

Background: Super-low-dose endotoxin causes mild but significant pro-inflammatory skewing.
Results: Inhibition of GSK3 or activation of CREB ablates preferential induction of pro-inflammatory genes by super-low-dose LPS.
Conclusion: GSK3 is necessary for pro-inflammatory gene induction in response to super-low-dose LPS, acting by activating FoxO1 and suppressing CREB.
Significance: Persistent, non-resolving inflammation is a characteristic of many chronic diseases, and this study points toward potential therapeutic targets.
Keywords: inflammation, Innate Immunity, Lipopolysaccharide (LPS), Monocyte, Systems Biology
Abstract
Innate monocytes and macrophages can be dynamically programmed into distinct states depending upon the strength of external stimuli. Innate programming may bear significant relevance to the pathogenesis and resolution of human inflammatory diseases. However, systems analyses with regard to the dynamic programming of innate leukocytes are lacking. In this study, we focused on the dynamic responses of human promonocytic THP-1 cells to lipopolysaccharide (LPS). We observed that varying dosages of LPS differentially modulate the expression of selected pro- and anti- inflammatory mediators such as IL-6 and IL-33. Super-low dosages of LPS preferentially induced the pro-inflammatory mediator IL-6, while higher dosages of LPS induced both IL-6 and IL-33. Mechanistically, we demonstrated that super-low and high doses of LPS cause differential activation of GSK3 and Akt, as well as the transcription factors FoxO1 and CREB. Inhibition of GSK3 enabled THP-1 cells to express IL-33 when challenged with super-low dose LPS. On the other hand, activation of CREB with adenosine suppressed IL-6 expression. Taken together, our study reveals a dynamic modulation of monocytic cells in response to varying dosages of endotoxin, and may shed light on our understanding of the dynamic balance that controls pathogenesis and resolution of inflammatory diseases.
Introduction
Increasing evidence from both clinical and laboratory studies indicate that innate immune cells can be programmed into diverse states with varying degrees of pro- and anti-inflammatory phenotypes (1, 2), with consequences for host defense and inflammation (3). Despites its clinical relevance, mechanistic studies with regard to innate cell programming are scant. To fill this critical void, we have examined the dynamic responses of human THP-1 monocytic cells challenged with a model stimulant, bacterial endotoxin lipopolysaccharide (LPS),2 a major component of the cell walls of Gram-negative bacteria. It is a ubiquitous environmental toxin (4, 5). High doses of LPS are responsible most prominently for septic shock (6, 7). On the other hand, low doses of circulating LPS are common in chronic disease settings, and may contribute to the development of persistent, low-grade, non-resolving inflammation (8–10). The distinct pathological effects of varying dosages of LPS may reflect differential programming of innate leukocytes.
At the biochemical level, LPS is recognized by the Toll-like receptor (TLR) 4. High dosages of LPS can activate multiple pathways capable of inducing both pro- and anti-inflammatory genes (11, 12). Of particular note, the phosphoinositide-3-kinase (PI3K)/Akt signaling pathway induced by high-dose LPS serves as a negative mechanism to down-regulate inflammatory processes, and is also responsible for the expression of anti-inflammatory mediators, partly through the activation of CREB (13). In contrast, super-low dose LPS fails to induce anti-inflammatory mediators, and preferentially induces low-grade inflammatory mediators (14). Studies from other groups in other cellular systems indicate that GSK3 and Akt may form a mutually inhibitory circuit (12, 15). Akt was shown to inhibit the function of GSK3 (16), while GSK3 may in turn inhibit Akt.
Other studies also suggest that GSK3 is a critical molecule involved in various inflammatory processes in vitro and in vivo. Inhibition, knockdown, or knock-out of GSK3 has been shown to inhibit the expression of pro-inflammatory mediators in response to LPS (17, 18). Inhibitory phosphorylation of GSK3 mediated by PI3K/Akt is necessary for the protective induction of anti-inflammatory IL-10 following ischemia/reperfusion injury (19), which triggers inflammation through TLR4. Pharmacological activation of PI3K also suppresses toxicity-induced apoptosis in neurons in a GSK3-dependent fashion (20). These findings suggest that the overall role of GSK3 in the LPS response consists of the promotion of pro-inflammatory cytokine production and suppression of anti-inflammatory mediators (21, 22).
Downstream, GSK3 appears to play a role in the regulation of the transcription factors forkhead box O1 (FoxO1) and cAMP response-element-binding protein (CREB). The anti-inflammatory effects of Akt on TLR4 signaling appear to be mediated through FoxO1 (23), suggesting that GSK3 may be important for the activation of FoxO1. GSK3 can potently suppress CREB (24). Recently, it was shown that the anti-inflammatory effects of JAK3 in the context of TLR4 stimulation are exerted by suppressing GSK3, which enables increased CREB activity (13). In cells stimulated by the TLR2 ligand zymosan, the activation status of CREB corresponds closely with heightened production of IL-10 (25). Suppression of CREB by GSK3 may result in the pro-inflammatory skewing of immune responses. Circumstantially, genes suppressed by GSK3 tend to be regulated by CREB (26), and the suppression of IL-10 production by interferon-γ is due to its activation of GSK3 and ensuing suppression of CREB (15). CREB seems to oppose the pro-inflammatory effects of GSK3 by augmenting anti-inflammatory gene expression.
