

Abstract
Ischaemic stroke patients treated with Selective Serotonin Reuptake Inhibitors (SSRI) show improved motor, cognitive and executive functions, but the underlying mechanism(s) are incompletely understood. Here, we report that cerebral arterioles in the rat brain superfused with therapeutically effective doses of the SSRI fluoxetine showed consistent, dose-dependent vasodilatation (by 1.2 to 1.6-fold), suppressible by muscarinic and nitric oxide synthase (NOS) antagonists [atropine, NG-nitro-l-arginine methyl ester (l-NAME)] but resistant to nicotinic and serotoninergic antagonists (mecamylamine, methylsergide). Fluoxetine administered 10–30 min. following experimental vascular photo-thrombosis increased arterial diameter (1.3–1.6), inducing partial, but lasting reperfusion of the ischaemic brain. In brain endothelial b.End.3 cells, fluoxetine induced rapid muscarinic receptor-dependent increases in intracellular [Ca2+] and promoted albumin- and eNOS-dependent nitric oxide (NO) production and HSP90 interaction. In vitro, fluoxetine suppressed recombinant human acetylcholinesterase (rhAChE) activity only in the presence of albumin. That fluoxetine induces vasodilatation of cerebral arterioles suggests co-promotion of endothelial muscarinic and nitric oxide signalling, facilitated by albumin-dependent inhibition of serum AChE.
Keywords: acetylcholinesterase, fluoxetine, Ischaemic stroke, muscarinic receptors, nitric oxide, vasodilatation
Introduction
Over 795,000 ischaemic stroke events occur per year world wide [1], causing irreversible long-lasting neurological injury and an increased risk of delayed neurological decline. Today, thrombolysis is the only proven treatment [2], highlighting the unmet need for new prevention and treatment strategies. Recent reports on SSRIs, the most common family of prescribed antidepressants, show beneficial effects on post-stroke cognitive [3], motor [4] and executive functions [5]. The underlying mechanisms are not fully understood. Initially it was thought that this effect is related to the SSRI's mood stabilizing properties through their suppression of neuronal serotonin uptake, which leads to elevated concentrations of 5-hydroxytryptamine (5-HT). However, the SSRI fluoxetine also mediates serotonin-dependent vasodilatation in ex vivo models, including small branched rat anterior cerebral arterioles [6]. Additionally, while activation of the 5-HT1 and 5-HT7 receptors mediates vaso-relaxation, activation of the 5-HT2 serotonin receptors on smooth muscle cells could also enhance vasoconstriction [7].
Fluoxetine also exerts chronic non-serotoninergic effects, and its potential neuroprotective role has been attributed to microglia inhibition and subsequently decreased release of multiple pro-inflammatory and cytotoxic factors including nitric oxide (NO) [8]. Fluoxetine alters the levels and composition of brain GABA(A) receptors and reduces the responsiveness of GABA(A)-R to GABA-mimetic drugs such as pentobarbital [9]. In addition, it reduces the conductance of several voltage-dependent Na+ and K+ channels in different tissues [10] and inhibits nicotinic muscle α1β1γδ acetylcholine receptors (nAChR) or neuronal α2β4 or α3β4nAChRs [11]. Fluoxetine was further reported to inhibit the hydrolytic activities of the ACh hydrolyzing protein acetylcholinesterase (AChE) [12], which could increase cholinergic signalling in the circulation.
Altered cholinergic signalling can modulate vascular tone, which involves release of several dilator and constrictor substances. These include prostacyclin, endothelium-derived hyperpolarizing factor, bradykinin and NO. Specifically, acetylcholine (ACh) released from parasympathetic fibres binds to muscarinic ACh receptors (mAChR) on endothelial cells, resulting in the release of calcium from intracellular stores [13,14]. This facilitates binding of calmodulin to eNOS which subsequently produces NO from l-arginine. NO diffuses to vascular smooth muscles, inducing the synthesis of cyclic guanosine monophosphate (cGMP) [15], cGMP-dependent protein kinase activation and dephosphorylation of myosin light chains and results in a subsequent relaxation of smooth muscle cells within the vessel walls. Together, these data raise the possibility that the beneficial outcome of SSRIs in stroke patients is dependent on a direct effect on cerebral vessels.
Materials and methods
Stroke induction
Focal cerebral ischaemia was induced according to an established model of photothrombosis [16]. Following craniotomy (see In vivo animal preparation) saline-diluted Rose Bengal (7.5 mg/ml) was administered intravenously (0.133 ml/100 g body weight) and the neocortex (ca. 1 mm diameter) was exposed to green laser light (532 nm, Laser 2000 CNI-532) for 15 min. to induce a focal photochemical reaction and subsequent endothelial damage and clot formation. Cerebral blood perfusion was investigated by fluorescent angiography using intravenous injection of sodium fluorescein (1 mg/ml, 0.1 ml/kg; M.W.; 376.27, for details see [17]).
In vivo animal preparation
The in vivo experiments were performed using established methods [17,18]. Briefly, adult male Sprague Dawley rats weighing 210–280 g were deeply anaesthetized by intraperitoneal injection of ketamine (100 mg/ml, 0.08 ml/100 g) and xylazine (20 mg/ml, 0.06 ml/100 g). The tail vein was catheterized and the animal was placed in a stereotactic frame under a fluorescence stereomicroscope (SteReOLumar V12; Zeiss, Oberkochen, Germany). Body temperature was continuously monitored and maintained at 38 ± 0.5°C using a heating pad. A bone window was drilled over the right motor-somatosensory cortex (3–6 mm size, located between 1 and 4 mm lateral, 2 frontal to 4 mm caudal from Bregma). The dura was opened and the cortex was continuously superfused with artificial cerebrospinal fluid (aCSF) containing (in mM): 129 NaCl, 21 NaHCO3, 1.25 NaH2PO4, 1.8 MgSO4, 1.6 CaCl2, 3 KCl and 10 glucose (pH 7.4) at 20 ml/min. Different pharmakons were added for short periods to the aCSF to investigate their effect on vascular tone: fluoxetine (1–500 μM), carbachol (50 μM), serotonin (1, 50 μM), bradykinin (100 μM), l-NAME (1 mM) and atropine (50 μM, 1 mM) in combination with fluoxetine (500 μM) or carbachol (50 μM) (all chemicals were purchased from Sigma-Aldrich, Rehovot, Israel). High concentrations were used to reach a rapidly saturating effect. Pial vessels were imaged at 0.5–1 Hz using an EMCCD camera (DL-658 M-TIL; Andor Technology, Belfast, UK) for at least 1 min. preceding drug application, during drug application and upon washout with aCSF. A binary image was created to segment vascular and extravascular compartments and automatically calculate vascular diameter at five points along a selected artery for each time-point. Image processing and analysis was performed in MATLAB.
AChE activity assays
AChE activities were determined in recombinant enzyme preparations and serum samples using a standard colorimetric assay adapted to a 96-well microtiter plate. Assays were performed in 0.1 M phosphate buffer, pH 7.4, 0.5 mM dithio-bis-nitrobenzoic acid and 1 mM acetylthiocholine substrate at room temperature [19]. Optical density at 405 nm was monitored for 20 min. at 3 to 5-min. intervals.
