

Abstract
Sarcolemmal Na+/H+ exchanger 1 (NHE1) activity is essential for the intracellular pH (pHi) homeostasis in cardiac myocytes. Emerging evidence indicates that sarcolemmal NHE1 dysfunction was closely related to cardiomyocyte death, but it remains unclear whether defective trafficking of NHE1 plays a role in the vital cellular signalling processes. Dynamin (DNM), a large guanosine triphosphatase (GTPase), is best known for its roles in membrane trafficking events. Herein, using co-immunoprecipitation, cell surface biotinylation and confocal microscopy techniques, we investigated the potential regulation on cardiac NHE1 activity by DNM. We identified that DNM2, a cardiac isoform of DNM, directly binds to NHE1. Overexpression of a wild-type DNM2 or a dominant-negative DNM2 mutant with defective GTPase activity in adult rat ventricular myocytes (ARVMs) facilitated or retarded the internalization of sarcolemmal NHE1, whereby reducing or increasing its activity respectively. Importantly, the increased NHE1 activity associated with DNM2 deficiency led to ARVMs apoptosis, as demonstrated by cell viability, terminal deoxynucleotidyl transferase–mediated dUTP nick-end labelling assay, Bcl-1/Bax expression and caspase-3 activity, which were effectively rescued by pharmacological inhibition of NHE1 with zoniporide. Thus, our results demonstrate that disruption of the DNM2-dependent retrograde trafficking of NHE1 contributes to cardiomyocyte apoptosis.
Keywords: Na+/H+ exchanger 1, dynamin, trafficking, apoptosis, cardiomyocytes
Introduction
Na+/H+ exchangers (NHEs), a ubiquitously expressed family of integral membrane proteins, play important roles in the regulation of intracellular pH (pHi) via H+ extrusion driven by transmembrane Na+ gradient [1]. To date, 12 mammalian isoforms of NHE have been identified, although NHE1 is the primary acid extrusion system in cardiac cells [2]. Several studies have demonstrated that sarcolemmal NHE1 dysfunction was closely related to cardiomyocyte death under pathophysiological conditions such as ischaemia-reperfusion injury, ventricular hypertrophy and myocardial infarction, underlying the pathophysiological importance of the fine-tuned NHE1 activity [3–6].
NHE1 activity is dynamically changed in response to the fluctuation of pHi, and is also subject to the modulation by various extracellular stimuli, such as growth factors, hormones, integrin engagement [7–9], and the intracellular messengers such as protein kinase A, mitogen-activated protein kinase and Ca2+/calmodulin complex [10–12]. The enhancement of NHE1 activity in parallel with the increase in its membrane expression implicated the involvement of a protein trafficking process in the regulation of NHE1 activity [13]. However, the molecular mechanism underlying this process remains undefined.
Dynamin (DNM) is a superfamily of large guanosine triphosphatases (GTPase) that hydrolyse GTP to form GDP [14]. Mammals express three DNM isoforms, of which DNM2 is ubiquitously expressed; DNM1 and DNM3 are predominantly distributed in neurons and testis respectively [15]. DNM is best known for its roles in membrane-trafficking processes, for example, caveolae-mediated and clathrin-dependent endocytosis, trafficking from the trans-Golgi network and membrane invagination/fission [16–19]. A recent study reported the regulatory effects of DNM on sarcolemmal L-type calcium channel [20], suggesting the potential role of DNM in the cardiomyocyte membrane-related protein trafficking. In this study, we aimed to determine whether cardiac DNM can modulate the trafficking and activity of sarcolemmal NHE1, and if so, to explore the related physiological implication.
Materials and methods
Construction of adenoviral vectors
The wild-type (WT) and the mutated form (K44A, defective in GTP binding) of DNM2 were used in the manipulation of DNM2 activity. Briefly, the cDNAs of rat DNM2WT/DNM2K44A (obtained from ATCC; DNM2WT: MBA-94, DNM2K44A: MBA-95; http://www.atcc.org) were cloned into the pDONR221 vector using BP Clonase® enzyme mix (Invitrogen, Grand Island, NY, USA). The pDONR221 vectors containing the DNM2WT/DNM2K44A sequences were subsequently recombined with the pAd/CMV/V5-DEST vector using LR Clonase® enzyme mix (Invitrogen) to yield the pAd/CMV/V5-DEST-DNM2-WT or pAd/CMV/V5-DEST-DNM2-K44A construct. The recombinant adenoviruses harbouring the DNM2WT/DNM2K44A gene were generated in HEK293 cells and titrated as described previously [21].
Isolation of ventricular myocytes and adenoviral gene transfer
The protocol for animal experiments was approved by the Institutional Animal Care and Use Committee of Tongji University. Adult rat ventricular myocytes (ARVMs) were isolated from male Sprague-Dawley rats weighing 250–300 g, plated on laminin-coated dishes and cultured according to a protocol described previously [22]. Adenovirus-mediated gene transfer was performed by adding the vectors encoding DNM2WT or DNM2K44A into culture medium at the desired multiplication of infection (MOI). GFP- or β-gal-expressing viruses were used as a mock control. In some sets of the experiments, the infected cells were treated with zoniporide (Tocris, Minneapolis, MN, USA) at 1 μM for 36 hrs. The viability of the infected ARVMs was evaluated by Trypan Blue exclusion, as described elsewhere [23].
Cell-surface biotinylation
The ARVMs were washed with cold PBS and incubated with 2 mg/ml sulfo-NHS-LC-Biotin (Pierce, Rockford, IL, USA) for 30 min. at 4°C. Unreacted biotin was quenched by a Quenching solution containing 100 mM glycine in PBS. The cells were subsequently lysed in RIPA buffer (150 mM NaCl, 50 mM Tris–HCl, pH 7.4, 1% sodium deoxycholate, 1% NP-40, 1 mM PMSF and 1 mM EDTA). The lysates containing biotinylated proteins were incubated with pre-washed streptavidin-agarose beads (Pierce) at 4°C overnight. After three washes, bound surface proteins were eluted and separated by SDS-PAGE.
