

Background: The HbN of Mycobacterium tuberculosis carries a potent nitric-oxide dioxygenase activity despite lacking a reductase domain.
Results: The NADH-ferredoxin reductase system acts as an efficient partner for the reduction of HbN.
Conclusion: The interactions of HbN with the reductase are modulated by its CD loop and the Pre-A region.
Significance: The present study provides new insights into the mechanism of electron transfer during nitric oxide detoxification by HbN.
Keywords: Electron Transfer, Hemoglobin, Hemoglobin Myoglobin, Microbial Pathogenesis, Molecular Biology, Mycobacterium Tuberculosis, Nitric Oxide
Abstract
Many pathogenic microorganisms have evolved hemoglobin-mediated nitric oxide (NO) detoxification mechanisms, where a globin domain in conjunction with a partner reductase catalyzes the conversion of toxic NO to innocuous nitrate. The truncated hemoglobin HbN of Mycobacterium tuberculosis displays a potent NO dioxygenase activity despite lacking a reductase domain. The mechanism by which HbN recycles itself during NO dioxygenation and the reductase that participates in this process are currently unknown. This study demonstrates that the NADH-ferredoxin/flavodoxin system is a fairly efficient partner for electron transfer to HbN with an observed reduction rate of 6.2 μm/min−1, which is nearly 3- and 5-fold faster than reported for Vitreoscilla hemoglobin and myoglobin, respectively. Structural docking of the HbN with Escherichia coli NADH-flavodoxin reductase (FdR) together with site-directed mutagenesis revealed that the CD loop of the HbN forms contacts with the reductase, and that Gly48 may have a vital role. The donor to acceptor electron coupling parameters calculated using the semiempirical pathway method amounts to an average of about 6.4 10−5 eV, which is lower than the value obtained for E. coli flavoHb (8.0 10−4 eV), but still supports the feasibility of an efficient electron transfer. The deletion of Pre-A abrogated the heme iron reduction by FdR in the HbN, thus signifying its involvement during intermolecular interactions of the HbN and FdR. The present study, thus, unravels a novel role of the CD loop and Pre-A motif in assisting the interactions of the HbN with the reductase and the electron cycling, which may be vital for its NO-scavenging function.
Introduction
Hemoglobins (Hbs) and flavohemoglobins (flavoHbs) are employed as the first line of defense by many pathogenic organisms to disarm the toxicity of macrophage-generated nitric oxide (NO) during intracellular infection (1–3). Truncated hemoglobin, HbN, of Mycobacterium tuberculosis (Mtb)2 protects its host from the toxic effects of NO due to its potent O2-dependent NO dioxygenase (NOD) activity (4–6). The HbN-deficient strain of Mycobacterium bovis displays extremely low NOD activity and lacks respiratory protection from NO as compared with the native strain (5), suggesting that the presence of HbN contributes to the survival ability of Mtb in the NO-enriched environment of macrophages (6). Raman spectroscopy revealed that heme iron coordination in MtbHbN is well suited for performing O2/NO chemistry for NO dioxygenation (7), which is modulated by two unusual structural features of the HbN: a 12-residue long highly flexible N-terminal Pre-A region that is required for the optimal NOD activity (8), and a protein tunnel system composed of short and long branches (9) that facilitates ligand entry to the distal heme site (9, 10). The functional role of the tunnel has been underlined in a dual path mechanism for ligand access to the heme cavity (11), in which binding of O2 to the heme triggers dynamical alterations in the protein backbone that regulate the opening of the long branch via a phenylalanine gate (Phe(E15)) (11, 12), thus facilitating access of NO to the O2-bound heme.
Bacterial and yeast flavoHbs, acting as NO-dioxygenase enzymes, are endowed with heme and flavin binding domains where (i) in a first step the oxyferrous heme interacts with NO to make nitrate and ferric heme, and (ii) subsequently the flavin domain shuttles an electron from NADH to the heme, which is converted to the ferrous state, thus enabling the protein to start the cycle again (1, 13, 14). The efficiency of the NOD function depends on both oxidative and reductive reactions (13, 14). The structural elements that determine NOD function are related to the stabilization of O2 into the deoxygenated heme, a fast migration of NO to the heme cavity, and its interactions with heme-bound O2, and a rapid electron transfer from an electron donor to recover the active ferrous state to start the fresh cycle (1, 15, 16). Although other members of the Hb superfamily can catalyze NO dioxygenation when paired with a suitable electron donor (17–19), all of them are not equally efficient (13). The lack of an integrated reductase diminishes the ability of many single domain Hbs and myoglobins (Mbs) to drive the NO dioxygenation cycle (16, 17), as noted in the poor NO oxidation activity of human Hb and sperm whale Mb (NOD rate constants, kNOD, of ∼89 and 43 μm−1 s−1, respectively) (16, 20). Furthermore, the partner reductases compatible to various oxidized Hbs and Mbs and their mechanisms for the electron transfer remain poorly defined. Unlike many single domain Hbs, the catalytic efficiency of NO dioxygenation (oxidative half) by MtbHbN is significantly high (kNOD ∼ 745 μm−1 s−1) and compares well with two-domain flavoHbs that include a reductase domain (1, 5). Therefore, it is intriguing how MtbHbN carries out the NOD function so efficiently, because the electron transfer event, which depends on the structural and functional compatibility with a cognate reductase, might limit the overall rate of NO dioxygenation. Any information on a partner reductase that can efficiently catalyze the reduction of the MtbHbN and the molecular determinants of the interaction between the MtbHbN and reductase is not available so far.
Because MtbHbN is functionally active in Escherichia coli, Salmonella enterica Typhimurium, and Mycobacterium smegmatis and allow these hosts to metabolize NO and resist nitrosative stress (4, 6, 8), it must be able to utilize heterologous reductases to carry out NO dioxygenation, although this process may not be specific to its native reductase system. Indeed, it can be argued whether the crucial role played by MtbHbN for the survival of the bacillus requires the expression of a specific reductase partner, which to the best of our knowledge has not been identified as yet. For efficient electron cycling, the reductase might simply require a precise coordination and structural alignment with the globin domain, so that the cofactor (heme and FAD) binding regions lie at a permissible distance for facile electron flow. Accordingly, it seems reasonable to hypothesize that MtbHbN may establish transient interactions with one or more partners depending on their structural and functional compatibility. In this context, it is important to identify reductases that carry out the reductive process of MtbHbN and the amino acid residues or regions involved in recognition and binding to a reductase partner.
In the absence of any information on the specific reductase partner involved in NOD function in Mtb and hence the mechanism of electron cycling, this study addresses the challenging question of the enzymatic reduction of MtbHbN by examining the feasibility of the reductive half-reaction for a set of reductases. Here we demonstrate that NADH-ferredoxin reductase (FdR) catalyzes efficiently the reduction of ferric MtbHbN required for its NOD function. Detailed spectroscopic and structural studies have unraveled the mechanism by which MtbHbN may establish the intermolecular alliance with a compatible reductase partner. The interaction involves its flexible CD loop region and is dependent on the dynamical motion of the protein backbone, which is modulated by its N-terminal Pre-A region.
EXPERIMENTAL PROCEDURES
Strains, Plasmids, and Culture Conditions
E. coli strains JM109 and BL21DE3 were used routinely for cloning and expression of recombinant genes. Bacterial cultures were grown in Luria-Bertani (LB) or Terrific Broth (containing 24 g of yeast extract, 12 g of Bacto-tryptone, 12.54 g of K2HPO4, 2.31 g of KH2PO4) medium at 37 °C at 180 rpm. When required, ampicillin and kanamycin (Sigma) were added at concentrations of 100 and 30 μg/ml, respectively. Plasmids, pBluescript (Stratagene), and pET28c (Novagene) were used for cloning and expression of the HbN encoding gene of Mtb and its site-directed mutagenesis as described earlier (4, 8). Construction of the Pre-A-deleted mutant of HbN (HbNΔPre-A) has been reported earlier (8). The oligonucleotides were custom synthesized by Essence Life Sciences, India.
Cloning, Expression, and Purification of Reductases and HbN Reductase Chimeras
Reductase encoding genes of Mtb and E. coli were amplified from the genomic DNA by a standard PCR method using gene-specific forward and reverse primers, designed on the basis of their known coding sequences. Genes for the Mtb reductases TrxB (Rv3913), KshB (Rv3571), as well as the E. coli NADH-flavodoxin reductase (FdR, Ec948414) were amplified by PCR, authenticated by nucleotide sequencing, and expressed under the T7 promoter of pET28c. Wild type and HbN mutants were purified by ion exchange chromatography (DEAE-Sepharose, Sigma), equilibrated with 10 mm Tris·Cl (pH 8.0) and eluted using NaCl gradient, as mentioned previously (13).