Photo-induced cross-linking
Cross-linking was triggered by long wavelength light as described [20]. Briefly, proteins were brought to a concentration of 10 μM, diluted 1:10 into 50 mM Tris-Cl pH 7.5 and 150 mM NaCl in 20μl to reach a working concentration of 1 μl Tris(2,2′bipyridyl)dichlororuthenium(II) hexahydrate (Ru(II)bpy32+, Sigma-Aldrich) and 1 μl ammonium persulphate (BioRad, Hercules, CA, USA) were added to final concentrations of 125 μM and 2.5 mM respectively. Samples were mixed gently, illuminated for 30–60 sec., and the reaction terminated by adding SDS-PAGE loading buffer containing 1.4 μl β-mercaptoethanol. Samples were then heated (5 min., 95°C), separated by standard SDS-PAGE and transferred to nitrocellulose membranes. Proteins were visualized using specific primary antibodies against AChE (Santa Cruz Biotechnology, Santa Cruz, CA, USA, sc-6431) followed by HRP-conjugated secondary antibodies (Jackson ImmunoResearch Inc., West Grove, PA, USA) and enhanced chemiluminescence (EZ-ECL; Biological Industries, Beit-HaEmek, Israel). Recombinant human AChE-R was produced and provided by Protalix Biotherapeutics (Carmiel, Israel).
Protein extraction and immunoblot analysis
Homogenates were prepared in a High salt/detergent buffer [0.01 M Tris, 1 M NaCl, 1% Triton X-100 and 1 mM EGTA (pH-7.4)], 200 μl per sample. For co-immunoprecipitation, cell extracts were separated, incubated with anti-HSP90 antibodies (sc-1055; Santa Cruz Biotechnology) and blotted as previously described [21]. Immunodetection (4°C, overnight) was with mouse monoclonal anti α-eNOS diluted 1:1000 (sc-654; Santa Cruz). Development (room temp, 2 hrs) was with horseradish peroxidase-conjugated goat anti-mouse antibodies diluted 1:10,000 (Jackson Laboratories) and enhanced chemiluminescence (ECL) kit (Amersham Pharmacia Biotech, Buckinghamshire, UK).
Calcium imaging
b. End.3 cells are endothelial cells from cerebral cortex origin of BALB/c mice (ATCC Washington, DC, USA). To monitor changes in intracellular Ca2+, b.End.3 cell cultures were plated on eight-well microscopy chambers (Applied BioPhysics, Troy, NY, USA) and incubated (1 hr, 37°C) in 300 μl Dulbecco Modified Eagle Medium (DMEM) containing 5 μM of the Ca2+ fluorescent dye, fluo 4-AM (Molecular Probes, Eugene, OR, USA). Cells were then washed twice in DMEM. Live cell imaging was performed using an FV-1000 confocal microscope (Olympus, Tokyo, Japan), equipped with an IX81 inverted microscope. A 60×/1.4 oil immersion objective was used. The FV-1000 system is equipped with an incubator (LIS, Basle, Switzerland), which controls temperature, CO2 and humidity.
Scans were performed using the ZDC application (Z drift compensation). Fluo 4-AM was excited using a 488 nm laser line and the emission filter was 505–525 nm. The images were continuously acquired before and after addition of the noted reagents, at time intervals of 200 msec., for a total of 600 sec. DIC images were also taken. Data analysis was performed using ImageJ.
Immunocytochemistry
For standard immunocytochemistry, b.End.3 cell cultures were fixed in 4% paraformaldehyde in PBS for 20 min., permeabilized in 0.1%Triton X-100 for 4 min., blocked in 5% horse serum for 1 hr, and then incubated with primary antibodies overnight at 4°C. Cy3-or Cy2-conjugated secondary antibodies were then added for 1 hr at room temp. Cover slips were rinsed twice with PBS and once with DDW, then mounted for confocal microscopy (FluoroMount-G; Electron Microscopy Sciences, Washington, PA, USA). The antibodies were anti-eNOS and anti-HSP90 (both at 1:200, Santa Cruz). Slides were scanned using the Olympus FV-1000 confocal microscope as above. Image Analysis was performed using ImageJ (National Institute of Health, Bethesda, MD, USA).
Results
Fluoxetine induces a serotonin-independent, muscarinic/eNOS-mediated vasodilatation
In the circulation of treated patients, fluoxetine reaches levels of 0.95 μM [22]. In experimental studies using the open cranial window method, a concentration gradient between the perfusion solution (aCSF) and the brain parenchyma of a factor 10 has been reported. [23]. To directly measure the effect of clinically relevant doses of fluoxetine on cerebral vessels and on blood flow, we used direct fluorescence imaging to follow arteriolar diameter in experimental rats (Fig. 1 and SF 1). Within 60 sec. of fluoxetine superfusion, we observed a dose-dependent, robust increase in arteriolar diameter (by about 1.2-fold, n = 3 rats at 5 μM and 1.5-fold at 50 μM, n = 6, SF. 1A). The only effect of increasing fluoxetine concentration further was that the onset of the vasodilatation occurred earlier, presumably due to faster equilibration of the tissue with the required concentrations of fluoxetine. We therefore frequently used high concentrations of drugs to overcome potential desensitization.
Fig. 1.