Akt (also known as protein kinase B) is regulated by the PI3K and mammalian target of rapamycin (mTOR) pathways (12, 27). Activation of Akt by rapamycin blunts the sensitivity of pro-inflammatory genes to LPS and increases CREB activity (28), and mTOR/Akt signaling additionally suppresses inflammation by inactivating the transcription factor forkhead box O1 (FoxO1) (23). The role of FoxO1 in TLR4 signaling is predominantly pro-inflammatory. Overexpression of FoxO1 results in increased expression of TLR4 and pro-inflammatory cytokine genes in response to LPS, and knockdown or removal blunts the ability of TLR4 stimulation to induce these genes (29). FoxO1 is also necessary for pro-inflammatory cytokine production by memory T cells (30), pointing to a generalized role for FoxO1 in the regulation of inflammatory gene transcription. Blocking of inhibitory phosphorylation of FoxO1 results in increased pro-inflammatory cytokine production by macrophages upon challenge with a TLR2 ligand (31). Defective Akt activation results in increased FoxO1 activity and pro-inflammatory cytokine production (23). The broadly anti-inflammatory effects of PI3K signaling in TLR4 stimulation (32) can thus be ascribed to the tandem activation of CREB and suppression of FoxO1 by Akt, but the competition between GSK3 and Akt has not been well characterized in the context of LPS challenge.
In this study, we tested the hypothesis that monocytes may be dynamically programmed by varying dosages of LPS through the competing circuits of GSK3 and Akt. To test this, we examined the expression profiles of selected pro- and anti- inflammatory mediators in human monocytic THP-1 cells challenged with varying dosages of LPS. We observed distinct expression patterns of IL-6 and IL-33 in THP-1 cells treated with super-low or high dose LPS, and this pattern correlated with distinct activation statuses of GSK3 and Akt, as well as the transcription factors FoxO1 and CREB. We further demonstrated that pharmacological intervention by either GSK3 inhibition or CREB activation is sufficient to reverse the pro-inflammatory skewing characteristic of cells stimulated by super-low-dose LPS. Our study reveals a unique network responsible for the dynamic programming of innate leukocytes by varying dosages of bacterial endotoxin.
EXPERIMENTAL PROCEDURES
Cell Culture and Reagents
THP-1 cells were grown in RPMI 1640 (Invitrogen 11875–119) supplemented with 2 mm l-glutamine, 1% penicillin/streptomycin (Invitrogen 15140-122), and 1% fetal bovine serum (FBS) (Seradigm 1600-500). Before performing experiments, cells were seeded overnight at a density of 106 cells/ml in RPMI containing 2 mm l-glutamine, 1% penicillin/streptomycin, and 1% FBS. LPS from Escherichia coli O114:B4 (Sigma L2630) was dissolved in PBS. The GSK3 inhibitors SB216763 (Sigma S3442) and indirubin-3′-oxime (Sigma I0404) were dissolved in DMSO. Adenosine (Sigma A4036) was prepared in ddH2O.
Real-time PCR
RNA was extracted using TRIzol (Invitrogen 15596–026). Samples were then treated with 20% chloroform by volume at room temperature for 3 min, followed by centrifugation at 12,000 rpm for 15 min at 4 °C. Aqueous phase was transferred to new tubes and isopropanol (50% initial volume) was added, then samples were incubated for 10 min at room temperature. Samples were then centrifuged at 12,000 rpm for 10 min at 4 °C, and the supernatant was discarded. The RNA pellet was then washed with 75% ethanol in DEPC-treated water (100% initial volume), and centrifuged for an additional 10 min at 12,000 rpm, 4 °C. Supernatant was discarded and RNA pellets were resuspended in 30 μl of DEPC-treated water. DNA digestion was performed at 37 °C for 30 min (Invitrogen AM2222), followed by 5 min treatment with 85 °C to degrade DNase. 1500 ng of RNA was reverse-transcribed using a high-capacity kit (Invitrogen 4368813) at 37 °C for 2 h, followed by enzyme inactivation for 5 min at 85 °C. Real-time PCR was performed on a Bio-Rad CFX96 machine using a 2× SYBR Green mix (Bio-Rad 172-5271); the PCR protocol was denaturation at 95 °C for 3 min, followed by 45 cycles of 95 °C for 15 s and 60 °C for 60 s. Primers for human IL-6 (F 5′-AGCCACTCACCTCTTCAGAACGAA, R 5′-AGTGCCTCTTTGCTGCTTTCACAC), TNFα (F 5′-TCAATCGGCCCGACTATCTC, R 5′-CAGGGCAATGATCCCAAAGT), IL-10 (F 5′-TCCTTGCTGGAGGACTTTAAGGGT, R 5′-TGTCTGGGTCTTGGTTCTCAGCTT), and IL-33 (F 5′-GGAAGAACACAGCAAGCAAAGCCT, R 5′-TAAGGCCAGAGCGGAGCTTCATAA) were purchased from IDT. Readouts were analyzed by the ΔΔCQ method.