Co-immunoprecipitation and western blotting
The protein extracts from freshly isolated ARVMs were pre-cleaned by Protein G Sepharose (Sigma-Aldrich, St. Louis, MO, USA) and then incubated with 5 μg of primary antibodies for 2 hrs, followed by Protein G Sepharose for 1 hr. The Sepharose were washed five times and boiled in Laemmli buffer for analyses of the associated proteins by SDS–PAGE. Western blotting was performed with the primary antibodies against DNM2 (Abcam, Cambridge, MA, USA), NHE1 (Santa Cruz, Dallas, TX, USA), Bcl-2 (Beyotime, Jiangsu, China), Bax (Cell signaling, Boston, MA, USA), cleaved caspase-3 (Cell signaling, Boston, MA, USA) and GAPDH (Cell signaling, Boston, MA, USA).
NHE1 trafficking experiments in HEK293 cells
NHE1 trafficking was studied in HEK293 cells, as reported previously [24]. The plasmid encoding human NHE1 and GFP (hNHE1-GFP) fusion protein was a gift from Dr. Chi-Hung Lin (National Yang-Ming University, Taiwan). One day before transfection, the cells were plated on coverslips in 35-mm dishes at 40–50% confluence. Transfection was performed with 1.6 μg of hNHE1-GFP plasmid and Lipofectamine 2000™ transfection reagent (Invitrogen) according to the manufacturer's instructions. After 24 hrs, Adv-DNM2WT, -DNM2K44A or a mock vector was added for another 24 hrs prior to imaging analyses.
Measurement of Na+/H+ exchanger activity
The activity of NHE1 was measured as the rate of Na+-dependent pHi recovery following an acid load (NH4Cl pre-pulse) as described previously [25]. Briefly, cultured ARVMs were loaded with the membrane-permeable form of a pHi indicator 2′, 7′-bis (carboxyethyl)-5 (6)-carboxyfluorescein ester (BCECF-AM) (1 μM, Dojindo Laboratories, Rockville, MD, USA) for 30 min. at 37°C. The cells were then rinsed with HEPES-buffered Tyrode's solution (in mM: NaCl 140, KCl 5, Hepes 10, CaCl2 2, MgCl2 1; pH 7.4 with NaOH). After the baseline pHi was recorded, the cells were perfused with an acid-loaded solution based on the Tyrode's composition with the following substitutions: NaCl and KCl were, respectively, replaced with 20 mM NH4Cl and 120 mM N-methyl-D-glucamine (NMDG/Cl). Subsequently, normal Tyrode's solution was added to yield the NHE activity as the pHi recovery rate. Throughout the experiment, the pHi was monitored using a Leica SP5 inverted microscope. The cells were excited successively at 490 and 450 nm. The 490/450 emission ratio obtained from the intracellular BCECF-AM was converted to a linear pH scale using in situ data calibration, which was performed at the end of each experiment using the nigericin technique described elsewhere [26].
TUNEL and Hoechst staining
In situ cell death was evaluated in the ARVMs with a terminal deoxynucleotidyl transferase–mediated dUTP nick-end labelling (TUNEL) assay kit (Roche Diagnostics, Indianapolis, IN, USA) according to the manufacturer's instructions. Hoechst 33258 (Beyotime) staining was subsequently performed on the same ARVMs. The percentage of the TUNEL-positive cells was determined by counting 200–300 cells over 10 randomly chosen fields (200×) in each coverslip.
Measurement of caspase-3 activity
Caspase-3 activity was measured using a colorimetric assay kit (Beyotime) according to the manufacturer's instructions.
Statistical analysis
Data are presented as means ± SEM. Statistical significance was determined by a one-way anova or unpaired Student's t-test where appropriate. A value of P < 0.05 was considered to be statistically significant.
Results
Sarcolemmal NHE1 trafficking is dependent on the DNM2 GTPase activity in the cardiomyocyte
DNM2 is a ubiquitous DNM other than the neuron-specific DNM1 and DNM3 [15], and our previous study identified DNM2 as the single isoform of DNMs in the rat heart (J Li, L Xu, JC Ye, X Li, DS Zhang, DD Liang, XR Xu, M Qi, CM Li, H Zhang, J Wang, Y Liu, YH Chen, unpublished data). To determine whether the GTPase activity of DNM2 affects sarcolemmal NHE1 trafficking, we specifically enhanced or inhibited the DNM2 activity by overexpressing a WT DNM2 (DNM2WT) or a dominant-negative DNM2 mutant (DNM2K44A, defective for GTPase activity) in the cardiomyocytes respectively. The transfection efficiency was evaluated by fluorescent microscopy and western blotting at 24 hrs after infection. As shown in Figure 1A, the expression level of DNM2 was markedly up-regulated (three- to fourfold) by the overexpression of DNM2WT or DNM2K44A. The expression of membrane-bound NHE1 was analysed by cell-surface biotinylation to detect the retrograde trafficking of NHE1 from the sarcolemma to the cytosol. Compared with the mock control, DNM2WT overexpression reduced the surface NHE1 level, suggesting an enhanced internalization of sarcolemmal NHE1; whereas overexpression of DNM2K44A led to an increase in NHE1 expression in the surface, indicating an impaired retrograde trafficking of NHE1 (Fig. 1B). We then investigated whether DNM2 regulated the NHE1 trafficking via a direct interaction. The protein extracts from freshly isolated ARVMs were immunoprecipitated with a specific antibody against NHE1 or DNM2. Immunoprecipitation with the anti-NHE1 antibody revealed the complex formation between NHE1 and DNM2. Similar results were obtained from the reciprocal experiments (Fig. 1C).
Fig. 1.