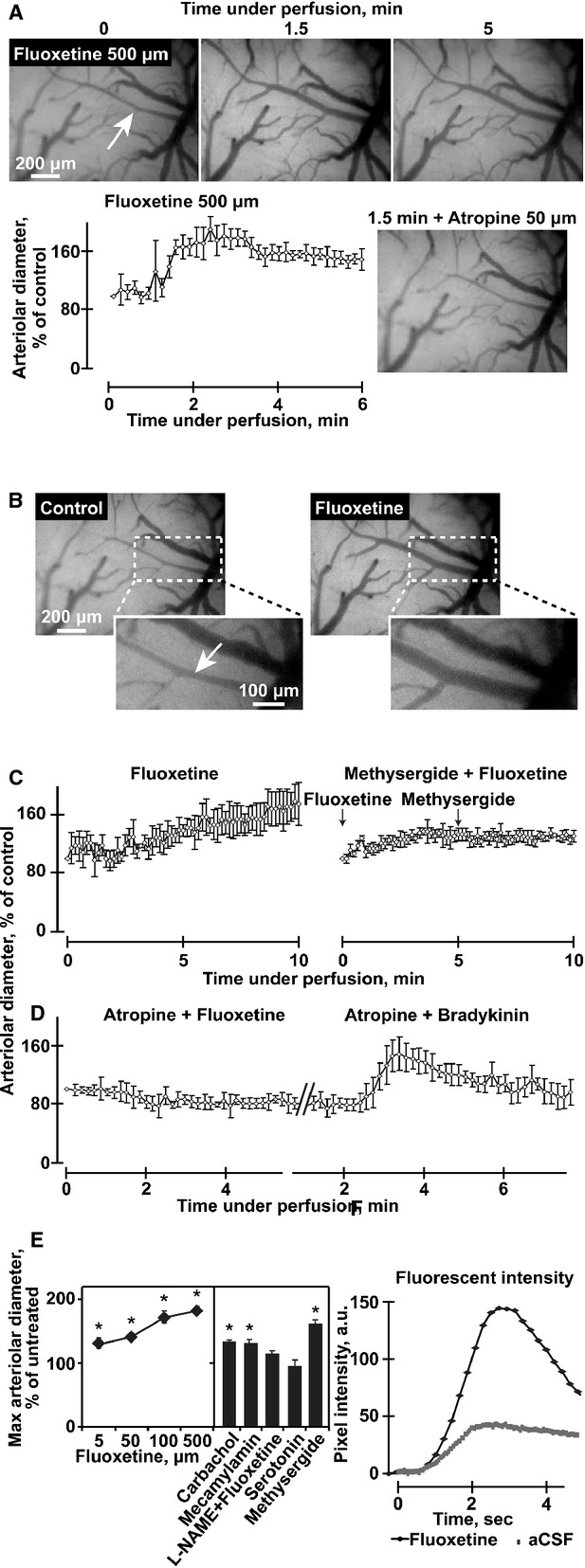
Fluoxetine induces a rapid, robust and long-lasting muscarinic/eNOS-mediated vasodilatation. (A) Representative images of pial arterioles in the rat cerebral cortex. Shown are photographs of the cranial windows at the noted time-points in minutes and administered pharmakons. Note rapid response and inhibition of 500 μM fluoxetine-mediated vasodilatation inhibition by 50 μM atropine. Arterioles and venules were identified visually and arterioles identity confirmed by their earlier filling with the fluorescent dye in the angiography experiments. B. Representative images of pial arterioles in the rat cerebral cortex. Shown are photographs before (Control) and 1.5 min. under 100 μM fluoxetine perfusion (fluoxetine). Below: enlarged dashed frames. As the focus is not similar, we performed diameter analysis for multiple consecutive images acquired at 0.5–1 Hz. (C and D) Arteriolar diameter presented as percentage from control (averages ± S.D. for 5 points along an arteriole) following perfusion of 100 μM fluoxetine without or with 50 μM atropine, 5 μM fluoxetine with 100 μM methysegrid, or 100 μM bradykinin with 50 μM atropine-each presenting one out of four experimental animals with similar outcome, except for one animal in the bradikinin + atropine test (anova P < 0.001). (E) Left: Maximal arterial diameter following perfusion of the noted compounds, presented as average ± S.D. at ± 10 sec. around the peak. The aterioles we investigated had a mean diameter of 24.35 ± 9.72 μm before pharmacological manipulation. Right: Carbachol, mecamylamine and methysergide (50 μM), but not serotonin (50 mM) enlarges arteriolar diameter, and l-NAME (1 mM) blocks the 500 μM fluoxetine-induced enlargement in arteriolar diameter (anova P < 0.001). (F) Arterial fluorescent intensity representing cerebral blood flow, in pixel intensity following bolus injection of fluorescein sodium salt of fluoxetine versus ‘control angiography’. (anova P < 0.001, N = 3).
This effect was selective to the arterial compartment as no vasodilatation was found in morphologically identified veins. The non-degradable ACh analogue carbachol at 50 μM induced a similar effect (SF. 1B). Given that activation of endothelial muscarinic receptors modulates the diameter of cerebral blood vessels [24], we also co-applied fluoxetine together with the non-specific muscarinic antagonist, atropine. 50 μM atropine blocked the fluoxetine-mediated vasodilatation (Fig. 1C). Perfusion with artificial cerebrospinal fluid (aCSF) containing serotonin was performed so that the final serotonin levels would correspond to those levels of serotonin that were measured 1 day following fluoxetine treatment (50 μM, Fig. 1A). This caused no change in arteriolar diameter, and co-superfusion of 5 μM fluoxetine together with the serotonin receptor blocker, methylsergide (100 μM) did not block the fluoxetine-induced vasodilatation. (n = 5 rats, Fig. 1B:right). Additionally, 50–100 μM atropine applied alone had no significant effect on vessel diameter. However, when co-administered with fluoxetine it consistently prevented its dilatation effect (Fig. 1C). Subsequent addition of the potent vasodilator bradykinin induced significant vasodilatation, excluding the possibility of lost vessels reactivity in the presence of atropine (Fig. 1D). Carbachol, the non-degradable ACh analogue, increased arteriolar diameter to an extent similar to that obtained by fluoxetine (Fig. 1E). Finally, the fluoxetine-mediated vasodilatation could be suppressed by the NOS inhibitor l-NAME (Fig. 1E) and fluoxetine (50 μM) increased the peak intensity of peripherally injected fluorescein sodium salt in all tested arterioles by 1.4 ± 0.3-fold (Fig. 1E), suggesting increased regional cerebral blood flow.
Fluoxetine increases blood flow in a rat ischaemic stroke model
Given that events within microvessels are linked to the behaviour of the entire neurovascular unit [25], we induced local photothrombosis [16] to test for fluoxetine effects on blood flow following experimental stroke due to photothrombosis. Using fluorescent angiography complete occlusion of the photothrombotic vessels could be confirmed. Superfusion of the brain with 50 μM fluoxetine within 10–30 min. following photothrombosis significantly increased the diameter (1.3, n = 4 rats) of both thrombosed (occluded) and adjacent cerebral arterioles (Fig. 2A and B). Fluorescein angiography demonstrated increased blood flow and partial reperfusion through the thrombosed vessels for the whole duration of the experiment (4 hrs, n = 4 rats, Fig. 2C), both in the core and the peri-ischaemic regions of the occluded site (Fig. 2D).
Fig. 2.
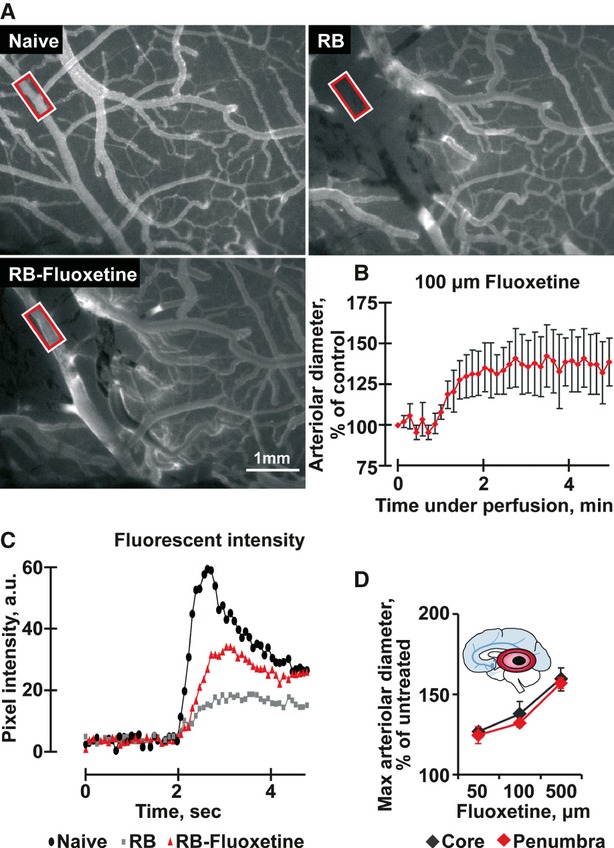
Fluoxetine increases blood flow after laser-induced small artery ischaemia. (A) Representative images of the naïve pial vasculature, before (left) and after photothrombosis (Rose Bengal – RB, right) and after 100 μM fluoxetine treatment (RB + Fluoxetine, below) following i.v. injection of fluorescein sodium salt. Red-framed rectangles indicate areas of blood flow measurement. (B) Arteriolar diameter presented as percentage of control (average ± S.D. for 5 different points within the arteriole) following perfusion of 100 μM fluoxetine in an ischaemic rat. (C) Fluorescent intensity representing cerebral blood flow and the arteriolar peak in the above samples. (anova P < 0.001). (D) Maximal arteriolar diameter following perfusion of fluoxetine in the noted concentrations. Presented are average ± S.D. at 10 sec. around the peak within the core (black) and the penumbra (red). (anova P < 0.001). Inset: Scheme of brain involvement in ischaemic stroke. A core of infarcted tissue (black zone) is surrounded by a peripheral region referred to as the penumbra (red).