Western Blots
Whole-cell lysates were harvested using lysis buffer consisting of 2% SDS, 5% Tris-HCl, pH 6.8, and 10% glycerol, placed on ice for 20 min, boiled for 5 min, then centrifuged at 12,000 rpm for 4 min at room temperature for removal of intracellular debris. Protein concentration was assessed by Bradford assay. Protein samples were run on 10% acrylamide gels at 100 V, followed by transfer at 110 V before blocking in 5% milk in TBS-T. Antibodies against pCREB-S133 (Cell Signaling 9191) were used at a concentration of 1:4000 in 5% milk in TBS-T, and CREB (Cell Signaling 9197S), pFoxO1-S256 (Cell Signaling 9461S), FoxO1 (Cell Signaling 9454S), Akt (Santa Cruz Biotechnology sc-8312), GSK3β (Santa Cruz Biotechnology sc-9166), and GAPDH (Santa Cruz Biotechnology sc-25778) at 1:1000. pAkt-S473 (Cell Signaling 9271S) and pGSK3β-Y216 (Santa Cruz Biotechnology sc-135653) antibodies were diluted 1:4000 in 5% BSA in TBS-T.
Statistics
Statistical analysis by Holm-Sidak pairwise comparison or Student's t test was performed using SigmaPlot 11 software (SigmaPlot) as detailed in the figure legends. Results were considered to be statistically significant at p < 0.05.
RESULTS
Differential Regulation of Inflammatory Genes by LPS
The phenomena of endotoxin tolerance and priming, in which stimulation with LPS alters the nature of the inflammatory response to a subsequent challenge, have been extensively documented (33–36). We therefore sought to characterize the differences between the inflammatory response to super-low (<1 ng/ml) and high (>10 ng/ml) doses of LPS. Stimulation of THP-1 cells with varying dosages of LPS for 4 h reveals that the pro-inflammatory genes IL-6 and tumor necrosis factor α (TNFα) are more sensitive to LPS than IL-33, as transcription of the latter is significantly up-regulated only by high doses of LPS (Fig. 1). In a separate experiment, closer analysis employing different statistical methods reveals that IL-6 transcription is mildly but significantly up-regulated by LPS doses as low as 50 pg/ml, while IL-33 is not (Fig. 2), suggesting that stimulation with super-low-dose LPS results in skewing of the TLR4 response toward pro-inflammatory gene expression.
FIGURE 1.

Relative induction of pro- and anti-inflammatory genes in THP-1 cells by 4 h stimulation with different concentrations of LPS. Total RNAs were harvested from THP-1 cells challenged as indicated. Real-time RT-PCR was performed to determine the expression levels of IL-6 (A) TNFα (B), and IL-33 (C). Different letters denote statistically significant differences between groups (p < 0.05, Holm-Sidak).
FIGURE 2.

Relative induction of pro-and anti-inflammatory genes in human THP-1 cells by low- and high-dose LPS. Total RNA was isolated from THP-1 cells treated with either 100 ng/ml LPS (A) or 50 pg/ml LPS (B) for 4 h. Real-time RT-PCR was performed to determine the expression levels of both IL-6 and IL-33. Data are representative of three separate experiments (*, p < 0.05; Student's t test).
Mechanistic Effects of Different LPS Dosages
The signaling kinases GSK3 and Akt are known to compete in the regulation of inflammation (22, 37). This led us to explore whether the differential regulation of pro- and anti-inflammatory genes by varying dosages of LPS could be explained by differential effects on signal transduction through GSK3 and Akt. Stimulation of THP-1 cells with 100 ng/ml LPS for 60 min resulted in robust phosphorylation of Akt at Ser-473, a marker of its activation (38). In contrast, super-low-dose LPS failed to increase, but rather decreased Akt-Ser-473 phosphorylation (Fig. 3A).
FIGURE 3.

Differential regulation of GSK3 and Akt in THP-1 cells by LPS. THP-1 cells were treated with either 50 pg/ml or 100 ng/ml LPS for various time periods as indicated. Whole cell lysates were separated on SDS-PAGE. The levels of pAkt-S473 and total Akt (A) pGSK3β-Y216 and total GSK3β (B), and pPyk2-Y402 and total Pyk2 (C) were determined by Western blot with specific antibodies. Data are representative of three separate experiments.
Our data suggest that GSK3 may be differentially modulated by super-low and high doses LPS. Given that GSK3 activation is facilitated by tyrosine 216 phosphorylation, we further probed the pY216 GSK3 levels in cells treated with super-low dose LPS. As shown in Fig. 3B, 50 pg/ml LPS caused a rapid increase of pY216-GSK3 in THP-1 cells. Since Pyk2 is the upstream kinase responsible for the activating Tyr-216 phosphorylation of GSK3 (39), we proceeded to test the activation status of Pyk2 by Western blot. The tyrosine kinase activity of Pyk2 is inhibited by phosphorylation at Tyr-402 (40). We found that 50 pg/ml LPS caused a rapid and dramatic drop in the levels of inhibitory Tyr-402 phosphorylation of Pyk2, indicating an increase in its activity (Fig. 3C). In contrast, higher dose LPS (100 ng/ml) caused a robust elevation of Tyr-402 phosphorylation, indicating its inhibition (Fig. 3C). This correlates with our above finding that super-low dose LPS activates GSK3 through increasing its Tyr-216 phosphorylation.
