

Abstract
Microparticles (MP) are generated during a vast number of biological processes such as inflammation, cell activation and apoptosis. Increasing evidence points towards an important role of MP as intercellular messengers of biological information. During atherogenesis, monocytes infiltrate the vascular wall and foster inflammation, accompanied by the release of monocytic MP (mono-MP). To date, only little is known about the biological function of mono-MP in the vascular wall. Here, we investigated the role of mono-MP during atherogenesis. Mono-MP were generated by starvation of THP-1 monocytes and isolated by ultracentrifugation. To investigate whether mono-MP influence atherogenesis, ApoE−/− mice were fed a high-fat, cholesterol-rich diet for 8 weeks and simultaneously treated with mono-MP or vehicle twice a week. Mice treated with mono-MP showed significantly increased monocyte and T-cell infiltration into the vessel wall, as assessed by Moma-2 and CD3 staining, and enhanced plaque formation, as assessed by oil-red-O staining. However, atherosclerotic plaque composition was not influenced by mono-MP application. In vitro, incubation of mono-MP with murine macrophages and endothelial cells resulted in the uptake of calcein-labelled mono-MP. Mono-MP uptake initiated the generation of intracellular reactive oxygen species. Murine macrophages pre-treated with mono-MP showed significantly enhanced expression of CCR2, migration to MCP-1 and increased release of pro-inflammatory interleukin-6. Co-incubation of mono-MP with endothelial cells resulted in significantly increased expression of ICAM-1, as assessed by RT-PCR and ELISA. Mono-MP act as paracrine messengers that intensify inflammation during atherogenesis by stimulating vascular-bound and inflammatory cells in their vicinity.
Keywords: monocytic microparticles atherosclerosis, vascular inflammation
Introduction
Atherosclerosis is a chronic inflammatory disease and represents the leading cause of heart failure and death in the industrialized world [1]. Onset and progression are closely connected with damage of the endothelium, subsequent vascular inflammation and accumulation of monocytes and macrophages. During these inflammatory processes, cell activation and apoptosis occur that are accompanied by the release of membrane-derived MP. MP carry surface molecules and cytoplasmic constituents of their parent cells and, therefore, have been considered epiphenomena and biomarkers in several cardiovascular disorders have been considered as such as atherosclerosis, hypertension and diabetes [2,3]. However, growing evidence suggests that MP also possess functional features as they act as small messenger molecules transferring biological information [2]. Autocrine and paracrine effects on inflammation and coagulation of MP have been described [4–7]. In the context of vascular biology, it has been shown that MP derived from endothelial cells (EMP) contribute to endothelial regeneration e.g. via the delivery of micro-RNA-126 that induces CXCL-12-dependent vascular protection [8]. In contrast, MP isolated from human atherosclerotic plaques mediate rather detrimental effects by promoting endothelial ICAM-1-dependent monocyte adhesion and transendothelial migration [9]. These studies point towards a differential role of MP during atherogenesis depending on their cellular origin. Like endothelial cells, monocytes shed monocytic MP (mono-MP) during activation and apoptosis as well. Interestingly, tobacco smoke induces the release of pro-coagulant MP from human monocytes, suggesting a direct association of a classical cardiovascular risk factor with the release of mono-MP [10]. On the cellular level, it has been shown that MP derived from THP-1 monocytes activate endothelial cells in an interleukin-1ß-dependent mechanism with subsequent up-regulation of adhesion molecules in vitro [11]. Furthermore, THP-1 mono-MP enhance nitrosative stress in endothelial cells [12] indicating pro-inflammatory effects on the endothelium as well. However, the molecular role and function of mono-MP during vascular inflammation in vivo and their effects on monocytic cells in vitro are largely unknown, although monocytes play a pivotal role during vascular inflammatory processes. Here, we have investigated the role of mono-MP during vascular inflammation in vivo and their influence on murine macrophages as well as endothelial cells in vitro.