Supporting this notion, cultured mouse b.End.3 brain endothelial cells loaded with the Ca2+ fluorescent dye, fluo 4-AM showed that fluoxetine (10 μM) induced a rapid and robust increase in intracellular calcium ([Ca2+]i, 1.5 ± 0.3 mmol, Fig. 3A). Because ACh has been shown to increase [Ca2+]i in endothelium [15] and to enhance NO production in peripheral blood vessels, [26] we further tested for [Ca2+]i increases in response to high ACh levels (10 mM). The fluoxetine effect was more robust than that of the rapidly degradable ACh (data not shown) and was prevented by atropine (Fig. 3B), suggesting that fluoxetine directly activates muscarinic receptors on endothelial cells. When treated with the NO fluorescent dye DAF-2DA, cultured b.End.3 cells displayed atropine-sensitive NO elevation in the presence of 50 μM fluoxetine (Fig. 3C), and NO labelling was particularly intense in cellular regions surrounding the nucleus (Fig. 3D).
Fig. 3.
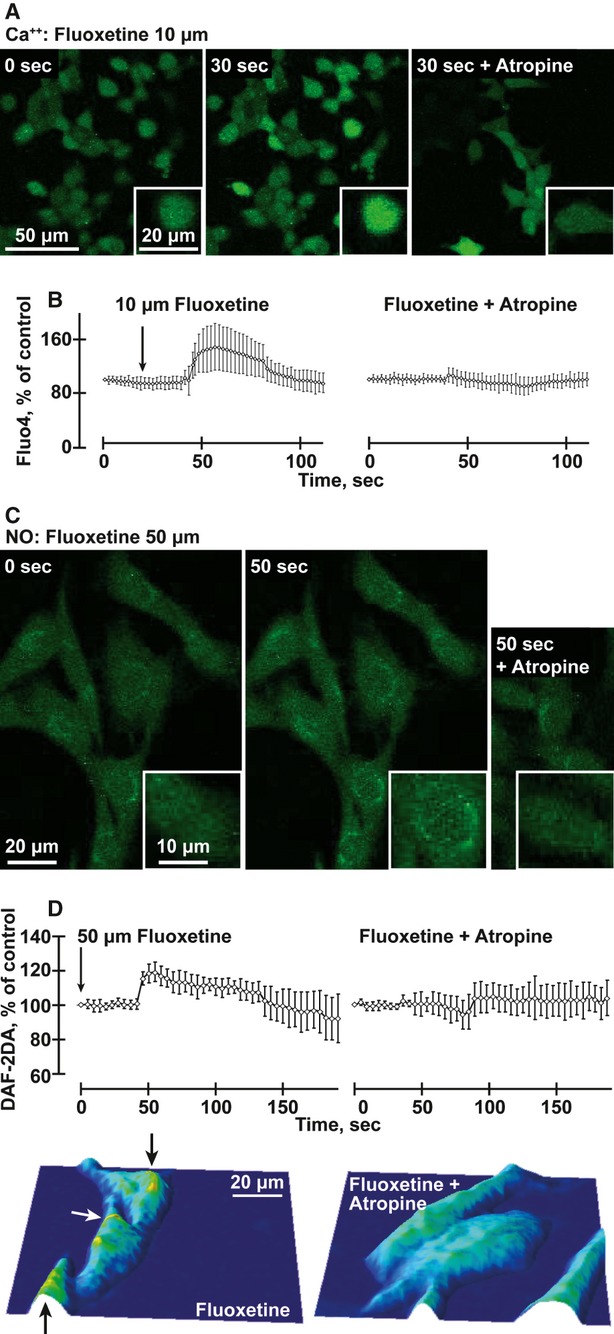
Fluoxetine induces atropine-suppressible intracellular Ca2+ and NO release in mouse b.End.3 cells (A) Ca++: b.End.3 cells stained with Fluo-4 and treated with 10 μM fluoxetine (FL) for 30 sec. with or without atropine. (B) Quantification of average changes in Ca++ signals in 40 cells under the conditions noted above. (anova P < 0.001). (C) NO: b.End.3 cells stained with DAF-2DA and treated with 50 μM fluoxetine with or without atropine. (D) Quantification of changes in NO signals in 30 cells under the conditions noted above. (anova P < 0.001). Below: Typographical map of the NO signal represents the field of the image above, following fluoxetine treatment of b.End.3 cells with or without atropine. Note high levels near the nucleus (yellow, marked by arrows).
Fluoxetine enhances long-lasting eNOS-HSP90 interactions in cultured b.End.3 cells
Binding of the chaperone HSP90 to eNOS increases NO synthesis [27,28]. This likely occurs through protection of eNOS from dephosphorylation by HSP90 limitation of eNOS accessibility to protein phosphatases or by protein kinase–mediated eNOS phosphorylation and facilitated calmodulin association with eNOS [29,30]. To study if fluoxetine affects eNOS interactions with its activator HSP90, we treated b.End.3 cells for 24 hrs with 10 μM fluoxetine and measured eNOS-HSP90 interaction and co-localization by co-immunoprecipitation and immunocytochemistry in un-stimulated and treated cells [28]. Both HSP90 and eNOS showed significant up-regulation following treatment with fluoxetine (Fig. 4A and B; Student's t-test, P < 0.00 and P < 0.02 reciprocally). HSP90-eNOS colocalization was also increased in cells treated with fluoxetine (by 27%; Fig. 4B). Finally, increased HSP90-eNOS association following fluoxetine was detected by co-immunoprecipitation (Fig. 4B). Thus, both in vivo and ex vivo findings supported relevance of NO signalling to the fluoxetine effect.
Fig. 4.