Downstream of GSK3 and Akt lie two competing transcription factors: FoxO1 and CREB (13, 29). While FoxO1 contributes to the expression of pro-inflammatory mediators (23, 29), CREB is largely involved in the expression of anti-inflammatory mediators (25, 41). Pharmacological activation of Akt corresponds with suppression of FoxO1 (42), which, in light of GSK3 suppression by Akt (26), points to a connection between GSK3 and FoxO1 activity. Akt is also important for the activation of CREB (13, 15).
Since we observed opposing effects of super-low and high dose LPS on GSK3 and Akt, we further tested the activation status of FoxO1 and CREB. We found that treatment with 50 pg/ml LPS caused an increase in total FoxO1 proteins levels, while treatment with 100 ng/ml LPS led to a decrease in the total FoxO1 level (Fig. 4A). Since the decrease in FoxO1 may be attributed to its inhibitory phosphorylation by Akt at Ser-256 (43), we also tested the level of Ser-256 phosphorylation of FoxO1. As shown in Fig. 4, only the high dose LPS triggered FOXO1 phosphorylation at Ser-256, while super-low dose LPS had no effect. The effects of LPS on the activation of CREB followed an opposite pattern. Phosphorylation of CREB at Ser-133 results in its activation (24), and this phosphorylation was only induced by high-dose LPS, with low-dose LPS not triggering appreciable Ser-133 phosphorylation of CREB (Fig. 4B).
FIGURE 4.

Differential regulation of FoxO1 and CREB by LPS. THP-1 cells were treated with either 50 pg/ml or 100 ng/ml LPS for various time periods as indicated. Whole cell lysates were separated on SDS-PAGE. The levels of pFoxO1 and total FoxO1 (A) and pCREB-S133 and total CREB (B) were determined by Western blot with specific antibodies. Data are representative of three separate experiments.
Inhibition of GSK3 Reverses Pro-inflammatory Skewing
In vivo studies indicate that GSK3 is a key kinase controlling chronic inflammation (12, 13, 44). Given our above findings, we tested whether selective inhibition of GSK3 may alter the inflammatory skewing of innate monocytes by super-low dose LPS. We first tested indirubin, a GSK3 inhibitor derived from natural compounds (18), and found that costimulation of THP-1 cells with 50 pg/ml LPS and 10 μm indirubin abolished the preferential IL-6 induction typically triggered by this dosage (Fig. 5A). Mechanistically, we observed that the suppression of IL-6 by indirubin corresponds with prevention of FoxO1 up-regulation (Fig. 5B). We observed that SB216763, a synthetic compound which potently and selectively inhibits GSK3 (45), sensitized IL-33 to super-low dose LPS, resulting in significant IL-33 induction by 50 pg/ml LPS (Fig. 5C). The effect of SB216763 on IL-33 correlated with increased CREB activation (Fig. 5D).
FIGURE 5.

Effect of GSK3 inhibition on cellular responses to LPS stimulation. A, THP-1 cells were treated with the combination of 50 pg/ml LPS alone or together with 10 μm indirubin for 4 h. Total RNA was harvested and used for real-time RT-PCR analysis for IL-6. B, total protein lysates were harvested from THP-1 cells treated as indicated and used for Western blot analyses for FoxO1 and GAPDH. C, THP-1 cells were treated with the combination of 50 pg/ml LPS alone or together with 10 μm SB216763 for 4 h. Total RNA was harvested and used for real-time RT-PCR analysis for IL-33. D, total protein lysates were harvested from THP-1 cells treated as indicated, and used for Western blot analyses for CREB and GAPDH. Data are representative of three separate experiments (*, p < 0.05, Student's t test).
Activation of CREB Reverses Pro-inflammatory Skewing
We hypothesized that the pro-inflammatory effects of GSK3 may be caused by its inhibition of CREB. We tested this with adenosine, an Akt/CREB agonist known to exert anti-inflammatory effects in macrophages (46–48). Stimulation of THP-1 cells for 4 h with a range of LPS concentrations in the presence of 500 nm adenosine significantly suppressed the transcription of IL-6 and TNFα while promoting the transcription of IL-10 and IL-33 (Fig. 6, A–D). We further tested the levels of CREB in cell nucleus. Co-stimulation of cells with 50 pg/ml LPS and adenosine dramatically elevated the nuclear levels of pCREB-S133 (Fig. 6E). Taken together, our data reveal a potential competitive circuit responsible for the dynamic sensing of varying dosages of endotoxin by THP-1 cells (Fig. 7).
FIGURE 6.

Activation of CREB suppresses pro-inflammatory gene induction and promotes anti-inflammatory gene induction by LPS in THP-1 cells. THP-1 cells were treated with various dosages of LPS with 500 nm adenosine for 4 h. The levels of IL-6 (A), TNFα (B), IL-10 (C), and IL-33 mRNA (D) were measured by real-time RT-PCR (*, p < 0.05; Student's t test). E, THP-1 cells were treated with various dosages of LPS with 500 nm adenosine for 2 h. Nuclear protein lysates were separated on SDS-PAGE and probed with specific antibodies against pCREB-S133, CREB, or Lamin-B1. Data are representative of three separate experiments.
FIGURE 7.
