

Abstract
Rationale
Abdominal aortic aneurysms (AAAs) are a chronic inflammatory vascular disease for which pharmacological treatments are not available. A mouse model of AAA formation involves chronic infusion of angiotensin II (AngII) and previous studies indicated a primary role for the angiotensin II type 1a (AT1a) receptor, in AAA formation. β-arrestin-2 (βarr2) is a multifunctional scaffolding protein that binds G-protein coupled receptors such as AT1a, and regulates numerous signaling pathways and pathophysiological processes. However, a role for βarr2 in AngII-induced AAA formation is currently unknown.
Objective
To determine if βarr2 played a role in AngII-induced AAA formation in mice.
Methods and Results
Treatment of βarr2+/+ and βarr2-/- mice on the hyperlipidemic ApoE-/- background or normolipidemic C57BL/6 background with AngII for 28 days indicated that βarr2 deficiency significantly attenuated AAA formation. βarr2 deficiency attenuated AngII-induced expression of cyclooxygenase-2 (COX-2), monocyte chemoattractant protein-1 (MCP-1), macrophage inflammatory protein 1α (MIP1α), and macrophage infiltration. AngII also increased the levels of phosphorylated-extracellular signal-regulated kinase 1/2 (p-ERK1/2) in ApoE-/-/βarr2+/+ aortas, whereas βarr2 deficiency diminished this increase. Furthermore, inhibition of ERK1/2 activation with CI1040 (100mg/kg/day) reduced the level of AngII-induced COX-2 expression in ApoE-/-/βarr2+/+ mice to the level observed in ApoE-/-/βarr2-/- mice. AngII treatment also increased matrix metalloproteinase (MMP) expression and disruption of the elastic layer in ApoE-/-/βarr2+/+ aortas and βarr2-deficiency reduced these effects.
Conclusions
βarr2 contributes to AngII-induced AAA formation in mice by p-ERK1/2-mediated COX-2 induction and increased inflammation. These studies suggest that for the AT1a receptor, G-protein-independent, βarr2-dependent signaling plays a major role in AngII-induced AAA formation.
Keywords: Aneurysm, β-Arrestin-2, cyclooxygenase-2, angiotensin
Introduction
Abdominal aortic aneurysms (AAAs) are a common vascular condition associated with several risk factors including advanced age, male gender, smoking and hypercholesterolemia.1 AAA formation begins as an abnormal dilation of the aorta, which may gradually expand over a period of years followed by eventual weakening and rupture of the vessel wall, resulting in mortality in about 90% of cases.2-4 The pathophysiology of AAAs can be roughly categorized into two processes: inflammation and extracellular matrix degeneration.5 Inflammation is exemplified by the release of inflammatory mediators such as monocyte chemoattractant protein 1 (MCP-1) and interleukin 6 and the resultant infiltration of inflammatory cells, particularly macrophages, into the vessel wall.6-9 Inflammatory cells are a major source of proteolytic enzymes such as matrix metalloproteinase 2 (MMP2) and MMP9 that are known to disrupt the structural integrity of the vessel wall and degrade the components of the extracellular matrix such as elastin and collagen.3, 10, 11 This extracellular matrix degeneration and vascular remodeling contribute to the progression and severity of the disease. Currently, there are no pharmacological treatments for AAAs with endovascular or surgical repair being the only options.
A widely used mouse model of AAAs involves chronic infusion of angiotensin II (AngII) and this model displays multiple characteristics of human AAAs and has provided mechanistic insight into the formation and progression of the disease.12, 13 AngII mediates many of its physiological and pathological effects by activating the G protein coupled receptor (GPCR) AT1 in most species, and the ‘a’ subtype of AT1 (AT1a) in rodents.14-17 Genetic ablation of the AT1a receptor in mice or treatment of mice with the AT1 receptor antagonist, losartan, attenuates AngII-induced AAA formation, indicating a critical role for this receptor in AngII-induced AAA pathology.17, 18
The traditional signaling pathway employed by the AT1a receptor involves G-protein mediated activation of phospholipase Cβ and the liberation of the second messengers, diacylglycerol and inositol-triphosphate, resulting in downstream signaling events.16 However, in recent years it has become increasingly evident that AT1a is also involved in G-protein-independent signaling by binding to scaffolding proteins known as β-arrestin-1 (βarr1) and β-arrestin-2 (βarr2).19, 20 βarr1 and βarr2 are ubiquitously expressed multifunctional proteins, which were previously thought to be important in termination of GPCR signaling.19, 20 However, studies over the past decade have demonstrated that βarr1 and βarr2 can mediate G-protein-independent signaling, and contribute to various physiological and pathological conditions.21-23 The coupling of βarr1 and βarr2 with GPCRs is known to recruit signaling molecules such as the c-Src family kinases, protein kinase B (AKT), phosphotidylinositol 3 kinase, as well as key players in the mitogen activated protein kinase (MAPK) signaling pathway, including p38, c-Jun N terminal kinase 3 and extracellular signal-regulated kinase 1/2 (ERK1/2).19, 24 The ERK1/2 cascade is the best-studied example of βarr-activated signaling for the AT1a receptor. Studies have shown that βarr1 and βarr2 bind with equal affinity to AT1a, however both isoforms demonstrate differential roles in ERK1/2 signaling, wherein βarr2 is thought to be the primary isoform responsible for ERK1/2 activation.25
Cyclooxygenase-2 (COX-2) is an enzyme important in the synthesis of inflammatory mediators such as prostaglandin (PG) E2, and has been shown to contribute to human AAA formation.9, 26 Using the AngII-induced AAA model, we previously reported that genetic or pharmacological inactivation of COX-2 attenuated AAA incidence and severity in mice.8, 27 Furthermore, these studies showed that AngII treatment significantly induced COX-2 expression in the abdominal aorta during the initiation and progression of the disease.8 These studies also suggested that the increased COX-2 expression in smooth muscle cells may contribute to AAA formation by initiating macrophage infiltration into the aorta via the release of inflammatory chemokines including MCP-1 and macrophage inflammatory protein 1α (MIP1α).8 Additional studies have shown that the genetic deficiency of microsomal PGE synthase 1 (mPGES1), an enzyme known to couple with COX-2 and convert COX-2-generated PGH2 into PGE2, or antagonism of the PGE2 receptor, EP4, ablates AngII-induced AAA formation in mice.6, 28, 29 These studies provide further evidence for the role of COX-2-derived PGE2 in AAA formation.
COX-2 induction in response to AngII has been shown to occur via the activation of several signaling molecules including components of the MAPK pathway such as ERK1/2.30-32 Furthermore, ERK1/2 activation has also been shown to be important in the formation of AngII-induced AAAs, as inhibition of ERK1/2 activation impairs AAA formation in mice.33 These findings underscore the importance of COX-2 and ERK1/2 in AAA formation and suggest that activated ERK1/2 (p-ERK1/2) may contribute to COX-2 induction during AAA formation. Because βarr2 is important in AngII-AT1a-mediated ERK1/2 activation, in the present study we investigated whether βarr2 played a role in AngII-induced AAA formation. Using both normolipidemic βarr2-deficient mice and hyperlipidemic ApoE-/-/βarr2-/- mice, we demonstrate that βarr2 deficiency significantly attenuates AAA formation. Furthermore, mechanistically it appears that βarr2 mediates AngII-induced COX-2 expression via a p-ERK1/2-dependent pathway and increases the expression of inflammatory markers, thereby contributing to AAA formation.
Methods
Mice
Male, hyperlipidemic ApoE-/-/βarr2+/+ and ApoE-/-/βarr2-/- mice at 8-10 weeks of age were use for measuring AngII-induced AAA incidence and severity and for mechanistic experiments. AngII-induced AAA incidence was also examined in 8-month old, male, βarr2+/+ and βarr2-/- mice34 on a normolipidemic C57BL/6 background. Please refer to the supplemental methods section for details.
AAA quantitation
Mice were treated with AngII (1000ng/kg/min) or saline via subcutaneously implanted osmotic mini-pumps and euthanized after 28 days for AAA quantification. A >50% increase in external diameter of the abdominal aorta was used to define the occurrence of an AAA. AAA severity was determined as using a classification scheme described previously8 and by measuring the wet weights of the abdominal aortas. The presence of an AAA as well as the scoring of AAA pathology was determined by an investigator who was blinded to the mouse genotypes. Upon determination of the AAA incidence and classification, another investigator matched the scored AAAs to the genotypes of the mice.
Blood pressure measurements
Blood pressure was measured in conscious, unrestrained, male ApoE-/-/βarr2+/+ and ApoE-/-/βarr2-/- mice by radio-telemetry, before and after treatment with AngII (1000ng/kg/min, osmotic mini-pumps), using methods similar to those described previously.35, 36 For more details, see the supplemental methods section.
