

Background: High affinity human plasminogen (hPg) binding to Group A Streptococcus (GAS) is mediated by the coiled-coil M-like protein, PAM.
Results: Amino acid residues in the a1a2 domain of PAM direct high affinity PAM/hPg binding regardless of oligomerization status of PAM.
Conclusion: The a1a2 domain defines its hPg binding site, regardless of PAM dimerization.
Significance: The virulence of GAS is altered by mutations in PAM(a1a2).
Keywords: Bacteria, Bacterial Pathogenesis, Protein Domain, Protein Engineering, Protein Folding
Abstract
A emm53 subclass of Group A Streptococcus pyogenes (GAS) interacts tightly with human plasma plasminogen (hPg) and plasmin (hPm) via the kringle 2 (K2hPg) domain of hPg/hPm and the N-terminal a1a2 regions of a GAS coiled-coil M-like protein (PAM). Previous studies have shown that a monomeric PAM fragment, VEK30 (residues 97–125 + Tyr), interacted specifically with isolated K2hPg. However, the binding strength of VEK30 (KD = 56 nm) was ∼60-fold weaker than that of full-length dimeric PAM (KD = 1 nm). To assess whether this attenuated binding was due to the inability of VEK30 to dimerize, we defined the minimal length of PAM required to dimerize using a series of peptides with additional PAM residues placed at the NH2 and COOH termini of VEK30. VEK64 (PAM residues 83–145 + Tyr) was found to be the smallest peptide that adopted an α-helical dimer, and was bound to K2hPg with nearly the same affinity as PAM (KD = 1–2 nm). However, addition of two PAM residues (Arg126-His127) to the COOH terminus of VEK30 (VEK32) maintained a monomeric peptidic structure, but exhibited similar K2hPg binding affinity as full-length dimeric PAM. We identified five residues in a1a2 (Arg113, His114, Glu116, Arg126, His127), mutation of which reduced PAM binding affinity for K2hPg by ∼1000-fold. Replacement of these critical residues by Ala in the GAS genome resulted in reduced virulence, similar to the effects of inactivating the PAM gene entirely. We conclude that rather than dimerization of PAM, the five key residues in the binding domain of PAM are essential to mediate the high affinity interaction with hPg, leading to increased GAS virulence.
Introduction
More than 200 M-protein-based serotypes of Group A Streptococcus pyogenes (GAS)2 exist that cause >700 million cases of human pharyngeal and dermal infections worldwide each year. These infections range from non-invasive pharyngitis and impetigo, to severe and treatment-resistant forms of infectious diseases, e.g. necrotizing fasciitis and streptococcal toxic shock syndrome, as well as postinfectious nonpyogenic diseases, e.g. rheumatic fever and glomuleronephritis. Because of the existence of these latter highly virulent strains of GAS, which lead to high morbidity and oftentimes death, virulence factors of GAS have been sought for vaccine development as a global health issue. All GAS strains contain a form of a surface-resident M-protein, which is housed in a virulence operon of GAS under control of the multiple gene activator, mga (1). A subset of these M-proteins present in skin-tropic highly virulent strains of GAS bind with high affinity to host human plasma plasminogen (hPg), and this interaction is a critical determinant of the virulence of these strains (2). All GAS strains secrete a hPg activator, streptokinase, which, through a series of steps, converts GAS-bound hPg to the serine protease, plasmin (hPm) (3). The hPm remains bound to GAS, thus providing GAS to a proteolytic surface. This feature enables GAS to degrade encapsulating fibrin and, directly or indirectly, extracellular matrix proteins, thereby encouraging higher infectivity and systemic dissemination of the bacteria (4).
The noncatalytic chain of hPm and corresponding regions of hPg contain five ∼80 residue triple disulfide-linked kringle (K) domains (5). The main functions of kringle motifs are to mediate the binding of hPg to activation effectors and bind to relevant receptors and other proteins. Chief among the ligands for kringles is Lys and Lys analogues, with K1hPg, K4hPg, and K5hPg showing highest affinity for Lys. K2hPg possesses the lowest Lys binding strength for Lys binding kringles (6–9) and K3hPg does not possess a binding site for Lys (10). The physiological role for the kringle-based Lys binding sites (LBS) is to promote functional interactions of hPg and hPm with both cell-bound receptors and with other proteins that contain COOH-terminal Lys residues, e.g. enolase, annexin, PgR(KT) (11). One exception to this general rule is the binding of PAM to hPg. In this case, PAM uses the LBS of the weakest Lys-binding hPg kringle, K2hPg, for its tight (KD ∼ 1 nm) and specific interaction with hPg (12). This interaction does not require a COOH-terminal Lys (13).
The locus of PAM that interacts with hPg has been identified as the a1a2 N-terminal region (PAMa1a2) (14). A small peptide, VEK30 (PAM residues 97–125), which spans the PAMa1a2 domain, has been shown to bind in a unique manner to K2hPg. VEK30 does not possess a COOH-terminal Lys residue, yet requires the LBS for its interaction with K2hPg. To define the structural basis for VEK30/K2hPg binding, both x-ray (15, 16) and NMR structural analyses (13) have been employed. The data reveal that VEK30 forms an isosteric Lys utilizing the side chains of Arg113, His114, and Glu116 (PAM numbering) that is conformationally presented around one-turn of an α-helix in VEK30. Whereas this binding modality is novel and important, the strength of binding of VEK30 to K2hPg is ∼60-fold weaker than that displayed by PAM toward K2hPg.
The structure of PAM, like other M-proteins (17), is best represented by an α-helical irregular coiled-coil. However, VEK30 has been determined to be an α-helical monomer (15), and the question thus raised is whether the binding energy of PAM to K2hPg contains a significant contribution from the dimeric structure of PAM. To resolve this issue, we have expressed a number of extended recombinant VEK30 peptides and determined their conformational properties, dimerization states, binding strengths to K2hPg, and abilities to stimulate hPg activation. Herein, we show that strong hPg binding by PAM is independent of its oligomerization status and internal residues within the PAM a1a2 domain are major contributors to hPg binding.
EXPERIMENTAL PROCEDURES
Gene Expression and Protein Purification
Human recombinant (r) K2Pg (C4G/E56D/L72Y; kringle numbering from C1 of K2hPg), a triple variant of wild-type (WT) K2Pg that possessed an ∼5-fold enhanced affinity for ω-amino acids and VEK30, without any loss of binding specificity, and avoids purification complications from a non-LBS free Src homology group, was expressed and purified from transfected Pichia pastoris GS115 cells (9). rPAMAP53 (residues 42–392, lacking the 41-residue amino-terminal PAM signal peptide, as well as the 35-residue COOH-terminal transmembrane insertion and intracellular regions), was expressed in Escherichia coli BL21/DE3 cells and purified according to the method detailed earlier (18). rPAM[5A] was generated by mutation of Arg113, His114, Glu116, Arg126, and His127 (numbered from Met1 of PAMAP53) to Ala, using the QuikChange II XL site-directed mutagenesis kit (Invitrogen). rPAM[8A] was generated by replacing Asp103, Lys110, and Lys123 with Ala in rPAM[5A]. Both rPAM[5A] and rPAM[8A] were expressed and purified in the same manner as WT-PAMAP53.