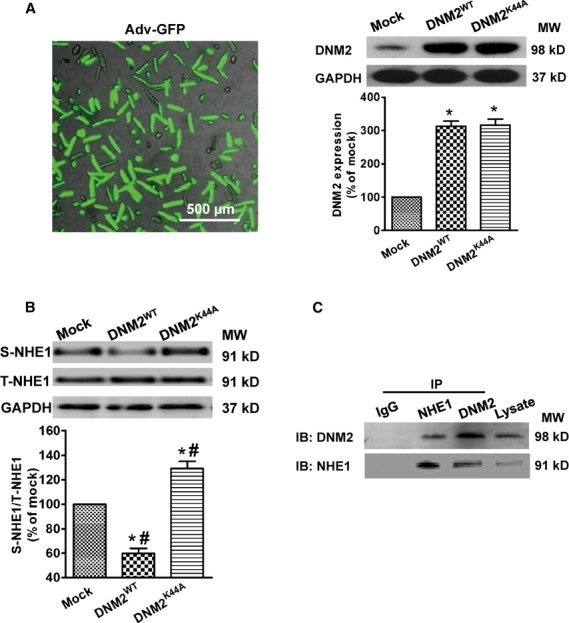
Sarcolemmal NHE1 trafficking is DNM2 activity dependent in ARVMs. (A) The transfection efficiency and overexpression of DNM2WT or DNM2K44A in ARVMs were detected by fluorescent microscopy and western blotting. Left: microscopic imaging of adenovirus-mediated GFP expression. Right: western bloting analysis of DNM2WT and DNM2K44A overexpression. GAPDH was used as a loading control. (B) Overexpression of DNM2WT or DNM2K44A induced a decrease or an increase in NHE1 protein expression in the cell surface respectively. A typical example of a western blot (upper panel) and the pooled data (lower panel) are shown. (C) A representative example of co-immunoprecipitation experiments for DNM2 and NHE1 proteins from freshly isolated ARVMs. Cell lysates were incubated with anti-NHE1, -DNM2 or -IgG (as a control) antibodies. The precipitates were then immunoblotted with the antibodies of DNM2 or NHE1. Data are expressed as means ± SEM of five independent experiments. *P < 0.05 compared with mock; #P < 0.05 compared with groups other than mock. S-NHE1: surface NHE1; T-NHE1: total NHE1; IB: immunoblot; IP: immunoprecipitation.
The DNM2-dependent regulation of NHE1 trafficking was further tested in HEK293 cells. In the absence of exogenous DNM2WT or DNM2K44A, NHE1 was mostly located on the cell surface with only a small fraction in the cytoplasm. Overexpression of DNM2WT (Fig. 2A) caused a significant translocation of membrane NHE1 into the cytoplasm. In contrast, DNM2K44A overexpression detained NHE1 protein on the cell surface (Fig. 2B).
Fig. 2.
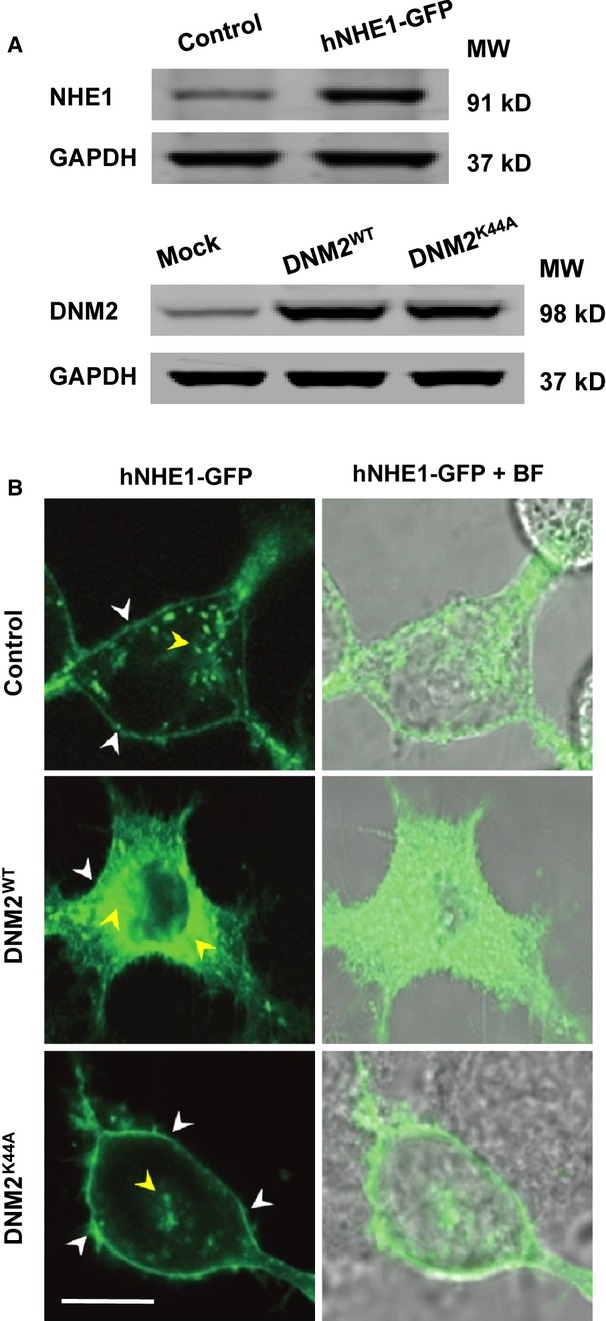
Verification of DNM2-dependent NHE1 trafficking in HEK293 cells. (A) The expression of exogenous NHE1 and overexpression of DNM2WT or DNM2K44A in HEK293 cells. (B) The confocal imaging of NHE1 distribution in transfected HEK293 cells. In mock cells that expressed NHE1 alone, the NHE1 was mainly located on the cell surface (white arrows) and only slightly distributed in the cytoplasm (as clusters demonstrated by yellow arrows). Overexpression of DNM2WT induced the translocation of NHE1 from the cell surface to the cytoplasm, whereas DNM2K44A overexpression impaired the retrograde trafficking of NHE1, resulting in the retention of NHE1 on the membrane surface. BF: bright field; bar indicates 10 μm.