Inhibition of ERK1/2 activation
AngII-treated ApoE-/-/βarr2+/+ mice were treated with the MAPK kinase 1(MEK1) inhibitor, CI1040, as previously described.33
Histology and immunohistochemistry
Mice were euthanized at the designated time-points (7 or 28 days) and abdominal aortic sections were stained with hematoxylin and eosin (H&E) or Verhoeff-Van Geisen (VVG, elastin) for histological analysis or with antibodies against COX-2, Mac-3 or p-ERK1/2, for immunohistochemical analysis. Quantitation of immunohistochemistry was performed by determining the ratio of the number of COX-2, p-ERK1/2 or Mac-3 positive cells to the total number of hematoxylin positive nuclei per field (at 400× magnification). For more details, please refer to the supplemental methods section.
Quantitation of mRNA expression
Real-time quantitative PCR analysis for COX-2, CD68, MCP-1, MIP1α, MMP2 and MMP9 was performed using Taqman gene expression assays and the ΔΔCt method with hypoxanthine phosphoribosyl transferase (HPRT) as the endogenous control. For more details, see the supplemental methods section.
Statistics
Data are shown as the mean ± SEM of independent experiments. Fisher's exact test was used to compare AAA incidence among different groups. Two-way ANOVA was use to examine changes in blood pressure between genotypes. One-way ANOVA followed by Bonferroni's multiple comparison tests were used to determine the statistical significance of the data in all other experiments. Values with P ≤ 0.05 were considered statistically significant.
Results
βarr2 deficiency attenuates the incidence and severity of AngII-induced AAAs
The effect of βarr2 deficiency on AAA development in hyperlipidemic mice was determined by crossing βarr2-/- mice with hyperlipidemic ApoE-/- mice. AngII infusion for 28 days induced a 61.8% incidence (21 of 34 mice) of AAAs in ApoE-/-/βarr2+/+ mice, whereas the AAA incidence in ApoE-/-/βarr2-/- mice was significantly reduced to 9.5% (2 of 21 mice) (Figure 1A). As expected, AAAs were not observed in the saline-infused ApoE-/-/βarr2+/+ or ApoE-/-/βarr2-/- mice (Figure 1A). The deficiency of βarr2 also attenuated the severity of AngII-induced AAAs, as determined by a classification scheme based on the external diameter of the abdominal aortas (Figure 1B) as well as by measuring the wet weights of the abdominal aortas (Figure 1C). Histological examination of cross-sections of the abdominal aortas from ApoE-/-/βarr2+/+ mice showed severe thickening and prominent remodeling with thrombi present in the vessel wall (Figure 1D). In contrast, the abdominal aortas from AngII-treated ApoE-/-/βarr2-/- showed minimal thickening and remodeling (Figure 1E), and appeared to be histologically similar to abdominal aortas from saline-treated ApoE-/-/βarr2+/+ mice (Figure 1F). Aortas from saline-treated ApoE-/-/βarr2-/- mice were histologically indistinguishable from saline-treated ApoE-/-/βarr2+/+ mice (data not shown). These data demonstrate that the deficiency of βarr2 attenuates both AngII-induced AAA incidence and severity in mice on a hyperlipidemic background.
Figure 1. AngII-induced AAA formation in hyperlipidemic ApoE-/-/βarr2+/+ and ApoE-/-/βarr2-/- mice and normolipidemic C57BL/6 βarr2+/+ and βarr2-/- mice.
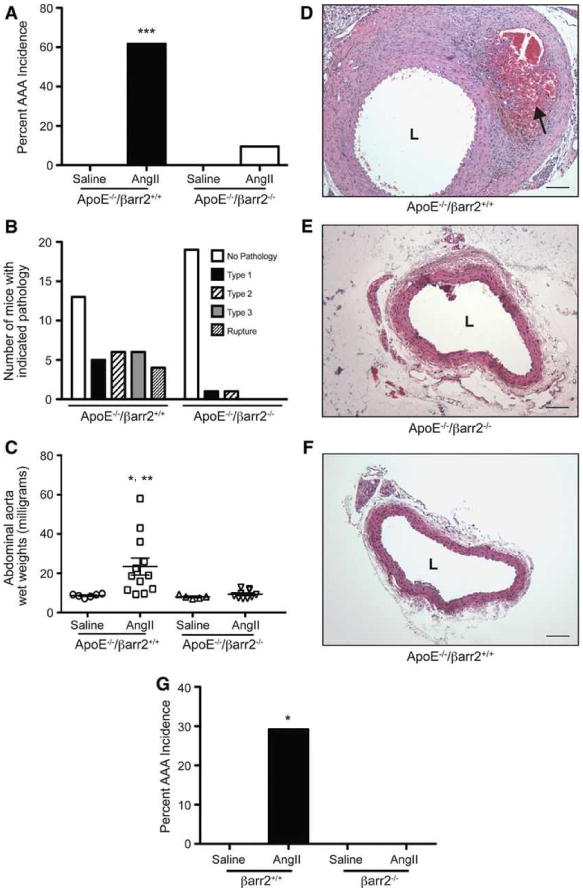
A) Percentage of AAA incidence following 28 days of AngII infusion in ApoE-/-/βarr2+/+ and ApoE-/-/βarr2-/- mice. ***, significantly different from saline treated ApoE-/-/βarr2+/+ mice and AngII-treated ApoE-/-/βarr2-/- mice, P<0.001, N ≥10 per group. B) AAAs in ApoE-/-/βarr2+/+ and ApoE-/-/βarr2-/- mice were scored from Type 1 to Type 4 pathology, based on the external diameter using a classification scheme similar to the one described previously18 C) AAA severity in in ApoE-/-/βarr2+/+ and ApoE-/-/βarr2-/- mice as determined by abdominal aorta wet weights. Each symbol represents an individual animal. *, significantly different from saline-treated ApoE-/-/βarr2+/+ mice, P<0.05. **, significantly different from AngII-treated ApoE-/-/βarr2-/- mice, P<0.01, N ≥5 per group. Representative images of H&E-stained aortic sections from D) AngII-treated ApoE-/-/βarr2+/+ mice E) AngII-treated ApoE-/-/βarr2-/- and F) saline-treated ApoE-/-/βarr2+/+ mice. Arrow indicates thrombus formation. Scale bars, 0.1mm. L, lumen. G) Percentage of AAA incidence in normolipidemic C57BL/6 βarr2+/+ and -βarr2-/- mice following 28 days of AngII infusion. *, significantly different from saline-treated βarr2+/+ mice and AngII-treated βarr2-/- mice, P<0.05, N ≥10 per group.
Two previous studies have shown that aged, normolipidemic wild-type C57BL/6 mice (7-11 months) also develop an increased incidence of AAAs when infused with AngII for 28 days; however, the AAA incidence is significantly lower than that observed in hyperlipidemic ApoE-/- mice.37, 38 Therefore, in addition to studies in the hyperlipidemic ApoE-/- strain, we examined the effect of βarr2 deficiency on AAA incidence in 8-month old, normolipidemic C57BL/6 mice. AngII infusion resulted in a 29.2% incidence (7 of 24 mice) of AAAs in the normolipidemic C57BL/6 βarr2+/+ mice (Figure 1G). In contrast, none of the normolipidemic C57BL/6 βarr2-/- mice (0 of 15 mice) displayed any pathology (Figure 1G). Of the 7 C57BL/6 βarr2+/+ mice that did develop AAAs, 2 mice displayed a Type 2 aneurysm, 3 mice had a Type 3 aneurysm, and 2 mice displayed aneurysmal rupture. Furthermore, similar to previous studies,38 the AAAs in the normolipidemic C57BL/6 βarr2+/+ mice were grossly similar to those that developed in the hyperlipidemic ApoE-/- mice following AngII infusion. Thus, the deficiency of βarr2 attenuates AngII-induced AAA incidence in both hyperlipidemic and normolipidemic mouse strains. Because the AngII-induced AAA incidence in the C57BL/6 βarr2+/+ mice on the normolipidemic background is markedly lower than that observed in the hyperlipidemicApoE-/-/βarr2+/+ mice (29.2% versus 61.8%), the mechanistic studies described below were performed in the ApoE-/-/βarr2+/+ and ApoE-/-/βarr2-/- mice.