HbN reductase chimeras HbN-KshB and HbN-FdR were also generated. To this end, the reductase domain was attached to the C-terminal portion of Mtb HbN with the help of a 12-residue long linker (-GTASVLKAIDTG-) carrying two glycine residues to allow sufficient flexibility for the interactions between HbN and the reductase. To generate the chimera, first the HbN and the reductase encoding genes were amplified separately by PCR. Three overlapping primers (5′-TGATGCTGTTCCGACTGGTGCCGTGGT-3′; 5′-CGCTTTCAGTACTGATGCTGTTCC-3′; 5′-ACCGTAGAATTCTCCCGTGTCAAT-3′) encoding the linker region and carrying a EcoRI restriction site at the 3′-end were incorporated at the C terminus of the glbN gene by standard overlapping PCR. The linker carrying the glbN gene was then joined with different reductase encoding genes via the EcoRI site and expressed under the T7 promoter of pET28c. Reductases and HbN chimera were purified by nickel-nitrilotriacetic acid affinity chromatography using the manufacturer's protocol. Expressions of recombinant proteins were visualized on a 15% SDS-PAGE. Redox states of proteins were checked spectrophotometrically by recording the Soret α and β peaks.
Isolation of Glycosylated HbN from the HbN Overexpressing Mtb
The HbN was overexpressed as a His-tagged protein in M. tuberculosis H37Ra and purified as described previously (21). Briefly, HbN expressing cells of Mtb were collected by centrifugation and suspended in 10 mm Tris·Cl (pH 7.8) with lysozyme (10 μg/ml) to incubate at 37 ºC for 30 min. The cell suspension was then lysed with French Press and HbN was purified by metal affinity chromatography following standard procedures.
Measurements of Kinetics of the HbN Reduction
The assay developed by Hayashi et al. (18) was used here with a slight modification as described elsewhere (13). Briefly, the reaction was set up in a sealed cuvette chamber carrying a reaction mixture consisting of different concentrations of oxidized HbN and NADH in a 0.1 m phosphate buffer (pH 7.2) saturated with CO. The reaction was initiated by adding the reductase with the help of a gas tight Hamilton syringe. The time course for the shift of the Soret peak from 406 nm (oxidized form) to the reduced form of heme (resulting binding of CO) was recorded at 420 nm and reduction of FAD was followed at 460 nm. The difference in absorbance between ferric and CO-bound species of HbN was plotted with respect to time to calculate the initial velocity of the reaction.
Measurement of Cofactors and NO Oxidation Profile of HbN
The heme content of cells was measured by the procedure described earlier (4, 22). Briefly, heme proteins were re-suspended in 2.2 m pyridine, in 0.1 n NaOH. Heme concentrations were determined by measuring the difference in absorption spectra at 556 and 539 nm for dithionite-reduced and ferricyanide-oxidized protein, respectively. FAD content of proteins was checked as described elsewhere (23). Briefly, the flavin cofactor of the protein was extracted after boiling for 5 min and centrifugation at 14,000 × g. FAD content was determined through fluorescence at 520 nm with excitation at 460 nm using an extinction coefficient of 11.6 mm−1 cm−1. Titration of oxygenated MtbHbN and its mutants by NO was checked essentially as described earlier (8, 12), and profiles of NO-induced oxidation were recorded after each addition of NO for the conversion of oxygenated HbN into the oxidized state.
Structural Docking of MtbHbN and FdR
The x-ray structure of FdR from E. coli (PDB code 1FDR; 1.7-Å resolution) (24) and MtbHbN (PDB code 1IDR, chain A; 1.9-Å resolution) (11) were used to generate the three-dimensional models of the protein-protein complex with ClusPro2.0 (25). First, a rigid body docking was performed with PIPER (26), a program that relies on the Fast Fourier Transform-based approach used with pairwise interaction potentials. The top 1000 ranked solutions obtained from PIPER were then processed with ClusPro, a clustering algorithm that groups the solutions using the root mean square deviation between solutions with a cut-off of 9 Å. Finally, the generated models were classified according to the weight of different coefficients as balanced, electrostatic favored, hydrophobic favored, and van der Waals +electrostatic favored. Choice of the final model was made on the basis of the lowest energy using the balanced ClusPro score (25, 26), the solution with the highest cluster members, and the lowest distance between the heme prosthetic group in MtbHbN with respect to the flavin cofactor in FdR, and further validated by visual inspection of the contacts at the protein-protein interface.
Molecular Dynamics Simulations
The three-dimensional structural model of the HbN-FdR complex was refined by means of extended molecular dynamics (MD) simulation (0.5 μs) performed with the PMEMD.cuda module of the AMBER12 suite (27) using the parm99SB force field (28). The heme parameters were developed and thoroughly tested in previous works (29, 30). Parameters for the FAD cofactor were taken from Antony et al. (31), although the atomic charges were assigned by fitting the atomic point charges to the B3LYP/6–31G(d) quantum mechanical electrostatic potential using the RESP method (32).
To set up the systems for MD simulations, the complex was immersed in a pre-equilibrated octahedral box of TIP3P (33) water molecules and counterions were added to maintain the neutrality of the simulated system. The final system contained the protein, 6 Na+ cations, and around 23,400 water molecules (∼76,200 atoms). The equilibration protocol consisted of performing an optimization of the initial structures, followed by slow heating to the target temperature (298 K). The heating was performed in 2-ns MD at constant volume, followed by 2-ns MD at constant pressure (1 bar). Once the equilibration was completed, the system was simulated in the NPT ensemble using periodic boundary conditions, the SHAKE algorithm (34) to constrain the bonds connecting hydrogen, and heavy atoms in conjunction with a 1-fs time step for the integration of Newton's equations with a Langevin collision frequency of 2 ps−1 (35), particle mesh Ewald for electrostatic interactions (36), and a cutoff of 10 Å for noncovalent interactions. Frames were collected at 10-ps intervals for subsequent analysis of the trajectory.
Electron Transfer Pathway
The semiempirical pathway method was used to identify the most plausible pathway for electron transfer (ET) between the heme group and the FAD isoalloxazine ring, as estimated using the algorithm developed by Beratan et al. (37, 38). Briefly, the method looks for the path that exhibits the highest donor to acceptor electron coupling (HAB). For a given path, HAB is computed as the product of a number of steps, each with a given coupling value, that defines the pathway. A step can either be through atom, when two orbitals share an atom, which are assumed to have a coupling value of 0.6, and through space, when orbitals are separated by empty space, for which the coupling is assumed to decay exponentially with the orbital-to-orbital distance by using a decay factor of −1.7. The HAB value (in eV) is finally obtained by multiplying the obtained decay factor for the best ET path by 1 eV, which is the default value in the algorithm. This value represents in a qualitative way the feasibility for the electron transfer process for a given path.
To obtain a proper description of the system, minor adjustments of the parameters used in the pathway algorithm were required. First, all orbitals of the heme iron were considered to be equivalent. Second, couplings between all porphyrin and FAD isoalloxazine ring located orbitals were set to one to reproduce their resonant character. Calculations were performed for snapshots taken at 10-ps intervals during the whole MD simulation. This strategy has been successfully used in previous works to study the role of protein fluctuations on the ET pathways in several systems, including heme and FAD containing proteins (39–41).
Peptide Synthesis
To design the peptide for the CD loop region, the interface between globin and the reductase was examined, paying attention to the placement of the hydrophobic and hydrophilic residues at the interaction site. On the basis of this information, a database of venom peptides (42) was scanned to identify peptides that can be effective in disrupting the interactions of the CD loop due to its composition of hydrophobic and hydrophilic residues. One of the peptides (ASMSECDPAEHCTGQSSECPADVFHK) was selected to check its specificity for masking the CD loop and interrupting the ET, whereas keeping unaffected both the oxygen binding and NO oxidation properties of the HbN. Furthermore, a nonspecific peptide with a randomly generated amino acid sequence (EDAQIQVTYYDKAEKDYVTKS) was used as a control. The peptides were synthesized using the IMTECH synthesis facilities by automated solid phase peptide synthesis using the 9-fluorenylmethoxycarbonyl-tert-butyl (Fmoc/tBu) strategy. The peptides were purified through high performance liquid chromatography (HPLC) and their identity was confirmed by matrix-assisted laser desorption ionization (MALDI) mass spectrometry.
RESULTS
Interactions of Mtb HbN with Native and Heterologous Flavoreductases
Because NADP-ferredoxin/flavodoxin reductase systems are known to reduce met-Hb into the ferrous state (43) and similar reductase modules are found naturally fused with globins in two-domain flavoHbs, a series of flavoreductase systems were selected for testing the enzymatic reduction of MtbHbN. Two different flavoreductases of Mtb, e.g. KshB, and TrxB and NADH-ferredoxin reductase (FdR) of E. coli that exhibits sequence/structural similarity to the reductase domain of flavoHbs were taken up for testing their potential for the reduction of HbN. This assay exploits the ability of Hbs to form a CO-adduct in its ferrous state, giving a specific Soret peak at 420 nm after conversion of Fe3+ to Fe2+ in the presence of a compatible redox partner. Accordingly, the efficiency of MtbHbN reduction was monitored spectrophotometrically by the shift and increase in the Soret peak from 406 to 420 nm due to formation of the CO adduct of HbN in the presence of different reductases.