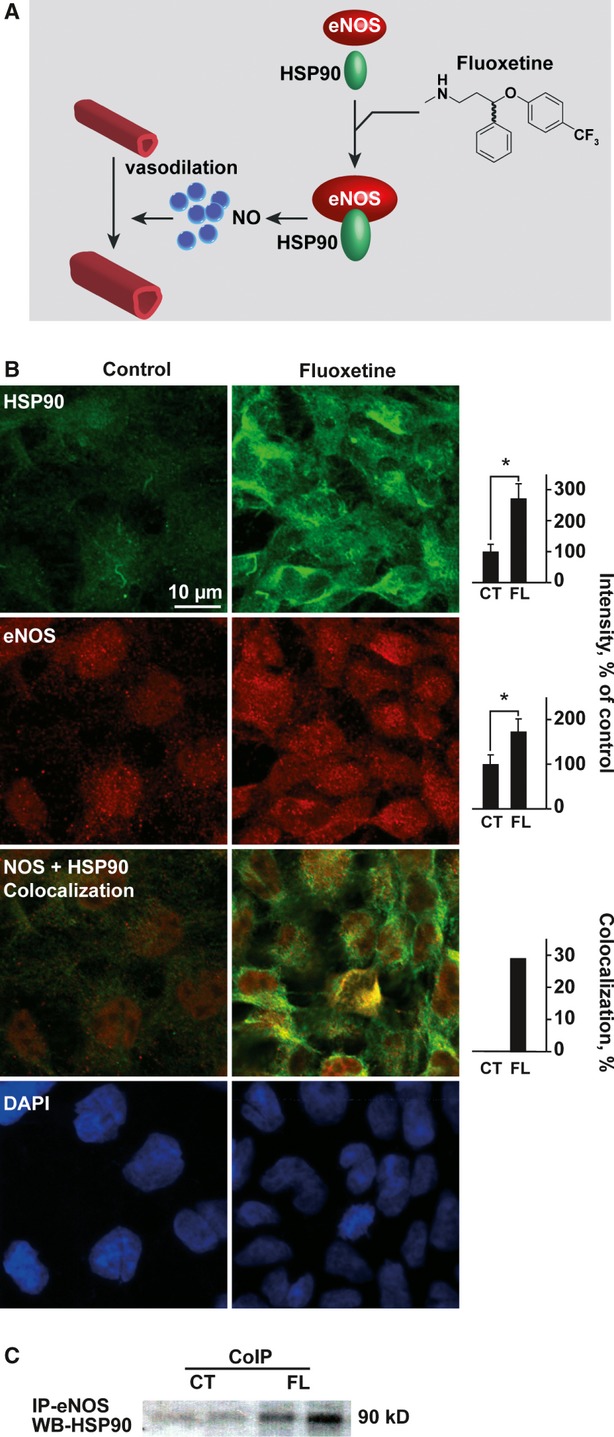
Fluoxetine increases the levels of eNOS and its enhancer HSP90 in B.End.3 endothelial cells. (A) Top scheme: Fluoxetine induction of eNOS-HSP90-mediated vasodilatation involves facilitation of NO production by eNOS. (B) Top row: Enhanced HSP90 under fluoxetine. Columns: Quantification (Student's t-test CT-FL P < 0.00). Second row: Enhanced eNOS immunolabeling in b.End.3 cells pretreated for 24 hrs with 10 μM fluoxetine. Columns: Quantification (Student's t-test P < 0.00).Third row: Enhanced HSP90-eNOS colocalizaion under fluoxetine. Columns: Quantification (Student's t-test P < 0.00). Fourth row: DAPI staining showed sustained cell density. (C) B.End.3 cell extract was incubated with anti-eNOS antibodies and precipitates electroblotted. Immunolabeling showed HSP90-induced elevation of eNOS in the fluoxetine-treated cell lysate compared to untreated cells.
Fluoxetine inhibits ACh hydrolysis by albumin-AChE complexes
SSRIs were reported to inhibit AChE activity when added to serum samples [12], suggesting that apart from direct activation of muscarinic receptors (as demonstrated above), fluoxetine may enhance cholinergic signalling by elevating the levels of ACh through AChE inhibition. To test for this possibility we incubated either serum or highly purified human recombinant (hr) AChE with fluoxetine (1–100 μM) and measured rates of ACh hydrolysis. Compatible with the previous report, fluoxetine exerted a dose-dependent inhibition of serum AChE. However, fluoxetine incubation with purified hrAChE yielded no inhibition (Fig. 5A and B). This raised the possibility that the presence of serum albumin was necessary for enabling the inhibition of ACh hydrolysis by fluoxetine. Correspondingly, fluoxetine (10 μM) inhibited the hydrolysis activity of purified enzyme incubated with but not without serum levels (70 mg/ml) of purified albumin by 50% (Fig. 5B, P < 0.0001, Student's t-test). A photo-induced cross-linking assay (Fig. 5C) followed by immunoblot analyses demonstrated rapid formation of slowly migrating multimeric AChE conjugates, formation of albumin-AChE complexes and that albumin interfered with the multimerization of AChE (Fig. 5D). Taken together, these findings suggest that albumin allows improved fluoxetine accessibility to AChE monomers by preventing their multimerization, which enables inhibition of AChE's hydrolytic activity by fluoxetine. This indicated that SSRIs could also modulate cholinergic signalling by elevating the levels of endogenously produced ACh.
Fig. 5.
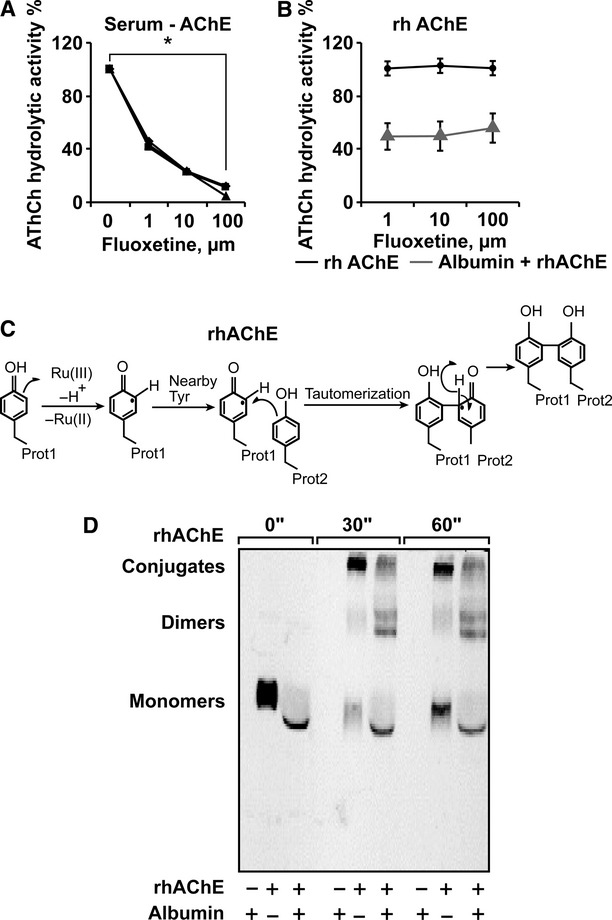
Fluoxetine reduces rhAChE hydrolytic activity when interacting with albumin (A) AChE activity following 15 min. incubation of human serum samples with the noted μM doses of fluoxetine. Presented are residual percentages of hydrolytic activity from control values of serum samples from three different individuals. Average ± S.D. serum AChE activity without fluoxetine: 146.4 ± 10.8; N = 3. (anova P < 0.001). (B) hrAChE activity following incubation of 15 min. with the noted doses of fluoxetine with and without 70 mg/ml Albumin. (anova P < 0.001). (C) Scheme of photo-induced cross-linking by ruthenium ions. (D) Albumin attenuates the formation of AChE multimeric conjugates in the photo-induced cross-linking assay. Note the photo-induction, within 30 sec. of slowly migrating AChE multimeric conjugates from rhAChE monomers and the attenuated production of these multimers, yielding higher dimer levels in the presence of albumin.