βarr2 deficiency does not alter AngII-induced increases in blood pressure
Previous studies have demonstrated that AngII infusion rapidly increases systolic blood pressure in mice.39-41 Therefore, to determine whether βarr2-deficiency affected AngII-mediated increases in blood pressure, we measured the systolic blood pressure in ApoE-/-/βarr2+/+ and ApoE-/-/βarr2-/- mice. As shown in Table 1, AngII treatment significantly increased the mean systolic blood pressure in both ApoE-/-/βarr2+/+ and ApoE-/-/βarr2-/- mice as compared to basal blood pressure. However, there was no significant difference in the systolic blood pressure between ApoE-/-/βarr2+/+ and ApoE-/-/βarr2-/- mice at baseline or with AngII treatment (Table 1). These results indicate that the reduction in the AngII-induced AAA incidence in βarr2-deficient mice is not dependent on alterations in blood pressure.
Table 1. Systolic Blood Pressure.
| ApoE-/-βarr2+/+ | ApoE-/-/βarr2-/- | |
|---|---|---|
| Baseline (mmHg) | 123.5 ± 7.1 | 122.9 ± 3.9 |
| AngII (mmHg) | 153.3 ± 7.2 * | 154.9 ± 10.2 * |
Data represent the mean ± SEM,
, significantly different from baseline, P < 0.0001, N ≥ 4 per group.
AngII-induced COX-2 expression is attenuated in aortas from βarr2-deficient mice
AngII is known to be a potent inducer of COX-2 expression30, 31, 42 and our previous studies have indicated that COX-2 contributes to AngII-induced AAA formation.8, 27 Therefore, to determine whether βarr2 played a role in AngII-induced COX-2 expression, ApoE-/-/βarr2+/+ and ApoE-/-/βarr2-/- mice were treated with saline or AngII for 7 or 28 days and analyzed for COX-2 expression. AngII infusion significantly increased the levels of COX-2 mRNA in the aortas of ApoE-/-/βarr2+/+ mice at experimental days 7 (Figure 2A) and 28 (Figure 2B), as compared to saline infusion. In contrast, there was a significant reduction in AngII-induced COX-2 expression in the aortas of βarr2-deficient mice at both time-points. In addition, numerous COX-2 expressing cells were detected in the medial and adventitial layers of the abdominal aorta from ApoE-/-/βarr2+/+ mice after 7 days (Figure 2C) and throughout the aneurysmal tissue after 28 days of AngII infusion (Figure 2D). In contrast, significantly fewer COX-2 positive cells were observed in the abdominal aortas from ApoE-/-/βarr2-/- mice at both time-points (Figure 2E, 2F and 2G). Thus, βarr2 plays a key role in the AngII-induced COX-2 expression during AAA development.
Figure 2. COX-2 expression is attenuated in the aortas of βarr2-deficient mice.
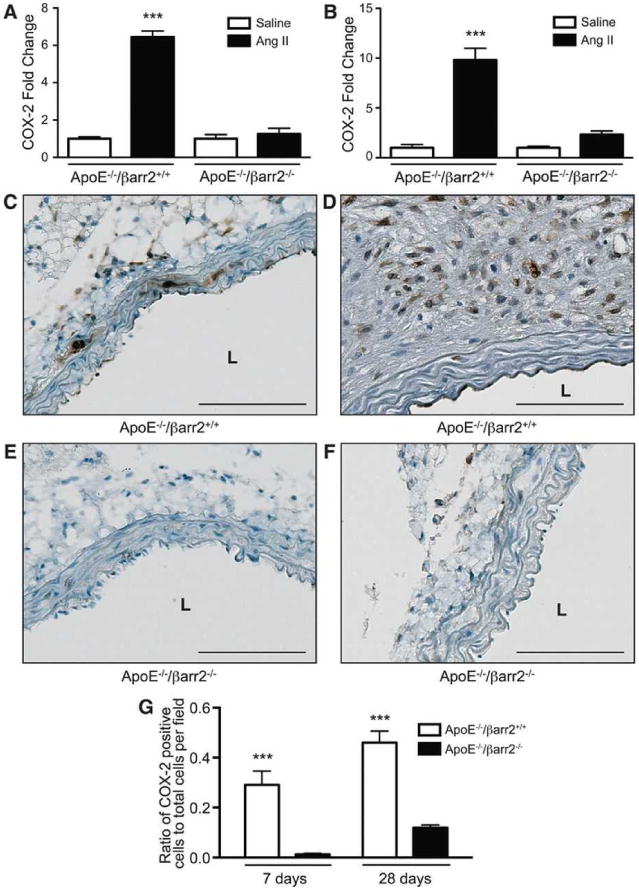
Quantitative PCR analysis of COX-2 mRNA expression in the aortas of ApoE-/-/βarr2+/+ and ApoE-/-/βarr2-/- mice at A) 7 days and B) 28 days following AngII or saline infusion. ***, significantly different from saline-treated ApoE-/-/βarr2+/+ and AngII-treated ApoE-/-/βarr2-/- mice. Data represent mean ± SEM, P<0.001, N ≥ 6 per group. Representative photomicrographs of COX-2 immunostaining in abdominal aortic segments from ApoE-/-/βarr2+/+ mice at C) 7 and D) 28 days after AngII infusion and ApoE-/-/βarr2-/- mice at E) 7 and F) 28 days following AngII infusion. Brown staining (DAB) indicates COX-2 expression and sections are counterstained with hematoxylin (blue). Scale bars, 0.1mm. L, lumen. G) Quantitation of COX-2 immuno-positive cells. ***, significantly different from ApoE-/-/βarr2-/- mice. Data represent mean ± SEM, P<0.001, N = 5 per group.
βarr2 contributes to ERK1/2 activation in AngII-induced AAAs and p-ERK1/2 contributes to COX-2 induction
ERK1/2 signaling is an extensively studied pathway activated by the AT1a-βarr2 complex and p-ERK1/2 is also known to contribute to COX-2 induction in several cell types.19, 30-32, 43 As shown in Figure 3A, immunohistochemical analysis indicated the presence of p-ERK1/2 in the medial layer of aortas from ApoE-/-/βarr2+/+ mice after 7 days of AngII infusion. When analyzed at 28 days after AngII infusion, numerous p-ERK1/2 containing cells were observed throughout the aneurysmal aortas from ApoE-/-/βarr2+/+ mice (Figure 3B). In contrast, in the abdominal aortas from ApoE-/-/βarr2-/- mice, significantly reduced p-ERK1/2 was observed at both time-points (Figures 3C, 3D and 3E).
Figure 3. βarr2 deficiency results in attenuated ERK1/2 activation.
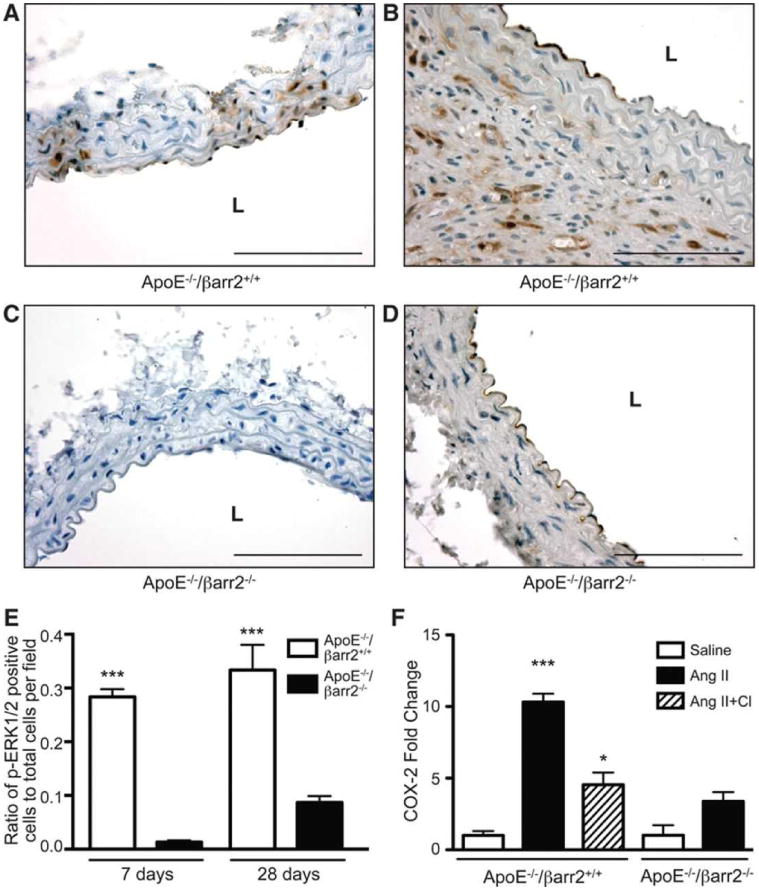
Representative photomicrographs of p-ERK1/2 immunostaining in abdominal aortic segments from ApoE-/-/βarr2+/+ mice at A) 7 and B) 28 days after AngII infusion and ApoE-/-/βarr2-/- mice at C) 7 and D) 28 days following AngII infusion. Brown staining indicates p-ERK1/2 and sections are counterstained with hematoxylin (blue). Scale bars, 0.1mm. L, lumen E) Quantitation of p-ERK1/2 immuno-positive cells. ***, significantly different from ApoE-/-/βarr2-/- mice. Data represent mean ± SEM, P<0.001, N = 5 per group F) Quantitative PCR analysis of COX-2 mRNA expression in abdominal aortas from ApoE-/-/βarr2+/+ and ApoE-/-/βarr2-/- mice following treatment with saline, AngII or AngII plus CI1040 (CI, MEK1 inhibitor) for 7 days. ***, significantly different from saline-treated ApoE-/-/βarr2+/+ mice, P<0.001, *, significantly different from AngII-treated ApoE-/-/βarr2+/+ mice, P<0.05, Data represent mean ± SEM, N ≥ 5 per group. COX-2 mRNA levels in AngII-treated ApoE-/-/βarr2-/- mice were not significantly different from saline-treated ApoE-/-/βarr2-/- mice or AngII plus CI treated ApoE-/-/βarr2+/+ mice.