The efficiency of the redox state change in MtbHbN varied in the presence of different reductases. Among these, FdR of E. coli was found to be the most effective (Fig. 1A), whereas the HbN reduction by TrxB and KshB was very slow in the presence of NADH. The rate of heme iron reduction in the HbN by FdR was estimated as 6.2 μm/min−1 (Table 1). At 250 μm NADH, the optical spectrum of the flavin changed very fast and nearly reached the fully reduced state in less than 30 s, whereas it remains partially reduced at lower concentrations (Fig. 1B). Therefore, reactions of the HbN with different reductases were checked at the steady state level of reduced flavin during the phase when the absorbance at 460 nm reached the plateau. In the presence of 250 μm NADH, a rapid decrease occurred at 460 nm due to FAD reduction in the presence of all three flavoreductases (Fig. 1B), indicating rapid reduction of FAD and initial production of FADH2. Despite that reduction of the HbN still occurred very slowly in the case of KshB and did not occur at all in the case of TrxB (Fig. 1A), suggests that the ET from FAD to the heme might be rate-limiting. These results indicated that FdR is an efficient redox partner for the enzymatic reduction of the MtbHbN.
FIGURE 1.
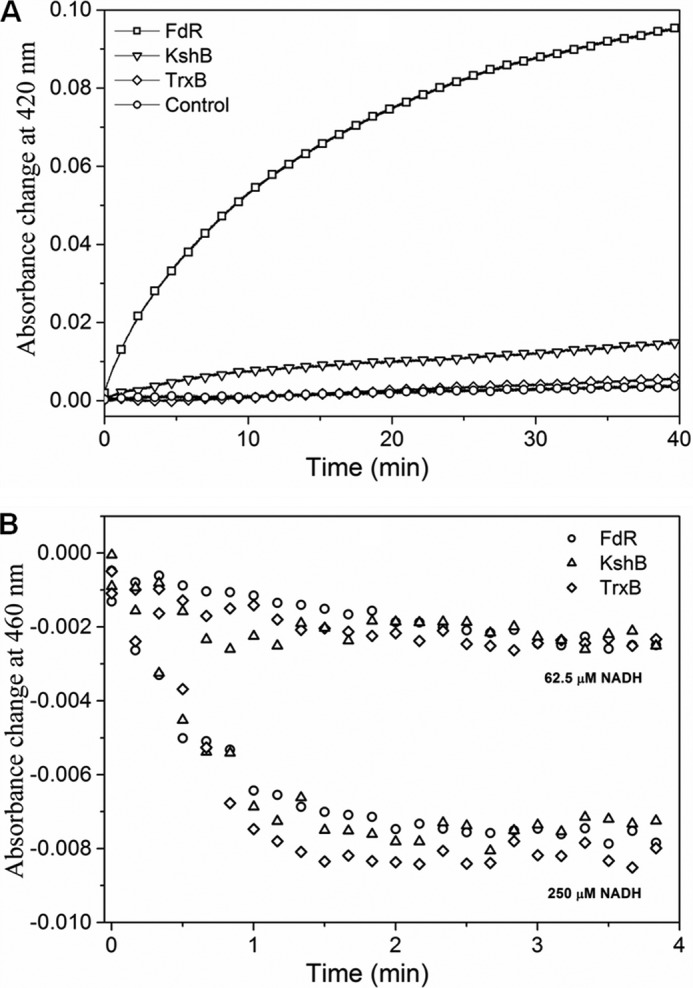
Reduction profile of oxidized HbN in the presence of different reductases. A, kinetic traces of the absorbance change at 420 nm for the reaction between ferric HbN with flavoreductases associated with binding of CO to ferrous HbN. Oxidized HbN (8 μm) was mixed with flavoreductases (1 μm) in the presence of 250 μm NADH and CO. Shift in the Soret peak of oxidized HbN (406 nm) to 420 nm (CO adduct formation) was recorded at different time intervals. B, profile of FAD reduction by NADH during interactions of ferric HbN with flavoreductases. Absorbance change at 460 nm was recorded for the reduction of FAD.
TABLE 1.
Kinetic parameters for in vitro heme reduction of MtbHbN and its mutants
| Protein complex | Km | kcat | Heme iron reduction | Ref. |
|---|---|---|---|---|
| μm | min−1 | μm/min | ||
| HbN-FdR | 19.9 | 13 | 6.2 | This study |
| HbN-KshBa | This study | |||
| HbNS47W-FdR | 20.9 | 9.9 | 5.1 | This study |
| HbNG48I-FdR | 19.4 | 8 | 4.2 | This study |
| HbNT49W-FdR | 20.0 | 13.2 | 6.2 | This study |
| HbNΔPre-A-FdR | 16.3 | 1.5 | 0.9 | This study |
| VHbb-FdR | 12.4 | 7 | 2.2 | This study |
| Mb-FdR | 15 | 3.5 | 1.3 | Our estimation |
| Mb-FdR | 17 | 5 | 2 | 13 |
a Not determined due to extremely slow reaction.
b VHb (Vitreoscilla hemoglobin) as reported elsewhere (19).
Reduction of Glycosylated HbN by FdR
Because HbN is post-translationally modified by glycosylation in its native host, Mtb (21), we further checked the reduction profile of glycan-linked HbN isolated from the HbN overexpressing strain of Mtb and compared its reduction profile with its unglycosylated counterpart along with other single domain Hbs, e.g. Vitreoscilla hemoglobin (VHb) and myoglobin (Mb), using FdR as a reductase partner. Traces of time course reduction of these Hbs by FdR are shown in Fig. 2. The difference in absorbance between ferric and CO-bound protein was plotted versus time to calculate the initial velocity of the reaction (under the conditions used in this study, the velocity was directly proportional to the concentration of FdR). The reduction efficiency of glycan-linked HbN was found comparable with its unglycosylated counterpart and nearly 3- and 5-fold higher than the reduction of VHb and Mb, respectively (Fig. 2, Table 1). Kinetic parameters of heme reduction by FdR for VHb and Mb are given in Table 1.
FIGURE 2.
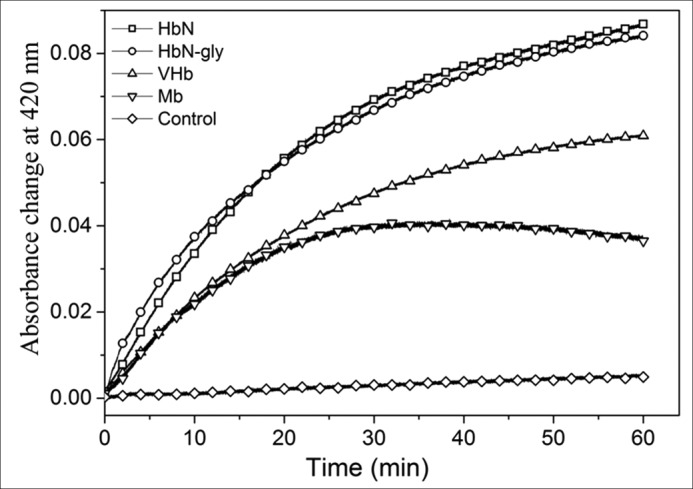
Comparison of reduction profile of glycosylated HbN with different single domain Hbs in the presence of FdR. Comparative reduction profile of HbN, glycosylated HbN (HbN-gly), Vitreoscilla hemoglobin (VHb), and Mb in the presence of FdR. The control reaction was set without adding FdR.
Integration of a Reductase Domain with HbN and Construction of HbN Reductase Chimeras
Because the efficiency of MtbHbN reduction varied significantly with different reductases, we selected Mtb KshB and E. coli FdR as representative slow and fast acting reductases, respectively, to gain insight into the mechanism of HbN reduction. To this end, the two reductases were integrated individually at the C terminus of HbN to mimic the two-domain flavoHb-like structure. These chimeras were created with the notion that covalent linkage between reductase and HbN will speed up the formation of close contacts between heme and FAD binding regions, thus facilitating the ET process. The HbN-KshB and HbN-FdR chimeras were cloned, expressed, and purified. The purified proteins exhibited expected masses of 54 and 42 kDa for HbN-KshB and HbN-FdR, respectively, and conferred reddish tinge to the cell when over-expressed in E. coli. The two chimeras were found associated with cofactors (heme and FAD) and their absorption spectra resembled the oxy-form of MtbHbN with the Soret peak at 414 nm and α and β peaks at 540 and 575 nm, respectively, indicating the presence of a functional heme domain.
The reduction profiles of the HbN chimeras were determined by monitoring the change in absorbance at 460 nm for the reduction of FAD, and 420 nm for the reduction of heme and CO adduct formation, and compared with the profile obtained for E. coli HMP. A fast decrease in the absorbance at 460 nm was observed for the two chimeras in the presence of NADH (250 μm), the reduction profiles being very similar to the decrease obtained for the separate reductases (data not shown). Moreover, they resembled the decrease observed for E. coli HMP, indicating a rapid reduction of FAD and formation of FADH2 in all the cases. However, no distinct change was observed at 420 nm for the HbN-KshB chimera, whereas a rapid increase in absorbance at 420 nm due to the binding of CO to the reduced heme was found for HbN-FdR (Fig. 3A). On the other hand, the reduction of the ferric HbN-KshB chimera was restored and the Soret peak shifted to 420 nm when FdR was added in the reaction mixture (Fig. 3B). It is thus likely that the heme and FAD domains are not in close contact with each other in the HbN-KshB chimera, thus allowing FdR to interact with the heme domain of HbN-KshB.