rVEK-based peptides were designed by progressively adding residues to the NH2 and COOH termini of VEK30 (PAM residues 97–125). The peptides were expressed in E. coli BL21/DE3 employing the (His)6-tagged streptococcal protein GB1 domain fusion expression system (13, 19). The final constructs, except for VEK32 (PAM residues 97–127), contained, sequentially from the 5′: an ATG initiation codon, a purification (His)6 tag, the GB1 domain for enhanced solubility, a 9-residue linker, and a thrombin cleavage site, LVPR↓GS (19). This cassette was sequentially followed by synthetic genes encoding VEK or VEK variants, all inserted into plasmid pET-15b (Novagen, Gibbstown, NJ). A translation stop codon was placed immediately downstream of the VEK30 open reading frame. Thus, all VEK-based peptides cleaved via the thrombin site possessed a GS dipeptide at their NH2 termini. In addition, a Tyr was placed at the COOH termini of each peptide for 280-nm absorption properties, as well as for radiolabeling, when needed. For VEK32, a FXa cleavage site (IEGR↓) was used instead of the thrombin-sensitive site. Thus, cleavage with FXa does not leave extra residues at the NH2 terminus. Furthermore, we did not include an artificial COOH-terminal Tyr residue in VEK75 (PAM residues 88–162), because Tyr156 was present within its natural sequence.
All rVEK-peptides were affinity purified from culture medium on a Ni2+-Sepharose affinity chromatography column (HisTrap HP; GE Healthcare), after induction of the bacterial culture with isopropyl β-d-1-thiogalactopyranoside. The protein obtained was tag-digested following the method described previously (13). For final purifications, the peptides were chromatographed on a Superdex-75 16/60 size exclusion column (GE Healthcare), equilibrated and eluted with 20 mm Hepes-NaOH, 150 mm NaCl, pH 7.4. The integrity of the purified peptides was determined by MALDI-TOF mass spectrometry on an Autoflex III (Bruker Daltonics, Bremen, Germany). All peptides provided masses within 0.03% of their expected values. Recombinant streptokinase was prepared following previously described methodology (18). Human Glu-Pg (hPg) was obtained from the Enzyme Research Laboratory (South Bend, IN).
Protein Purity and Concentration
The purities of recombinant WT-PAMAP53, rPAM[5A], and rPAM[8A], K2Pg, and all VEK-peptides, were further assessed with 15% (w/v) SDS-PAGE and/or Tris/Tricine-PAGE. Protein concentrations were measured by the absorbance at 280 nm using the following calculated molar extinction coefficients (m−1 cm−1): His6-tagged WT-rPAMAP53, rPAM[5A], and rPAM[8A], 8,250; K2Pg, 10,810; VEK + Tyr peptides and VEK75, 1,490.
Circular Dichroism (CD) Spectroscopy
Far-UV CD spectra were collected for PAMAP53 and VEK-related peptides on an Aviv model 202 SF Spectrometer (Aviv, Lakewood, NJ). CD spectral data were recorded from 190 to 240 nm in a 0.1-cm path length cell at 25 °C. The buffer used was 100 mm sodium phosphate, pH 7.4. Spectral data represented the average of three scans, collected at a 1.0-nm bandwidth at 1.0-nm intervals. A buffer reference scan was subtracted from each sample scan. The mean residue ellipticities ([θ]) were calculated using: [θ] = (θ × MRW)/(l × c), where θ is the CD signal in mdeg, MRW is the mean residue weight in g/mol, l is the path length in mm, and c is the protein or peptide concentration in mg/ml (20).
Surface Plasmon Resonance (SPR)
The binding kinetics of WT-PAMAP53, PAMa1a2 mutants, and VEK-peptides to rK2Pg were measured in real-time by SPR using a BIAcore X100 Biosensor system (GE Healthcare). All binding experiments were conducted at 25 °C employing HBS-EP (10 mm Hepes, 0.15 m NaCl, 3 mm EDTA, 0.005% polysorbate 20, pH 7.4) as the running buffer at a flow rate of 10 μl/min. K2Pg, diluted to a final concentration of 10 μm in 0.1 m NaOAc, pH 5.0, was injected into flow cell 2 for immobilization on the CM-5 sensor chip surface using the amine-coupling kit, to a level of ∼1000 resonance units. Nonbound sites on the sensor chip surface were afterward blocked by injection of 1 m ethanolamine, pH 8.5. All binding experiments were conducted by injecting various concentrations of analytes in HBS-EP buffer over the K2Pg-coupled CM5 chip surface. Each concentration was injected for an association time of 2 min followed by 5 min dissociation time. The chip surface was regenerated between cycles using 10 mm glycine, pH 2.0, which did not change the binding properties of K2Pg bound to the CM-5 chip. The binding data from these sensorgrams were subtracted from those obtained using a reference flow cell prepared by the same method, but without immobilizing K2Pg on the chip. Sensorgrams were analyzed using BIAevaluation software 4.1 (BiaCore Life Sciences). The apparent equilibrium dissociation constants (KD) were calculated from the instrument software by the ratio of the dissociation (koff) and association rates (kon). Nonlinear fitting of the association and dissociation curves with a 1:1 binding model was employed.
Analytical Ultracentrifugation
Sedimentation equilibrium experiments were performed employing a Beckman (Fullerton, CA) XL-I analytical ultracentrifuge at 20 °C. The UV absorption mode at 280 nm was used to monitor the data. Samples were dissolved in 100 mm sodium phosphate, pH 7.4, at final concentration of 100–300 μm. A six-sector cell loaded with 110 μl of samples and 125 μl of reference buffer was used. Samples were individually rotated at speeds of 45,000 and 48,000 rpm for VEK35; 36,000 and 40,000 rpm for both VEK59 and VEK64; 32,000 and 36,000 rpm for VEK75; 13,000 and 18,000 rpm for PAM; 12,000 and 18,000 rpm for rPAM[5A] and rPAM[8A], respectively. Radial scans were collected every 2 h. At a given rotor speed, equilibrium was considered to have been reached once the temporal scans were invariant. The partial specific volume of VEK-peptides, PAM, PAM mutants, and the buffer density, were calculated using Sednterp and were determined to be 0.721, 0.723, 0.724, 0.729, and 0.73 ml/g for VEK35, VEK59, VEK64, VEK75, PAM, rPAM[5A], and rPAM[8A], respectively. The buffer density was 1.0091 g/ml. The ultracentrifuge data were analyzed by the Optima XL-A/XL-I data analysis software (Beckman Coulter, Brea, CA), and apparent molecular weights were obtained. Non-linear fitting of the concentration gradients for obtaining the molecular weights obtained at equilibrium was accomplished by mathematically maximizing the randomness of the residuals (21).
Small Angle X-ray Scattering
For these experiments, hPg was prepared from pooled human plasma on a Lys-Sepharose column as previously described (22). hPg, and the ligands, ϵ-aminocaproic acid (EACA), t-aminomethylcyclohexane-1-carboxylic acid (tranexamic acid; TxA), VEK35, and VEK75 were suspended in a buffer containing 100 mm sodium phosphate, pH 7.4, with 5% glycerol. hPg (5 μm) was pre-equilibrated with different concentrations of the ligands at 16 °C. The data were collected at the Australian Synchrotron (Clayton, AU) small angle x-ray scattering/WAXS beamline. The samples were maintained at 16 °C throughout data collection.