To determine if ERK1/2 activation was involved in the AngII-induced COX-2 expression, we utilized the MEK1 inhibitor, CI1040 that has previously been shown to inhibit ERK1/2 phosphorylation and attenuate AngII-induced AAA formation in mice.33 In the absence of CI1040 treatment, AngII infusion for 7 days resulted in a significant induction in COX-2 mRNA in the aortas of ApoE-/-/βarr2+/+ mice (Figure 3F). However, in mice treated with CI1040 plus AngII, the increase in COX-2 expression was significantly reduced compared to AngII treatment alone. Furthermore, COX-2 mRNA levels following CI1040 plus AngII treatment were not statistically different from those observed in AngII-treated ApoE-/-/βarr2-/- mice (Figure 3F). Thus, the data indicate that βarr2 is important in AngII-induced ERK1/2 activation, which may subsequently increase COX-2 expression.
βarr2-deficiency attenuates AngII-induced macrophage infiltration into the abdominal aorta
Macrophage infiltration is an important early event during AAA formation.7 Therefore we examined the role of βarr2 in macrophage infiltration during AngII-induced AAA formation. Immunohistochemical staining for the macrophage marker Mac-3, in the abdominal aortas from AngII-treated ApoE-/-/βarr2+/+ mice indicated an abundant infiltration of macrophages into the media and adventitia by day 7 (Figure 4A), which continued through the 28-day AngII infusion (Figure 4B). However, Mac-3 staining was significantly reduced in the abdominal aortas of ApoE-/-/βarr2-/- mice at both time-points, indicating decreased macrophage infiltration (Figures 4C, 4D and 4E). In addition, we observed a robust increase in the mRNA expression for the macrophage marker, CD68, in the aortas of ApoE-/-/βarr2+/+ mice following AngII infusion (Figure 4F). In contrast, AngII infusion in ApoE-/-/βarr2-/- mice did not significantly increase CD68 mRNA expression when compared to saline controls (Figure 4F).
Figure 4. βarr2 contributes to macrophage infiltration into the aorta following AngII infusion.
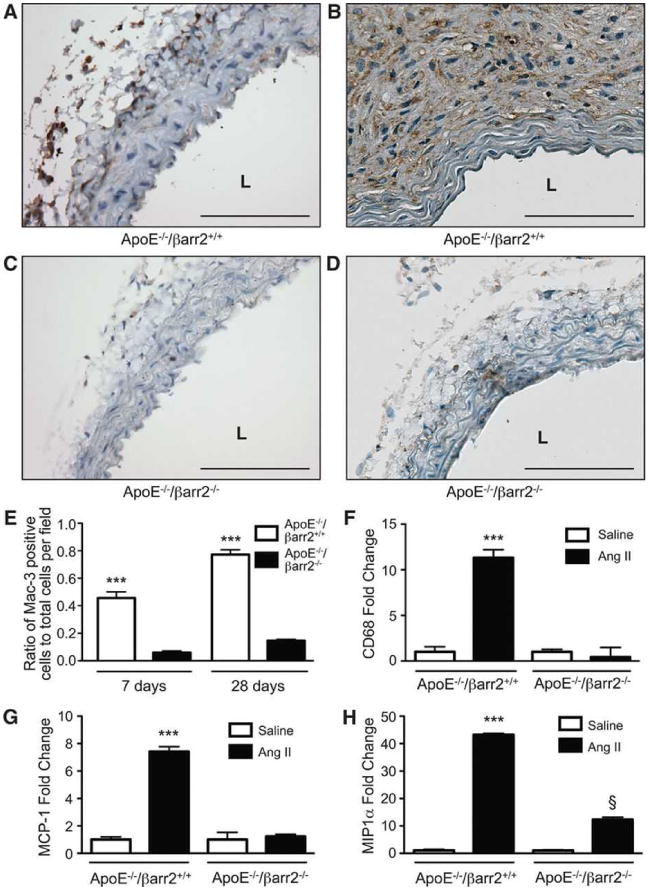
Representative photomicrographs of Mac-3 immunostaining in abdominal aortic segments from ApoE-/-/βarr2+/+ mice at A) 7 and B) 28 days after AngII infusion and ApoE-/-/βarr2-/- mice at C) 7 and D) 28 days after AngII infusion. Brown staining shows Mac-3 expression and sections are counter-stained with hematoxylin (blue). Scale bars, 0.1mm. L, lumen. E) Quantitation of Mac-3 immuno-positive cells. ***, significantly different from ApoE-/-/βarr2-/- mice. Data represent mean ± SEM, P<0.001, N = 5 per group. F) Quantitative PCR analysis CD68 mRNA expression in the abdominal aortas of ApoE-/-/βarr2+/+ and ApoE-/-/βarr2-/- mice following treatment with saline or AngII for 7 days. ***, significantly different from saline-treated ApoE-/-/βarr2+/+ and AngII-treated ApoE-/-/βarr2-/- mice, P<0.001. Data represent mean ± SEM, N ≥ 6 per group. G) Quantitative PCR analysis of MCP-1 mRNA expression in the abdominal aortas of ApoE-/-/βarr2+/+ and ApoE-/-/βarr2-/- mice following treatment with saline or AngII for 7 days. ***, significantly different from saline-treated ApoE-/-/βarr2+/+ and AngII-treated ApoE-/-/βarr2-/- mice, P<0.001. Data represent mean ± SEM, N ≥ 6 per group. H) Quantitative PCR analysis of MIP1α mRNA expression in the abdominal aortas of ApoE-/-/βarr2+/+ and ApoE-/-/βarr2-/- mice following treatment with saline or AngII for 7 days. ***, significantly different from saline-treated ApoE-/-/βarr2+/+ and AngII-treated ApoE-/-/βarr2-/- mice, P<0.001. §, significantly different from saline-treated ApoE-/-/βarr2-/- mice, P<0.001. Data represent mean ± SEM, N ≥ 6 per group.
AngII has been shown to stimulate the secretion of inflammatory chemokines such as MCP-1 and MIP1α, which are known to increase macrophage infiltration.8, 44-47 Therefore, the expression of MCP-1 and MIP1α was examined in the aortas from ApoE-/-/βarr2+/+ and ApoE-/-/βarr2-/- during the initial stage of AAA formation. AngII infusion for 7 days significantly increased the mRNA expression of both MCP-1 (Figure 4G) and MIP1α (Figure 4H) in the ApoE-/-/βarr2+/+ aortas as compared to saline infusion. However, the increases in MCP-1 and MIP1α expression in response to AngII were significantly lower in the aortas of ApoE-/-/βarr2-/- mice (Figures 4G and 4H). Therefore, the data suggest that attenuated chemokine production may be responsible for reduced macrophage infiltration and diminished AAA formation in ApoE-/-/βarr2-/- mice.