FIGURE 3.
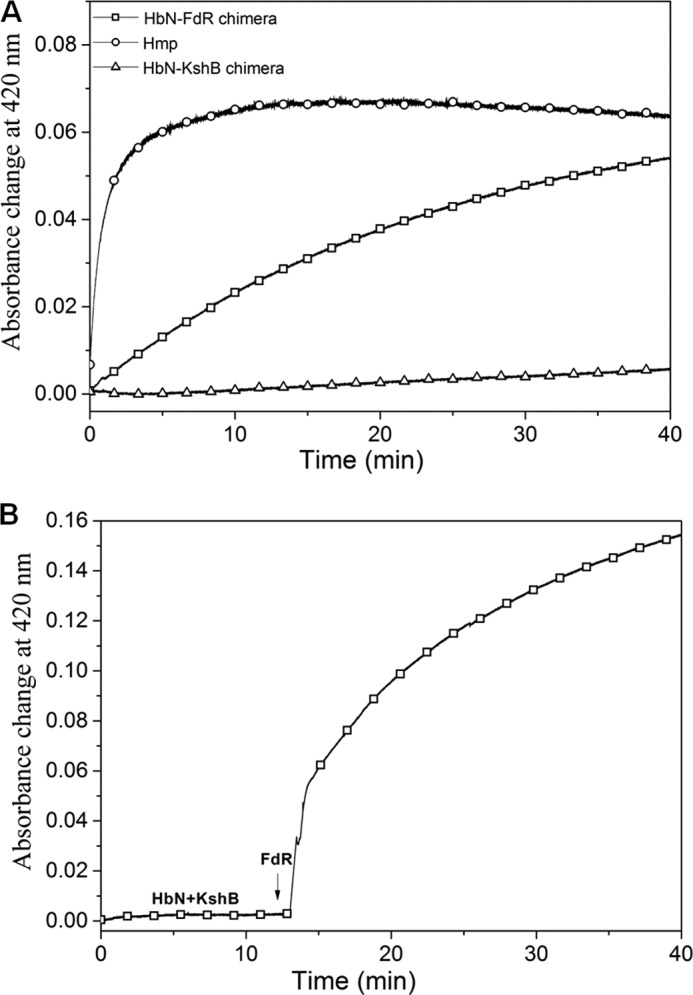
Reduction of HbN-reductase chimera. A, autoreduction profile of E. coli HMP, HbN-FdR, and HbN-KshB chimeras in the presence of 250 μm NADH. B, autoreduction of the HbN-KshB chimera in the presence of 250 μm NADH before and 10 min after the start of reaction when FdR was added.
Molecular Docking and Identification of Reactive Configuration of HbN-FdR
Due to the high efficiency in converting ferric HbN to the ferrous species, FdR was chosen as a model reductase to identify key molecular elements of the HbN structure involved in interaction with the reductase and electron flow to ferric HbN. Docking was used to explore the interactions between HbN and FdR that can place both heme and FAD cofactors in a permissible distance for the ET using ClusPro2.0. Of various possible protein-protein poses (Table 2), the most probable one was selected on the basis of the largest number of members in the cluster of docked solutions (120 poses), the lowest energy as determined by using the balanced ClusPro score (−907.4; score units) and the shortest distance from the geometrical centers of heme and isoalloxazine rings (18.3 Å). In the docked pose the interaction primarily involves the CD loop of HbN, which lies close to the C terminus of FdR. It is worth noting that the docked structure resembles the crystal structure of flavoHb (FHP) from Alcaligenes eutrophus (PDB entry 1CQX) with respect to the placement of two co-factor binding regions and the nature of residues at the protein-protein interface (data not shown).
TABLE 2.
Details of the docking results obtained using the balanced ClusPro score (E = 0.40Erep − 0.40Eatt + 600Eelec + 1.00EDARS (21)) for the 10 most populated protein-protein complex clusters indicating for each of them the distance between de centers of geometry of the heme group in MtbHbN and the flavin group in FdR
| Cluster | Members | Representative | Weighted Score | Distance |
|---|---|---|---|---|
| Å | ||||
| 1 | 120 | Center | −608.0 | 18.3 |
| Lowest energy | −907.4 | |||
| 2 | 117 | Center | −606.5 | 29.6 |
| Lowest energy | −761.6 | |||
| 3 | 113 | Center | −627.5 | 21.7 |
| Lowest energy | −827.5 | |||
| 4 | 79 | Center | −686.6 | 34.5 |
| Lowest energy | −744.5 | |||
| 5 | 71 | Center | −584.6 | 20.5 |
| Lowest energy | −703.9 | |||
| 6 | 55 | Center | −588.3 | 28.2 |
| Lowest energy | −728.5 | |||
| 7 | 52 | Center | −605 | 24.2 |
| Lowest energy | −640.6 | |||
| 8 | 49 | Center | −586.5 | 19.5 |
| Lowest energy | −675.8 | |||
| 9 | 43 | Center | −701.8 | 41.5 |
| Lowest energy | −701.8 | |||
| 10 | 34 | Center | −607.8 | 19.4 |
| Lowest energy | −668.5 |
Refinement of the docked pose was accomplished by means of an extended MD simulation, which confirmed the structural integrity of both HbN and FdR in the complex (Fig. 4). Thus, the root mean square deviation of the backbone atoms along the trajectory generally ranged from 1.0 to 2.0 Å for both FdR and HbN (note that the highly flexible Pre-A and loop F regions were excluded from the analysis for HbN). Nevertheless, the relative position of the two proteins varied significantly along the MD simulation, leading to a configuration that was stable during the last 200 ns of the trajectory (Fig. 5A), in which the distance between heme and flavin cofactors ranged from 6 to 9 Å, although contacts between the heme propionate and the FAD isoalloxazine ring as short as 5 Å were transiently achieved (Fig. 5B).
FIGURE 4.

Representative snapshot of the binding interface between HbN and FdR. A, representation of the relative orientation of HbN and FdR. B, detailed view of selected interactions formed at the interface between the two proteins. The backbone of HbN and FdR is shown as cyan and yellow schematics, respectively, and the heme and FAD units are shown as green and magenta sticks. Residues that participate in salt bridge or hydrophobic contacts between the proteins are shown as red and orange spheres, respectively. The part of the CD loop that partially fills the region between heme and FAD is shown in blue.
FIGURE 5.
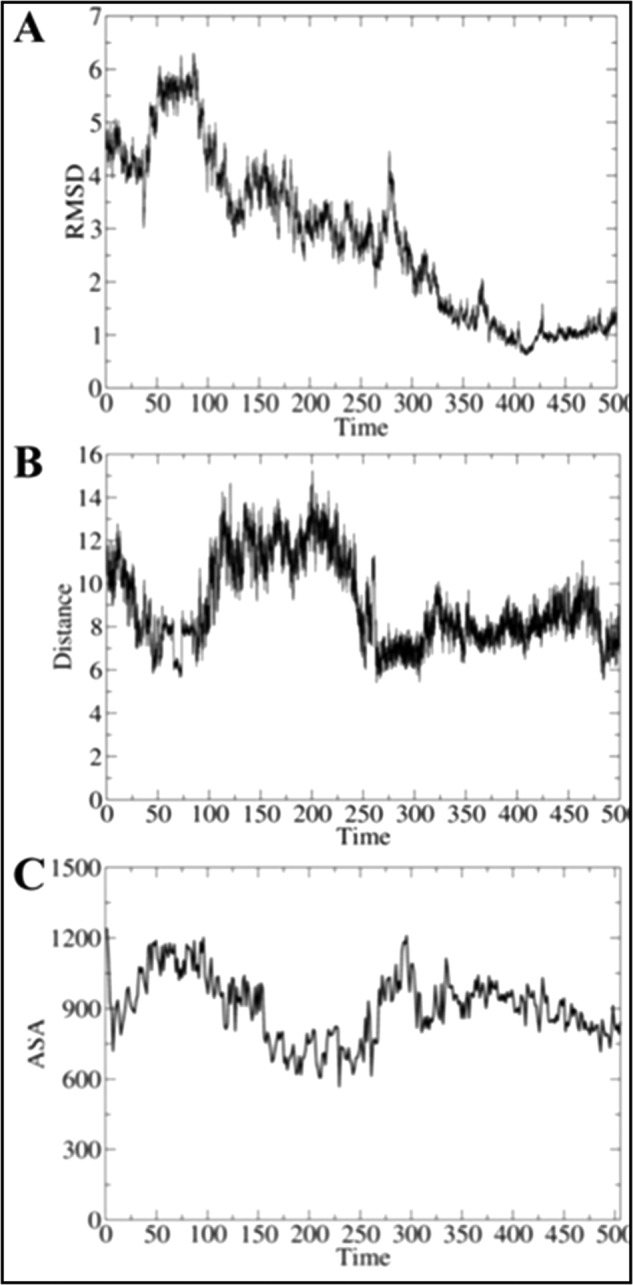
Time evolution of selected structural parameters of the HbN-FdR complex. Time evolution (ns) of (A) the root mean square deviation (RMSD; Å) determined for the backbone atoms of the HbN-FdR complex, (B) the distance (Å) between the isoalloxazine ring (methyl group at position C8) and one of the heme propionate groups, and (C) the accessible solvent area (ASA; Å2) of the interface for the HbN-FdR complex.