DNM2-dependent NHE1 trafficking is coupled with the change in NHE1 activity
To determine whether the alteration in the DNM2-dependent NHE1 translocation modified NHE1 activity, we assayed the activity of NHE1 in the ARVMs overexpressing DNM2WT or DNM2K44A. The response of these cells to an acute acid load induced by the application of NH4Cl in the extracellular solution was assessed. As shown in Figure 3A through C, when NH4Cl was removed and the Na+-Tyrode's solution was reintroduced, the pHi began to recover because of the NHE1-mediated H+ extrusion. The rate of this Na+-dependent intracellular alkalinization, which was calculated from the point at which the pHi started to recover and is indicated by the dotted line in each trace, represents the NHE1 activity. The Na+-dependent pHi recovery was slower in the ARVMs overexpressing DNM2WT than in the ARVMs receiving the mock vector (0.041 ± 0.001, n = 57 cells versus 0.068 ± 0.002 pHi units/min., n = 50 cells), whereas a markedly accelerated pHi recovery was observed in the ARVMs overexpressing DNM2K44A (0.142 ± 0.001 pHi units/min., n = 60 cells; Fig. 3D). In addition, compared with mock control or DNM2WT overexpression, DNM2K44A overexpression resulted in an elevated resting pHi (Fig. 3E).
Fig. 3.
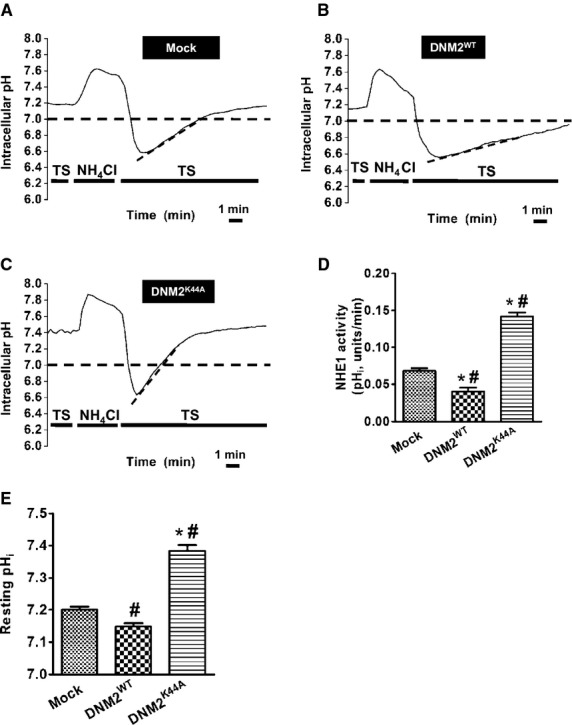
DNM2-dependent NHE1 trafficking is coupled to the change in NHE1 activity in ARVMs. (A–C) Representative pHi traces of individual ventricular myocyte receiving the mock vector (A), DNM2WT (B) or DNM2K44A (C), showing a Na+-dependent pHi recovery after exposure to NH4Cl. ARVMs were perfused with HEPES-buffered solution followed by 20 mM NH4Cl. The acute exposure to NH4Cl resulted in the acidification of the cytosol after removing NH4Cl. When the extracellular normal HEPES-buffered solution was reintroduced, the pHi started to increase in response to the intracellular alkalinization, which occurred at a certain rate (dotted line) that reflected the NHE1 activity. TS: Tyrode's solution. (D) The activity of NHE1, indicated by the mean rates of the Na+-dependent pHi recovery, differed among the cells receiving the mock, DNM2WT or DNM2K44A vector. (E) The effects of DNM2WT or DNM2K44A overexpression on the resting intracellular pHi. Data are expressed as means ± SEM of five independent experiments. *P < 0.05 compared with mock; #P < 0.05 compared with groups other than mock.
Increased NHE1 activity induced by impaired retrograde trafficking contributes to apoptotic death in DNM2-defective ARVMs
We next explored the potential effect of the DNM2 deficiency–induced NHE1 activation on the viability of ARVMs. As shown in Figure 4, the viability of the DNM2K44A-ARVMs (45.2 ± 1.3%, n = 5) was markedly lower than that in mock control (92.5 ± 1.6%, n = 5) or the DNM2WT-ARVMs (91.8 ± 1.4%, n = 5). The treatment with zoniporide, a selective NHE1 inhibitor [27], improved the viability of DNM2K44A-ARVMs (61.6 ± 1.8%, n = 5), and had no effect on the cell viability in either the DNM2WT-ARVMs or the mock control, suggesting the involvement of NHE1 activation in the ARVM death.
Fig. 4.