ScatterBrain IDL-based software (Australian Synchrotron) was used for data analysis and buffer corrections. PRIMUS (23) was used to determine the radius of gyration (Rg) based on the modified Guinier approximation (24). Rg values were normalized (showing the fraction of open conformation) within a titration series and plotted against the ligand concentrations. From the titration curve, Kopen, the concentration of ligand required to induce 50% of the maximal conformational change in hPg, was calculated using Prism software.
hPg Binding and Activation Assays with GAS Cells
The hPg binding abilities of individual GAS strains were examined by ELISA following the procedure described earlier (18). Individual wells of 96-well NUNC Maxisorb plates (NUNC Thermo, Rochester, NY) were coated with 50 μl of the cell suspensions (∼2 × 107 cfu) of individual GAS strains and incubated overnight at 4 °C. Nonspecific binding was blocked with 1% BSA. After washing 3 times with PBS, 100 μl of 20 μg/ml of hPg was added to the sample wells. Following a 2-h incubation at room temperature, the plate was washed 3 times and incubated for 2 h at room temperature, first with mouse anti-hPg (ERL, South Bend, IN), and, second, with horseradish peroxidase (HRP)-conjugated rabbit anti-mouse IgG (AbD Serotec, Oxfordshire, UK), diluted 1:1000. Color development was accelerated by adding 3,3′,5,5′-tetramethylbenzidine substrate (R&D Systems, Minneapolis, MN). This reaction was terminated after 20 min using an equal volume of 2 m H2SO4. The absorbance (A) was measured at 450 nm using a plate reader (Molecular Devices, Sunnyvale, CA). Each sample was tested in triplicate and the assay was repeated 3 times. Negative controls were prepared following the same procedure with the exception of PBS in place of cell suspensions. The data are presented as the mean ± S.E.
For measurements of hPg activation by GAS strains, 50 μl of cell suspensions of individually grown GAS strains were premixed with 0.25 mm chromogenic substrate, H-d-Val-Leu-Lys-p-nitroanilide (S2251; Chromogenix, Milan, Italy) and 0.2 μm Glu-hPg in 200 μl of 10 mm Hepes, 150 mm NaCl, pH 7.4. The reaction was accelerated with the addition of 5 nm recombinant streptokinase. The A405 nm was measured continuously at 37 °C for 4 h and recorded at 1-min intervals.
Isogenic Variants of AP53
For this work, we generated isogenic variants of AP53. The original AP53 clinical isolate used in this study contains an inactivating mutation, ΔThr1404, followed by a frameshift (25) in the cluster of virulence (Cov) sensor component of the two-component Cov responder (R)-sensor (S) transcriptional regulator, yielding CovR+S− (25). Because this mutation does not affect PAM expression or hPg binding to AP53 cells (25), we expressed the pam mutants in this CovR+S− strain for this in vitro work. To construct the targeting vector for the AP53/ΔPAM mutant, the chloramphenicol acetyltransferase (cat) gene was 5′-flanked by 300–400 bp of genomic DNA upstream of the ATG for pam and 3′-flanked by 300–400 bp downstream of the TAG stop codon for pam. The construct was inserted into the temperature-sensitive plasmid pHY304, which was transformed into AP53 cells. Single-double crossover (SCO-DCO) was then accomplished as described earlier (25), leading to generation of AP53/ΔPAM. To construct AP53/PAM[8A], the pam gene, with the same genomic flanking regions as above, was inserted in plasmid pHY304 and Asp103, Lys110, Arg113, His114, Glu116, Lys123, Arg126, and His127 were all mutated to Ala by routine methods. SCO-DCO was accomplished as above, thus generating the desired construct, AP53/PAM[8A].
Mouse Survival Studies
For survival studies, C57Bl/6 male mice, 6–10 weeks of age, containing the hPg transgene (26), were used as described earlier (27). A bacterial load of 6.5–7.2 × 109 GAS cells/mouse were injected subcutaneously and monitored for 10 days, with observations twice daily. For these comparative survival studies, we employed AP53/CovR+S− as the highly virulent form of AP53, viz. AP53CovR+S−/PAM+, and AP53/CovR+S−/ΔPAM and AP53/CovR+S−/PAM[8A] as the PAM inactivated samples.
RESULTS
The PAM-derived internal peptide, VEK30, has been used as a surrogate for PAM in defining its critical functional role in hPg binding. Although important structure-function relationships have been obtained using this peptide, VEK30 nonetheless exhibits ∼60-fold weaker interaction with K2hPg than full-length PAM. The goal of this study was to understand whether the weaker binding of VEK30 was due to the linear sequence selected for the peptide, the α-helical conformation, and/or its dimerization state. This latter issue is most relevant because intact PAM, like all M-proteins (17, 28, 29), exists on GAS as an α-helical coiled-coil dimer, and the ability of M-proteins to dimerize is critical to their ability to bind other ligands (30–32). Thus, we generated a series of recombinant peptides from PAM to assess their ability to dimerize and interact with K2hPg. The peptides prepared and purified are identified and characterized in Table 1. Their linear molecular weights have been determined by MALDI-TOF, and are within 0.03% of their calculated weights, thus affirming their integrities (Table 1).
TABLE 1.
Molecular weights of recombinant proteins and peptides
| Peptides/proteins | Molecular mass calculateda | Molecular mass MALDI-TOFb | Molecular mass AUCc | Kd (K2hPg) |
|---|---|---|---|---|
| rPAMd | 41,143 | 41,012 | 81,507 | 1.10 nm |
| VEK30e | 3,772 | 3,774 | 3,210f | 56 nm |
| VEK32g | 3,921 | 3,920 | NDh | 1.11 nm |
| VEK35i | 4,433 | 4,432 | 4,851 | 1.90 nm |
| VEK59j | 7,187 | 7,183 | 7,193 | 1.08 nm |
| VEK64k | 7,843 | 7,841 | 15,050 | 1.09 nm |
| VEK75l | 9,019 | 8,998 | 17,300 | 1.35 nm |
| r-PAM[5A]m | 40,782 | 40,204 | 78,559 | 1100 nm |
| r-PAM[8A]n | 40,624 | 40,645 | 72,248 | 1500 nm |
a Calculated molecular mass of the linear sequence of the peptide.
b Molecular mass of the linear sequence of the peptide from MALDI-TOF.
c Molecular mass in solution from analytical ultracentrifugation. The values represent the averages of duplicate runs at two different rotor speeds.
d rPAM comprises residues Asn42 to 392 and lacks the 41-residue signal peptide and the 35-residue COOH-terminal membrane insertion and intracellular domains.
e GS(V97EKLTADAELQRLK110NERHEEAELE120RLKSE125)Y. Residues outside the parentheses in all cases are exogenous to PAM and are products of the expression casettes.
f (V97EKLTADAELQRLK110NERHEEAELE120RLKSERH127)Y.
g From Ref. 46.
h ND, not determined.
i GS(V97EKLTADAELQRLK110NERHEEAELE120RLKSERHDHD130)Y.
j GS(E88ELQGLKDDVEKL100TADAELQRLK110NERHEEAELE120RLKSERHDHD130KKEAERKALE140DKLAD145)Y.
k GS(L83REKEEEL90QGLKDDVEKL100TADAELQRLK110NERHEEAELE120RLKSERHDHD130KKEAERKALE140DKLAD145)Y.
l GS(E88ELQGLKDDVEKL100TADAELQRLK110NERHEEAELE120RLKSERHDHD130KKEAERKALE140DKLADKQEHL150NGALRYINEKEA162).
m GS(Asn42-Gln392/R113A,H114A,E116A,R126A,H127A)H6.
n GS(Asn42-Gln392/K99A,K110A,R113A,H114A,E116A,K123A,R126A,H127A)H6.