βarr2 contributes to increased MMP2 and MMP9 expression during AAA formation
MMPs have been mechanistically implicated in the pathogenesis of AAAs and MMP2 and MMP9 in particular are known to have a concerted role in AAA initiation and progression.48, 49 As shown in Figures 5A and 5B, AngII treatment significantly increased MMP2 mRNA expression at days 7 and 28 in ApoE-/-/βarr2+/+ mice. However, in ApoE-/-/βarr2-/- mice, the increase in MMP2 expression was significantly lower when analyzed at both the time-points following AngII infusion (Figures 5A and 5B). In contrast to MMP2 expression, AngII treatment resulted in only a modest increase in MMP9 mRNA expression at 7 days in ApoE-/-/βarr2+/+ mice, and βarr2 deficiency did not appear to significantly alter MMP9 mRNA expression at this time-point (Figure 5C). However, after 28 days of AngII infusion, MMP9 expression was significantly increased in ApoE-/-/βarr2+/+ mice and βarr2 deficiency attenuated this increase (Figure 5D). Increased MMP expression has been associated with the proteolytic degradation and elastin breakage that occurs during aneurysmal expansion. Verhoeff-Van Gieson (VVG) staining of abdominal aortic segments from ApoE-/-/βarr2+/+ mice after 28 days of AngII infusion showed regions of breakage and discontinuity of the medial elastin layer (Figure 5E). In contrast, the elastin fibers in the aortas of AngII-treated ApoE-/-/βarr2-/- mice appeared intact (Figure 5F), similar to the aortas of saline-treated ApoE-/-/βarr2+/+ mice (Figure 5G). Aortas from saline-treated ApoE-/-/βarr2-/- mice were histologically indistinguishable from saline-treated ApoE-/-/βarr2+/+ mice (data not shown).
Figure 5. MMP expression is attenuated in the aortas of βarr2-deficient mice.
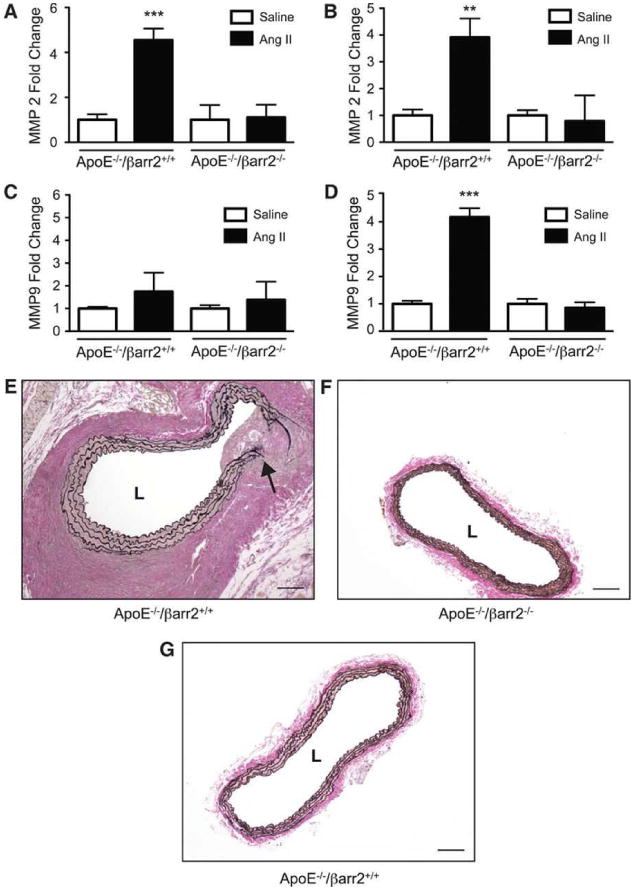
Quantitative PCR analysis of MMP2 mRNA expression in the abdominal aortas of ApoE-/-/βarr2+/+ and ApoE-/-/βarr2-/- mice at A) 7 days or B) 28 days following AngII or saline infusion. ***, significantly different from saline-treated ApoE-/-/βarr2+/+ and AngII-treated ApoE-/-/βarr2-/- mice, P<0.001, **, significantly different from saline-treated ApoE-/-/βarr2+/+ and AngII-treated ApoE-/-/βarr2-/- mice, P<0.01. Data represent mean ± SEM, N ≥ 6. Quantitative PCR analysis of MMP9 mRNA expression in the abdominal aortas of ApoE-/-/βarr2+/+ and ApoE-/-/βarr2-/- mice at C) 7 days or D) 28 days following AngII or saline infusion. ***, significantly different from saline-treated ApoE-/-/βarr2+/+ and AngII-treated ApoE-/-/βarr2-/- mice, P<0.001. Data represent mean ± SEM, N ≥ 6. Representative images of VVG stained abdominal aortic sections from E) AngII-treated ApoE-/-/βarr2+/+ mice. Arrow indicates breakage in the elastin layer F) AngII-treated ApoE-/-/βarr2-/- and G) saline-treated ApoE-/-/βarr2+/+ mice. Scale bars, 0.1mm. L, lumen.
Discussion
Several studies have demonstrated that AngII infusion leads to the formation of AAAs in mice.12, 13, 50 AngII exerts its diverse bioactive effects primarily by activating the AT1a receptor in mice, which plays a critical role in AngII-induced AAA development.17 In addition to mediating traditional G-protein-dependent signaling, the AT1a receptor is also involved in G-protein-independent signaling by forming a complex with the multifunctional scaffolding protein βarr2.19, 24 In the present study, we investigated a role for βarr2 in AngII-induced AAA formation, and found that βarr2 deficiency significantly attenuates AAA formation in mice in both a hyperlipidemic ApoE background and a normolipidemic C57BL/6 background (Figure 1). These studies suggest that G-protein-independent, βarr2-dependent signaling for the AT1a receptor plays a major role in AngII-induced AAA formation.
Although our current studies showed that βarr2 deficiency attenuated AngII-induced AAA formation in mice on the hyperlipidemic ApoE-/- background, it was not clear whether hyperlipidemia was required for this observed phenotype. Therefore, we examined the effect of βarr2 deficiency on AngII-induced AAA formation in mice on a normolipidemic C57BL/6 background. We have previously reported that 8-10-week old, normolipidemic C57BL/6 mice develop only a 5% incidence of AngII-induced AAAs;27 however, two recent reports have utilized aged (7-11 month old) C57BL/6 mice and have shown that these mice develop an approximately 40% incidence of AAAs in response to AngII.37, 38 Therefore, we performed experiments using 8-month old, normolipidemic βarr2+/+ and βarr2-/- mice on a C57BL/6 background and found that similar to the effect in ApoE-/- mice, the deficiency of βarr2 also significantly attenuated AngII-induced AAA incidence in the normolipidemic strain (Figure 1G). Thus, the effect of βarr2 deficiency on attenuating AAA incidence is independent of the hyperlipidemic environment. Because we observed only a low AAA incidence (29.2%) in the C57BL/6 βarr2+/+ strain, the mechanistic studies in our current report were performed in mice on the ApoE-/- background.
Our previous studies have indicated an important role for COX-2 in AAA formation as genetic deficiency of COX-2 or treatment of mice with the COX-2 selective inhibitor celecoxib, significantly attenuated AngII-induced AAA incidence and severity.8, 27 Because our present study shows that βarr2 deficiency also attenuates AngII-induced AAA formation, we investigated a possible link between COX-2 and βarr2 in this pathology. Although previous studies have shown that AngII-mediated COX-2 induction can occur via G-protein-dependent AT1a receptor signaling,30-32 the results in the present study show that the deficiency of βarr2 attenuates AngII-mediated COX-2 induction. This suggests the involvement of a βarr2-dependent, G-protein-independent mechanism for AngII-mediated COX-2 induction. Our findings are supported by a recent study that showed that treatment of rat vascular SMCs with a ‘biased’ agonist for the AT1a receptor, which activates G-protein-independent, βarr2-dependent signaling,20, 51 increased COX-2 expression.52 Based on these studies, together with the results from our current study, we conclude that AngII-mediated COX-2 induction can indeed occur via a G-protein-independent pathway involving βarr2.
AngII-induced COX-2 expression can occur via activation of components of the MAPK pathway such as ERK1/2,30-32 and ERK1/2 activation is also known to be involved in AngII-induced AAA formation.33 Our data show that βarr2 deficiency prevented the AngII-mediated ERK1/2 activation in ApoE-/-/βarr2-/- mice as compared to ApoE-/-/βarr2+/+ mice, when examined at either the incipient (7 days) or advanced stage (28 days) of AAA development. Furthermore, similar to the deficiency of βarr2, inhibition of ERK1/2 activation significantly abrogated the AngII-mediated increase in COX-2 expression. Thus, AngII-AT1a-βarr2-mediated COX-2 induction appears to occur, in part, by a mechanism involving activated ERK1/2.
A hallmark of AAA pathology is macrophage infiltration into the aorta, which is an early cellular event following AngII infusion that continues throughout the development of the disease.7, 53 Our previous studies using the COX-2-deficient mice and more recent studies using an EP4-selective antagonist have suggested that COX-2-derived PGE2 stimulates the production of inflammatory chemokines such as MCP-1 and MIP1α, which contribute to macrophage infiltration during AAA development.6, 8, 29 Because βarr2 has been shown to contribute to pro-inflammatory gene expression in other models of inflammation,22, 54 we examined the role of βarr2 in the inflammatory response that occurs during AAA formation. The observations that βarr2 deficiency attenuated the expression of COX-2 and the inflammatory chemokines, MCP-1 and MIP1α, together with reducing macrophage infiltration, suggest that βarr2 positively regulates inflammation in AngII-induced AAAs through the induction of COX-2 expression.