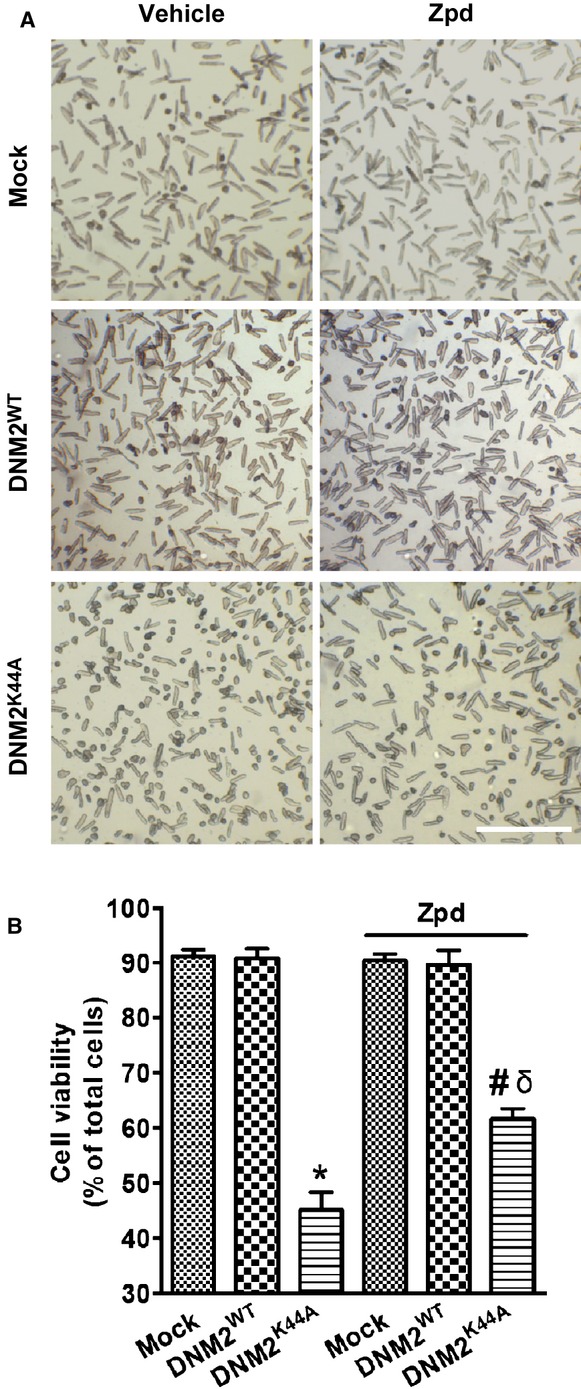
The enhanced activation of NHE1 induced by DNM2 deficiency reduces cell viability in ARVMs. Cell viability was measured by staining with trypan blue to confirm cell membrane integrity. (A) Representative micrographs of the ARVMs infected with the mock, DNM2WT, or DNM2K44A vector at 36 hrs in the presence or absence of the NHE1 inhibitor zoniporide. (B) Data collection from A. Similar results were observed in five independent experiments. *P < 0.05 compared with mock; #P < 0.05 compared with DNM2K44A; δP < 0.05 compared with mock plus zpd. Zpd: zoniporide; bar indicates 500 μm.
Hoechst33258 staining and TUNEL assay were used to determine the contribution of apoptosis to the loss of cell viability in DNM2K44A-ARVMs. The percentage of TUNEL-positive nuclei was greater in the ARVMs infected with Adv-DNM2K44A (23.2 ± 2.1%, n = 5) than in the ARVMs infected with the mock vector or Adv-DNM2WT (mock: 4.3 ± 0.6%, n = 5; DNM2WT: 3.4 ± 0.6%, n = 5). The application of zoniporide reduced the percentage of TUNEL-positive cells in the DNM2K44A-ARVMs (12.5 ± 0.8%, n = 5; Fig. 5A and B). These results implicated that sarcolemmal NHE1 activation induced by the defective DNM2-dependent trafficking contributes to the apoptotic death of ARVMs.
Fig. 5.
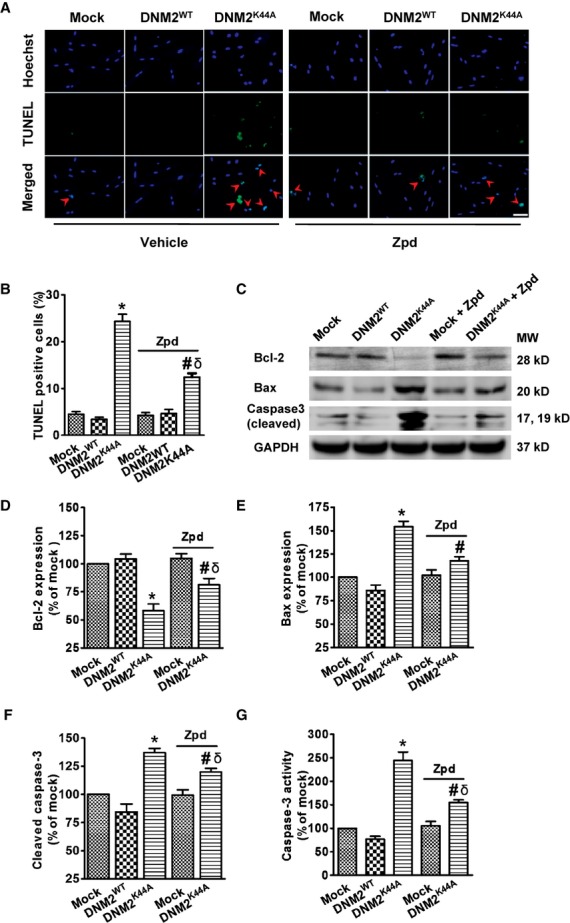
NHE1 activation induced by trafficking impairment induces apoptotic death in DNM2-defective ARVMs. (A) Representative images of ARVMs transfected with mock vector, DNM2WT and DNM2K44A in the presence or absence of zoniporide. Apoptotic cells were visualized by fluorescence microscopy using the TUNEL method (green), Hoechst33258 staining of nuclei (blue) and merged images (cyan blue) showing DNA fragmentation and the condensation of nuclei (red arrow). (B) Pooled data from A. (C) A typical western bloting analysis showing the expression levels of Bcl-2, Bax and cleaved caspase-3 in transfected ARVMs. GAPDH was used as a loading control. (D–F) Data collection from C. (G) Caspase-3 activity in transfected ARVMs. Data are presented as means ± SEM of five independent experiments. *P < 0.05 compared with mock; #P < 0.05 compared with DNM2K44A; δP < 0.05 compared with mock plus zpd. Zpd: zoniporide; bar indicates 50 μm.
The DNM2- and NHE1-associated apoptosis in ARVMs was further substantiated by the finding that the DNM2K44A overexpression reduced the level of Bcl-2 protein, which functions at the contact sites of the mitochondrial membrane to inhibit apoptosis [28]. Moreover, the level of pro-apoptotic Bax was increased by the overexpression of DNM2K44A. When zoniporide was applied, the alteration in Bcl-2 or Bax level was significantly alleviated (Fig. 5C–E).