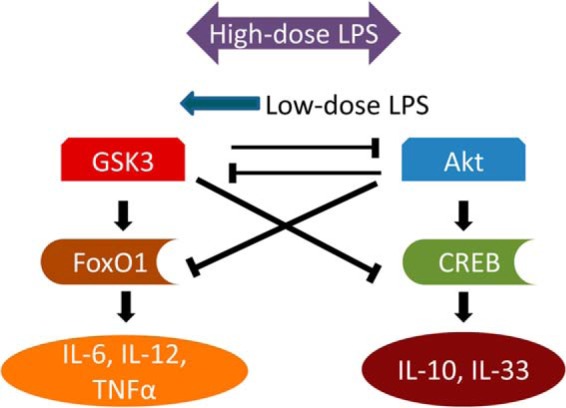
The proposed signaling network contributing to the pro-inflammatory skewing of innate immunity by super-low dose LPS. GSK3 and Akt are engaged in competitive inhibition while simultaneously driving the activity of downstream transcription factors, ultimately promoting the expression of pro- or anti-inflammatory genes. High-dose LPS robustly activates both arms of this response, while super-low dose LPS preferentially causes mild activation of GSK3.
DISCUSSION
We investigated the pro-inflammatory skewing effects of super-low-dose LPS and found that the mild and preferential induction of pro-inflammatory genes by 50 pg/ml LPS. This phenomenon was correlated with preferential activation of GSK3 and inhibition of Akt. This was in contrast to the effect of high dose LPS, which induced both pro- and anti-inflammatory mediators. The proper regulation and resolution of inflammation requires activation of both pro- and anti-inflammatory mediators (49, 50), and the non-responsiveness of these resolving circuits to very low doses of LPS may be key to the pro-inflammatory skewing effects of these dosages, contributing to chronically dysregulated low-grade inflammation. By inhibiting GSK3, we were able to abolish the mild, preferential induction of pro-inflammatory IL-6 usually caused by super-low-dose LPS, and confer sensitivity to IL-33, which is not ordinarily induced by super-low dose LPS. The balanced expression of IL-6 and IL-33 could also be achieved by the addition of adenosine, an agonist capable of augmenting Akt and CREB. In the context of TLR4 activation, IL-6 and TNFα are both activated by FoxO1 (29), while CREB drives the transcription of IL-10 and IL-33 (15, 41). Our finding that adenosine potentiated the response of IL-10 and IL-33 to LPS while blunting induction of IL-6 and TNFα is therefore strongly suggestive of a competitive link between FoxO1 and CREB.
There is increasing appreciation for the role of GSK3 in inflammation (17, 44, 51). Inhibition of GSK3, in particular, is becoming an appealing strategy for the alleviation of inflammatory symptoms (51–53). Our finding that pharmacological inhibition of GSK3 reverses the pro-inflammatory skewing effects of super-low-dose LPS is thus in line with recent developments in the field. The inhibitors we employed are not necessarily selective for the β or α isoform of GSK3 (45). In particular, indirubin may have inhibitory effects on other kinases such as cyclin-dependent kinases (18). These pleiotropic effects may explain the failure of indirubin to significantly alter the expression of IL-33 (data not shown). Likewise, SB216763 may have differential effects on various isoforms of GSK3, and this may explain its selective effects on IL-33 instead of IL-6. Further study is necessary to determine the precise contribution of the different GSK3 isoforms to the pro-inflammatory skewing of monocytes. The anti-inflammatory effects of GSK3 inhibition appear to be mediated through differential activation of FoxO1 and CREB. This relationship is further buttressed by our observation that CREB activation by adenosine recapitulates the suppression of pro-inflammatory skewing in monocytes brought about by GSK3 inhibition.
We posit that super-low-dose LPS preferentially activates pro-inflammatory genes through selective activation of GSK3, leading to heightened activation of FoxO1 at the expense of CREB (Fig. 7). Competitive inhibition between these kinases may be a compelling explanation for the phenomena herein described, as we were able to reverse it either by inhibiting GSK3 or activating Akt/CREB. This is consistent with previous observations that reveal an anti-inflammatory role of Akt in the context of TLR4 stimulation (12, 54).
We realize that further biochemical studies are needed to tease out the detailed mechanisms responsible for the competitive circuitry between GSK3 and Akt, as well as the consequences of this relationship. Missing links include membrane receptor combinations, distinct adaptor molecules (e.g. MyD88, TRIF, etc), and a myriad of kinases and phosphatases, as well as intracellular trafficking of signaling molecules responsible for the sensing of varying doses of LPS. Nevertheless, this study is among the first to elucidate a fundamental principle and a key functional motif that may be responsible for the dynamic balance of pro- and anti-inflammatory responses in innate leukocytes.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 HL115835 (to L. L.).
- LPS
- lipopolysaccharide
- TLR
- Toll-like receptor
- FoxO1
- forkhead box O1
- CREB
- cAMP response element-binding protein
- IL
- interleukin
- TNF
- tumor necrosis factor.