Proteolytic degradation within the vessel wall mediated largely by MMPs is a common feature of AAAs, wherein elastin fragmentation in the medial and adventitial layers is believed to contribute to the initial dilation, and increased collagenase activity may be responsible for aneurysmal rupture.55 A preponderance of evidence in experimental models as well as in humans supports a role for MMPs, particularly MMP2 and MMP9, in AAA initiation and progression.10, 11, 48, 56 Our studies show that the deficiency of βarr2 attenuated the increase in MMP2 when analyzed at 7 and 28 days following AngII infusion. However, the effect of βarr2 deficiency on MMP9 expression was prominent only at the late stages of aneurysm development (Figure 5D). These results may be explained by previous studies suggesting an important role for MMP2 during the formative stage of AAAs involving macrophage infiltration, and a dominant role for MMP9 during the progressive stage of AAAs.5
AngII is known to be a potent inducer of hypertension in mice.39-41 However, it has also been shown that administration of hydralazine to reduce AngII-induced hypertension does not affect the formation of AAAs, indicating that AngII-induced AAA formation is independent AngII-induced hypertension.57 Our studies show that βarr2 deficiency had no effect on the hypertensive response to AngII (Table 1). Thus, these results suggest that our observed effect of βarr2 deficiency on reducing AAA formation is independent of alterations in blood pressure.
In summary, these studies demonstrate that βarr2 plays a crucial role in the development of AngII-induced AAAs in mice via the induction of COX-2 expression. Based on our current data and previous studies, we propose the following mechanism for βarr2-dependent AAA formation. Activation of AT1a by AngII leads to βarr2 recruitment and AT1a-βarr2 complex formation, which results in the activation of ERK1/2 and COX-2 induction. The AT1a-βarr2-dependent increase in COX-2 expression contributes to AAA initiation via the induction of inflammatory chemokines and resultant recruitment of macrophages, and this combined with increased MMP expression exacerbates AAA pathology. Thus, our studies identify βarr2 as a novel target for the design of pharmacotherapies for AAAs. The fact that βarr2-dependent and G-protein-dependent signaling pathways are pharmacologically distinguishable has given rise to the concept of ‘biased agonism’, whereby a ligand may selectively trigger one or the other pathway.24, 58 Based on this emerging concept, future studies using ‘biased’ ligands to target βarr2-mediated signaling may provide further mechanistic insight into the contribution of βarr2 to AAA formation.
Supplementary Material
Novelty and Significance.
What Is Known?
Angiotensin II (AngII) infusion results in the formation of abdominal aortic aneurysms (AAAs) in mice by activating the AT1a receptor and inducing an inflammatory response.
Inflammatory mediators produced by cyclooxygenase-2 (COX-2) are key contributors to AngII-induced AAA pathology.
β-arrestin 2 (βarr2) activates extracellular signal-regulated kinase 1/2 (ERK1/2) signaling by coupling with AT1a, and ERK1/2 activation has been shown independently to be important in AAA development.
What New Information Does This Article Contribute?
βarr2 is critical for the development of AngII-induced AAAs in mice.
βarr2 mediates AngII-induced AAA formation by inducing COX-2 expression via the ERK1/2 pathway and by increasing inflammation.
AAAs are a life-threatening vascular condition for which no pharmacological treatments are currently available. Hence, a basic understanding of the mechanisms that contribute to AAA formation is required to develop better treatments for this condition. A commonly used mouse model to study AAA formation involves infusion of AngII, which leads to adverse cardiovascular effects primarily by activating the G-protein coupled receptor (GPCR), AT1a. Recent studies have shown that the multifunctional scaffolding protein βarr2 forms a complex with GPCRs such as AT1a to initiate G-protein-independent signaling and contribute to many pathologies. Herein, we show that βarr2 deficiency attenuates AngII-induced AAAs in mice, suggesting a role for G-protein-independent signaling by the AT1a receptor in AAA formation. Our work provides evidence that βarr2 contributes to AngII-induced AAA development by activating ERK1/2 signaling andinducing the pro-inflammatory enzyme - COX-2. These results suggest that βarr2 may be a novel target in designing pharmacological treatments for AAAs.
Acknowledgments
The authors thank Natasha Clayton, BS, HT and Pamela Ovwigho, BS, of the Histology core at the NIEHS for their assistance with the H&E and VVG staining techniques, and Lois Wyrick, BFA, of the NIEHS Arts and Graphics Core for her assistance in preparation of digital images.
Sources of Funding: This work was supported by the Division of Intramural Research of the National Institute of Environmental Health Sciences (NIEHS) at the National Institutes of Health (NIH).
Nonstandard Abbreviations
- AAA
abdominal aortic aneurysm
- AngII
angiotensin II
- ApoE
apolipoprotein E
- AT1a
angiotensin II type 1a
- βarr1
β-arrestin-1
- βarr2
β-arrestin-2
- COX-2
cyclooxygenase-2
- DAB
di-amino benzidine
- ERK1/2
extracellular signal-regulated kinase 1/2
- GPCR
G protein coupled receptor
- H&E
hematoxylin and eosin
- MAPK
mitogen activated protein kinase
- MCP-1
monocyte chemoattractant protein 1
- MEK1
MAPK kinase 1
- MIP1α
macrophage inflammatory protein 1α
- MMP
matrix metalloproteinase
- mPGES1
microsomal prostaglandin E synthase 1
- p-ERK1/2
phosphorylated ERK1/2
- PGE2
prostaglandin E2
- VVG
Verhoeff-Van Geisen
Footnotes
In February 2013, the average time from submission to first decision for all original research paper submitted to Circulation Research was 11.98 days.
Disclosures: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Golledge J, Muller J, Daugherty A, Norman P. Abdominal aortic aneurysm: pathogenesis and implications for management. Arterioscler Thromb Vasc Biol. 2006;26:2605–2613. doi: 10.1161/01.ATV.0000245819.32762.cb. [DOI] [PubMed] [Google Scholar]
- 2.Wassef M, Baxter BT, Chisholm RL, Dalman RL, Fillinger MF, Heinecke J, Humphrey JD, Kuivaniemi H, Parks WC, Pearce WH, Platsoucas CD, Sukhova GK, Thompson RW, Tilson MD, Zarins CK. Pathogenesis of abdominal aortic aneurysms: a multidisciplinary research program supported by the National Heart, Lung, and Blood Institute. J Vasc Surg. 2001;34:730–738. doi: 10.1067/mva.2001.116966. [DOI] [PubMed] [Google Scholar]
- 3.Hellenthal FA, Buurman WA, Wodzig WK, Schurink GW. Biomarkers of AAA progression. Part 1: extracellular matrix degeneration. Nat Rev Cardiol. 2009;6:464–474. doi: 10.1038/nrcardio.2009.80. [DOI] [PubMed] [Google Scholar]
- 4.Hellenthal FA, Buurman WA, Wodzig WK, Schurink GW. Biomarkers of abdominal aortic aneurysm progression. Part 2: inflammation. Nat Rev Cardiol. 2009;6:543–552. doi: 10.1038/nrcardio.2009.102. [DOI] [PubMed] [Google Scholar]
- 5.Freestone T, Turner RJ, Coady A, Higman DJ, Greenhalgh RM, Powell JT. Inflammation and matrix metalloproteinases in the enlarging abdominal aortic aneurysm. Arterioscler Thromb Vasc Biol. 1995;15:1145–1151. doi: 10.1161/01.atv.15.8.1145. [DOI] [PubMed] [Google Scholar]