Caspase-3 activation is the terminal step in the apoptotic pathway [29]. Using western blotting and caspase activity measurements, we detected the endogenous levels of the large fragment of activated caspase-3 and the caspase-3 protease activity. As shown in Figure 5C, F and G, overexpression of DNM2K44A but not DNM2WT induced an increase in caspase-3 activity, which was partially inhibited by zoniporide.
Discussion
In this study, we demonstrate for the first time that in ARVMs, sarcolemmal NHE1 activity is regulated by a DNM2-dependent trafficking process. DNM2 up-regulation decreased the NHE1-mediated H+ extrusion, whereas its deficiency led to an augment in the NHE1 activity. Interestingly, NHE1 activation elicited by DNM2 deficiency induced cardiomyocyte apoptosis, which can be partially rescued by pharmacological inhibition of NHE1 activity.
NHE1 has been primarily considered to be a membrane ‘housekeeping’ resident protein, yet the regulation of sarcolemmal NHE1 translocation in cardiomyocytes has not been explored. Recent studies indicate that membrane NHE1 has a half-life of ∼24 hrs [30] and that increased metabolic activity or ischemia can modify the translocation of NHE1 [31], which suggests the existence of dynamic trafficking of membrane NHE1. In this study, we have investigated whether a net translocation of NHE1 to the sarcolemma of cardiomyocytes was regulated by DNM2, a key player in intracellular internalization, and we confirmed the occurrence of this process through cell-surface biotinylation and complementary techniques of immunofluorescence microscopy. Our results indicate that the membrane expression of NHE1 was changed in response to the alteration of DNM2 activity (Fig. 1B), and this finding was corroborated in HEK293 cells overexpressing exogenous NHE1 (Fig. 2). Moreover, co-immunoprecipitation revealed a tight association between NHE1 and DNM2 in ARVMs (Fig. 1C). Taken together, these data suggest that the trafficking of sarcolemmal NHE1 between cell surface and cytoplasm can be regulated by DNM2 activity.
Internalization of plasmalemmal transporters is not only involved in their catabolism but also in the regulation of the number of the membrane-bound transporters, thereby affecting their activity [32]. The activation of intrinsic catalytic activity of NHE1 has been extensively studied and established experimentally [33]. We hereby report a DNM2-mediated endocytic regulation of sarcolemmal NHE1 activity in cardiomyocytes. The confocal microscopy of the ARVMs loaded with BCECF-AM showed that the NHE1 activity was decreased in the DNM2WT-overexpressing cells, although it was raised by the overexpression of DNM2K44A, accompanied by the upregulation of resting pHi (Fig. 3). The increase in the surface amount of NHE1 as a result impaired endocytosis may contribute to the activation of NHE1 and lead to enhanced H+ extrusion, resulting in the intracellular alkalinization. In contrast, the reduction in surface NHE1 expression caused by excessive internalization decreased the activity of NHE1. The relatively small change in the NHE1 activity and the lack of a significant drop of the resting pHi in the DNM2WT-overexpressing cells was probably attributed to the minimal activity of sarcolemmal NHE1 under basal condition [3] and/or the involvement of mitochondrial NHE activity [34].
The activation of NHE1 has been convincingly demonstrated to regulate cardiomyocyte apoptosis and to be deleterious in a number of pathophysiological conditions, including left ventricular hypertrophy, congestive heart failure and myocardial infarction [35, 36]. This study demonstrates that the deficiency of DNM2 leads to a decreased cell viability and promotes apoptotic cell death, which is partially reversed by the inhibition of NHE1 activity (Figs 4, 5A and B), suggesting an important role for NHE1 activation in the regulation of cardiomyocyte apoptosis. Several lines of evidence support the deleterious effects of the enhanced NHE1 activation in the DNM2 deficiency–mediated cardiomyocyte death. On one hand, excessive Na+ influx through the activated NHE1 may activate the sarcolemmal Na+/Ca2+ exchanger, leading to the augmentation of Ca2+ influx into the cytosol and the ensuing cell death [37, 38]. On the other hand, the intracellular alkalinization by the overactivation of NHE1 may be an early event in the apoptotic cascade [39], occurring prior to caspase activation and DNA fragmentation. Moreover, a reduced expression of DNM2 and an activation of NHE1 were also found in ischaemic myocardium of rat (data not shown), further underscoring the potential pathological importance of DNM2 deficiency–mediated NHE1 activation in the heart.
Caspase-3 represents the common pathway of the caspase cascade and has been implicated in the apoptosis of cardiomyocytes [40]. The results that the caspase-3 activity was elevated by the overexpression of DNM2K44A (Fig. 5C, F and G) confirmed the DNM2 deficiency–mediated apoptosis. Bax contributes to the progression of apoptosis by disrupting the function of mitochondrial membranes, whereas Bcl-2 binds to and inactivates Bax to prevent apoptosis [41]. A robust increase in Bax expression and a reduction in Bcl-2 level were observed in the DNM2-defective ARVMs (Fig. 5C–E), suggesting the activation of the mitochondrial-dependent apoptosis pathway. The use of zoniporide partially alleviated the alteration in the expression levels of Bcl-2, Bax and cleaved caspase-3, underlying the involvement of NHE1 activation. The lack of full rescue from apoptosis by zoniporide suggests that there may be targets other than NHE1 that can be regulated by DNM2 and also contribute to the maintenance of cell survival. These issues remains to be addressed in future studies.
In summary, this study demonstrates that the activity of sarcolemmal NHE1 can be regulated by a DNM2-dependent trafficking. The enhanced activation of NHE1 induced by DNM2 deficiency leads to cardiomyocyte apoptosis. These findings provide a novel mechanism by which the aberrant NHE1 trafficking affects cardiomyocyte survival and may offer new insights regarding the pathogenesis and management of myocardial injury.