Discussion
We found that the SSRI fluoxetine induces direct, rapid and robust vasodilatation of surface arterioles of experimental rats in vivo, and restores blood flow in thrombotic arterioles. The fluoxetine effect was serotonin-independent and atropine- and l-NAME-sensitive, suggesting muscarinic/NO regulation. Correspondingly, fluoxetine elevated intracellular Ca2+ levels in cultured brain endothelial cells in a manner suppressible by atropine, and induced muscarinic-mediated release of NO, augmenting eNOS and its activator protein HSP90 levels and facilitating HSP90-eNOS interaction in b.End. 3 cells. In the presence of physiological albumin concentrations, fluoxetine bound to highly purified recombinant AChE and suppressed the activity of serum AChE, and of highly purified recombinant AChE mixed with albumin, but not the activity of highly purified recombinant AChE without albumin, together suggesting that fluoxetine can increase ACh levels and cholinergic transmission within blood vessels both by facilitating mAChR activation and by inhibiting AChE complexed with albumin.
Four different routes through which fluoxetine could potentially modulate the vascular tone were considered and investigated. (1) Direct serotonin-dependent vasodilatation was excluded as serotonin and serotonin antagonists failed to affect the fluoxetine-mediated vasodilatation, even when high doses were applied. (2) Inhibition of nicotinic receptors was excluded because mecamylamine failed to affect the fluoxetine-mediated vasodilatation. (3) In contrast, mAChR and eNOS- activation were involved in fluoxetine-mediated vasodilatation as both atropine and L-NAME blocked the fluoxetine effect on rat brain arterioles and the fluoxetine-inducible augmentation of eNOS-HSP90 interaction in brain endothelial cells. (4) Likewise, ACh elevation through AChE inhibition was supported by fluoxetine inhibition of AChE under albumin-AChE interaction, which would be exclusively enabled within arterioles, as shown by cross-linking and enzyme assays. To act from both sides of the endothelial cells, fluoxetine should also cross the blood-brain barrier when applied by superfusion. This diffusion pathway may explain the high levels required in our experiments. Also, high concentrations may be required to overcome desensitization [31]. High concentrations (1 mM) of atropine were used to address the specificity of atropine antagonism, Atropine blocked muscarinic, but not bradykinin (100 μM) receptor-mediated vasodilatation, even when fluoxetine was applied at 500 μM (SF. 2A). This is compatible with the hypothesis that intracellular Ca2+ elevation and eNOS activation, but not COX1 activation were involved in the fluoxetine effect (SF. 2B). Chronic fluoxetine treatment has been reported to elevate CREB levels [32], which would predictably enhance neuronal excitation and correspondingly mitigate ischaemia-induced spatial cognitive deficits [3]. Compatible with the improved executive function under fluoxetine treatment [5], our findings call for testing for possible association of this response with enhanced cholinergic tone in the treated brain.
Our data exclude the possibility that the vasodilatation effect of fluoxetine was mediated by increasing serotonin levels. Rather, we support vasodilatation-associated muscarinic reactions in brain vessels [33]. Extending previous reports of inhibition of serum AChE by SSRI's [12], our results attribute the fluoxetine-mediated AChE inhibition within brain vessels, to the presence of albumin which would increase the levels of ACh in the micro-environment of endothelial cells. Therefore, as long as the blood-brain barrier (BBB) is intact, our findings preclude such effects within the brain tissue itself. By operating through both direct activation of mAChR and elevation of ACh due to AChE inhibition, the vasodilatation observed in surface arterioles thus involves both direct and indirect muscarinic effect [13]. This double effect may explain why fluoxetine was more effective than carbachol in its atropine-suppressible vasodilatative function. Smooth muscle nicotinic receptor blockade by fluoxetine [34] could possibly add upon these effects, but was not tested in the current study.
Recent studies showed that 14 days of treatment with fluoxetine and sertraline, starting 1hr after the ischaemic insult significantly reduced photothrombosis-induced infarct size in mice [35], and significantly increased the expression of haem oxygenase-1 (HO-1) and hypoxia-inducible factor-1α in the ischaemic region [35]. Compatible with these reports, we found the long-lasting vasodilatation of brain arterioles induced by clinically relevant concentrations of fluoxetine to associate with increased blood flow through the ischaemic and per-ischaemic regions. Further supporting this concept, improved cerebral perfusion in the peri-ischaemic region strongly influences the prognosis of stroke patients [36]. At the molecular level, the up-regulation of HSP90 and fluoxetine treatment suggests that fluoxetine, perhaps by elevating the cholinergic tone, imposes cellular stress conditions over the endothelial cell. Of note, the chaperon HSP90 is one of the most abundant proteins expressed in all cell types and is involved in numerous cell pathways [29,30] (e.g. assisting in protein folding, maintaining the tertiary structure of the proteasome and its ATPase activity and participating in glucocorticoid receptor translocation from the cytoplasm into the nucleus) [29,30,37]. Thus, the fluoxetine-induced elevation of HSP90, and its tightened interaction and co-localization with eNOS likely affect multiple cellular pathways.
Fluoxetine-mediated vasodilatation was shown in arterioles, where the majority of occlusion events occur but not in the venous circulation. The richer innervations by cholinergic afferents of arteries and arterioles compared to veins and venules [38] may underlie this observation. Also, the walls of arteries and arterioles consist of interchanging layers of endothelium, smooth muscle and connective tissue whereas cerebral veins lack smooth muscles, suggesting that smooth muscle relaxation may participate in the fluoxetine-mediated vasodilatation. Blockage of Na+ channels by antidepressant drugs [10] may also underlie relaxation of brain vessel smooth muscles, thus promoting vasodilatation and increasing blood flow.
An important hallmark of ischaemic (and other) brain injury is a marked promiscuity of the BBB [39], which would make it permeable to serum albumin. Our current findings predict that once BBB malfunction would expose extracellular AChE within the brain tissue to albumin, fluoxetine should suppress its catalytic activity as well. This may explain the enhanced cholinergic signalling in the ischaemic brain [40]. Stroke patients show reduced serum AChE levels and those patients with minimally declined AChE are at additional risk of poor neurological outcome [19]. It is tempting to speculate that under BBB breakdown, SSRIs will act both as efficient vasodilators and as neuronal cholinergic stimulators, as they will also block brain AChE [12,19,41]. This may further predict added value to SSRIs in other diseases with loss of BBB function, such as neurodegenerative diseases (e.g. Alzheimer's disease) and traumatic brain injury.
Together, our observations suggest that fluoxetine induces vasodilatation by activating the muscarinic-NO pathway, tightening HSP90/eNOS potentiating cholinergic signalling thanks to and albumin/AChE interactions. This may both limit the risk of stroke and underlie the post-stroke beneficial effect of fluoxetine. Future animal and clinical studies would be required to clarify to what extent early post-stroke administration of SSRIs could maximize their beneficial effect as vasodilators to efficiently enhance blood flow to the ischaemic region.