In the final structure (Fig. 4), close contacts in the complex involve residues in helix B, the CD loop, and the beginning of helix E and F-G regions of HbN with the last 40 residues in the N-terminal segment of FdR, leading to an interface characterized by a solvent-accessible area of around 900 Å2 (Fig. 5C). The protein-protein interface is stabilized by hydrophobic contacts between residues Leu36 and Met50 (HbN) with Pro339, Val342, and Ala368 (FdR). Furthermore, additional stabilization is afforded by up to five salt bridge interactions (Glu25–Arg362, Glu29–Lys357, Asp39–Arg343, Arg52–Glu226, and Arg83 with the phosphate α of FAD), which are distributed around the central hydrophobic cluster. Finally, a series of polar residues in the CD loop (Ser47, Thr49, and Asn50) fill the space between the heme propionates and the isoalloxazine ring of FAD.
As a last remark, it is worth noting that the three-dimensional model of the HbN-FdR complex locates the C terminus of HbN and the N terminus of FdR pointing toward the same region. Keeping in mind both the intrinsic flexibility of the last eight residues in HbN (not seen in the x-ray structure) and the length of the 12-residue long linker used to build up the chimeras, molecular modeling analysis (data not shown) showed that the spatial proximity of the C and N terminus of HbN and FdR, respectively, is consistent with the efficient ET rate of HbN-FdR chimera, which should be capable of adopting the same three-dimensional structure as obtained for the separate proteins.
An estimation of the electron transfer efficiency, given by the electron coupling determined from the snapshots sampled along the whole trajectory was found to be about 6.4 × 10−5 eV, the highest value being 9.0 × 10−4. It is worth noting that these values compare well with the electron coupling reported for the E. coli flavoHb (7.1 × 10−5 eV for the x-ray structure, and 8.0 × 10−4 eV from the snapshots sampled in MD simulations) (37), a system that is known to cycle electrons very efficiently. The slightly lower average value obtained for the HbN-FdR complex possibly reflects the fact that ET involves a protein-protein complex, whereas in E. coli flavoHb it involves the intramolecular ET between domains in the same protein.
The polar residues in the CD loop, together with water molecules, have a major contribution to the ET pathways (Fig. 6A). Thus, Ser47, Gly48, and water residues contribute to define the ET pathway during the whole trajectory, they being involved in around 28, 42, and 29% of the ET pathways. A series of representative ET pathways sampled at the end of the trajectory are shown in Fig. 6B. A detailed analysis of the pathways reveals that the ET from the FAD isoalloxazine ring to the heme follows three major pathways (Fig. 6B): (i) a direct transfer through the backbone of Ser47 and/or Gly48 residues, (ii) assisted by a water molecule prior to transfer to residues in the CD loop, and (iii) directly through a water molecule. This latter pathway agrees with the trends found for the ET in E. coli flavoHb (38), where a water molecule was found to assist the ET from the isoalloxazine ring to the heme (37). Nevertheless, it is worth noting that calculations predict a large electron coupling even in the absence of water molecules (about 5.0 × 10−5 eV), which highlights the critical role of the CD loop in assisting ET in the HbN-FdR complex.
FIGURE 6.
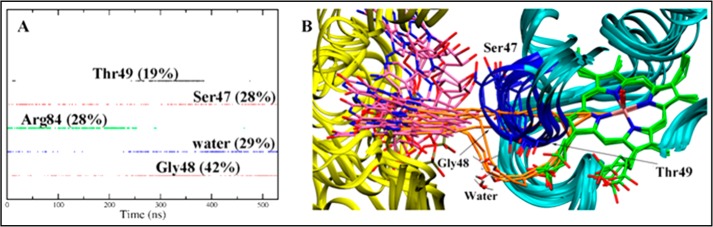
Representation of the major ET pathways. A, contribution of selected residues to the ET pathways. B, superposition of representative snapshots together with the associated ET pathways (shown in “orange”) from the FAD isoalloxazine ring to the heme in the process of MtbHbN reduction. Heme and FAD are shown as green and magenta sticks. Residues in the CD loop are shown in blue.
Role of CD Loop of Mtb HbN in Protein-Protein Interactions and Electron Transfer Pathway
To further confirm the role of the CD loop in defining the interface between MtbHbN and FdR, we designed a short peptide against the CD loop region, anticipating that binding of the peptide at the interface region of FdR will alter the electron flow. To check the specificity of this peptide against the CD loop, a randomly designed peptide was also used as a control to check any nonspecific effect on the reduction of ferric HbN in the presence of FdR. In the presence of the CD loop-specific peptide, the Soret peak of ferric HbN at 406 nm remained unchanged as compared with the control reaction, in which this peptide was not added. In contrast, when a nonspecific random peptide was added in the reaction mixture, ferric HbN was readily reduced in the presence of FdR as observed by the shift in the Soret peak to 420 nm due to the formation of the CO-adduct of HbN (Fig. 7A), signifying the reduction of HbN.
FIGURE 7.
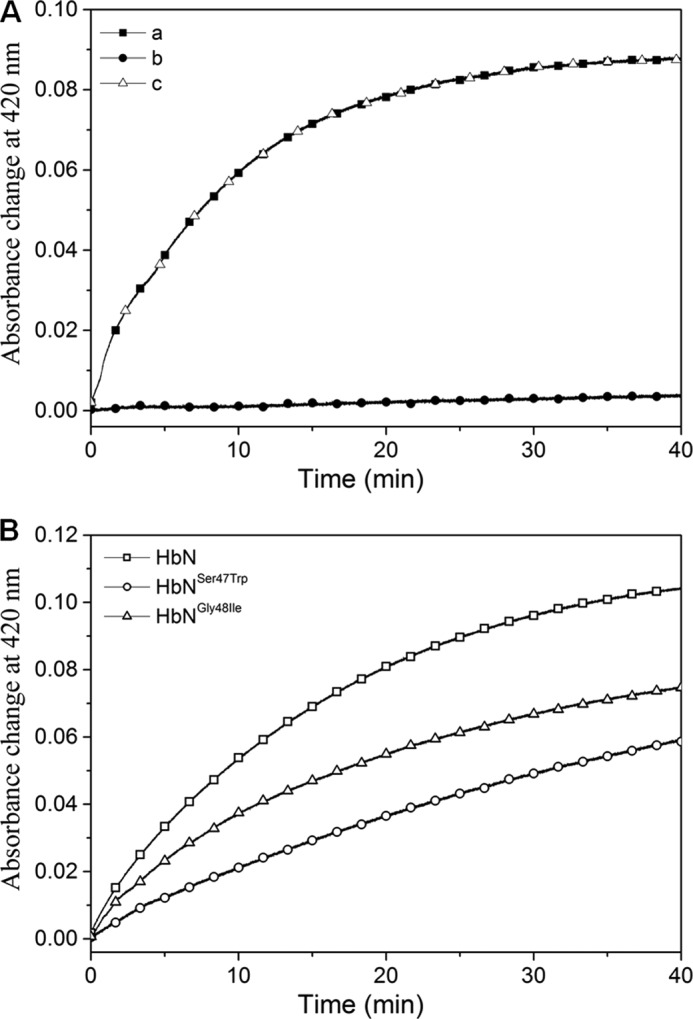
Effect of CD-loop specific peptide and CD-loop mutation on reduction of MtbHbN by FdR. A, reduction of MtbHbN (8 μm) in the presence of CD loop-specific and nonspecific peptide (40 μm) by FdR (1 μm). A, control reaction with HbN-FdR; B, HbN-FdR with CD loop peptide; C, HbN-FdR with nonspecific peptide. B, reduction profile of HbN CD loop mutants by FdR recorded as absorbance change at 420 nm due to conversion of ferric HbN into ferrous state and binding of CO.
Site-directed Mutagenesis of CD Loop of MtbHbN
Because structural and MD simulation studies supported the involvement of CD loop residues Ser47, Gly48, and Thr49 in defining the ET pathway, the role of these residues was further checked by site-directed mutagenesis. Thus, Ser47, Gly48, and Thr49 were mutated to Trp, Ile, and Trp, respectively, to generate HbN mutants, HbNS47W, HbNG48I, and HbNT49W, and the efficiency of these mutants for reduction of heme in the presence of FdR was determined and compared with the wild type protein. As shown in Fig. 7B, all mutants were less efficient than MtbHbN in formation of the CO adduct, this effect being specially relevant for the HbNG48I mutant (see also Table 1). At this point, it is worth noting that reduction of FAD remained unaltered during interactions of the mutants with FdR (data not shown), thus suggesting that the lower efficiency in promoting the reduction of ferric HbN reflects a less efficient ET from the reductase.