Acknowledgments
This study was supported by the National Key Basic Research Program of China (2013CB531100, to Yi-Han Chen), the Foundation for Innovative Research Groups of China (81221001, to Yi-Han Chen), the Major International Joint Research Program Fund of China (81120108004, to Yi-Han Chen), the National Natural Science Foundation of China (81170224 and 81270313, to Jun Li; 31271214, to Yi-Han Chen) and the Shanghai Natural Science Fund (10JC1414700, to Yi-Han Chen).
Conflicts of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Casey JR, Grinstein S, Orlowski J. Sensors and regulators of intracellular pH. Nat Rev Mol Cell Biol. 2010;11:50–61. doi: 10.1038/nrm2820. [DOI] [PubMed] [Google Scholar]
- 2.Brett CL, Donowitz M, Rao R. Evolutionary origins of eukaryotic sodium/proton exchangers. Am J Physiol Cell Physiol. 2005;288:223–39. doi: 10.1152/ajpcell.00360.2004. [DOI] [PubMed] [Google Scholar]
- 3.Schelling JR, Abu JB. Regulation of cell survival by Na+/H+ exchanger-1. Am J Physiol Renal Physiol. 2008;295:625–32. doi: 10.1152/ajprenal.90212.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang Y, Meyer JW, Ashraf M, et al. Mice with a null mutation in the NHE1 Na+-H+ exchanger are resistant to cardiac ischemia-reperfusion injury. Circ Res. 2003;93:776–82. doi: 10.1161/01.RES.0000094746.24774.DC. [DOI] [PubMed] [Google Scholar]
- 5.Karmazyn M, Liu Q, Gan XT, et al. Aldosterone increases NHE-1 expression and induces NHE-1-dependent hypertrophy in neonatal rat ventricular myocytes. Hypertension. 2003;42:1171–6. doi: 10.1161/01.HYP.0000102863.23854.0B. [DOI] [PubMed] [Google Scholar]
- 6.Chen L, Chen CX, Gan XT, et al. Inhibition and reversal of myocardial infarction-induced hypertrophy and heart failure by NHE-1 inhibition. Am J Physiol Heart Circ Physiol. 2004;286:H381–7. doi: 10.1152/ajpheart.00602.2003. [DOI] [PubMed] [Google Scholar]
- 7.Jenkins EC, Jr, Debnath S, Gundry S, et al. Intracellular pH regulation by Na+/H+ exchanger-1 (NHE1) is required for growth factor-induced mammary branching morphogenesis. Dev Biol. 2012;365:71–81. doi: 10.1016/j.ydbio.2012.02.010. [DOI] [PubMed] [Google Scholar]
- 8.Kilić A, Javadov S, Karmazyn M. Estrogen exerts concentration-dependent pro-and anti-hypertrophic effects on adult cultured ventricular myocytes. Role of NHE-1 in estrogen-induced hypertrophy. J Mol Cell Cardiol. 2009;46:360–9. doi: 10.1016/j.yjmcc.2008.11.018. [DOI] [PubMed] [Google Scholar]
- 9.Karydis A, Jimenez-Vidal M, Denker SP, et al. Mislocalized scaffolding by the Na-H exchanger NHE1 dominantly inhibits fibronectin production and TGF-β activation. Mol Biol Cell. 2009;20:2327–36. doi: 10.1091/mbc.E08-08-0842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bertrand B, Wakabayashi S, Ikeda T, et al. The Na+/H+ exchanger isoform 1 (NHE1) is a novel member of the calmodulin-binding proteins. Identification and characterization of calmodulin-binding sites. J Biol Chem. 1994;269:13703–9. [PubMed] [Google Scholar]
- 11.Sabri A, Byron KL, Samarel AM, et al. Hydrogen peroxide activates mitogen-activated protein kinases and Na+-H+ exchange in neonatal rat cardiac myocytes. Circ Res. 1998;82:1053–62. doi: 10.1161/01.res.82.10.1053. [DOI] [PubMed] [Google Scholar]
- 12.Kandasamy RA, Yu FH, Harris R, et al. Plasma membrane Na+/H+ exchanger isoforms (NHE-1, −2, and −3) are differentially responsive to second messenger agonists of the protein kinase A and C pathways. J Biol Chem. 1995;270:29209–16. doi: 10.1074/jbc.270.49.29209. [DOI] [PubMed] [Google Scholar]
- 13.Simonin A, Fuster D. Nedd4-1 and β-arrestin-1 are key regulators of Na+/H+ exchanger 1 ubiquitylation, endocytosis, and function. J Biol Chem. 2010;285:38293–303. doi: 10.1074/jbc.M110.115089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heymann JAW, Hinshaw JE. Dynamins at a glance. J Cell Sci. 2009;122:3427–31. doi: 10.1242/jcs.051714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Praefcke GJ, McMahon HT. The dynamin superfamily: universal membrane tubulation and fission molecules? Nat Rev Mol Cell Biol. 2004;5:133–47. doi: 10.1038/nrm1313. [DOI] [PubMed] [Google Scholar]
- 16.Le PU, Nabi IR. Distinct caveolae-mediated endocytic pathways target the Golgi apparatus and the endoplasmic reticulum. J Cell Sci. 2003;116:1059–71. doi: 10.1242/jcs.00327. [DOI] [PubMed] [Google Scholar]
- 17.Puri V, Watanabe R, Singh RD, et al. Clathrin-dependent and -independent internalization of plasma membrane sphingolipids initiates two Golgi targeting pathways. J Cell Biol. 2001;154:535–48. doi: 10.1083/jcb.200102084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maier O, Knoblich M, Westermann P. Dynamin II binds to the trans-golgi network. Biochem Bioph Res Co. 1996;223:229–33. doi: 10.1006/bbrc.1996.0876. [DOI] [PubMed] [Google Scholar]