Materials and methods
Mono-MP Generation
THP-1 monocytes were cultured under standard cell culture conditions (37°C, 5% CO2) in RPMI 1640 medium (Invitrogen, CA, USA) containing 10% FBS. After cells proliferated to confluency, they were subjected to starvation for 48 hrs to induce apoptosis. Supernatant was removed and centrifuged at 201g for 10 min. The supernatant was centrifuged again (18,000g, 45 min.) to obtain an adequate MP pellet. The pellet was dissolved in RPMI 1640 medium without supplements. To analyse the number of MP gained from cultured THP-1 monocytes, a sample was stained with Annexin V-FITC (BD Biosciences, NJ, USA) and 20 μl of sample, combined with 300 μl Annexin V binding buffer (BD Biosciences), was added to TruCount tubes (BD Biosciences). Annexin V positive mono-MP were counted using TruCount beads (BD Bioscience, San Jose, CA, USA). A monocytic origin was assured by detecting CD14 on MP by means of FACS analysis.
For FACS analysis, a linear scale was used for forward scatter signal (FSC) and side scatter signal (SSC) and a logarithmic scale for each fluorescent channel. The MP population was gated FSC versus SSC, after size calibration was performed with 0.9 μm beads and FSC (BioCytex, Marseille, France). (For an example of the flow cytometric dot plots see Fig. S1B). Upper and lower limits were adjusted using an unstained control before each measurement (Fig. S1A). The threshold used in the FSC channel was 30. For calculating the absolute mono-MP numbers, the Annexin V positive population and the beads were each gated and the following formula used: (number of events for Annexin V/number of events in TruCount bead region) × (number of TruCount beads per test/test volume). Control experiments were carried out with heat inactivated mono-MP (hi mono-MP). For inactivation, mono-MP were exposed to 95°C for 10 min. and subsequently to ultrasound for 5 min. MP as well as exosomes are also known as microvesicles. Differentiating between both molecules is essential because both vesicle types are membrane-shed particles, but possess different properties. To exclude contamination and assure that we did not generate exosomes, Western blot experiments were carried out. TSG-101 is a marker for exosomes and particularly inducible by endotoxin stimulation such as LPS [13]. Whereas microvesicles stemming from LPS-stimulated THP-1 cells were positive for TSG-101, microvesicles that were taken from starved THP-1 cells did not express this marker (data not shown). Therefore, we concluded that microvesicles used for our experiments negative for TSG-101 are indeed MP and were not contaminated with other sub-cellular components that occur during apoptosis.
Animals
The impact of mono-MP on atherosclerosis in vivo was analysed in apolipoprotein-E-deficient mice (ApoE−/−) (C57BL/6 genetic background; Charles River, Sulzfeld, Germany). Mice were housed in a 22°C room with a 12-hrs light/dark cycle and received water ad libitum. For the assessment of early diet-induced atherosclerosis, 10-week-old ApoE−/− mice were fed a high-fat, cholesterol-rich diet [21% fat, 19.5% casein and 1.25% cholesterol (Ssniff, Soest, Germany)]. Simultaneously, mice were treated with 1 × 106 mono-MP (n = 7) or vehicle (RPMI-1640 medium) (n = 5) by intravenous injection twice a week. After 8 weeks of treatment, mice were killed, blood was drawn and organs were immediately collected. Plasma cholesterol levels were analysed using gas chromatography-flame ionization detection as previously described [14]. Systolic blood pressure and heart rate were determined in conscious animals using a computerized tail-cuff system (BP-2000; Visitech System, Apex, NC, USA). After 3 days of habituation to the pre-warmed tail-cuff device, systolic blood pressure and heart rate were measured for 3 days. All experiments were performed in accordance with institutional guidelines and the German animal protection law.
Assessment of atherosclerotic plaque formation
Murine hearts and aortas were removed, immediately fixed in tissue tec (OCT embedding mediums; Miles Laboratories Inc., IL, USA), snap frozen and stored at –80°C. Cryosections of the aortic sinus were prepared using a Leica cryostat (Leica microsystems, Wetzlar, Germany) (9 μm) and transferred on slides. Oil-red-O staining was used to determine plaque formation. Samples were fixed with 3.7% formaldehyde for 1 hr, embedded in oil-red-O working solution (0.5%) (Sigma–Aldrich, St. Louis, MO, USA) for 15 min. and nuclear and cytoplasmic structures were counterstained with haematoxylin (Merck, Darmstadt, Germany). To quantify the extent of vascular atherosclerosis, images were recorded using a Zeiss microscope (Carl Zeiss, Jena, Germany) and macroscopic analysis was performed with axiovision software. Plaque formation is expressed as mean plaque cross-sectional area (mm2).