- 6.Yokoyama U, Ishiwata R, Jin MH, Kato Y, Suzuki O, Jin H, Ichikawa Y, Kumagaya S, Katayama Y, Fujita T, Okumura S, Sato M, Sugimoto Y, Aoki H, Suzuki S, Masuda M, Minamisawa S, Ishikawa Y. Inhibition of EP4 Signaling Attenuates Aortic Aneurysm Formation. PLoS One. 2012;7:e36724. doi: 10.1371/journal.pone.0036724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saraff K, Babamusta F, Cassis LA, Daugherty A. Aortic dissection precedes formation of aneurysms and atherosclerosis in angiotensin II-infused, apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2003;23:1621–1626. doi: 10.1161/01.ATV.0000085631.76095.64. [DOI] [PubMed] [Google Scholar]
- 8.Gitlin JM, Trivedi DB, Langenbach R, Loftin CD. Genetic deficiency of cyclooxygenase-2 attenuates abdominal aortic aneurysm formation in mice. Cardiovascular Res. 2007;73:227–236. doi: 10.1016/j.cardiores.2006.10.015. [DOI] [PubMed] [Google Scholar]
- 9.Walton LJ, Franklin IJ, Bayston T, Brown LC, Greenhalgh RM, Taylor GW, Powell JT. Inhibition of prostaglandin E2 synthesis in abdominal aortic aneurysms: implications for smooth muscle cell viability, inflammatory processes, and the expansion of abdominal aortic aneurysms. Circulation. 1999;100:48–54. doi: 10.1161/01.cir.100.1.48. [DOI] [PubMed] [Google Scholar]
- 10.Thompson RW, Holmes DR, Mertens RA, Liao S, Botney MD, Mecham RP, Welgus HG, Parks WC. Production and localization of 92-kilodalton gelatinase in abdominal aortic aneurysms. An elastolytic metalloproteinase expressed by aneurysm-infiltrating macrophages. J Clin Invest. 1995;96:318–326. doi: 10.1172/JCI118037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davis V, Persidskaia R, Baca-Regen L, Itoh Y, Nagase H, Persidsky Y, Ghorpade A, Baxter BT. Matrix metalloproteinase-2 production and its binding to the matrix are increased in abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 1998;18:1625–1633. doi: 10.1161/01.atv.18.10.1625. [DOI] [PubMed] [Google Scholar]
- 12.Daugherty A, Cassis LA. Mouse models of abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2004;24:429–434. doi: 10.1161/01.ATV.0000118013.72016.ea. [DOI] [PubMed] [Google Scholar]
- 13.Daugherty A, Manning MW, Cassis LA. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J Clin Invest. 2000;105:1605–1612. doi: 10.1172/JCI7818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sasamura H, Hein L, Krieger JE, Pratt RE, Kobilka BK, Dzau VJ. Cloning, characterization, and expression of two angiotensin receptor (AT-1) isoforms from the mouse genome. Biochem Biophys Res Commun. 1992;185:253–259. doi: 10.1016/s0006-291x(05)80983-0. [DOI] [PubMed] [Google Scholar]
- 15.Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol. 2007;292:C82–97. doi: 10.1152/ajpcell.00287.2006. [DOI] [PubMed] [Google Scholar]
- 16.Oro C, Qian H, Thomas WG. Type 1 angiotensin receptor pharmacology: signaling beyond G proteins. Pharmacol Ther. 2007;113:210–226. doi: 10.1016/j.pharmthera.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cassis LA, Rateri DL, Lu H, Daugherty A. Bone marrow transplantation reveals that recipient AT1a receptors are required to initiate angiotensin II-induced atherosclerosis and aneurysms. Arterioscler Thromb Vasc Biol. 2007;27:380–386. doi: 10.1161/01.ATV.0000254680.71485.92. [DOI] [PubMed] [Google Scholar]
- 18.Daugherty A, Manning MW, Cassis LA. Antagonism of AT2 receptors augments angiotensin II-induced abdominal aortic aneurysms and atherosclerosis. Br J Pharmacol. 2001;134:865–870. doi: 10.1038/sj.bjp.0704331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.DeWire SM, Ahn S, Lefkowitz RJ, Shenoy SK. Beta-arrestins and cell signaling. Annu Rev Physiol. 2007;69:483–510. doi: 10.1146/annurev.physiol.69.022405.154749. [DOI] [PubMed] [Google Scholar]
- 20.Lefkowitz RJ, Rajagopal K, Whalen EJ. New roles for beta-arrestins in cell signaling: not just for seven-transmembrane receptors. Mol Cell. 2006;24:643–652. doi: 10.1016/j.molcel.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 21.Chun KS, Lao HC, Trempus CS, Okada M, Langenbach R. The prostaglandin receptor EP2 activates multiple signaling pathways and beta-arrestin1 complex formation during mouse skin papilloma development. Carcinogenesis. 2009;30:1620–1627. doi: 10.1093/carcin/bgp168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Walker JK, Fong AM, Lawson BL, Savov JD, Patel DD, Schwartz DA, Lefkowitz RJ. Beta-arrestin-2 regulates the development of allergic asthma. J Clin Invest. 2003;112:566–574. doi: 10.1172/JCI17265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim J, Zhang L, Peppel K, Wu JH, Zidar DA, Brian L, DeWire SM, Exum ST, Lefkowitz RJ, Freedman NJ. Beta-arrestins regulate atherosclerosis and neointimal hyperplasia by controlling smooth muscle cell proliferation and migration. Circ Res. 2008;103:70–79. doi: 10.1161/CIRCRESAHA.108.172338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shukla AK, Xiao K, Lefkowitz RJ. Emerging paradigms of beta-arrestin-dependent seven transmembrane receptor signaling. Trends Biochem Sci. 2011;36:457–469. doi: 10.1016/j.tibs.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ahn S, Wei H, Garrison TR, Lefkowitz RJ. Reciprocal regulation of angiotensin receptor-activated extracellular signal-regulated kinases by beta-arrestins 1 and 2. J Biol Chem. 2004;279:7807–7811. doi: 10.1074/jbc.C300443200. [DOI] [PubMed] [Google Scholar]
- 26.Holmes DR, Wester W, Thompson RW, Reilly JM. Prostaglandin E2 synthesis and cyclooxygenase expression in abdominal aortic aneurysms. J Vasc Surg. 1997;25:810–815. doi: 10.1016/s0741-5214(97)70210-6. [DOI] [PubMed] [Google Scholar]
- 27.King VL, Trivedi DB, Gitlin JM, Loftin CD. Selective cyclooxygenase-2 inhibition with celecoxib decreases angiotensin II-induced abdominal aortic aneurysm formation in mice. Arterioscler Thromb Vasc Biol. 2006;26:1137–1143. doi: 10.1161/01.ATV.0000216119.79008.ac. [DOI] [PubMed] [Google Scholar]
- 28.Wang M, Lee E, Song W, Ricciotti E, Rader DJ, Lawson JA, Pure E, FitzGerald GA. Microsomal prostaglandin E synthase-1 deletion suppresses oxidative stress and angiotensin II-induced abdominal aortic aneurysm formation. Circulation. 2008;117:1302–1309. doi: 10.1161/CIRCULATIONAHA.107.731398. [DOI] [PubMed] [Google Scholar]
- 29.Cao RY, St Amand T, Li X, Yoon SH, Wang CP, Song H, Maruyama T, Brown PM, Zelt DT, Funk CD. Prostaglandin Receptor EP4 in Abdominal Aortic Aneurysms. Am J Pathol. 2012 doi: 10.1016/j.ajpath.2012.03.016. [DOI] [PubMed] [Google Scholar]
- 30.Ohnaka K, Numaguchi K, Yamakawa T, Inagami T. Induction of cyclooxygenase-2 by angiotensin II in cultured rat vascular smooth muscle cells. Hypertension. 2000;35:68–75. doi: 10.1161/01.hyp.35.1.68. [DOI] [PubMed] [Google Scholar]
- 31.Hu ZW, Kerb R, Shi XY, Wei-Lavery T, Hoffman BB. Angiotensin II Increases Expression of Cyclooxygenase-2: Implications for the Function of Vascular Smooth Muscle Cells. J Pharmacol Exp Ther. 2002;303:563–573. doi: 10.1124/jpet.102.037705. [DOI] [PubMed] [Google Scholar]
- 32.Galan M, Miguel M, Beltran AE, Rodriguez C, Garcia-Redondo AB, Rodriguez-Calvo R, Alonso MJ, Martinez-Gonzalez J, Salaices M. Angiotensin II differentially modulates cyclooxygenase-2, microsomal prostaglandin E2 synthase-1 and prostaglandin I2 synthase expression in adventitial fibroblasts exposed to inflammatory stimuli. J Hypertens. 2011;29:529–536. doi: 10.1097/HJH.0b013e328342b271. [DOI] [PubMed] [Google Scholar]
- 33.Zhang Y, Naggar JC, Welzig CM, Beasley D, Moulton KS, Park HJ, Galper JB. Simvastatin inhibits angiotensin II-induced abdominal aortic aneurysm formation in apolipoprotein E-knockout mice: possible role of ERK. Arterioscler Thromb Vasc Biol. 2009;29:1764–1771. doi: 10.1161/ATVBAHA.109.192609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG, Lin FT. Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science. 1999;286:2495–2498. doi: 10.1126/science.286.5449.2495. [DOI] [PubMed] [Google Scholar]