- 19.Pucadyil TJ, Schmid SL. Real-time visualization of dynamin-catalyzed membrane fission and vesicle release. Cell. 2008;135:1263–75. doi: 10.1016/j.cell.2008.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hong T, Smyth JW, Gao D, et al. BIN1 localizes the L-type calcium channel to cardiac T-tubules. PLoS Biol. 2010;8:e1000312. doi: 10.1371/journal.pbio.1000312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lusky M, Christ M, Rittner K, et al. In vitro and in vivo biology of recombinant adenovirus vectors with E1, E1/E2A, or E1/E4 deleted. J Virol. 1998;72:2022–32. doi: 10.1128/jvi.72.3.2022-2032.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Communal C, Huq F, Lebeche D, et al. Decreased efficiency of adenovirus-mediated gene transfer in aging cardiomyocytes. Circulation. 2003;107:1170–5. doi: 10.1161/01.CIR.0000051467.31874.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Das A, Smolenski A, Lohmann SM, et al. Cyclic GMP-dependent protein kinase Iα attenuates necrosis and apoptosis following ischemia/reoxygenation in adult cardiomyocyte. J Biol Chem. 2006;281:38644–52. doi: 10.1074/jbc.M606142200. [DOI] [PubMed] [Google Scholar]
- 24.Yan W, Nehrke K, Choi J, et al. The Nck-interacting kinase (NIK) phosphorylates the Na+-H+ exchanger NHE1 and regulates NHE1 activation by platelet-derived growth factor. J Biol Chem. 2001;276:31349–56. doi: 10.1074/jbc.M102679200. [DOI] [PubMed] [Google Scholar]
- 25.Silver RB, Mackins CJ, Smith NCE, et al. Coupling of histamine H3 receptors to neuronal Na+/H+ exchange: a novel protective mechanism in myocardial ischemia. Proc Natl Acad Sci USA. 2001;98:2855–9. doi: 10.1073/pnas.051599198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thomas JA, Buchsbaum RN, Zimniak A, et al. Intracellular pH measurements in Ehrlich ascites tumor cells utilizing spectroscopic probes generated in situ. Biochemistry. 1979;18:2210–8. doi: 10.1021/bi00578a012. [DOI] [PubMed] [Google Scholar]
- 27.Knight DR, Smith AH, Flynn DM, et al. A novel sodium-hydrogen exchanger isoform-1 inhibitor, zoniporide, reduces ischemic myocardial injury in vitro and in vivo. J Pharmacol Exp Ther. 2001;297:254–9. [PubMed] [Google Scholar]
- 28.Korsmeyer SJ. BCL-2 gene family and the regulation of programmed cell death. Cancer Res. 1999;59:1693s–700s. [PubMed] [Google Scholar]
- 29.Porter AG, Janicke RU. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999;6:99–104. doi: 10.1038/sj.cdd.4400476. [DOI] [PubMed] [Google Scholar]
- 30.Cavet ME, Akhter S, Murtazina R, et al. Half-lives of plasma membrane Na+/H+ exchangers NHE1–3: plasma membrane NHE2 has a rapid rate of degradation. Am J Physiol Cell Physiol. 2001;281:C2039–48. doi: 10.1152/ajpcell.2001.281.6.C2039. [DOI] [PubMed] [Google Scholar]
- 31.Lawrence SP, Holman GD, Koumanov F. Translocation of the Na+/H+ exchanger 1 (NHE1) in cardiomyocyte responses to insulin and energy-status signalling. Biochem J. 2010;432:515–23. doi: 10.1042/BJ20100717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chow C, Khurana S, Woodside M, et al. The epithelial Na+/H+ exchanger, NHE3, is internalized through a clathrin-mediated pathway. J Biol Chem. 1999;274:37551–8. doi: 10.1074/jbc.274.53.37551. [DOI] [PubMed] [Google Scholar]
- 33.Putney LK, Denker SP, Barber DL. The changing face of the Na+/H+ exchanger, NHE1: structure, regulation, and cellular actions. Annu Rev Pharmacol Toxicol. 2002;42:527–52. doi: 10.1146/annurev.pharmtox.42.092001.143801. [DOI] [PubMed] [Google Scholar]
- 34.Murphy E, Eisner DA. Regulation of intracellular and mitochondrial sodium in health and disease. Circ Res. 2009;104:292–303. doi: 10.1161/CIRCRESAHA.108.189050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Olivetti G, Abbi R, Quaini F, et al. Apoptosis in the failing human heart. N Engl J Med. 1997;336:1131–41. doi: 10.1056/NEJM199704173361603. [DOI] [PubMed] [Google Scholar]
- 36.Yaoita H, Ogawa K, Maehara K, et al. Apoptosis in relevant clinical situations. Cardiovasc Res. 2000;45:630–41. doi: 10.1016/s0008-6363(99)00349-1. [DOI] [PubMed] [Google Scholar]
- 37.Saini HK, Dhalla NS. Modification of intracellular calcium concentration in cardiomyocytes by inhibition of sarcolemmal Na+/H+ exchanger. Am J Physiol Heart Circ Physiol. 2006;291:H2790–800. doi: 10.1152/ajpheart.00535.2006. [DOI] [PubMed] [Google Scholar]
- 38.Kintner DB, Chen X, Currie J, et al. Excessive Na+/H+ exchange in disruption of dendritic Na+ and Ca2+ homeostasis and mitochondrial dysfunction following in vitro ischemia. J Biol Chem. 2010;285:35155–68. doi: 10.1074/jbc.M110.101212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lagadic-Gossmann D, Huc L, Lecureur V. Alterations of intracellular pH homeostasis in apoptosis: origins and roles. Cell Death Differ. 2004;11:953–61. doi: 10.1038/sj.cdd.4401466. [DOI] [PubMed] [Google Scholar]
- 40.Cook SA, Poole-Wilson PA. Cardiac myocyte apoptosis. Eur Heart J. 1999;20:1619–29. doi: 10.1053/euhj.1999.1548. [DOI] [PubMed] [Google Scholar]
- 41.Crow MT, Mani K, Nam Y, et al. The mitochondrial death pathway and cardiac myocyte apoptosis. Circ Res. 2004;95:957–70. doi: 10.1161/01.RES.0000148632.35500.d9. [DOI] [PubMed] [Google Scholar]