells to H2O2. A, dose-response curves of the P-gp-expressing sublines KB-V1 and the parental KB-3-1 human adenocarcinoma cell line treated with H2O2 (without or with pyruvate supplemented in the cell growth medium) for 72 h. The supplementation of 110 mg/liter pyruvate to the growth medium, which scavenges H2O2, inhibited the cytotoxicity of hydrogen peroxide to both cell lines by an order of magnitude. Catalase could not be demonstrated to reverse H2O2-mediated cytotoxicity for technical reasons, because the catalase-supplemented culture medium instantly produces thick foam on addition of H2O2 that makes it impossible to conduct the cytotoxicity assay. As expected, generation of H2O2 in cell cultures by glucose oxidase also showed no synergism with tiopronin, presumably because the tiopronin depletes the H2O2 as it is generated. Other ROS-generating agents such as paraquat likewise did not elicit CS and did not synergize with tiopronin (data not shown). B, consumption of 250 μm H2O2 by KB-3-1 and KB-V1 cells, measured by xylenol orange assay. C, consumption of H2O2 in the presence of increasing concentrations of tiopronin. Although H2O2 did not reproduce the activity of tiopronin, we hypothesized that subtoxic H2O2 might synergize with tiopronin activity. This is difficult to assess, because we found that H2O2 directly oxidizes tiopronin (and therefore tiopronin reduces H2O2) in solution. For example, incubation of increasing concentrations of tiopronin with 1 mm H2O2 for 1 h results in a corresponding decrease in the levels of H2O2. D, cytotoxicity of tiopronin in the presence of 1 mm H2O2. When cells were incubated with a highly cytotoxic level of H2O2 (1 mm), very little cell viability could be detected. Increasing concentrations of tiopronin allowed KB-3-1 cells to recover viability, because H2O2 and tiopronin react and are depleted before concentrations of tiopronin greater than 1,000 μm prove cytotoxic in a fashion similar to tiopronin alone (Fig. 2a). This depletion of H2O2 by tiopronin (2RSH → RSSR + 2H+ + 2e−) is consistent with the exploratory utilization of tiopronin as a thiol to scavenge ROS associated with reperfusion injury and the reactive species 2-amino propanal involved in neuroprotection.
DISCUSSION
Tiopronin is an orphan drug used to treat patients with diverse conditions such as cystinuria and rheumatoid arthritis and has been used in clinical trials as a ROS scavenger. The redox chemistry of thiol compounds is complicated, mainly associated with ROS scavenger activity (via oxidation of the thiol), but thiols such as cysteamine (β-mercaptoethylamine) (24) and d-penicillamine (38) have been shown to directly generate H2O2. It has been reported that tiopronin can scavenge ROS (via its thiol) over short periods of time (shown for tiopronin in Fig. 12). However, tiopronin also acts as an inhibitor of GPx, which may lead to chronic exposure of cells to ROS over time and may account for confounding data in the literature showing that tiopronin, despite being considered an antioxidant, has been shown to elevate oxidative stress (39). As such, there would appear to be a contradiction between the acute ROS scavenging effects of tiopronin reported over 1 h and the involvement of ROS in eliciting collateral sensitivity of KB-V1 cells during a 72-h assay. This can be accounted for by tiopronin inhibition of GPx in long term assays.
We have shown here that the collateral sensitivity demonstrated by KB-V1 cells to tiopronin requires the generation of reactive oxygen species, and this may be mediated by inhibition of one or more glutathione peroxidases. GPx1–4 are selenocysteine-containing proteins that utilize glutathione as a co-factor to reduce H2O2 and organic hydroperoxides. Each possesses separate localized functions in the cellular cytosol (GPx1), in the gastrointestinal tract (GPx2), as an extracellular protein in blood plasma (GPx3), and through membrane phospholipid association (GPx4) (40, 41). In the human, GPx6 has also been demonstrated to incorporate selenocysteine (42). Therefore, 5 of 25 known Sec-incorporating human proteins are members of the GPx family. Chaudiere et al. (15) demonstrated that mercaptosuccinate and tiopronin do not inhibit other ROS-related enzymes such as catalase and glutathione reductase. However, it is possible that other Sec-containing proteins such as the thioredoxin reductases, selenoprotein P and W, and thyroid hormone deiodinases may be inhibited, and this remains to be examined (43).