Sedimentation equilibrium ultracentrifugation analysis was performed at two different rotor speeds. Examples of the primary data obtained are shown in Fig. 1, and the molecular weight data for the peptides examined are summarized in Table 1. Monomeric species are observed throughout the equilibrium concentration gradient created at two different rotor speeds for VEK30, VEK32, VEK35, and VEK59. The longer peptides, VEK64 and VEK75, as well as PAM, presented dimeric species throughout their equilibrium concentration gradients at two different rotor speeds. Thus, VEK30–59 clearly are monomeric, and peptides VEK >64 are dimers.
FIGURE 1.

Sedimentation equilibrium ultracentrifugation analysis of VEK-peptides. A, VEK64. The data were collected at a rotor speed of 36,000 rpm. B, VEK35. The data were collected at a rotor speed of 48,000 rpm. The partial specific volume of the VEK64 was calculated from its amino acid composition was 0.723 ml/g and for VEK35 was 0.721 ml/g. In both cases, the buffer was 0.1 m phosphate, pH 7.4 (density, 1.005 g/ml). The temperature of the experiments was 20 °C. The lines drawn through the data points were best fits for molecular masses of 15,050 Da for VEK64 and 4,851 Da for VEK35. The random residuals for the non-linear fits of the concentration gradients show that a single molecular weight species is present throughout the cell (21) of the molecular weight determined from the best-fit obtained by minimizing the sum of the residuals.
CD spectra were also obtained for these same peptides, examples of which are provided in Fig. 2 for PAM, VEK35, and VEK64. In all cases, spectra typical of α-helices are obtained, with ellipticity minima at 208 and 222 nm. The comparative spectra are very similar, suggesting that each peptide has a very high α-helical content. Using an ellipticity value of approximately −30,300fH-2340 (fH = fractional helix) at 222 nm as a comparative measure of α-helical content (20), all peptides contain ∼60–70% α-helical content, with VEK75 displaying the highest α-helicity among the VEK-peptides tested.
FIGURE 2.

Far-UV CD spectra of VEK-peptides. A, VEK35, 15 μm (dotted line); B, VEK64, 12 μm (solid gray line); C, PAM, 3 μm (solid black line). The buffer was 0.1 m phosphate, pH 7.4, at 20 °C. Mean residue ellipticities are expressed as degrees cm2 dmol−1.
After peptide characterizations, SPR was used to investigate the binding affinities of VEK-based peptides to K2hPg. This kringle domain was used as the immobilized ligand and VEK-peptides and/or rPAM as soluble analyte(s). Sensorgrams of these interactions were recorded (Fig. 3) and the dissociation constants (KD) obtained are listed in Table 1. rPAM binds strongly to K2hPg (∼1 nm), which is nearly 60-fold higher than VEK30/K2hPg binding. The two dimeric VEK-peptides, VEK64 and VEK75, showed K2hPg binding efficiencies at nearly the same affinity as rPAM. Unexpectedly, all monomeric constructs, including VEK32, which contain two additional C-terminal residues on VEK30, showed equally strong binding to K2hPg (KD ∼ 1–2 nm), thereby indicating that dimerization is not the key determinant for effective Pg binding. Previously, the solution structure of VEK30 bound to K2hPg had shown that most of the VEK30/K2hPg interaction involved side chains of Arg113, His114, and Glu116 of VEK30 (13). Because addition of two residues, Arg126 and His127, fortified the binding efficiency to the level of rPAM, we investigated the contributions of Arg113, His114, Glu116, Arg126, and His127 on K2hPg binding in more detail.
FIGURE 3.
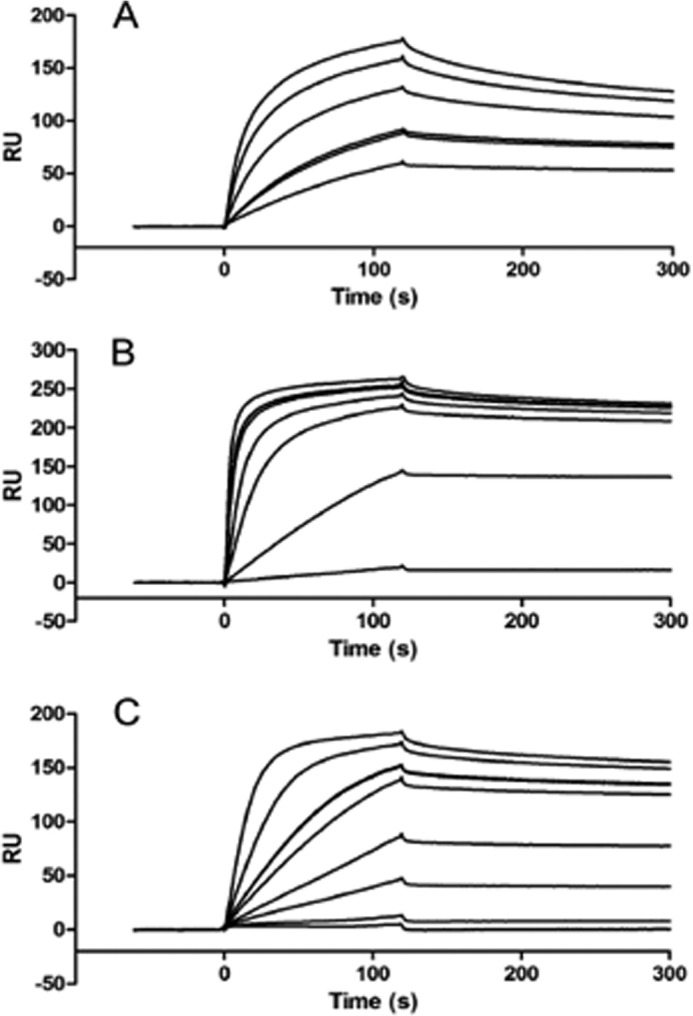
SPR of VEK-peptide binding to K2hPg. PAM (A), VEK64 (B), VEK35 (C). All binding experiments were conducted at 25 °C using 10 mm Hepes, 0.15 m NaCl, 3 mm EDTA, 0.005% polysorbate 20, pH 7.4, as the running buffer at a flow rate of 10 μl/min. The concentration range of PAM and VEK-peptides were progressively increased from 1 to 200 nm for the titrations. In cases where a double line is seen, the experiment was run twice at that concentration.
All five residues, i.e. Arg113, His114, Glu116, Arg126, and His127, were mutated to Ala in rPAM, generating rPAM[5A]. We also generated rPAM[8A] by replacing three additional residues, viz. Asp103, Lys110, and Lys123, with Ala because these residues had been shown to be involved in exosite interactions in the solution structure of VEK30 (13), thereby augmenting the affinity and facilitating docking of VEK30 to the K2hPg. Next we measured the binding affinities of rPAM[5A] and rPAM[8A] to K2hPg by SPR (Table 1). Mutations of these five residues in rPAM[5A] significantly reduced the affinity of this variant for K2hPg by 1000 times, relative to rPAM. rPAM[8A] also showed a similar binding affinity as rPAM[5A] for K2hPg, thereby indicating that three additional residues, i.e. Asp103, Lys110, and Lys123 do not contribute significantly to the hPg binding, and that Arg113, His114, Glu116, Arg126, and His127 are the major contributors to this binding energy. CD spectra of both mutants were found to be similar to that of rPAM (data not shown). Therefore, the decrease in binding affinity is not due to loss of secondary structures. These results support an important role for Arg113, His114, Glu116, Arg117, and His118, and indicate the indispensability of these residues in hPg binding.