- 35.Mills PA, Huetteman DA, Brockway BP, Zwiers LM, Gelsema AJ, Schwartz RS, Kramer K. A new method for measurement of blood pressure, heart rate, and activity in the mouse by radiotelemetry. J Appl Physiol. 2000;88:1537–1544. doi: 10.1152/jappl.2000.88.5.1537. [DOI] [PubMed] [Google Scholar]
- 36.Lee CR, Imig JD, Edin ML, Foley J, DeGraff LM, Bradbury JA, Graves JP, Lih FB, Clark J, Myers P, Perrow AL, Lepp AN, Kannon MA, Ronnekleiv OK, Alkayed NJ, Falck JR, Tomer KB, Zeldin DC. Endothelial expression of human cytochrome p450 epoxygenases lowers blood pressure and attenuates hypertension-induced renal injury in mice. FASEB J. 2010;24:3770–3781. doi: 10.1096/fj.10-160119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deng GG, Martin-McNulty B, Sukovich DA, Freay A, Halks-Miller M, Thinnes T, Loskutoff DJ, Carmeliet P, Dole WP, Wang YX. Urokinase-type plasminogen activator plays a critical role in angiotensin II-induced abdominal aortic aneurysm. Circ Res. 2003;92:510–517. doi: 10.1161/01.RES.0000061571.49375.E1. [DOI] [PubMed] [Google Scholar]
- 38.Uchida HA, Poduri A, Subramanian V, Cassis LA, Daugherty A. Urokinase-type plasminogen activator deficiency in bone marrow-derived cells augments rupture of angiotensin II-induced abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2011;31:2845–2852. doi: 10.1161/ATVBAHA.111.234997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cassis LA, Helton MJ, Howatt DA, King VL, Daugherty A. Aldosterone does not mediate angiotensin II-induced atherosclerosis and abdominal aortic aneurysms. Br J Pharmacol. 2005;144:443–448. doi: 10.1038/sj.bjp.0706098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Francois H, Athirakul K, Mao L, Rockman H, Coffman TM. Role for thromboxane receptors in angiotensin-II-induced hypertension. Hypertension. 2004;43:364–369. doi: 10.1161/01.HYP.0000112225.27560.24. [DOI] [PubMed] [Google Scholar]
- 41.Wang YX, Martin-McNulty B, Freay AD, Sukovich DA, Halks-Miller M, Li WW, Vergona R, Sullivan ME, Morser J, Dole WP, Deng GG. Angiotensin II increases urokinase-type plasminogen activator expression and induces aneurysm in the abdominal aorta of apolipoprotein E-deficient mice. Am J Pathol. 2001;159:1455–1464. doi: 10.1016/S0002-9440(10)62532-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Young W, Mahboubi K, Haider A, Li I, Ferreri NR. Cyclooxygenase-2 is required for tumor necrosis factor-alpha- and angiotensin II-mediated proliferation of vascular smooth muscle cells. Circ Res. 2000;86:906–914. doi: 10.1161/01.res.86.8.906. [DOI] [PubMed] [Google Scholar]
- 43.Kovacs JJ, Hara MR, Davenport CL, Kim J, Lefkowitz RJ. Arrestin development: Emerging roles for beta-arrestins in developmental signaling pathways. Dev Cell. 2009;17:443–458. doi: 10.1016/j.devcel.2009.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tham DM, Martin-McNulty B, Wang YX, Wilson DW, Vergona R, Sullivan ME, Dole W, Rutledge JC. Angiotensin II is associated with activation of NF-kappaB-mediated genes and downregulation of PPARs. Physiol Genomics. 2002;11:21–30. doi: 10.1152/physiolgenomics.00062.2002. [DOI] [PubMed] [Google Scholar]
- 45.Zhao L, Moos MP, Grabner R, Pedrono F, Fan J, Kaiser B, John N, Schmidt S, Spanbroek R, Lotzer K, Huang L, Cui J, Rader DJ, Evans JF, Habenicht AJ, Funk CD. The 5-lipoxygenase pathway promotes pathogenesis of hyperlipidemia-dependent aortic aneurysm. Nat Med. 2004;10:966–973. doi: 10.1038/nm1099. [DOI] [PubMed] [Google Scholar]
- 46.Ayabe N, Babaev VR, Tang Y, Tanizawa T, Fogo AB, Linton MF, Ichikawa I, Fazio S, Kon V. Transiently heightened angiotensin II has distinct effects on atherosclerosis and aneurysm formation in hyperlipidemic mice. Atherosclerosis. 2006;184:312–321. doi: 10.1016/j.atherosclerosis.2005.05.016. [DOI] [PubMed] [Google Scholar]
- 47.Bruemmer D, Collins AR, Noh G, Wang W, Territo M, Arias-Magallona S, Fishbein MC, Blaschke F, Kintscher U, Graf K, Law RE, Hsueh WA. Angiotensin II-accelerated atherosclerosis and aneurysm formation is attenuated in osteopontin-deficient mice. J Clin Invest. 2003;112:1318–1331. doi: 10.1172/JCI18141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Longo GM, Xiong W, Greiner TC, Zhao Y, Fiotti N, Baxter BT. Matrix metalloproteinases 2 and 9 work in concert to produce aortic aneurysms. J Clin Invest. 2002;110:625–632. doi: 10.1172/JCI15334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang YX, Martin-McNulty B, da Cunha V, Vincelette J, Lu X, Feng Q, Halks-Miller M, Mahmoudi M, Schroeder M, Subramanyam B, Tseng JL, Deng GD, Schirm S, Johns A, Kauser K, Dole WP, Light DR. Fasudil, a Rho-kinase inhibitor, attenuates angiotensin II-induced abdominal aortic aneurysm in apolipoprotein E-deficient mice by inhibiting apoptosis and proteolysis. Circulation. 2005;111:2219–2226. doi: 10.1161/01.CIR.0000163544.17221.BE. [DOI] [PubMed] [Google Scholar]
- 50.Daugherty A, Cassis LA, Lu H. Complex pathologies of angiotensin II-induced abdominal aortic aneurysms. J Zhejiang Univ Sci B. 2011;12:624–628. doi: 10.1631/jzus.B1101002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rajagopal K, Whalen EJ, Violin JD, Stiber JA, Rosenberg PB, Premont RT, Coffman TM, Rockman HA, Lefkowitz RJ. Beta-arrestin2-mediated inotropic effects of the angiotensin II type 1A receptor in isolated cardiac myocytes. Proc Natl Acad Sci U S A. 2006;103:16284–16289. doi: 10.1073/pnas.0607583103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Morinelli TA, Kendall RT, Luttrell LM, Walker LP, Ullian ME. Angiotensin II-induced cyclooxygenase 2 expression in rat aorta vascular smooth muscle cells does not require heterotrimeric G protein activation. J Pharmacol Exp Ther. 2009;330:118–124. doi: 10.1124/jpet.109.151829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rateri DL, Howatt DA, Moorleghen JJ, Charnigo R, Cassis LA, Daugherty A. Prolonged infusion of angiotensin ii in apoE(-/-) mice promotes macrophage recruitment with continued expansion of abdominal aortic aneurysm. Am J Pathol. 2011;179:1542–1548. doi: 10.1016/j.ajpath.2011.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Porter KJ, Gonipeta B, Parvataneni S, Appledorn DM, Patial S, Sharma D, Gangur V, Amalfitano A, Parameswaran N. Regulation of lipopolysaccharide-induced inflammatory response and endotoxemia by beta-arrestins. J Cell Physiol. 2010;225:406–416. doi: 10.1002/jcp.22289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thompson RW, Baxter BT. Mmp inhibition in abdominal aortic aneurysms. Rationale for a prospective randomized clinical trial. Ann N Y Acad Sci. 1999;878:159–178. doi: 10.1111/j.1749-6632.1999.tb07682.x. [DOI] [PubMed] [Google Scholar]
- 56.Manning MW, Cassis LA, Daugherty A. Differential effects of doxycycline, a broad-spectrum matrix metalloproteinase inhibitor, on angiotensin II-induced atherosclerosis and abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2003;23:483–488. doi: 10.1161/01.ATV.0000058404.92759.32. [DOI] [PubMed] [Google Scholar]
- 57.Cassis LA, Gupte M, Thayer S, Zhang X, Charnigo R, Howatt DA, Rateri DL, Daugherty A. Ang II infusion promotes abdominal aortic aneurysms independent of increased blood pressure in hypercholesterolemic mice. Am J Physiol Heart Circ Physiol. 2009;296:H1660–1665. doi: 10.1152/ajpheart.00028.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gesty-Palmer D, Luttrell LM. Refining efficacy: Exploiting functional selectivity for drug discovery. Adv Pharmacol. 2011;62:79–107. doi: 10.1016/B978-0-12-385952-5.00009-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.