A number of chemotherapeutics have been shown to generate H2O2 or lipid peroxides as part of their mechanism of action (44), and these ROS species are scavenged by glutathione peroxidases (45). Two separate reports of doxorubicin (adriamycin)-resistant cell lines identified elevated Se-dependent GPx activity (46, 47), and cells resistant to etoposide (VP-16) also demonstrated elevated activity (48). Clinical studies have shown that elevated GPx3 expression confers platinum insensitivity to ovarian clear cell carcinoma (49), that low GPx1 expression in patients with diffuse large B-cell lymphoma leads to improved chemotherapy response (50), and that increased levels of GPx3 are part of a signature in clinical samples of ovarian cancer that predicts poorer response to chemotherapy (51).
Although a small number of GPx inhibitor studies exist (15, 52), like the compounds studied here, their efficacy is poor (high micromolar). We demonstrated that this inhibition occurs via a covalent adduct of tiopronin or mercaptosuccinic acid, ultimately with a Lys residue. Our mass spectrometry data indicate this likely occurs via interaction between these two compounds and the Sec residue of GPx. Given that both inhibitors contain a negative charge and a thiol, it is possible they initially interact with the GSH binding site. Park et al. (53) have shown that methylglyoxal inhibits GPx via reacting covalently with Arg groups in the glutathione binding site at millimolar concentrations. Following submission of this manuscript, Yang et al. (54) reported that cells expressing oncogenic HRASV12 demonstrated ROS-mediated synthetic lethality as a consequence of inactivation of GPx4 by small molecules—either depletion of GSH by erastin or direct inhibition by RSL3. Although HeLa lines (from which the KB cells used in this study are derived) do not carry oncogenic RAS mutations (55), the observations directly support our findings.
The reversal of tiopronin selectivity by catalase was replicated using lysate from human erythrocytes (which contain high levels of catalase; Fig. 9D). One implication of this observation is that tiopronin and other drugs whose activity is mediated by ROS may not be effective in vivo. Catalase was employed here at a concentration of 1,000 units/ml. In the human, catalase and GPx are highly active in erythrocytes (56), and normal catalase activity is ∼110,000 units/ml (57), 100 times greater than the amount used to reverse tiopronin in this study.
However, it is not clear whether catalase is present at such levels in the intratumoral environment or whether it would be expected that catalase in the cytoplasm of circulating erythrocytes can remove ROS from tumor cells. No serious side effects from tiopronin are experienced by patients during treatment for cystinuria using daily dosing regimens on the order of hundreds of milligrams to gram quantities per patient per day for several months to years. Inhibition of cellular GPx in vivo may be compensated for by the large extracellular pool of other ROS-scavenging enzymes. Given the role of GPx in depleting both organic peroxides and hydrogen peroxide, it seems likely that these species are responsible for the oxidation of ROS-sensitive dyes. However, it is generally accepted that there are no ROS species-specific dyes, and on this basis we do not infer a specific ROS species as being responsible (29, 58).
We have shown here for the first time the molecular mechanism by which a small molecule elicits collateral sensitivity in an MDR cell line. Because tiopronin selectively kills other MDR lines that do not overexpress P-gp, dysregulation of ROS may be a common feature of cells in culture selected for drug resistance. Given the correlation between GPx expression and cellular killing, the availability of a potent GPx inhibitor that can sensitize cells is of great interest.
Acknowledgments
We thank George Leiman for editorial assistance. We thank Susan H. Garfield and Poonam Mannan of the NCI Confocal Microscopy Core Facility for technical assistance.
This work was supported, in whole or in part, by the National Institutes of Health Intramural Research Program.
- MDR
- multidrug resistance or multidrug-resistant
- GPx
- glutathione peroxidase
- ROS
- reactive oxygen species
- Tio
- tiopronin
- CS
- collateral sensitivity
- P-gp
- P-glycoprotein
- CS
- collateral sensitivity
- DHFDA
- dihydrofluorescein diacetate
- SR
- selectivity ratio
- Sec or U
- selenocysteine
- NT
- nontargeting
- RBC
- red blood cell(s).