To discover the effects of monomeric and dimeric VEK-peptides on hPg conformation we also performed a real-time SAX analysis of hPg using VEK35 and VEK75. Full-length hPg can exist in a closed (T), poorly activatable conformation, and an open (R), highly activatable, conformation (33, 34). The transition from the closed to the open conformation is induced by LBS binding effectors, such as EACA and its analogues (34). EACA primarily binds to the LBS of K1hPg and K4hPg, and more weakly binds to K5hPg (6, 35–37). Because we have identified in this study peptides that interact very strongly and specifically with the LBS of K2hPg, we have tested these peptides on the global conformation of hPg. We find that fully closed hPg becomes measurably relaxed upon incubation with a stoichiometric amount of VEK75 (Fig. 4A). Furthermore, the amount of ligand required to induce the midpoint transition from closed to open forms of hPg (Kopen) is 1.86 μm for VEK75, 26.7 μm for VEK 35, 120.4 μm for TxA, and 349 μm for EACA. Therefore, both VEK35 and VEK75 efficiently induce a change in the hPg conformation and significantly outperformed EACA and TxA in this regard (Fig. 4B), being effective at much lower concentrations. Thus, binding interactions of VEK-peptides, regardless of their oligomerization states, mediate the transformation of the tight (T) to the relaxed (R) conformations of hPg. Although binding of these peptides are specific for the LBS of K2hPg (14, 38), this does not rule out the possibility that exosites, especially in the longer peptides, interact with other regions of hPg to induce functionally different final conformations. This may explain the fact that whereas macroscopic binding of VEK35 and VEK75 binding to K2hPg is nearly the same (Fig. 3 and Table 1), differences are observed for VEK35 and VEK75 with regard to their relative efficiencies to induce a conformational change in hPg (Fig. 4). These low-resolution conformational techniques cannot discriminate subtle differences in conformations. However, major changes are induced via isosteric Lys interactions of the a1a2 region of PAM with the LBS of K2hPg to cause a large conformational alteration in hPg, a previously unrecognized finding. These results further suggest the importance of the presence ofthe hPg binding a1a2 domain of PAM and its internal residues.
FIGURE 4.
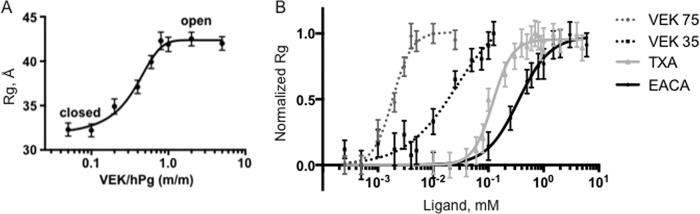
Conformational studies on hPg in the presence of VEK-peptides as determined by small angle x-ray scattering. A, hPg is fully open in the presence of a stoichiometric equivalent of VEK75. Plasminogen glycoform 2 was used for the experiments. B, the efficiency of VEK75 and VEK35 to induce conformational change in hPg is much higher than Lys analogues EACA and TxA.
To further investigate the role of these critical PAM residues in hPg binding, mutant GAS strains were generated and employed for cell-based assays. GAS strain AP53 was used as the WT PAM-producing strain, because AP53-PAM is a well characterized protein. For direct comparison with PAM, two mutant isogenic strains were generated, viz. AP53/PAM[8A], where eight residues in PAM (Asp103, Lys110, Arg113, His114, Glu116, Lys123, Arg126, and His127) were substituted by Ala, and GAS-AP53/ΔPAM, where the entire pam gene was replaced by an in-frame replacement of pam with the cat gene (25). We investigated the binding efficiency of hPg to these GAS cells by sandwich ELISA using mid-log phase cultures (A600 nm ∼ 0.6). As expected, AP53/PAM+ showed strong hPg binding (Fig. 5A), whereas AP53/PAM[8A], under the same conditions, showed a significantly reduced hPg binding relative to AP53PAM, undoubtedly because of the alteration of eight residues in PAM (Fig. 5A). The AP53/ΔPAM strain showed similar weak hPg binding to cells.
FIGURE 5.

hPg binding and activation by GAS cells. A, hPg binding by the indicated GAS strains examined by ELISA. Cells of the indicated strains were grown to mid-log phase (A600 nm ∼ 0.6) and resuspended in PBS to A600 nm ∼1.0. Cell suspensions of the indicated strains (∼2 × 107 cfu) were then added to the individual wells of a microtiter plate and incubated with Glu-hPg followed by appropriate antibody incubation with subsequent washes between. The color was developed with addition of HRP substrate 3,3′,5,5′-tetramethylbenzidine and detected at A450 nm. B, GAS cell suspensions of the indicated strains were mixed with Glu-hPg and chromogenic substrate S2251 in wells of microtiter plates. The reactions were accelerated by the addition of recombinant streptokinase and continually measured at A405 nm with 1-min intervals. Controls without cells were prepared in the same manner, except for the addition of GAS cells.
A similar observation was made when the activation of hPg by these strains was compared. Fig. 5B shows that hPg activation is highly stimulated by AP53/PAM+ cells, a finding that is in concurrence with the ELISA binding data. Comparatively, very little hPg is activated by other mutant GAS cell lines, including AP53/PAM[8A] and AP53/ΔPAM, both of which are only slightly higher than the basal level of activation without cells. Because we did not observe a significant difference in binding affinity between rPAM[5A] and rPAM[8A] to hPg using SPR, we conclude that the mutagenesis effect observed here is solely contributed by a 5-residue mutation. Therefore, these low activation properties of mutant AP53/PAM[8A] GAS cells especially demonstrate the requirement for the presence of all five PAM residues (Arg113, His114, Glu116, Arg126, and His127) for a fully active PAM, and in strains that contain PAM this protein is the dominant receptor for functional hPg binding.
Finally, we investigated the lethality of the PAM[8A] variant cell line on survival of hPg transgene containing mice (Fig. 6). Mice were subcutaneously injected with 6–8 × 108 cfu of WT-AP53/PAM+, AP53/ΔPAM, or AP53/PAM[8A]. All mice injected with WT-AP53/PAM+ expired within 4 days. Mutation of eight residues in the a1a2 domain of PAM, in AP53/PAM[8A], provided significant protection against lethality in mice, and deletion of the pam gene in GAS-AP53/ΔPAM offered similar protection from lethality in this model. It is possible that the presence of other hPg receptors and/or hPg-binding proteins, e.g. streptococcal enolase, Plr, contribute to the residual virulence in the mutant strains, as observed in our cell-based hPg binding and activation assays (Fig. 5), but this seems to be a more minor consideration in a PAM containing strain. These data suggest that AP53/PAM[8A] functions as a total PAM deletion with regard to hPg/hPm function, and eight critical residues of PAM in the a1a2 domain region are required for a fully functional and active PAM during GAS infections.