REFERENCES
- 1. Hanke M. L., Heim C. E., Angle A., Sanderson S. D., Kielian T. (2013) Targeting Macrophage Activation for the Prevention and Treatment of Staphylococcus aureus Biofilm Infections. J. Immunol. 190, 2159–2168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bunt S. K., Clements V. K., Hanson E. M., Sinha P., Ostrand-Rosenberg S. (2009) Inflammation enhances myeloid-derived suppressor cell cross-talk by signaling through Toll-like receptor 4. J. Leukoc. Biol. 85, 996–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Marrotte E. J., Chen D.-D., Hakim J. S., Chen A. F. (2010) Manganese superoxide dismutase expression in endothelial progenitor cells accelerates wound healing in diabetic mice. J. Clin. Invest. 120, 4207–4219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Perera P. Y., Mayadas T. N., Takeuchi O., Akira S., Zaks-Zilberman M., Goyert S. M., Vogel S. N. (2001) CD11b/CD18 acts in concert with CD14 and Toll-like receptor (TLR) 4 to elicit full lipopolysaccharide and taxol-inducible gene expression. J. Immunol. 166, 574–581 [DOI] [PubMed] [Google Scholar]
- 5. Beutler B., Rietschel E. T. (2003) Innate immune sensing and its roots: the story of endotoxin. Nat. Rev. Immunol. 3, 169–176 [DOI] [PubMed] [Google Scholar]
- 6. Huet O., Chin-Dusting J. P. (2014) Septic shock: desperately seeking treatment. Clin. Sci. 126, 31–39 [DOI] [PubMed] [Google Scholar]
- 7. Haziot A., Ferrero E., Köntgen F., Hijiya N., Yamamoto S., Silver J., Stewart C. L., Goyert S. M. (1996) Resistance to endotoxin shock and reduced dissemination of gram-negative bacteria in CD14-deficient mice. Immunity 4, 407–414 [DOI] [PubMed] [Google Scholar]
- 8. Erridge C. (2009) The roles of Toll-like receptors in atherosclerosis. J. Innat. Immun. 1, 340–349 [DOI] [PubMed] [Google Scholar]
- 9. Sivapalaratnam S., Farrugia R., Nieuwdorp M., Langford C. F., van Beem R. T., Maiwald S., Zwaginga J., Gusnanto A., Watkins N. A., Trip M. D., Ouwehand W. H. (2011) Identification of candidate genes linking systemic inflammation to atherosclerosis: results of a human in vivo LPS infusion study. BMC Med. Genom. 4, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Maitra U., Gan L., Chang S., Li L. (2011) Low-Dose Endotoxin Induces Inflammation by Selectively Removing Nuclear Receptors and Activating CCAAT/Enhancer-Binding Protein δ. J. Immunol. 186, 4467–4473 [DOI] [PubMed] [Google Scholar]
- 11. Sawa Y., Ueki T., Hata M., Iwasawa K., Tsuruga E., Kojima H., Ishikawa H., Yoshida S. (2008) LPS-induced IL-6, IL-8, VCAM-1, and ICAM-1 expression in human lymphatic endothelium. J. Histochem. Cytochem. 56, 97–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Antoniv T. T., Ivashkiv L. B. (2011) Interleukin-10-induced gene expression and suppressive function are selectively modulated by the PI3K-Akt-GSK3 pathway. J. Immunol. 132, 567–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang H., Brown J., Gao S., Liang S., Jotwani R., Zhou H., Suttles J., Scott D. A., Lamont R. J. (2013) The Role of JAK-3 in Regulating TLR-Mediated Inflammatory Cytokine Production in Innate Immune Cells. J. Immunol. 191, 1164–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yamaguchi Y., Hashimoto T., Sakurai H., Yoshimoto T., Ohmichi Y., Morimoto A., Harimoto K., Ohmichi M., Eguchi K., Kumazawa T. (2011) Low rather than high dose lipopolysaccharide 'priming' of muscle provides an animal model of persistent elevated mechanical sensitivity for the study of chronic pain. Eur. J. Pain 15, 724–731 [DOI] [PubMed] [Google Scholar]
- 15. Hu X., Paik P. K., Chen J., Yarilina A., Kockeritz L., Lu T. T., Woodgett J. R., Ivashkiv L. B. (2006) IFN-γ Suppresses IL-10 Production and Synergizes with TLR2 by Regulating GSK3 and CREB/AP-1 Proteins. Immunity 24, 563–574 [DOI] [PubMed] [Google Scholar]
- 16. Rodionova E., Conzelmann M., Maraskovsky E., Hess M., Kirsch M., Giese T., Ho A. D., Zöller M., Dreger P., Luft T. (2007) GSK-3 mediates differentiation and activation of proinflammatory dendritic cells. Blood 109, 1584–1592 [DOI] [PubMed] [Google Scholar]
- 17. Park S. H., Park-Min K.-H., Chen J., Hu X., Ivashkiv L. B. (2011) Tumor necrosis factor induces GSK3 kinase-mediated cross-tolerance to endotoxin in macrophages. Nat. Immunol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Babcock A. S., Anderson A. L., Rice C. D. (2013) Indirubin-3′-(2,3 dihydroxypropyl)-oximether (E804) is a potent modulator of LPS-stimulated macrophage functions. Toxicol. Appl. Pharmacol. 266, 157–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ren F., Duan Z., Cheng Q., Shen X., Gao F., Bai L., Liu J., Busuttil R. W., Kupiec-Weglinski J. W., Zhai Y. (2011) Inhibition of glycogen synthase kinase 3 beta ameliorates liver ischemia reperfusion injury by way of an interleukin-10-mediated immune regulatory mechanism. Hepatology 54, 687–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jang S., Jeong H.-S., Park J.-S., Kim Y.-S., Jin C.-Y., Seol M. B., Kim B.-C., Lee M.-C. (2010) Neuroprotective effects of (-)-epigallocatechin-3-gallate against quinolinic acid-induced excitotoxicity via PI3K pathway and NO inhibition. Brain Res. 1313, 25–33 [DOI] [PubMed] [Google Scholar]