REFERENCES
- 1. Gottesman M. M., Fojo T., Bates S. E. (2002) Multidrug resistance in cancer: role of ATP-dependent transporters. Nat. Rev. Cancer 2, 48–58 [DOI] [PubMed] [Google Scholar]
- 2. Gottesman M. M. (2002) Mechanisms of cancer drug resistance. Annu. Rev. Med. 53, 615–627 [DOI] [PubMed] [Google Scholar]
- 3. Fojo T. (2008) Novel therapies for cancer: why dirty might be better. Oncologist 13, 277–283 [DOI] [PubMed] [Google Scholar]
- 4. Hall M. D., Handley M. D., Gottesman M. M. (2009) Is resistance useless? Multidrug resistance and collateral sensitivity. Trends Pharmacol. Sci. 30, 546–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hall M. D., Salam N. K., Hellawell J. L., Fales H. M., Kensler C. B., Ludwig J. A., Szakács G., Hibbs D. E., Gottesman M. M. (2009) Synthesis, activity, and pharmacophore development for isatin-β-thiosemicarbazones with selective activity toward multidrug-resistant cells. J. Med. Chem. 52, 3191–3204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Szybalski W., Bryson V. (1952) Genetic studies on microbial cross resistance to toxic agents: I. cross resistance of Escherichia coli to fifteen antibiotics. J. Bacteriol. 64, 489–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ludwig J. A., Szakács G., Martin S. E., Chu B. F., Cardarelli C., Sauna Z. E., Caplen N. J., Fales H. M., Ambudkar S. V., Weinstein J. N., Gottesman M. M. (2006) Selective toxicity of NSC73306 in MDR1-positive cells as a new strategy to circumvent multidrug resistance in cancer. Cancer Res. 66, 4808–4815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Muller C., Bailly J. D., Goubin F., Laredo J., Jaffrézou J. P., Bordier C., Laurent G. (1994) Verapamil decreases P-glycoprotein expression in multidrug-resistant human leukemic cell lines. Int. J. Cancer 56, 749–754 [DOI] [PubMed] [Google Scholar]
- 9. Chan D. A., Giaccia A. J. (2011) Harnessing synthetic lethal interactions in anticancer drug discovery. Nat. Rev. Drug Discov. 10, 351–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Marks K. M., Park E. S., Arefolov A., Russo K., Ishihara K., Ring J. E., Clardy J., Clarke A. S., Pelish H. E. (2011) The selectivity of austocystin D arises from cell-line-specific drug activation by cytochrome P450 enzymes. J. Nat. Prod. 74, 567–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Minagawa Y., Kigawa J., Itamochi H., Terakawa N. (2001) The role of topoisomerase I inhibitor in cisplatin-resistant ovarian cancer. Hum. Cell 14, 237–243 [PubMed] [Google Scholar]
- 12. Pluchino K. M., Hall M. D., Goldsborough A. S., Callaghan R., Gottesman M. M. (2012) Collateral sensitivity as a strategy against cancer multidrug resistance. Drug Resist. Updat. 15, 98–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Goldsborough A. S., Handley M. D., Dulcey A. E., Pluchino K. M., Kannan P., Brimacombe K. R., Hall M. D., Griffiths G., Gottesman M. M. (2011) Collateral sensitivity of multidrug-resistant cells to the orphan drug tiopronin. J. Med. Chem. 54, 4987–4997 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 14. de Angelis L. (1978) Tiopronin. Drugs Today 14, 37–40 [Google Scholar]
- 15. Chaudiere J., Wilhelmsen E. C., Tappel A. L. (1984) Mechanism of selenium-glutathione peroxidase and its inhibition by mercaptocarboxylic acids and other mercaptans. J. Biol. Chem. 259, 1043–1050 [PubMed] [Google Scholar]