Pre-A Motif of MtbHbN Is Required for the Intermolecular Electron Transfer in MtbHbN-FdR Complex
Previous studies have shown that the Pre-A motif modulates the dynamics of the MtbHbN backbone to carry out the NOD function, whereas its deletion compromises the NOD function due to the hindered backbone motion (8, 12). The main difference in the dynamics of protein structure was observed around B and E helices and the movement of CD and EF loop regions, suggesting that Pre-A deletion may affect the intermolecular interactions of HbN with FdR and the efficiency of HbN reduction. To check this possibility we compared the ability of wild type HbN and its Pre-A-deleted mutant to be reduced in the presence of FdR. Interestingly, the Pre-A-deleted mutant displayed extremely slow conversion from Fe3+ HbN to the Fe2+ species, as observed from assays of CO binding to the ferrous species (Fig. 8A). The Km (16.3 μm) of the Pre-A mutant appeared slightly lower than the wild type HbN but the heme reduction was drastically reduced, corresponding to 0.9 μm−1 s−1.
FIGURE 8.
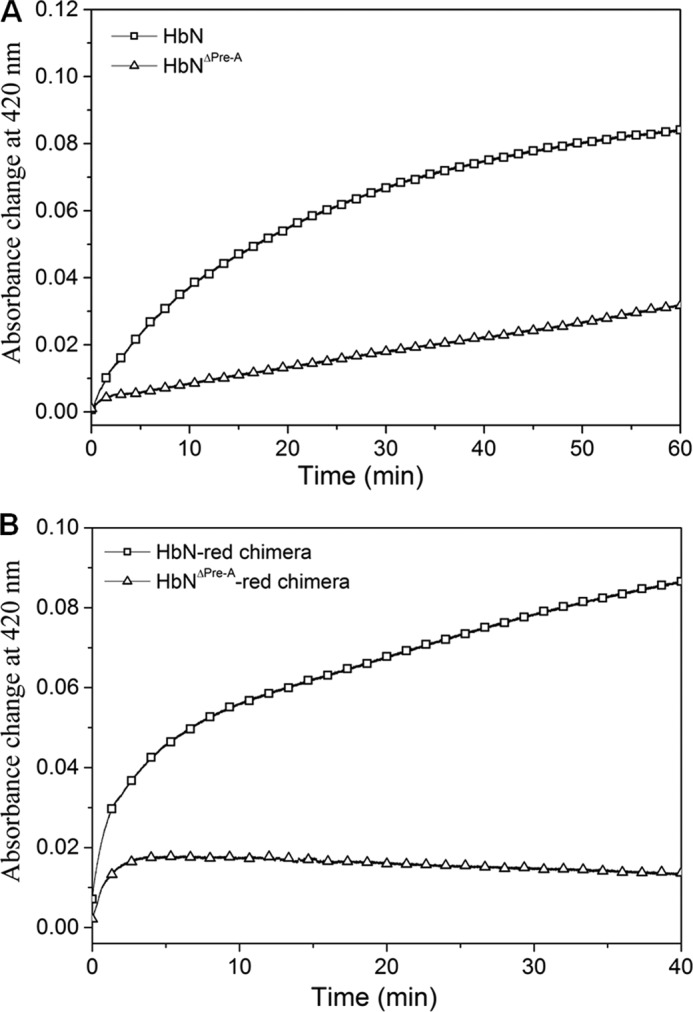
Reduction profile of (A) wild type and Pre-A mutant of HbN by FdR and (B) autoreduction of HbN-FdR and HbNΔPre-A-FdR chimeras. The graph depicts the efficiency of wild type and mutant HbN reduction as recorded by the increase in the absorbance at 420 nm (Soret peak) due to the formation of the CO-adduct after conversion of oxidized HbN into the reduced state of MtbHbN and Pre-A-deleted HbN (HbNΔPre-A) (A) and HbN-FdR chimera and Pre-A deleted HbN-FdR chimera (HbNΔPre-A-red) (B).
To further substantiate that the Pre-A motif is modulating the reduction ability of MtbHbN, we also deleted the Pre-A region in the chimera of HbN-FdR. Separately, we also created another chimera of HbN with the reductase domain of E. coli HMP that exhibited highly efficient ET ability (data not shown here) and then deleted the Pre-A region from the HbN-HMP chimera. In both cases, reduction of the globin domain was completely abrogated, indicating the requirement of Pre-A in regulating the CD loop of HbN for establishing the intermolecular contacts with the reductase for the catalytic electron cycling required for NOD function (Fig. 8B).
DISCUSSION
A large number of two-domain flavoHbs and Hbs in association with a suitable reductase are known to participate in NO detoxification (1, 2, 44, 45). Despite lacking an integrated reductase partner, MtbHbN displays an efficient NOD (kNOD = 745 μm−1 s−1) that compares well with flavoHbs (kNOD = 800–2400 μm−1 s−1) and is severalfold higher than Hbs and Mbs (1). The missing link in understanding the NOD function of the MtbHbN, which is integral to its uninterrupted NOD activity, is the molecular mechanism by which HbN associates with a reductase partner to replenish itself from a ferric to ferrous state for the next cycle of NOD activity. The reductase(s) compatible for the reductive half of the HbN-mediated NOD reaction is currently unknown.
To support the NOD function of MtbHbN, an efficient reduction is required to guarantee the survival of Mtb. Our results show that the reduction of MtbHbN is accomplished with a wide range of efficiency for distinct reductases, varying from the low enzymatic reduction triggered by Mtb flavoreductases KshB and TrxB to the high efficiency of E. coli FdR. This binary interaction may be similar to the mammalian membrane-bound microsomal NADPH-cytochrome 450 reductase, which is structurally related to ferredoxin reductase and participates in NOD function together with single domain Hbs displaying flavoHb-like properties (44). Thus, our results show that the NOD function of MtbHbN is not restricted to its native electron donating partner. Indeed, it is retained during its expression in various heterologous hosts (4, 6). Because ferredoxin/flavodoxin reductases are widely distributed in nature, they may complement with the HbN for sustaining its NOD function in a wide range of bacterial hosts.
During the reductive process, electrons are sequentially transferred from NADH to FAD and then to the heme iron, as has been demonstrated in the case of E. coli HMP (14) and binary interactions of metHb with cytochrome b5 reductase (16, 44, 45). When metHbN interacted with KshB and TrxB, the reduction of FAD occurred rapidly with NADH in a concentration-dependent manner, whereas reduction of the heme iron remained extremely slow. At 250 μm NADH, when FAD was completely bleached out, efficiency of heme reduction did not change in the case of KshB and TrxB, thereby indicating that the hydride transfer from NADH to FAD is not the limiting step. In contrast, reduction of FAD and ferric HbN occurred very efficiently in the presence of FdR. This process involves a single turnover of HbN molecules with limiting FdR in its reduced state that is cycled by NADH. Thus, the rate of HbN reduction may vary with the concentration of reduced flavin in each reaction mixture as a function of NADH concentration.
In an attempt to bring the heme and FAD cofactor binding sites closer, HbN was integrated with KshB. However, the integration did not speed up the reduction process. Interestingly, when FdR was added, it immediately enhanced the heme reduction in the HbN-KshB chimera, indicating that the heme and FAD binding regions are not in close contact and allowing FdR to interact with the heme binding region of HbN-KshB. In contrast, reduction of the heme iron occurred rapidly in the HbN-FdR chimera, signifying that FdR encompass suitable structural features compatible for the efficient reduction of the MtbHbN.
The three-dimensional structure obtained from MD simulations shows that the CD loop contributes to shape the protein-protein interface near the isoalloxazine ring of the FAD. The CD loop of HbN carries polar residues (Ser47, Thr49, and Asn50) that may participate in building transient contacts with the partner to facilitate ET from the FAD to the heme. At this point, it is worth noting that the distance between the propionate of heme to the nearest atom in the isoalloxazine ring remains in the range of 6–9 Å along the last 200 ns of the trajectory, which compares with the distance found between those cofactors in the flavoHb of A. eutropus (around 7 Å).
Because a dominant coupling pathway is expected to occur when donor and acceptor are located at short distances (46, 47), the large value of the electron coupling determined with the semiempirical pathway method supports the feasibility of this region in mediating the ET flow. Due to the presence of Gly48 the CD loop might afford the intrinsic flexibility to facilitate the adjustment of HbN at the protein-protein interface. The relevance of the CD loop is supported by inhibition of MtbHbN reduction by FdR in the presence of the CD loop peptide and by decrease in the formation of the CO-adduct in the CD loop-mutated variants of MtbHbN. In particular, Gly48 appears to be a major player in modulating the intrinsic flexibility of the CD loop for the adjustment of the co-factor binding sites at the interface for the smooth transfer. The flanking residues (Ser47 and Thr49) may also contribute in sustaining the appropriate environment for the electron flow. At this point, it is worth stressing that the NO oxidation profile of the HbN mutants remained unaffected (data not shown), signifying that the CD loop may not be affecting the gating role of Phe(E15) and the oxidative half of the HbN-mediated NOD function. It can then be concluded that the CD loop has a crucial role in mediating the reduction step between MtbHbN and FdR.