Assessment of leucocyte accumulation and plaque composition
To determine the accumulation of monocytes and macrophages in the atherosclerotic vessel wall, Moma-2 (anti-macrophage monoclonal antibody-2; Abcam, Cambridge, UK) staining was performed. First, sections were mounted in acetone for 45 min. and blocked with normal gout serum for 30 min. After incubation with Moma-2 for 1 hr, slides were carefully rinsed with 0.1 mol Tris (pH 6.8) and the second antibody (goat anti-rat IgG, AP-conjugated) (Sigma–Aldrich) was added for another hour. Sections were rinsed again and, for 2 min., incubated with AP reagent (Fast Red Tablets; Sigma–Aldrich). Nuclei and cytoplasm were counterstained with haematoxylin. The accumulation of monocytes and macrophages in the atherosclerotic plaque was quantified using a Zeiss microscope (Carl Zeiss). Axiovision software was used for macroscopic analysis. Monocyte and macrophage accumulation is expressed as Moma-2 positive area per total area of vessel wall in %. To assess T- cell infiltration into the atherosclerotic vessel wall, anti-CD3 staining (chemicals obtained from Dako, Hamburg, Germany) was carried out using an autostainer (Medac, Wedel, Germany). To analyse atherosclerotic plaque composition, collagen content was analysed using picrosirius red staining [Direct Red 80 (Sigma–Aldrich), picric acid solution 1,2% (Applichem, Darmstadt, Germany)] in accordance with standard protocols.
Circulating inflammatory cells
To investigate potential systemic immunomodulatory effects provoked by intravenous mono-MP application, inflammatory cells were analysed by means of flow cytometry. Wild-type mice were treated with 1 × 106 mono-MP (n = 5) or vehicle (n = 5) by intravenous injection for 3 days. On day 4, mice were killed and blood was drawn. A volume of 50 μl blood was incubated with Pharmlyse (BD Bioscience) for 10 min. at room temperature, washed with PBS and 1% FBS and the different fluorochrome-labelled antibodies were added for 30 min. at 4°C in the dark. CD3, CD4, CD8, CD11b and CD14 were analysed. All antibodies were obtained from BD Biosciences. Samples were diluted in 200 μl of PBS and 1% FBS before flow cytometric analysis was performed. Non-stained samples and mono-staining were used to discriminate true events from noise. Blood samples were measured using a BD FACS Calibur flow cytometer and BD CellQuestPro software (BD Biosciences).
Mono-MP staining and incorporation
Microparticles were isolated from starved THP-1 cells as described above and quantified by FACS analysis. Isolated mono-MP were stained with 1 μM calcein-AM (Sigma–Aldrich) in PBS and incubated for 40 min. at 37°C, as previously described [11]. Afterwards, mono-MP were washed and resuspended in PBS. A total of 2 × 104 murine bone marrow-derived macrophages were grown on a 96-well plate for 7 days as described below. Mono-MP and cells were finally incubated in DMEM (DMEM-F12; Invitrogen) in a 1:1-relation (mono-MP: cells) for 0.5–4 hrs at 37°C. Cells were washed once with PBS and fluorescence was measured using an ELISA-Reader (Tecan, Grödig/Salzburg, Austria) at 485 nm (excitation) and 530 nm (emission). Background fluorescence of cells alone was subtracted. A total of 1 × 105 murine bone marrow-derived macrophages were also grown on a 24-well plate and incubated with 1 × 105 calcein-AM-labelled mono-MP on day 7 for 4 hrs at 37°C. After washing for three times with PBS, cells were counterstained with the primary antibody CD68 (rat-anti-mouse; Abcam, Cambridge, UK), second antibody anti-rat Cy3 (Dianova, Hamburg, Germany) and 4,6-diamidino-2-phenylindole (DAPI; VectorLaboratories, Inc., Burlingame, USA). To confirm the uptake, mono-MP were also stained with PKH26 Red Fluorescent Cell Linker Kit (Sigma–Aldrich). Murine macrophages were seeded, cultivated and incubated with PKH-26-stained mono-MP as described above. Cells were now counterstained with CD68 (rat-anti-mouse; Abcam, Cambridge, UK), second antibody anti-rat Cy2 (Dianova) and DAPI (VectorLaboratories Inc.). In addition, uptake of mono-MP was confirmed in HUVEC. Therefore, 5 × 104 HUVEC were cultured in a 24 well-plate and incubated with calcein-labelled mono-MP for 4 hrs. HUVEC were counterstained with primary antibody CD31 (mouse-anti-human; Santa Cruz Biotechnology, CA, USA), second antibody anti-mouse Cy3 (Dianova) and 4,6-diamidino-2-phenylindole (DAPI; VectorLaboratories Inc.). Images were captured using a Zeiss Apotom microscope (Carl Zeiss).