- 16. Lubos E., Loscalzo J., Handy D. E. (2011) Glutathione peroxidase-1 in health and disease: from molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 15, 1957–1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Laberge R. M., Karwatsky J., Lincoln M. C., Leimanis M. L., Georges E. (2007) Modulation of GSH levels in ABCC1 expressing tumor cells triggers apoptosis through oxidative stress. Biochem. Pharmacol. 73, 1727–1737 [DOI] [PubMed] [Google Scholar]
- 18. Huff L. M., Lee J. S., Robey R. W., Fojo T. (2006) Characterization of gene rearrangements leading to activation of MDR-1. J. Biol. Chem. 281, 36501–36509 [DOI] [PubMed] [Google Scholar]
- 19. Shen D. W., Cardarelli C., Hwang J., Cornwell M., Richert N., Ishii S., Pastan I., Gottesman M. M. (1986) Multiple drug-resistant human KB carcinoma cells independently selected for high-level resistance to colchicine, adriamycin, or vinblastine show changes in expression of specific proteins. J. Biol. Chem. 261, 7762–7770 [PubMed] [Google Scholar]
- 20. Brimacombe K. R., Hall M. D., Auld D. S., Inglese J., Austin C. P., Gottesman M. M., Fung K. L. (2009) A dual-fluorescence high-throughput cell line system for probing multidrug resistance. Assay Drug Dev. Technol. 7, 233–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Danieli B., Giardini A., Lesma G., Passarella D., Peretto B., Sacchetti A., Silvani A., Pratesi G., Zunino F. (2006) Thiocolchicine-podophyllotoxin conjugates: dynamic libraries based on disulfide exchange reaction. J. Org. Chem. 71, 2848–2853 [DOI] [PubMed] [Google Scholar]
- 22. Barton D., Chen C., Wall G. M. (1991) Synthesis of disulfides via sulfenylation of alkyl and aryldithiopyridine N-oxides. Tetrahedron 47, 6127–6138 [Google Scholar]
- 23. Patrick T., Khazaeli S., Nadji S., Hering-Smith K., Reif D. (1993) Mechanistic studies of fluorodecarboxylation with xenon difluoride. J. Org. Chem. 58, 705–708 [Google Scholar]
- 24. Jeitner T. M., Lawrence D. A. (2001) Mechanisms for the cytotoxicity of cysteamine. Toxicol. Sci. 63, 57–64 [DOI] [PubMed] [Google Scholar]
- 25. Mix H., Lobanov A. V., Gladyshev V. N. (2007) SECIS elements in the coding regions of selenoprotein transcripts are functional in higher eukaryotes. Nucleic Acids Res. 35, 414–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Halliwell B., Whiteman M. (2004) Measuring reactive species and oxidative damage in vivo and in cell culture: how should you do it and what do the results mean? Br. J. Pharmacol. 142, 231–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Criddle D. N., Gillies S., Baumgartner-Wilson H. K., Jaffar M., Chinje E. C., Passmore S., Chvanov M., Barrow S., Gerasimenko O. V., Tepikin A. V., Sutton R., Petersen O. H. (2006) Menadione-induced reactive oxygen species generation via redox cycling promotes apoptosis of murine pancreatic acinar cells. J. Biol. Chem. 281, 40485–40492 [DOI] [PubMed] [Google Scholar]
- 28. Wang H., Joseph J. A. (1999) Quantifying cellular oxidative stress by dichlorofluorescein assay using microplate reader. Free Radic. Biol. Med. 27, 612–616 [DOI] [PubMed] [Google Scholar]
- 29. Hempel S. L., Buettner G. R., O'Malley Y. Q., Wessels D. A., Flaherty D. M. (1999) Dihydrofluorescein diacetate is superior for detecting intracellular oxidants: comparison with 2′,7′-dichlorodihydrofluorescein diacetate, 5(and 6)-carboxy-2′,7′-dichlorodihydrofluorescein diacetate, and dihydrorhodamine 123. Free Radic. Biol. Med. 27, 146–159 [DOI] [PubMed] [Google Scholar]