- 21. Beurel E., Jope R. S. (2009) Glycogen synthase kinase-3 promotes the synergistic action of interferon-γ on lipopolysaccharide-induced IL-6 production in RAW264.7 cells. Cell Signal. 21, 978–985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Martin M., Rehani K., Jope R. S., Michalek S. M. (2005) Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat. Immunol. 6, 777–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Brown J., Wang H., Suttles J., Graves D. T., Martin M. (2011) Mammalian target of rapamycin complex 2 (mTORC2) negatively regulates Toll-like receptor 4-mediated inflammatory response via FoxO1. J. Biol. Chem. 286, 44295–44305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mayr B., Montminy M. (2001) Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell Biol. 2, 599–609 [DOI] [PubMed] [Google Scholar]
- 25. Elcombe S. E., Naqvi S., Van Den Bosch M. W. M., MacKenzie K. F., Cianfanelli F., Brown G. D., Arthur J. S. C. (2013) Dectin-1 Regulates IL-10 Production via a MSK1/2 and CREB Dependent Pathway and Promotes the Induction of Regulatory Macrophage Markers. PLoS One 8, e60086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tullai J. W., Chen J., Schaffer M. E., Kamenetsky E., Kasif S., Cooper G. M. (2007) Glycogen synthase kinase-3 represses cyclic AMP response element-binding protein (CREB)-targeted immediate early genes in quiescent cells. J. Biol. Chem. 282, 9482–9491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Thomson A. W., Turnquist H. R., Raimondi G. (2009) Immunoregulatory functions of mTOR inhibition. Nat. Rev. Immunol. 9, 324–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rastogi R., Jiang Z., Ahmad N., Rosati R., Liu Y., Beuret L., Monks R., Charron J., Birnbaum M. J., Samavati L. (2013) Rapamycin Induces MAP Kinase Phosphatase (MKP)-1 Expression Through Activation of Protein Kinase B and Mitogen-activated Protein Kinase Kinase Pathways. J. Biol. Chem. 288, 33966–33977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fan W., Morinaga H., Kim J. J., Bae E., Spann N. J., Heinz S., Glass C. K., Olefsky J. M. (2010) FoxO1 regulates Tlr4 inflammatory pathway signalling in macrophages. EMBO J. 29, 4223–4236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tejera M. M., Kim E. H., Sullivan J. A., Plisch E. H., Suresh M. (2013) FoxO1 Controls Effector-to-Memory Transition and Maintenance of Functional CD8 T Cell Memory. J. Immunol. 191, 187–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Smolinska M. J., Horwood N. J., Page T. H., Smallie T., Foxwell B. M. J. (2008) Chemical inhibition of Src family kinases affects major LPS-activated pathways in primary human macrophages. Mol. Immunol. 45, 990–1000 [DOI] [PubMed] [Google Scholar]
- 32. Morris M., Li L. (2011) Molecular Mechanisms and Pathological Consequences of Endotoxin Tolerance and Priming. Arch. Immunol. Ther. Exp. 60, 13–18 [DOI] [PubMed] [Google Scholar]
- 33. Motegi A., Kinoshita M., Sato K., Shinomiya N., Ono S., Nonoyama S., Hiraide H., Seki S. (2006) An in vitro Shwartzman reaction-like response is augmented age-dependently in human peripheral blood mononuclear cells. J. Leukoc. Biol. 79, 463–472 [DOI] [PubMed] [Google Scholar]
- 34. Slofstra S. H., ten Cate H., Spek C. A. (2006) Low dose endotoxin priming is accountable for coagulation abnormalities and organ damage observed in the Shwartzman reaction. A comparison between a single-dose endotoxemia model and a double-hit endotoxin-induced Shwartzman reaction. Thromb. J. 4, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Porta C., Rimoldi M., Raes G., Brys L., Ghezzi P., Di Liberto D., Dieli F., Ghisletti S., Natoli G., De Baetselier P., Mantovani A., Sica A. (2009) Tolerance and M2 (alternative) macrophage polarization are related processes orchestrated by p50 nuclear factor κB. Proc. Natl. Acad. Sci. U.S.A. 106, 14978–14983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chen X., El Gazzar M., Yoza B. K., McCall C. E. (2009) The NF-kB factor RelB and histone H3 lysine methyltransferase G9a directly interact to generate epigenetic silencing in endotoxin tolerance. J. Biol. Chem. 284, 27857–27865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lochhead P. A., Coghlan M., Rice S. Q., Sutherland C. (2001) Inhibition of GSK-3 selectively reduces glucose-6-phosphatase and phosphatase and phosphoenolypyruvate carboxykinase gene expression. Diabetes 50, 937–946 [DOI] [PubMed] [Google Scholar]