Murine bone marrow-derived macrophages
Murine bone marrow-derived macrophages were generated through the use of monocyte-colony stimulating factor. Murine femurs and tibias were carefully prepared under sterile conditions, washed out with saline solution and cells were collected. Cells were grown in 6 cm dishes, 24-well plates or 96-well plates in a 5% CO2 atmosphere at 37°C in DMEM (DMEM-F12; Invitrogen) enriched with macrophage-colony stimulating factor (M-CSF) for 1 week. L-929 cells, a murine fibroblast cell line, were used as a source of M-CSF. L-929 cells were cultured in DMEM-F12 medium. L-929 conditioned medium was harvested at confluence; cells were removed by centrifugation and supernatant was stored at −80°C. For cultivating murine macrophages, 2/3 DMEM-F12 medium was mixed with 1/3 L-929 conditioned medium. Additional medium was added to cells on day 4 and differentiation had finished on day 6. FACS analysis was used to confirm the expression of macrophage markers, revealing that 97% of cells were positive for CD11b and Ly6C (BD Biosciences) (Fig. 3). For the experiments, cells were incubated with mono-MP or control for 24 hrs in a 1:1 relation (mono-MP: cells).
Fig. 3.
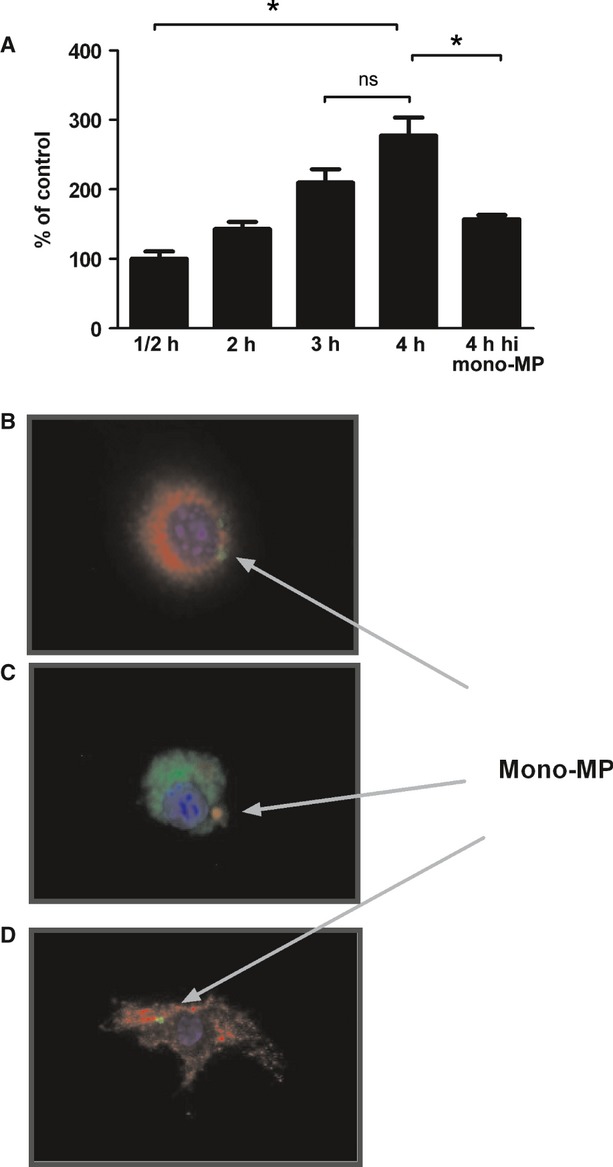
Mono-MP Uptake. (A) To investigate whether mono-MP were incorporated into target cells, cell culture experiments were performed. Calcein-labelled mono-MP were co-cultured with murine macrophages and engulfed in a time-dependent manner with a peak at 4 hrs of co-cultivation. Results were analysed using an ELISA reader at 485 nm (excitation) and 530 nm (emission). Heat denaturation of mono-MP was associated with a significant reduction in mono-MP uptake (4 hrs mono-MP: 277 ± 26% versus heat-inactivated mono-MP: 157 ± 6%, n = 4, *P < 0.05). (B) Calcein-labelled mono-MP uptake into macrophages. Calcein-labelled mono-MP are represented in green, CD68 and Cy3 staining of macrophages in red, the cell nucleus is coloured in blue by the use of Dapi. (C) PKH-26-labelled mono-MP uptake into macrophages. PKH-26 fluoresces in red, CD68 and Cy2-stained macrophages appear in green, cell nucleus in blue (Dapi). (D) Calcein-labelled mono-MP uptake into HUVEC. Calcein-labelled mono-MP appear in green, HUVEC counterstained with CD31 and Cy3 are demonstrated in red, the cell nucleus is represented in blue (Dapi).
Migration Assay