- 30. Raj L., Ide T., Gurkar A. U., Foley M., Schenone M., Li X., Tolliday N. J., Golub T. R., Carr S. A., Shamji A. F., Stern A. M., Mandinova A., Schreiber S. L., Lee S. W. (2011) Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature 475, 231–234 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 31. Nicholls P. (2012) Classical catalase: ancient and modern. Arch. Biochem. Biophys. 525, 95–101 [DOI] [PubMed] [Google Scholar]
- 32. Henzler T., Steudle E. (2000) Transport and metabolic degradation of hydrogen peroxide in Chara corallina: model calculations and measurements with the pressure probe suggest transport of H2O2 across water channels. J. Exp. Bot. 51, 2053–2066 [DOI] [PubMed] [Google Scholar]
- 33. Sawada T., Hashimoto S., Furukawa H., Tohma S., Inoue T., Ito K. (1997) Generation of reactive oxygen species is required for bucillamine, a novel anti-rheumatic drug, to induce apoptosis in concert with copper. Immunopharmacology 35, 195–202 [DOI] [PubMed] [Google Scholar]
- 34. Hall M. D., Brimacombe K. R., Varonka M. S., Pluchino K. M., Monda J. K., Li J., Walsh M. J., Boxer M. B., Warren T. H., Fales H. M., Gottesman M. M. (2011) Synthesis and structure-activity evaluation of isatin-β-thiosemicarbazones with improved selective activity toward multidrug-resistant cells expressing P-glycoprotein. J. Med. Chem. 54, 5878–5889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dringen R., Kussmaul L., Hamprecht B. (1998) Detoxification of exogenous hydrogen peroxide and organic hydroperoxides by cultured astroglial cells assessed by microtiter plate assay. Brain Res. Brain Res. Protoc. 2, 223–228 [DOI] [PubMed] [Google Scholar]
- 36. Jiang Z. Y., Woollard A. C., Wolff S. P. (1990) Hydrogen peroxide production during experimental protein glycation. FEBS Lett. 268, 69–71 [DOI] [PubMed] [Google Scholar]
- 37. Dringen R., Pawlowski P. G., Hirrlinger J. (2005) Peroxide detoxification by brain cells. J. Neurosci. Res. 79, 157–165 [DOI] [PubMed] [Google Scholar]
- 38. Gupte A., Mumper R. J. (2007) Copper chelation by d-penicillamine generates reactive oxygen species that are cytotoxic to human leukemia and breast cancer cells. Free Radic. Biol. Med. 43, 1271–1278 [DOI] [PubMed] [Google Scholar]
- 39. Udupi V., Rice-Evans C. (1992) Thiol compounds as protective agents in erythrocytes under oxidative stress. Free Radic. Res. Commun. 16, 315–323 [DOI] [PubMed] [Google Scholar]
- 40. Arthur J. R. (2000) The glutathione peroxidases. Cell. Mol. Life Sci. 57, 1825–1835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Flohe L., Brigelius-Flohe R. (2011) Selenoproteins of the glutathione peroxidase Family. In Selenium (Hatfield D. L., Berry M. J., Gladyshev V. N., eds) pp. 167–180, Springer, New York [Google Scholar]
- 42. Kryukov G. V., Castellano S., Novoselov S. V., Lobanov A. V., Zehtab O., Guigó R., Gladyshev V. N. (2003) Characterization of mammalian selenoproteomes. Science 300, 1439–1443 [DOI] [PubMed] [Google Scholar]
- 43. Gladyshev V. N., Hatfield D. L. (1999) Selenocysteine-containing proteins in mammals. J. Biomed. Sci. 6, 151–160 [DOI] [PubMed] [Google Scholar]
- 44. Cabello C. M., Bair W. B., 3rd, Wondrak G. T. (2007) Experimental therapeutics: targeting the redox Achilles heel of cancer. Curr. Opin. Investig. Drugs 8, 1022–1037 [PubMed] [Google Scholar]
- 45. Doroshow J. H., Akman S., Chu F. F., Esworthy S. (1990) Role of the glutathione-glutathione peroxidase cycle in the cytotoxicity of the anticancer quinones. Pharmacol. Ther. 47, 359–370 [DOI] [PubMed] [Google Scholar]