The structural information of the HbN-FdR complex also allows us to explain the similar reduction efficiency exhibited by glycosylated and unglycosylated HbN in the presence of FdR. Thus, the glycan moiety is linked at the C-terminal end of the HbN and should not interfere with the interactions between HbN and FdR. The present study has also unveiled a novel role of the Pre-A motif of HbN, which is required for its optimal NOD function by regulating the protein backbone motion and to facilitate diffusion of NO toward the active site (8). In the absence of Pre-A motif, the FdR was unable to reduce HbN individually as well as in the HbN-FdR chimera. However, the three-dimensional structure of the HbN-FdR complex does not support a direct interaction with the Pre-A segment. Rather, the large flexibility of the Pre-A region, which is modulated by the ability to form transient electrostatic contacts with the protein core (8, 9), influences the dynamics of HbN, and the motion of the protein backbone changes significantly when Pre-A is removed from the HbN. In particular, previous studies showed that the Pre-A-deleted HbN displayed a clear cut restriction in the movement of B and E helices as well as the EF loop region (16), which in conjunction with the CD loop provides key residues involved in the recognition of HbN and FdR. It can, therefore, be concluded that the Pre-A segment influences the NOD function by modulating the dynamics of the protein core, which in turn influences two distinct effects: (i) the relative motion between helices B and E favors the conformational changes of the Phe(E15) gate, thus favoring the transition between closed and open states (8, 12), and (ii) the enhanced flexibility of helices B and E regulates movement of the CD loop and facilitates the interactions between residues that modulate the appropriate recognition at the protein-protein interface. Thus, the results derived from MD simulations reveal the formation of salt bridges supported by residues located in helix B (Glu25, Glu29) and E (Arg52) as well as beginning of CD loop (Asp39) and loop F (Arg83), which contribute to the interaction between MtbHbN and FdR. The restricted motion of helices in the Pre-A-deleted mutant of MtbHbN might have affected the global flexibility of the protein matrix and abrogated its interactions with the reductase partner, leading to failure of the electron flow.
The HbN may have significant implications on the pathophysiology of Mtb due to its high oxygen affinity and NO detoxification ability, which may be crucial for sustaining its intracellular survival within the NO-enriched hypoxic environment of macrophages and granuloma. Although the HbN knockout strain of Mtb has not been generated so far, lack of respiratory protection from NO in the HbN-deleted strain of M. bovis (5) suggests that the loss of HbN may significantly affect intracellular survival of Mtb. We have demonstrated that Pre-A is required for NO oxidation by HbN by modulating the dynamical motion of the protein backbone required for ligand access (8). This study highlights that the Pre-A is also involved in regulating the interaction of HbN with the reductase partner for efficient cycling of NOD reaction. These results suggest that both HbN and its reductase partner might be well suited as targets for developing novel pharmacological strategies to fight tuberculosis.
The present study thus unravels a novel mechanism by which HbN interacts with a compatible redox partner to keep itself in the functional ferrous state for oxygen binding and NO scavenging. Taken together, this work presents three major findings: (i) enzymatic reduction of MtbHbN via native and heterologous flavoreductases occurs with varying efficiency and among these, reductase structurally similar to NADH-ferredoxin/FdR may be an efficient partner for the ET that may be vital for its oxygen binding and NO-scavenging function, (ii) the CD loop of MtbHbN plays an important role in establishing transient interactions with a partner reductase to facilitate the ET, and (iii) the Pre-A motif regulates interactions of the CD loop with the partner reductase and displays a larger effect on ET, thereby substantiating the relevance of the Pre-A motion on the dynamics of the protein backbone that may be essential for enzymatic reduction of MtbHbN.
Acknowledgments
We thank the DNA sequencing and peptide synthesis facilities of IMTECH, Computational facilities from the Barcelona Supercomputer Center, and the Center for Scientific and Academic Services of Catalonia.
This work was supported by CSIR Supra institutional project BSC0210, European Commission under FP7 program on health (NOstress), Spanish Ministerio de Ciencia e Innovación Grants SAF2011-27642 and PIB2010AR-00455 and Generalitat de Catalunya Grant 2014SGR1189, and fellowships from CSIR and the Dept. of Biotechnology, Government of India (to S. S., D. S., and H. D.).
- Mtb
- M. tuberculosis
- NO
- nitric oxide
- NOD
- nitric-oxide dioxygenase
- ET
- electron transfer
- MD
- molecular dynamics
- FdR
- flavodoxin reductase
- Mb
- myoglobin
- VHb
- Vitreoscilla hemoglobin.
REFERENCES
- 1. Gardner P. R. (2005) Nitric-oxide dioxygenase function and mechanism of flavohemoglobin, haemoglobin and myoglobin and their associated reductases. J. Inorg. Biochem. 99, 247–266 [DOI] [PubMed] [Google Scholar]
- 2. Forrester M. T., Foster M. W. (2012) Protection from nitrosative stress: a central role for microbial flavohemoglobin. Free Radic. Biol. Med. 52, 1620–1633 [DOI] [PubMed] [Google Scholar]
- 3. Gardner P. R., Gardner A. M., Brashear W. T., Suzuki T., Hvitved A. N., Setchell K. D., Olson J. S. (2006) Hemoglobins deoxygenate nitric oxide with high fidelity. J. Inorg. Biochem. 100, 542–550 [DOI] [PubMed] [Google Scholar]
- 4. Pathania R., Navani N. K., Gardner A. M., Gardner P. R., Dikshit K. L. (2002) Nitric oxide scavenging and detoxification by the Mycobacterium tuberculosis haemoglobin, HbN in Escherichia coli. Mol. Microbiol. 45, 1303–1314 [DOI] [PubMed] [Google Scholar]
- 5. Ouellet H., Ouellet Y., Richard C., Labarre M., Wittenberg B., Wittenberg J., Guertin M. (2002) Truncated hemoglobin HbN protects Mycobacterium bovis from nitric oxide. Proc. Natl. Acad. Sci. U.S.A. 99, 5902–5907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pawaria S., Rajamohan G., Gambhir V., Lama A., Varshney G. C., Dikshit K. L. (2007) Intracellular growth and survival of Salmonella enterica serovar Typhimurium carrying truncated hemoglobins of Mycobacterium tuberculosis. Microb. Pathog. 42, 119–128 [DOI] [PubMed] [Google Scholar]
- 7. Yeh S. R., Couture M., Ouellet Y., Guertin M., Rousseau D. L. (2000) A cooperative oxygen binding hemoglobin from Mycobacterium tuberculosis. J. Biol. Chem. 275, 1679–1684 [DOI] [PubMed] [Google Scholar]
- 8. Lama A., Pawaria S., Bidon-Chanal A., Anand A., Gelpí J. L., Arya S., Martí M., Estrin D. A., Luque F. J., Dikshit K. L. (2009) Role of Pre-A motif in nitric oxide scavenging by truncated hemoglobin, HbN, of Mycobacterium tuberculosis. J. Biol. Chem. 284, 14457–14468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Milani M., Pesce A., Ouellet Y., Ascenzi P., Guertin M., Bolognesi M. (2001) Mycobacterium tuberculosis hemoglobin N displays a protein tunnel suited for O2 diffusion to the heme. EMBO J. 20, 3902–3909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bidon-Chanal A., Martí M. A., Crespo A., Milani M., Orozco M., Bolognesi M., Luque F. J., Estrin D. A. (2006) Ligand-induced dynamical regulation of NO conversion in Mycobacterium tuberculosis truncated hemoglobin-N. Proteins 64, 457–464 [DOI] [PubMed] [Google Scholar]
- 11. Bidon-Chanal A., Martí M. A., Estrin D. A., Luque F. J. (2007) Dynamical regulation of ligand migration by a gate-opening molecular switch in truncated hemoglobin N from Mycobacterium tuberculosis. J. Am. Chem. Soc. 129, 6782–6788 [DOI] [PubMed] [Google Scholar]