- 38. Lee J. Y., Chiu Y.-H., Asara J., Cantley L. C. (2011) Inhibition of PI3K binding to activators by serine phosphorylation of PI3K regulatory subunit p85a Src homology-2 domains. Proc. Natl. Acad. Sci. U.S.A. 108, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lin C.-F., Tsai C.-C., Huang W.-C., Wang C.-Y., Tseng H.-C., Wang Y., Kai J.-I., Wang S.-W., Cheng Y.-L. (2008) IFN-γ synergizes with LPS to induce nitric oxide biosynthesis through glycogen synthase kinase-3-inhibited IL-10. J. Cell Biochem. 105, 746–755 [DOI] [PubMed] [Google Scholar]
- 40. Wu S. S., Jácamo R. O., Vong S. K., Rozengurt E. (2006) Differential regulation of Pyk2 phosphorylation at Tyr-402 and Tyr-580 in intestinal epithelial cells: Roles of calcium, Src, Rho kinase, and the cytoskeleton. Cell Signal. 18, 1932–1940 [DOI] [PubMed] [Google Scholar]
- 41. Polumuri S. K., Jayakar G. G., Shirey K. A., Roberts Z. J., Perkins D. J., Pitha P. M., Vogel S. N. (2012) Transcriptional regulation of murine IL-33 by TLR and non-TLR agonists. J. Immunol. 189, 50–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kim D. H., Park C. H., Park D., Choi Y. J., Park M. H., Chung K. W., Kim S. R., Lee J. S., Chung H. Y. (2013) Ginsenoside Rc modulates Akt/FoxO1 pathways and suppresses oxidative stress. Arch. Pharm. Res. 37, 813–820 [DOI] [PubMed] [Google Scholar]
- 43. Li J.-M., Wang W., Fan C.-Y., Wang M.-X., Zhang X., Hu Q.-H., Kong L.-D. (2013) Quercetin Preserves β-Cell Mass and Function in Fructose-Induced Hyperinsulinemia through Modulating Pancreatic Akt/FoxO1 Activation. eCAM 2013, 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Woodgett J. R., Ohashi P. S. (2005) GSK3: an in-Toll-erant protein kinase? Nat. Immunol. 6, 751–752 [DOI] [PubMed] [Google Scholar]
- 45. Coghlan M. P., Culbert A. A., Cross D. A., Corcoran S. L., Yates J. W., Pearce N. J., Rausch O. L., Murphy G. J., Carter P. S., Roxbee Cox L., Mills D., Brown M. J., Haigh D., Ward R. W., Smith D. G., Murray K. J., Reith A. D., Holder J. C. (2000) Selective small molecule inhibitors of glycogen synthase kinase-3 modulate glycogen metabolism and gene transcription. Chem. Biol. 7, 793–803 [DOI] [PubMed] [Google Scholar]
- 46. Livingston M., Heaney L. G., Ennis M. (2004) Adenosine, inflammation and asthma–a review. Inflamm. Res. 53, 171–178 [DOI] [PubMed] [Google Scholar]
- 47. Fredholm B. B., Chern Y., Franco R., Sitkovsky M. (2007) Aspects of the general biology of adenosine A2A signaling. Prog. Neurobiol. 83, 263–276 [DOI] [PubMed] [Google Scholar]
- 48. Ferrante C. J., Pinhal-Enfield G., Elson G., Cronstein B. N., Hasko G., Outram S., Leibovich S. J. (2013) The adenosine-dependent angiogenic switch of macrophages to an M2-like phenotype is independent of interleukin-4 receptor α (IL-4Rα) signaling. Inflammation 36, 921–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Das A., Ganesh K., Khanna S., Sen C. K., Roy S. (2014) Engulfment of Apoptotic Cells by Macrophages: A Role of MicroRNA-21 in the Resolution of Wound Inflammation. J. Immunol. 192, 1120–1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fredman G., Li Y., Dalli J., Chiang N., Serhan C. N. (2012) Self-Limited versus Delayed Resolution of Acute Inflammation: Temporal Regulation of Pro-Resolving Mediators and MicroRNA. Sci. Rep. 2, 639–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ramirez S. H., Fan S., Zhang M., Papugani A., Reichenbach N., Dykstra H., Mercer A. J., Tuma R. F., Persidsky Y. (2010) Inhibition of glycogen synthase kinase 3b (GSK3b) decreases inflammatory responses in brain endothelial cells. Am. J. Pathol. 176, 881–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shimizu T., Ueda J., Ho J. C., Iwasaki K., Poellinger L., Harada I., Sawada Y. (2012) Dual inhibition of Src and GSK3 maintains mouse embryonic stem cells, whose differentiation is mechanically regulated by Src signaling. Stem Cells 30, 1394–1404 [DOI] [PubMed] [Google Scholar]
- 53. Anderson R. M., Barger J. L., Edwards M. G., Braun K. H., O'Connor C. E., Prolla T. A., Weindruch R. (2008) Dynamic regulation of PGC-1a localization and turnover implicates mitochondrial adaptation in calorie restriction. Aging Cell 7, 101–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ni M., MacFarlane A. W., 4th, Toft M., Lowell C. A., Campbell K. S., Hamerman J. A. (2012) B-cell adaptor for PI3K (BCAP) negatively regulates Toll-like receptor signaling through activation of PI3K. Proc. Natl. Acad. Sci. U.S.A. 109, 267–272 [DOI] [PMC free article] [PubMed] [Google Scholar]