FIGURE 6.

Virulence of AP53 GAS strains evidenced by mice survival. C57BL/6 male mice were injected with 6.5–7.2 × 109 cfu of individual isogenic AP53 GAS strains, viz. WT-AP53 AP53/ΔPAM, and AP53/PAM[8A] (n = 5–7 mice for each strain). Survival was monitored for up to 10 days.
DISCUSSION
A key feature of certain pathogenic GAS strains is their ability to produce a variety of surface-bound and secreted virulence factors, which are known to contribute to the severity of their infection by interacting with host plasminogen activation system. Of four well studied hPg-binding proteins that have been described (39), hPg binding M and M-like protein (PAM) is the best characterized (40). PAM has long been recognized as a major virulence factor of skin-tropic GAS strains. Of all known receptors of hPg, PAM is the greatest contributor to direct hPg binding by GAS. Other hPg-binding proteins exist, such as streptococcal enolase and glyceralde-3-phosphate dehydrogenase (GAPDH), but these are less important in GAS strains wherein PAM and PAM-like high affinity hPg-binding proteins are present as the M-protein (25, 27, 41).
It has been demonstrated that PAM binding to K2hPg is mediated by an internal a1a2 repeat domain in the N terminus of PAM (14, 42). The importance of the a1a2 domain of PAM in hPg binding has also been demonstrated in Arp4, an M-like protein, which does not interact with hPg. Expression of a chimeric a1a2/PAM protein in Arp4 confers the ability of the isogenic Arp4 to bind hPg (43, 44). Structural studies of K2hPg complexed with VEK30, a 30-residue endopeptide derived from the a1a2 domain, demonstrated the manner in which internal charged residue side chains especially those of Arg113, His114, and Glu116 form a pseudo-Lys structure to occupy the LBS of K2hPg with a high affinity (KD ∼ 60 nm). Although this structure provides valuable insight into mechanisms of side chain arrangements in proteins that provide an isosteric Lys residue that interacts specifically with the LBS of K2hPg, it does not account for the full extent of binding of PAM to hPg. Because PAM is known to be a coiled-coil α-helical dimer, and the recent structure of a M1-protein revealed a dynamic coiled-coil dimer bound to fibrinogen (45), we examined whether the lack of a dimeric coiled-coil structure of isolated VEK30 might play a role in its weaker binding to hPg. Therefore, the purpose of this study was to investigate whether dimerization of PAM is an absolute requirement for its effective hPg binding.
To define the minimal length of PAM required for the retention of dimeric coiled-coil motif, we generated different constructs in which residues were added progressively to the N and C termini of the hPg binding core a1a2 domain. Using sedimentation equilibrium ultracentrifugation we determined that VEK64 contains the minimum number of residues required to form an α-helical dimer. Although both dimeric peptides tested for K2hPg binding showed a significant increase in binding affinity compared with VEK30 (Table 1), equally strong binding was displayed by several monomeric constructs, most notably, VEK32, which consists of two additional residues in VEK30 (Arg126 and His127). The Kd for VEK32 to K2hPg binding was equal to the same binding of full-length PAM to this kringle module (KD ∼ 1 nm). These data not only indicate that Arg126 and His127 are critical for hPg binding but also suggest that these residues may be more important for binding than the dimerization of PAM. This finding is in good agreement with small angle x-ray scattering analysis of monomeric VEK35 and dimeric VEK75, which showed that both peptides are far more efficient in inducing the conformational changes in hPg that occur upon occupancy of the LBS than TxA and EACA. Our data uniquely show that binding solely to K2hPg, the weakest LBS for small molecule Lys analogues, confers the conformational change in hPg that is known to occur by binding of Lys-like ligands to K1hPg, K4hPg, and K5hPg. Although monomeric VEK35 appears to bind very strongly and specifically to the LBS of K2hPg, dimeric VEK75 is superior in inducing changes in hPg conformation. One possible explanation for the lack of agreement between VEK75 and VEK35, with respect to hPg binding and activation characteristics may lie in the absence of residues in VEK35 that might contribute to exosite interactions between Pg and PAM, rendering the interaction tighter and thereby increasing the potency of Pg activation. This could also indicate a positive correlation between an increasing length of coiled-coils in PAM and increased potential for hPg activation, as dimeric VEK75 displays the highest α-helical content among all VEK-peptides tested. It has been reported previously that the presence of a 20-fold molar excess of VEK30 failed to induce any change in the closed conformation of hPg (46). Whereas we found that 5-fold molar excesses of VEK35 is sufficient to induce a closed to open form transition in hPg that probably highlights the importance of residues immediately after VEK30, possibly Arg126 and His127 in hPg binding and stimulation.
Thus, we shifted our attention to the internal residues of the a1a2 domain. Previous structural studies of VEK30 bound K2hPg have shown that Arg113, His114, and Glu116, all present in the one turn of the helix in the a1 repeat, form salt bridges and electrostatic interactions with the LBS of K2hPg thereby forming a pseudo-Lys structure. It is therefore anticipated that the corresponding residues in the a2 repeat, i.e. Arg126 and His127, would make a similar contribution to K2hPg binding. In fact, a strong hPg binding affinity reflected by a KD value of ∼1 nm as shown by VEK32, which included Arg126 and His127, supported this hypothesis. Indeed, when Arg113, His114, Glu116, Arg126, and His127 were replaced by Ala using site-directed mutagenesis, the resulting mutant showed drastically reduced K2hPg binding, as evidenced by a 1000-fold decrease in the Kd (∼1 μm) for this interaction. Mutation of three additional residues Asp103, Lys110, and Lys123, each to Ala, providing rPAM[8A], showed a similar binding efficiency (KD ∼ 1.5 μm) as PAM[5A]. These latter three residues were mutated to verify their effect on hPg binding as they were found to be involved in exosite interactions in K2hPg bound VEK30 structure (13). Additionally, in a previously published report, substitution of Lys110 was shown to reduce hPg binding by 80% (12). However, a recent study showed that mutation of Lys110 and Lys123 was not sufficient to abrogate hPg binding (41). Our results support this latter report and suggests that Arg113, His114, Glu116, Arg126, and His127 are the crucial residues for hPg binding, especially Arg126 and His127. In another study involving hPg binding M-like protein Prp, it was reported that mutagenesis of Arg107 and His108 (the corresponding PAM residues are Arg113 and His114) abolished hPg binding despite the presence of two Lys residues, equivalent to Lys110 and Lys123 of PAM (47). Although it is known that the hPg binding domain of PAM is highly variable (48), both Arg and His residues in the a1 and a2 repeats appear to be highly conserved in PAM variants (4). Dramatic reduction in hPg binding by simultaneous mutation of Arg113, His114, Arg126, and His127, as demonstrated by our study, therefore indicates that binding to hPg using Arg and His side chain residues might be a common binding mechanism for hPg-binding M proteins.
The importance of these residues is also illustrated in cell-based assays, which show hPg binding and activation efficiency of PAM mutant cells were significantly reduced to the level of a total pam gene inactivation when compared with WT cells. Whereas we used the PAM[8A] mutant strain for these studies, it could be safely assumed that the mutagenesis effect is concerted by the five crucial residues in rPAM[5A], because the hPg binding affinity for these mutants is similar between both rPAM[5A] and rPAM[8A] mutants as shown by their respective KD values (Table 1).