To test the migratory and chemotactic effect of monocytic MP on murine macrophages, 2 × 104 cells were loaded into the upper chamber of a modified Boyden Chamber with 8-μm pore sizes (VWR, Haasrode, Belgium). Therefore, 1 × 106 bone marrow-derived cells were seeded per 6 cm dish and grown as described above. On day 6, cells were stimulated with 1 × 106 mono-MP or vehicle for 24 hrs. The lower chambers were filled with either 20 × 104 mono-MP or heat-inactivated mono-MP, 10 ng/ml of MCP-1 (Sigma–Aldrich) in 750 μl medium or simply medium. After 2.5 hrs, migrated cells on the lower surface were fixed with 2% PFA and stained with DAPI (VectorLaboratories Inc.) and counted in at least four high-power fields by a blinded observer.
Real-time PCR
Quantitative RT-PCR experiments were performed to investigate the influence of mono-MP on the expression of CCR-2 in murine macrophages and ICAM-1 in HUVEC. Therefore, murine macrophages were cultivated for 6 days as described above, and HUVEC were cultured in 10 cm dishes using Endothelial Cell Growth medium MV (PromoCell GmbH, Heidelberg, Germany) with supplements. Both cell lines were stimulated for 24 hrs with mono-MP or heat-inactivated mono-MP (hi mono-MP) in a 10:1 relation (mono-MP: cells). RNA was extracted using peqGOLD RNAPure (Peqlab, Erlangen, Germany) according to the manufacturer's instructions. To analyse the CCR-2 expression in murine macrophages, RNA samples (1 μg) were reverse-transcribed to cDNA using the Reverse transcription System Kit (Promega, WI, USA), containing 1 μg RNA, deoxynucleotide triphosphates (dNTP), random primers and AMV Reverse Transcriptase. Primer sequences for CCR2 were: (forward 5′-AGAGGTCTCGGTTGGGTTGT-3′, reverse 5′-CACTGTCTTTGAGGCTTGTTGC-3′). RNA samples, containing 2 μg RNA, were reverse-transcribed to cDNA using Omniscript RT Kit (Qiagen, Hilden, Germany) to determine ICAM-1 levels. ICAM-1 (forward 5′-CGCAAGGTGACCGTGAATGT-3′, reverse 5′-CGTGGCTTGTGTGTTCGGTT-3′) and 18S (forward 5′-GTAACCCGTTGAACCCCATT-3′, reverse 5′-CCATCCAATCGGTAGTAGCG-3′) were obtained from Eurofins MWG Operon (Huntsville, AL, USA). 18S was used as housekeeping gene. In both cases, quantitative RT-PCR was performed in triplicates in a total volume of 20 μl containing SYBR Green Master Mix (Applied Biosystems, Carlsbad, CA, USA), DNAse/RNAse free water (Invitrogen), 1 μl of cDNA and 0.2 μl of each Primer. Reaction was measured using 7500 Fast Real-Time PCR-System and 7500 Fast System SDS Software (Applied Biosystems).