Acknowledgments
The authors are grateful to Dr. Estelle R. Bennett, Jerusalem for excellent technical assistance and to Protalix Biotherapeutics, Carmiel for rhAChE. This work was supported by a Legacy Heritage Biomedical Science Partnership Program of the Israel Science Foundation (Grant No. 1799/10, to HS) and the German Science Foundation (DFG-SFB/TR3 C8 to AF and UH). KO received a pre-doctoral Meidan fellowship.
Author contributions
KO and KS performed the research; analysed the data and wrote the paper; NB contributed essential reagents or tools and analysed the data; UH, AF and HS designed the research study; contributed essential reagents or tools and wrote the paper.
Conflicts of interest
The authors confirm that there are no conflicts of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Dose response, time curve and cholinergic involvement of the fluoxetine-mediated vasodilation effect (A) Arteriolar diameter presented as percentage from control (averages ± S.D. for 5 points along an arteriole) following perfusion of 5–100 µM fluoxetine. Note the faster response under 100 µM. (B) Arteriolar diameter presented as percentage from control (averages ± S.D. for 5 points along an arteriole) following perfusion of 50 µM carbachol (right structure).
Atropine suppresses fluoxetine-mediated vasodilation selectively but does not prevent the bradykinin effect (A) Representative images of pial arterioles in the rat cerebral cortex. Shown are photographs after applying the noted compounds. Note that atropine blocks the effects of 500 µM fluoxetine, but not of 100 µM bradikinin. (B) Schematic presentation of our working hypothesis; atropine selectively antagonizes endothelial muscarinic receptors and fluoxetine, but not bradikinin-mediated vasodilation is suppressed. Activation of either of these receptors increases intracellular Ca2+ levels and facilitates the production of NO from l-arginine by eNOS while bradikinin induces vasodilatation via cyclooxygenase-1 (COX1) as well.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Roger VL, Go AS, Lloyd-Jones DM, et al. Heart disease and stroke statistics–2011 update: a report from the American Heart Association. Circulation. 2011;123:e18–209. doi: 10.1161/CIR.0b013e3182009701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shuaib A, Butcher K, Mohammad AA, et al. Collateral blood vessels in acute ischaemic stroke: a potential therapeutic target. Lancet Neurol. 2011;10:909–21. doi: 10.1016/S1474-4422(11)70195-8. [DOI] [PubMed] [Google Scholar]
- 3.Li WL, Cai HH, Wang B, et al. Chronic fluoxetine treatment improves ischaemia-induced spatial cognitive deficits through increasing hippocampal neurogenesis after stroke. J Neurosci Res. 2009;87:112–22. doi: 10.1002/jnr.21829. [DOI] [PubMed] [Google Scholar]
- 4.Chollet F, Tardy J, Albucher JF, et al. Fluoxetine for motor recovery after acute ischaemic stroke (FLAME): a randomised placebo-controlled trial. Lancet Neurol. 2011;10:123–30. doi: 10.1016/S1474-4422(10)70314-8. [DOI] [PubMed] [Google Scholar]
- 5.Narushima K, Paradiso S, Moser DJ, et al. Effect of antidepressant therapy on executive function after stroke. Br J Psychiatry. 2007;190:260–5. doi: 10.1192/bjp.bp.106.025064. [DOI] [PubMed] [Google Scholar]
- 6.Ungvari Z, Pacher P, Kecskemeti V, et al. Fluoxetine dilates isolated small cerebral arteries of rats and attenuates constrictions to serotonin, norepinephrine, and a voltage-dependent Ca(2+) channel opener. Stroke. 1999;30:1949–54. doi: 10.1161/01.str.30.9.1949. [DOI] [PubMed] [Google Scholar]
- 7.Frishman WH, Grewall P. Serotonin and the heart. Ann Med. 2000;32:195–209. doi: 10.3109/07853890008998827. [DOI] [PubMed] [Google Scholar]
- 8.Hashioka S, Klegeris A, Monji A, et al. Antidepressants inhibit interferon-gamma-induced microglial production of IL-6 and nitric oxide. Exp Neurol. 2007;206:33–42. doi: 10.1016/j.expneurol.2007.03.022. [DOI] [PubMed] [Google Scholar]
- 9.Matsumoto K, Puia G, Dong E, et al. GABA(A) receptor neurotransmission dysfunction in a mouse model of social isolation-induced stress: possible insights into a non-serotonergic mechanism of action of SSRIs in mood and anxiety disorders. Stress. 2007;10:3–12. doi: 10.1080/10253890701200997. [DOI] [PubMed] [Google Scholar]
- 10.Pancrazio JJ, Kamatchi GL, Roscoe AK, et al. Inhibition of neuronal Na+ channels by antidepressant drugs. J Pharmacol Exp Ther. 1998;284:208–14. [PubMed] [Google Scholar]
- 11.Maggi L, Palma E, Miledi R, et al. Effects of fluoxetine on wild and mutant neuronal alpha 7 nicotinic receptors. Mol Psychiatry. 1998;3:350–5. doi: 10.1038/sj.mp.4000392. [DOI] [PubMed] [Google Scholar]
- 12.Muller TC, Rocha JB, Morsch VM, et al. Antidepressants inhibit human acetylcholinesterase and butyrylcholinesterase activity. Biochim Biophys Acta. 2002;1587:92–8. doi: 10.1016/s0925-4439(02)00071-6. [DOI] [PubMed] [Google Scholar]
- 13.Beny JL, Nguyen MN, Marino M, et al. Muscarinic receptor knockout mice confirm involvement of M3 receptor in endothelium-dependent vasodilatation in mouse arteries. J Cardiovasc Pharmacol. 2008;51:505–12. doi: 10.1097/FJC.0b013e31816d5f2f. [DOI] [PubMed] [Google Scholar]
- 14.Wess J, Eglen RM, Gautam D. Muscarinic acetylcholine receptors: mutant mice provide new insights for drug development. Nat Rev Drug Discov. 2007;6:721–33. doi: 10.1038/nrd2379. [DOI] [PubMed] [Google Scholar]
- 15.Sessa WC. eNOS at a glance. J Cell Sci. 2004;117:2427–9. doi: 10.1242/jcs.01165. [DOI] [PubMed] [Google Scholar]
- 16.Watson BD, Dietrich WD, Busto R, et al. Induction of reproducible brain infarction by photochemically initiated thrombosis. Ann Neurol. 1985;17:497–504. doi: 10.1002/ana.410170513. [DOI] [PubMed] [Google Scholar]
- 17.Prager O, Chassidim Y, Klein C, et al. Dynamic in vivo imaging of cerebral blood flow and blood-brain barrier permeability. Neuroimage. 2010;49:337–44. doi: 10.1016/j.neuroimage.2009.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seiffert E, Dreier JP, Ivens S, et al. Lasting blood-brain barrier disruption induces epileptic focus in the rat somatosensory cortex. J Neurosci. 2004;24:7829–36. doi: 10.1523/JNEUROSCI.1751-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ben Assayag E, Shenhar-Tsarfaty S, Ofek K, et al. Serum cholinesterase activities distinguish between stroke patients and controls and predict 12-month mortality. Mol Med. 2010;16:278–86. doi: 10.2119/molmed.2010.00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fancy DA, Kodadek T. Chemistry for the analysis of protein-protein interactions: rapid and efficient cross-linking triggered by long wavelength light. Proc Natl Acad Sci USA. 1999;96:6020–4. doi: 10.1073/pnas.96.11.6020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gilboa-Geffen A, Lacoste PP, Soreq L, et al. The thymic theme of acetylcholinesterase splice variants in myasthenia gravis. Blood. 2007;109:4383–91. doi: 10.1182/blood-2006-07-033373. [DOI] [PubMed] [Google Scholar]