In summary, we sought to determine whether dimerization of PAM is a determining factor for the high affinity hPg binding affinity of PAM. This study highlights the relative contribution to hPg binding by residues within the a1a2 domain of PAM. The high affinity binding of peptides that do not dimerize, which nonetheless is equivalent to that of PAM, suggests that PAM dimerization is not an essential feature of the ability of PAM to tightly interact with hPg. These findings could have important implications for the design of therapeutics that target this essential protein-protein interaction for highly virulent GAS strains.
This work was supported, in whole or in part, by National Institutes of Health Grant HL03423.
- GAS
- Group A Streptococcus pyogenes
- hPm
- human plasmin
- hPg
- human plasminogen
- K
- kringle motifs
- LBS
- Lys binding site
- SPR
- surface plasmon resonance
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- EACA
- ϵ-aminocaproic acid
- Cov
- cluster of virulence
- TxA
- tranexamic acid
- rPAM
- recombinant PAM
- PAM
- plasminogen-binding group A streptococcal M-like protein.
REFERENCES
- 1. Caparon M. G., Scott J. R. (1987) Identification of a gene that regulates expression of M protein, the major virulence determinant of group A streptococci. Proc. Natl. Acad. Sci. U.S.A. 84, 8677–8681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sanderson-Smith M. L., Dinkla K., Cole J. N., Cork A. J., Maamary P. G., McArthur J. D., Chhatwal G. S., Walker M. J. (2008) M protein-mediated plasminogen binding is essential for the virulence of an invasive Streptococcus pyogenes isolate. FASEB J. 22, 2715–2722 [DOI] [PubMed] [Google Scholar]
- 3. Castellino F. J., Ploplis V. A. (2003) Human plasminogen: structure, activation, and function. Plasminogen Structure, activation, and regulation, pp. 3–17, Kluwer Academic/Plenum Publishers, Norwell, MA [Google Scholar]
- 4. McKay F. C., McArthur J. D., Sanderson-Smith M. L., Gardam S., Currie B. J., Sriprakash K. S., Fagan P. K., Towers R. J., Batzloff M. R., Chhatwal G. S., Ranson M., Walker M. J. (2004) Plasminogen binding by group A streptococcal isolates from a region of hyperendemicity for streptococcal skin infection and a high incidence of invasive infection. Infect. Immun. 72, 364–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Magnusson S., Petersen T. E., Sottrup-Jensen L., Claeys H. (1975) Complete primary structure of prothrombin: Isolation and reactivity of ten carboxylated glutamic residues and regulation of prothrombin activation by thrombin. in Proteases and Biological Control (Reich E., Rifkin D. B., Shaw E., eds) pp. 123–149, Cold Spring Harbor Laboratories, Cold Spring Harbor, NY [Google Scholar]
- 6. Menhart N., Sehl L. C., Kelley R. F., Castellino F. J. (1991) Construction, expression and purification of recombinant kringle 1 of human plasminogen and analysis of its interaction with ω-amino acids. Biochemistry 30, 1948–1957 [DOI] [PubMed] [Google Scholar]
- 7. Menhart N., McCance S. G., Sehl L. C., Castellino F. J. (1993) Functional independence of the kringle 4 and kringle 5 regions of human plasminogen. Biochemistry 32, 8799–8806 [DOI] [PubMed] [Google Scholar]
- 8. Menhart N., Castellino F. J. (1995) The importance of the hydrophobic components of the binding energies in the interaction of ω-amino acid ligands with isolated kringle polypeptide domains of human plasminogen. Int. J. Pept. Protein Res. 46, 464–470 [DOI] [PubMed] [Google Scholar]
- 9. Nilsen S. L., Prorok M., Castellino F. J. (1999) Enhancement through mutagenesis of the binding of the isolated kringle 2 domain of human plasminogen to ω-amino acid ligands and to an internal sequence of a Streptococcal surface protein. J. Biol. Chem. 274, 22380–22386 [DOI] [PubMed] [Google Scholar]
- 10. Christen M. T., Frank P., Schaller J., Llinás M. (2010) Human plasminogen kringle 3: solution structure, functional insights, phylogenetic landscape. Biochemistry 49, 7131–7150 [DOI] [PubMed] [Google Scholar]
- 11. Godier A., Hunt B. J. (2013) Plasminogen receptors and their role in the pathogenesis of inflammatory, autoimmune and malignant disease. J. Thromb. Haemost. 11, 26–34 [DOI] [PubMed] [Google Scholar]
- 12. Berge A., Sjöbring U. (1993) PAM, a novel plasminogen-binding protein from Streptococcus pyogenes. J. Biol. Chem. 268, 25417–25424 [PubMed] [Google Scholar]
- 13. Wang M., Zajicek J., Geiger J. H., Prorok M., Castellino F. J. (2010) Solution structure of the complex of VEK-30 and plasminogen kringle 2. J. Struct. Biol. 169, 349–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wistedt A. C., Kotarsky H., Marti D., Ringdahl U., Castellino F. J., Schaller J., Sjöbring U. (1998) Kringle 2 mediates high affinity binding of plasminogen to an internal sequence in streptococcal surface protein PAM. J. Biol. Chem. 273, 24420–24424 [DOI] [PubMed] [Google Scholar]
- 15. Rios-Steiner J. L., Schenone M., Mochalkin I., Tulinsky A., Castellino F. J. (2001) Structure and binding determinants of the recombinant kringle-2 domain of human plasminogen to an internal peptide from a group A Streptococcal surface protein. J. Mol. Biol. 308, 705–719 [DOI] [PubMed] [Google Scholar]
- 16. Cnudde S. E., Prorok M., Castellino F. J., Geiger J. H. (2006) X-ray crystallographic structure of the angiogenesis inhibitor, angiostatin, bound to a peptide from the group A streptococcal surface protein PAM. Biochemistry 45, 11052–11060 [DOI] [PubMed] [Google Scholar]
- 17. McNamara C., Zinkernagel A. S., Macheboeuf P., Cunningham M. W., Nizet V., Ghosh P. (2008) Coiled-coil irregularities and instabilities in group A Streptococcus M1 are required for virulence. Science 319, 1405–1408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang Y., Liang Z., Hsueh H. T., Ploplis V. A., Castellino F. J. (2012) Characterization of streptokinases from Group A streptococci reveals a strong functional relationship that supports the coinheritance of plasminogen-binding M-protein and cluster 2b streptokinase. J. Biol. Chem. 287, 42093–42103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cheng Y., Patel D. J. (2004) An efficient system for small protein expression and refolding. Biochem. Biophys. Res. Commun. 317, 401–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chen Y.-H., Yang J. T., Martinez H. M. (1972) Determination of secondary structure of proteins by circular dichroism and optical rotary dispersion. Biochemistry 11, 4120–4131 [DOI] [PubMed] [Google Scholar]
- 21. Fujita D., Suzuki K., Sato S., Yagi-Utsumi M., Yamaguchi Y., Mizuno N., Kumasaka T., Takata M., Noda M., Uchiyama S., Kato K., Fujita M. (2012) Protein encapsulation within synthetic molecular hosts. Nat. Commun. 3, 1093. [DOI] [PubMed] [Google Scholar]