Apoptosis
Caspase activation was analysed using caspase-3 assay. In brief, 2 × 105 murine bone marrow-derived macrophages were cultured in 24-well plates and stimulated with 2 × 105 mono-MP or vehicle for 24 hrs on day 6 of differentiation. Next, to induce apoptosis, the quinoline alkaloid Camptothecin (CPT) was added for 4 hrs (1 μM, 5 μM) that inhibits topoisomerase I and thereby acts cytotoxically. Afterwards, cells were lysed in 150 μl of CHAPS lysis buffer (10 mM HEPES, pH 7.4, 42 mM KCl, 5 mM MgCl2, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM PMSF, 1 mM DTT, 0.5% CHAPS, Roche Complete Mini protease inhibitors according to the manufacturer′s instructions) on ice. A volume of 150 μl reaction buffer (25 mM HEPES, pH 7.5, 1 mM EDTA, 0.1% CHAPS, 10% sucrose, 3 mM DTT) supplemented with 10 μM of the specific fluorigenic caspase substrate Ac-DEVD-AMC (Bachem, Torrance, CA, USA) was added to 50 μl of lysate and stored for 2 hrs at room temperature in the dark. The accumulation of fluorescent AMC was detected using a fluorescence 96-well plate reader at 380 nm (excitation) and 465 nm (emission). Arbitrary fluorescence units were normalized for incubation time and protein content, determined by Lowry protein assays (Biorad).
Proliferation
Proliferation of murine macrophages was assessed using BrdU-FACS protocol. For this purpose, murine bone marrow-derived macrophages were isolated and cultivated as described above. A total of 2 × 106 cells were seeded in each 6 cm dish. On day 5, cells were starved in DMEM (DMEM-F12; Invitrogen) with 1% FBS and 5% L-929 conditioned medium for 12 hrs. Afterwards, macrophages were incubated with DMEM-F12 with 10% FBS and 2 μl Bromodeoxyuridine (BrdU; BD Bioscience) per millilitre medium and stimulated with mono-MP or heat-inactivated mono-MP in a 1:1 relation for 24 hrs. Macrophages were then removed from dishes using Cellstripper non-enzymatic cell dissociation solution (Sigma–Aldrich) and centrifuged. Pellets were resuspended in 500 μl PBS and fixed with 1 ml 80% EtOH and 20% 50 μM Glycine (pH = 2). After 18 hrs incubation at −20°C, cells were centrifuged and washed twice with washing buffer (1× PBS with 10% FBS). Macrophages were denatured with 4N HCl, washed and incubated with FITC mouse anti-BrdU (BD Bioscience) for 2 hrs. After two more washing steps, Propidium iodide (Sigma–Aldrich) and RNAse A (Roche, Basel, Switzerland) were added for 30 min. and samples were measured using a BD FACSCalibur flow cytometer and BD CellQuestPro (BD Biosciences).
ELISA
ELISA experiments were carried out to determine IL-6 release of murine macrophages and soluble ICAM-1 levels in HUVEC supernatant. A total of 1 × 105 macrophages were seeded in each well of a 24-well plate, as described above. After 6 days, medium was changed and cells were stimulated with 1 × 105 mono-MP or vehicle in 500 μl DMEM/F12 medium (Invitrogen) for 24 hrs at 37°C. Second, 5 × 104 HUVEC were cultured in a 24-well plate and incubated with mono-MP or heat-inactivated mono-MP for 3 days, each day in a 1:1 relation (mono-MP: cells). Samples or standards (diluted as noted) were added to a 96-well plate (IL-6-ELISA; eBioscience, San Diego, CA, USA; sICAM-1 ELISA; Bender MedSystems, Burlingame, CA, USA), coated with capture antibody in accordance with the manufacturer's instructions and incubated for 2 hrs at room temperature. To analyse IL-6 release, wells were washed and detection antibody and Avidin-HRP were added. Finally, substrate solution was filled in each well for 10 min. Reaction was stopped using stop solution. Absorbance of both plates was read in a fluorescence 96-well plate reader using 450 nm as wavelength. IL-6 and sICAM-1 levels were determined by means of calculated standard curves.