- 22.Koran LM, Cain JW, Dominguez RA, et al. Are fluoxetine plasma levels related to outcome in obsessive-compulsive disorder? Am J Psychiatry. 1996;153:1450–4. doi: 10.1176/ajp.153.11.1450. [DOI] [PubMed] [Google Scholar]
- 23.Petzold GC, Haack S, von Bohlen Und Halbach O, et al. Nitric oxide modulates spreading depolarization threshold in the human and rodent cortex. Stroke. 2008;39:1292–9. doi: 10.1161/STROKEAHA.107.500710. [DOI] [PubMed] [Google Scholar]
- 24.Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288:373–6. doi: 10.1038/288373a0. [DOI] [PubMed] [Google Scholar]
- 25.O'Brien FE, Dinan TG, Griffin BT, et al. Interactions between antidepressants and P-glycoprotein at the blood-brain barrier: clinical significance of in vitro and in vivo findings. Br J Pharmacol. 2012;165:289–312. doi: 10.1111/j.1476-5381.2011.01557.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Drouin A, Thorin-Trescases N, Hamel E, et al. Endothelial nitric oxide synthase activation leads to dilatory H2O2 production in mouse cerebral arteries. Cardiovasc Res. 2007;73:73–81. doi: 10.1016/j.cardiores.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 27.Fleming I, Busse R. Signal transduction of eNOS activation. Cardiovasc Res. 1999;43:532–41. doi: 10.1016/s0008-6363(99)00094-2. [DOI] [PubMed] [Google Scholar]
- 28.Garcia-Cardena G, Fan R, Shah V, et al. Dynamic activation of endothelial nitric oxide synthase by Hsp90. Nature. 1998;392:821–4. doi: 10.1038/33934. [DOI] [PubMed] [Google Scholar]
- 29.Brouet A, Sonveaux P, Dessy C, et al. Hsp90 and caveolin are key targets for the proangiogenic nitric oxide-mediated effects of statins. Circ Res. 2001;89:866–73. doi: 10.1161/hh2201.100319. [DOI] [PubMed] [Google Scholar]
- 30.Takahashi S, Mendelsohn ME. Synergistic activation of endothelial nitric-oxide synthase (eNOS) by HSP90 and Akt: calcium-independent eNOS activation involves formation of an HSP90-Akt-CaM-bound eNOS complex. J Biol Chem. 2003;278:30821–7. doi: 10.1074/jbc.M304471200. [DOI] [PubMed] [Google Scholar]
- 31.Muller W, Misgeld U, Heinemann U. Carbachol effects on hippocampal neurons in vitro: dependence on the rate of rise of carbachol tissue concentration. Exp Brain Res. 1988;72:287–98. doi: 10.1007/BF00250251. [DOI] [PubMed] [Google Scholar]
- 32.Nibuya M, Nestler EJ, Duman RS. Chronic antidepressant administration increases the expression of cAMP response element binding protein (CREB) in rat hippocampus. J Neurosci. 1996;16:2365–72. doi: 10.1523/JNEUROSCI.16-07-02365.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Badaut J, Moro V, Seylaz J, et al. Distribution of muscarinic receptors on the endothelium of cortical vessels in the rat brain. Brain Res. 1997;778:25–33. doi: 10.1016/s0006-8993(97)00999-2. [DOI] [PubMed] [Google Scholar]
- 34.Garcia-Colunga J, Awad JN, Miledi R. Blockage of muscle and neuronal nicotinic acetylcholine receptors by fluoxetine (Prozac) Proc Natl Acad Sci USA. 1997;94:2041–4. doi: 10.1073/pnas.94.5.2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shin TK, Kang MS, Lee HY, et al. Fluoxetine and sertraline attenuate postischaemic brain injury in mice. Korean J Physiol Pharmacol. 2009;13:257–63. doi: 10.4196/kjpp.2009.13.3.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Astrup J, Siesjo BK, Symon L. Thresholds in cerebral ischaemia – the ischaemic penumbra. Stroke. 1981;12:723–5. doi: 10.1161/01.str.12.6.723. [DOI] [PubMed] [Google Scholar]
- 37.Csermely P, Schnaider T, Soti C, et al. The 90-kDa molecular chaperone family: structure, function, and clinical applications. A comprehensive review. Pharmacol Ther. 1998;79:129–68. doi: 10.1016/s0163-7258(98)00013-8. [DOI] [PubMed] [Google Scholar]
- 38.Harald Breivik MJC. Sympathetic Neural Blockade of upper and lower Extremity. In: Cousins MichaelJ, PO B, Carr DanielB, Horlocker TereseT., editors. Cousins and Bridenbaugh's neural blockade in clinical anesthesia and pain medicine. 4th ed. Philadelphia: Lippincott Williams & Wilkins; 2008. pp. 484–886. [Google Scholar]
- 39.Stoll G, Kleinschnitz C, Meuth SG, et al. Transient widespread blood-brain barrier alterations after cerebral photothrombosis as revealed by gadofluorine M-enhanced magnetic resonance imaging. J Cereb Blood Flow Metab. 2009;29:331–41. doi: 10.1038/jcbfm.2008.129. [DOI] [PubMed] [Google Scholar]
- 40.Kulik T, Kusano Y, Aronhime S, et al. Regulation of cerebral vasculature in normal and ischaemic brain. Neuropharmacology. 2008;55:281–8. doi: 10.1016/j.neuropharm.2008.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sandoval KE, Witt KA. Blood-brain barrier tight junction permeability and ischaemic stroke. Neurobiol Dis. 2008;32:200–19. doi: 10.1016/j.nbd.2008.08.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Dose response, time curve and cholinergic involvement of the fluoxetine-mediated vasodilation effect (A) Arteriolar diameter presented as percentage from control (averages ± S.D. for 5 points along an arteriole) following perfusion of 5–100 µM fluoxetine. Note the faster response under 100 µM. (B) Arteriolar diameter presented as percentage from control (averages ± S.D. for 5 points along an arteriole) following perfusion of 50 µM carbachol (right structure).
Atropine suppresses fluoxetine-mediated vasodilation selectively but does not prevent the bradykinin effect (A) Representative images of pial arterioles in the rat cerebral cortex. Shown are photographs after applying the noted compounds. Note that atropine blocks the effects of 500 µM fluoxetine, but not of 100 µM bradikinin. (B) Schematic presentation of our working hypothesis; atropine selectively antagonizes endothelial muscarinic receptors and fluoxetine, but not bradikinin-mediated vasodilation is suppressed. Activation of either of these receptors increases intracellular Ca2+ levels and facilitates the production of NO from l-arginine by eNOS while bradikinin induces vasodilatation via cyclooxygenase-1 (COX1) as well.