- 22. Brockway W. J., Castellino F. J. (1972) Measurement of the binding of antifibrinolytic amino acids to various plasminogens. Arch. Biochem. Biophys. 151, 194–199 [DOI] [PubMed] [Google Scholar]
- 23. Konarev P. V., Volkov V. V., Sokolova A. V., Koch M. H. J., Svergun D. I. (2003) PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 36, 1277–1282 [Google Scholar]
- 24. Shang C., Rice J. A. (2001) Interpretation of small-angle x-ray scattering data from dilute montmorillonite suspensions using a modified Guinier approximation. Phys. Rev. 64E, 021401 [DOI] [PubMed] [Google Scholar]
- 25. Liang Z., Zhang Y., Agrahari G., Chandrahas V., Glinton K., Donahue D. L., Balsara R. D., Ploplis V. A., Castellino F. J. (2013) A natural inactivating mutation in the CovS component of the CovRS regulatory operon in a pattern D Streptococcal pyogenes strain influences virulence-associated genes. J. Biol. Chem. 288, 6561–6573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sun H., Ringdahl U., Homeister J. W., Fay W. P., Engleberg N. C., Yang A. Y., Rozek L. S., Wang X., Sjöbring U., Ginsburg D. (2004) Plasminogen is a critical host pathogenicity factor for Group A streptococcal infection. Science 305, 1283–1286 [DOI] [PubMed] [Google Scholar]
- 27. Agrahari G., Liang Z., Mayfield J. A., Balsara R. D., Ploplis V. A., Castellino F. J. (2013) Complement-mediated opsonization of invasive Group A Streptococcus pyogenes strain AP53 is regulated by the bacterial two-component cluster of virulence responder/sensor (CovRS) system. J. Biol. Chem. 288, 27494–27504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fischetti V. A. (1989) Streptococcal M protein: molecular design and biological behavior. Clin. Microbiol. Rev. 2, 285–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Navarre W. W., Schneewind O. (1999) Surface proteins of Gram-positive bacteria and mechanisms of their targeting to the cell wall envelope. Microbiol. Mol. Biol. Rev. 63, 174–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Akerström B., Lindahl G., Björck L., Lindqvist A. (1992) Protein Arp and protein H from group A streptococci. Ig binding and dimerization are regulated by temperature. J. Immunol. 148, 3238–3243 [PubMed] [Google Scholar]
- 31. Sandin C., Linse S., Areschoug T., Woof J. M., Reinholdt J., Lindahl G. (2002) Isolation and detection of human IgA using a streptococcal IgA-binding peptide. J. Immunol. 169, 1357–1364 [DOI] [PubMed] [Google Scholar]
- 32. Persson J., Lindahl G. (2005) Single-step purification of human C4b-binding protein (C4BP) by affinity chromatography on a peptide derived from a streptococcal surface protein. J. Immunol. Methods 297, 83–95 [DOI] [PubMed] [Google Scholar]
- 33. Violand B. N., Sodetz J. M., Castellino F. J. (1975) The effect of ϵ-aminocaproic acid on the gross conformation of plasminogen and plasmin. Arch. Biochem. Biophys. 170, 300–305 [DOI] [PubMed] [Google Scholar]
- 34. Violand B. N., Byrne R., Castellino F. J. (1978) The effect of α-, ω-amino acids on human plasminogen structure and activation. J. Biol. Chem. 253, 5395–5401 [PubMed] [Google Scholar]
- 35. Sehl L. C., Castellino F. J. (1990) Thermodynamic properties of the binding of α-, ω-amino acids to the isolated kringle 4 region of human plasminogen as determined by high sensitivity titration calorimetry. J. Biol. Chem. 265, 5482–5486 [PubMed] [Google Scholar]
- 36. Sehl L. C. (1991) Thermodynamic and structural considerations for the interaction of kringle domains with ω-amino acids. Ph.D. Dissertation, University of Notre Dame, Notre Dame, IN [Google Scholar]
- 37. Chang Y., Mochalkin I., McCance S. G., Cheng B., Tulinsky A., Castellino F. J. (1998) Structure and ligand binding determinants of the recombinant kringle 5 domain of human plasminogen. Biochemistry 37, 3258–3271 [DOI] [PubMed] [Google Scholar]
- 38. Schenone M. M., Warder S. E., Martin J. A., Prorok M., Castellino F. J. (2000) An internal histidine residue from the bacterial surface protein, PAM, mediates its binding to the kringle-2 domain of human plasminogen. J. Pept. Res. 56, 438–445 [DOI] [PubMed] [Google Scholar]
- 39. Bhattacharya S., Ploplis V. A., Castellino F. J. (2012)Bacterial plasminogen receptors utilize host plasminogen system for effective invasion and dissemination. J. Biomed. Biotechnol. 2012, 482096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Svensson M. D., Sjöbring U., Bessen D. E. (1999) Selective distribution of a high-affinity plasminogen-binding site among group A streptococci associated with impetigo. Infect. Immun. 67, 3915–3920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sanderson-Smith M. L., Walker M. J., Ranson M. (2006) The maintenance of high affinity plasminogen binding by group A streptococcal plasminogen-binding M-like protein is mediated by arginine and histidine residues within the a1 and a2 repeat domains. J. Biol. Chem. 281, 25965–25971 [DOI] [PubMed] [Google Scholar]
- 42. Wistedt A. C., Ringdahl U., Müller-Esterl W., Sjøbring U. (1995) Identification of a plasminogen-binding motif in PAM, a bacterial surface protein. Mol. Microbiol. 18, 569–578 [DOI] [PubMed] [Google Scholar]
- 43. Ringdahl U., Svensson H. G., Kotarsky H., Gustafsson M., Weineisen M., Sjöbring U. (2000) A role for the fibrinogen-binding regions of streptococcal M proteins in phagocytosis resistance. Mol. Microbiol. 37, 1318–1326 [DOI] [PubMed] [Google Scholar]
- 44. Ringdahl U., Svensson M., Wistedt A. C., Renn T., Kellner R., Müller-Esterl W., Sjöbring U. (1998) Molecular co-operation between protein PAM and streptokinase for plasmin acquisition by Streptococcus pyogenes. J. Biol. Chem. 273, 6424–6430 [DOI] [PubMed] [Google Scholar]
- 45. Macheboeuf P., Buffalo C., Fu C. Y., Zinkernagel A. S., Cole J. N., Johnson J. E., Nizet V., Ghosh P. (2011) Streptococcal M1 protein constructs a pathological host fibrinogen network. Nature 472, 64–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Figuera-Losada M., Ranson M., Sanderson-Smith M. L., Walker M. J., Castellino F. J., Prorok M. (2010) Effects on human plasminogen conformation and activation rate caused by interaction with VEK-30, a peptide derived from the group A Streptococcal M-like protein (PAM). Biochim. Biophys. Acta 1804, 1342–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sanderson-Smith M. L., Dowton M., Ranson M., Walker M. J. (2007) The plasminogen-binding group A streptococcal M protein-related protein Prp binds plasminogen via arginine and histidine residues. J. Bacteriol. 189, 1435–1440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sanderson-Smith M., Batzloff M., Sriprakash K. S., Dowton M., Ranson M., Walker M. J. (2006) Divergence in the plasminogen-binding group a streptococcal M protein family: functional conservation of binding site and potential role for immune selection of variants. J. Biol. Chem. 281, 3217–3226 [DOI] [PubMed] [Google Scholar]