- 12. Oliveira A., Singh S., Bidon-Chanal A., Forti F., Martí M. A., Boechi L., Estrin D. A., Dikshit K. L., Luque F. J. (2012) Role of Phe(E15) gate in ligand entry and nitric oxide detoxification function of Mycobacterium tuberculosis truncated hemoglobin N. PLoS One 7, e49291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Smagghe B. J., Trent J. T., 3rd, Hargrove M. S. (2008) NO dioxygenase activity in hemoglobins is ubiquitous in vitro, but limited by reduction in vivo. PLoS One 3, e2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gardner A. M., Martin L. A., Gardner P. R., Dou Y., Olson J. S. (2000) Steady-state and transient kinetics of Escherichia coli nitric-oxide dioxygenase (flavohemoglobin): the B10 hydroxyl is essential for dioxygen binding and catalysis. J. Biol. Chem. 275, 12581–12589 [DOI] [PubMed] [Google Scholar]
- 15. Gardner P. R., Gardner A. M., Martin L. A., Dou Y., Li T., Olson J. S., Zhu H., Riggs A. F. (2000) Nitric-oxide dioxygenase actity and function of flavohemoglobins. J. Biol. Chem. 275, 31581–31587 [DOI] [PubMed] [Google Scholar]
- 16. Eich R. F., Li T., Lemon D. D., Doherty D. H., Curry S. R., Aitken J. F., Mathews A. J., Johnson K. A., Smith R. D., Phillips G. N., Jr., Olson J. S. (1996) Mechanism of NO-induced oxidation of myoglobin and hemoglobin. Biochemistry 35, 6976–6983 [DOI] [PubMed] [Google Scholar]
- 17. Fago A., Mathews A. J., Moens L., Dewilde S., Brittain T. (2006) The reaction of neuroglobin with potential redox protein partners of cytochrome b5 and cytochrome c. FEBS Lett. 580, 4884–4888 [DOI] [PubMed] [Google Scholar]
- 18. Hayashi A., Suzuki T., Shin M. (1973) An enzymic reduction system for metmyoglobin and methemoglobin and its application to functional studies of oxygen carriers. Biochim. Biophys. Acta 310, 309–316 [DOI] [PubMed] [Google Scholar]
- 19. Dikshit K. L., Webster D. A. (1988) Cloning, expression and characterization of bacterial globin gene from Vitreoscilla in Escherichia coli. Gene 70, 377–386 [DOI] [PubMed] [Google Scholar]
- 20. Doyle M. P., Hoekstra J. W. (1981) Oxidation of nitrogen oxides by bound dioxygen in hemoproteins. J. Inorg. Biochem. 14, 351–358 [DOI] [PubMed] [Google Scholar]
- 21. Arya S., Sethi D., Singh S., Hade M. D., Singh V., Raju P., Chodisetti S. B., Verma D., Varshney G. C., Agrewala J. N., Dikshit K. L. (2013) Truncated hemoglobin N is post-translationally modified in Mycobacterium tuberculosis and modulates host-pathogen interactions during intracellular infection. J. Biol. Chem. 288, 29987–29999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Appleby C. A. (1978) Purification of Rhizobium cytochromes P-450. Methods Enzymol. 52, 157–166 [DOI] [PubMed] [Google Scholar]
- 23. Thomas M. T., Shepherd M., Poole R. K., van Vliet A. H., Kelly D. J., Pearson B. M. (2011) Two respiratory enzyme systems in Campylobacter jejuni NCTC 11168 contributes to growth on L-lactate. Environ. Microbiol. 13, 48–61 [DOI] [PubMed] [Google Scholar]
- 24. Ingelman M., Bianchi V., Eklund H. (1997) The three-dimensional structure of flavodoxin reductase from Escherichia coli at 1.7-Å resolution. J. Mol. Biol. 268, 147–157 [DOI] [PubMed] [Google Scholar]
- 25. Kozakov D., Hall D. R., Beglov D., Brenke R., Comeau S. R., Shen Y., Li K., Zheng J., Vakili P., Paschalidis I. Ch., Vajda S. (2010) Achieving reliability and high accuracy in automated protein docking: Cluspro, PIPER, SDU, and stability analysis in CAPRI rounds 13–19. Proteins 78, 3124–3130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kozakov D., Brenke R., Comeau S. R., Vajda S. (2006) PIPER: An FFT-based protein docking program with pairwise potentials. Proteins 65, 392–406 [DOI] [PubMed] [Google Scholar]
- 27. Case D. A., Darden T. A., Cheatham T. E., 3rd, Simmerling C. L., Wang J., Duke R. E., Luo R., Walker R. C., Zhang W., Merz K. M., Robert B., Havik S., Roitberg A., Seabra G., Swalis J., Goetz A. W., Kolossváry I., Wong K. F., Paesani F., Vanicek J., Wolf R. M., Liu J., Wu X., Brozell S. R., Steinbrecher T., Gohlke H., Cai Q., Ye X., Wang J., Hsieh M. J., Cui G., Roe D. R., Mathews D. H., Seetin M. G., Salomon-Ferrer R., Sagui C., Babin V., Luchko T., Gusarov S., Kovalenko A., Kollman P. A. (2012) AMBER 12, University of California, San Francisco, CA [Google Scholar]
- 28. Hornak V., Abel R., Okur A., Strockbine B., Roitberg A., Simmerling C. (2006) Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 65, 712–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Capece L., Boechi L., Perissinotti L. L., Arroyo-Mañez P., Bikiel D. E., Smulevich G., Marti M. A., Estrin D. A. (2013) Small ligand-globin interactions: reviewing lessons derived from computer simulation. Biochim. Biophys. Acta 1834, 1722–1738 [DOI] [PubMed] [Google Scholar]
- 30. Marti M. A., Capece L., Bidon-Chanal A., Crespo A., Guallar V., Luque F. J., Estrin D. A. (2008) NO reactivity with globins as investigated through computer simulation. Methods Enzymol. 437, 477–498 [DOI] [PubMed] [Google Scholar]
- 31. Antony J., Medvedev D. M., Stuchebrukhov A. A. (2000) Theoretical study of electron transfer between the photolyase catalytic cofactor FADH− and DNA thymine dimer. J. Am. Chem. Soc. 122, 1057–1065 [Google Scholar]
- 32. Bayly C. I., Cieplak P., Cornell W., Kollman P. A. (1993) A well-behaved electrostatic potential based method using charge restraints for determining atom-centered charges: the RESP model. J. Phys. Chem. 97, 10269–10280 [Google Scholar]
- 33. Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., Klein M. L. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 [Google Scholar]
- 34. Ryckaert J.-P., Ciccotti G., Berendsen H. J. C. (1977) Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341 [Google Scholar]
- 35. Wu X., Brooks B. R. (2003) Self-guided Langevin dynamics simulation method. Chem. Phys. Lett. 381, 512–518 [Google Scholar]
- 36. Darden T., York D., Pedersen L. (1993) Particle mesh Ewald: an N. log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 [Google Scholar]
- 37. Beratan D. N., Onuchic J. N., Winkler J. R., Gray H. B. (1992), Electron-tunneling pathways in proteins. Science 258, 1740–1741 [DOI] [PubMed] [Google Scholar]
- 38. Beratan D. N., Betts J. N., Onuchic J. N. (1991) Protein electron transfer rates are predicted to be set by the bridging secondary and tertiary structure. Science 252, 1285–1288 [DOI] [PubMed] [Google Scholar]
- 39. Alvarez-Paggi D., Martín D. F., DeBiase P. M., Hildebrandt P., Martí M. A., Murgida D. H. (2010) Molecular basis of coupled protein and electron transfer dynamics of cytocrome c in biomimetic complexes. J. Am. Chem. Soc. 132, 5769–5778 [DOI] [PubMed] [Google Scholar]
- 40. Petruk A. A., Bartesaghi S., Trujillo M., Estrin D. A., Murgida D., Kalyanaraman B., Marti M. A., Radi R. (2012) Molecular basis of intermolecular electron transfer in proteins during simulation studies in model tyrosine-cysteine peptides in solution. Arch. Biochem. Biophys. 525, 82–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ferreiro D. N., Boechi L., Estrin D. A., Martí M. A. (2013) The key role of water in the dioxygenase function of Escherichia coli flavohemoglobin. J. Inorg. Biochem. 119, 75–84 [DOI] [PubMed] [Google Scholar]
- 42. Kohlhoff M., Borges M. H., Yarleque A., Cabezas C., Richardson M., Sanchez E. F. (2012) Exploring the proteome of the venome of the peruvian pit vipers Bothrops atrox, B. barnetti, and B. pictus. J. Proteomics 75, 2181–2195 [DOI] [PubMed] [Google Scholar]
- 43. Nagai M., Tomoda A., Yoneyama Y. (1981) Reduction of methemoglobin by ferredoxin and ferredoxin-NADP reductase system. J. Biol. Chem. 256, 9195–9197 [PubMed] [Google Scholar]
- 44. Frey A. D., Kallio P. T. (2005) Nitric oxide detoxification: a new era for bacterial globins in biotechnology. Trends Biotechnol. 23, 69–73 [DOI] [PubMed] [Google Scholar]
- 45. Hallstrom C. K., Gardner A. M., Gardner P. R. (2004) Nitric oxide metabolism in mammalian cells: substrate and inhibitor profiles of a NADPH-cytochrome P450 oxidoreductase coupled microsomal nitric-oxide dioxygenase. Free Radic. Biol. Med. 37, 216–228 [DOI] [PubMed] [Google Scholar]
- 46. Jasaitis A., Johansson M. P., Wikstrom M., Vos M. H., Vekhovsky I. (2007) Nanosecond electron tunnelling between the hemes in cytochrome bo3. Proc. Natl. Acad. Sci. U.S.A. 104, 20811–20814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Beratan D. N., Balabin I. A. (2008) Heme-copper oxidases use tunneling pathways. Proc. Natl. Acad. Sci. U.S.A. 105, 403–404 [DOI] [PMC free article] [PubMed] [Google Scholar]