- 46. Batist G., Tulpule A., Sinha B. K., Katki A. G., Myers C. E., Cowan K. H. (1986) Overexpression of a novel anionic glutathione transferase in multidrug-resistant human breast cancer cells. J. Biol. Chem. 261, 15544–15549 [PubMed] [Google Scholar]
- 47. Samuels B. L., Murray J. L., Cohen M. B., Safa A. R., Sinha B. K., Townsend A. J., Beckett M. A., Weichselbaum R. R. (1991) Increased glutathione peroxidase activity in a human sarcoma cell line with inherent doxorubicin resistance. Cancer Res. 51, 521–527 [PubMed] [Google Scholar]
- 48. Lock R. B., Hill B. T. (1988) Differential patterns of anti-tumour drug responses and mechanisms of resistance in a series of independently-derived VP-16-resistant human tumour cell lines. Int. J. Cancer 42, 373–381 [DOI] [PubMed] [Google Scholar]
- 49. Saga Y., Ohwada M., Suzuki M., Konno R., Kigawa J., Ueno S., Mano H. (2008) Glutathione peroxidase 3 is a candidate mechanism of anticancer drug resistance of ovarian clear cell adenocarcinoma. Oncol. Rep. 20, 1299–1303 [PubMed] [Google Scholar]
- 50. Andreadis C., Gimotty P. A., Wahl P., Hammond R., Houldsworth J., Schuster S. J., Rebbeck T. R. (2007) Members of the glutathione and ABC-transporter families are associated with clinical outcome in patients with diffuse large B-cell lymphoma. Blood 109, 3409–3416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gillet J. P., Calcagno A. M., Varma S., Davidson B., Bunkholt Elstrand M., Ganapathi R., Kamat A. A., Sood A. K., Ambudkar S. V., Seiden M. V., Rueda B. R., Gottesman M. M. (2012) Multidrug resistance-linked gene signature predicts overall survival of patients with primary ovarian serous carcinoma. Clin. Cancer Res. 18, 3197–3206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schulz R., Emmrich T., Lemmerhirt H., Leffler U., Sydow K., Hirt C., Kiefer T., Link A., Bednarski P. J. (2012) Identification of a glutathione peroxidase inhibitor that reverses resistance to anticancer drugs in human B-cell lymphoma cell lines. Bioorg. Med. Chem. Lett. 22, 6712–6715 [DOI] [PubMed] [Google Scholar]
- 53. Park Y. S., Koh Y. H., Takahashi M., Miyamoto Y., Suzuki K., Dohmae N., Takio K., Honke K., Taniguchi N. (2003) Identification of the binding site of methylglyoxal on glutathione peroxidase: methylglyoxal inhibits glutathione peroxidase activity via binding to glutathione binding sites Arg 184 and 185. Free Radic. Res. 37, 205–211 [DOI] [PubMed] [Google Scholar]
- 54. Yang W. S., SriRamaratnam R., Welsch M. E., Shimada K., Skouta R., Viswanathan V. S., Cheah J. H., Clemons P. A., Shamji A. F., Clish C. B., Brown L. M., Girotti A. W., Cornish V. W., Schreiber S. L., Stockwell B. R. (2014) Regulation of ferroptotic cancer cell death by GPx4. Cell 156, 317–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Omerovic J., Hammond D. E., Clague M. J., Prior I. A. (2008) Ras isoform abundance and signalling in human cancer cell lines. Oncogene 27, 2754–2762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gaetani G. F., Galiano S., Canepa L., Ferraris A. M., Kirkman H. N. (1989) Catalase and glutathione peroxidase are equally active in detoxification of hydrogen peroxide in human erythrocytes. Blood 73, 334–339 [PubMed] [Google Scholar]
- 57. Vitai M., Góth L. (1997) Reference ranges of normal blood catalase activity and levels in familial hypocatalasemia in Hungary. Clin. Chim. Acta 261, 35–42 [DOI] [PubMed] [Google Scholar]
- 58. Dikalov S. I., Harrison D. G. (2014) Methods for detection of mitochondrial and cellular reactive oxygen species. Antioxid. Redox. Signal. 20, 372–382 [DOI] [PMC free article] [PubMed] [Google Scholar]