Intracellular reactive oxygen species
A total of 1 × 106 murine macrophages were grown on 6-well plates and stimulated with 1 × 106 mono-MP or vehicle on day 7 for 4 hrs at 37°C. Intracellular ROS in murine macrophages were analysed by means of C2′,7′-dichlorodihydrofluorescin-diacetate (H2DCFDA 10 μmol/l; Invitrogen) using confocal laser-scanning microscopy techniques in accordance with standard protocols. In brief, dishes of sub-confluent cells, stimulated with mono-MP or vehicle, were washed and incubated in the dark for 30 min. in the presence of 10 mM DCF-diacetate. Culture dishes were transferred to a Zeiss Axiovert 200 M microscope (Carl Zeiss) and reactive oxygen species generation was detected as a consequence of the oxidation of dichlorodihydrofluorescein (excitation, 488 nm; emission long-pass LP515-nm filter set). Images were collected by single rapid scans and identical parameters, such as contrast and brightness, for all samples. The relative fluorescence intensity is an average of values of all experiments.
Statistical analysis
Data are presented as mean ± standard error of the mean (SEM). Normal distribution was assessed using Kolmogorov–Smirnov with Dallal–Wilkinson–Lillie for P-value and D'Agostino and Pearson omnibus normality testing. For pairwise comparison, unpaired Students’ t-test was used and for multiple comparison anova with a Tukey post hoc analysis was applied. Statistical significance was assumed when a null hypothesis could be rejected at P < 0.05.
Results
Blood pressure, heart rate, cholesterol level
Analysis of blood pressure, heart rate and cholesterol levels of ApoE−/− mice were carried out after 8 weeks of a high-fat, cholesterol-rich diet. No differences were observed between both groups (Fig. 1).
Fig. 1.
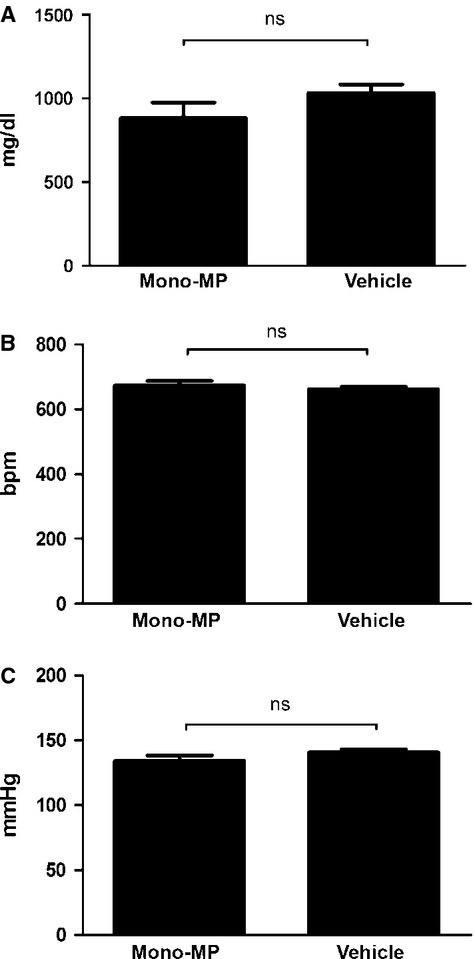
Blood pressure, heart rate, total cholesterol. Blood pressure, heart rate and cholesterol levels were determined after 8 weeks of a high-fat, cholesterol-rich diet. No differences were observed between both groups. (A) Cholesterol [mono-MP: 882.8 ± 92.42 mg/dl (n = 7) versus vehicle: 1032 ± 50.97 mg/dl (n = 5), P > 0.05] (B) Heart rate [mono-MP: 673.8 ± 13.54 bpm (n = 7) versus vehicle: 662.4 ± 7.3 bpm (n = 5), P > 0.05] (C) Blood pressure [mono-MP: 134.2 ± 4.1 mmHg (n = 7) versus vehicle: 140.7 ± 2.4 mmHg (n = 5), P > 0.05].
Mono-MP treatment aggravates atherosclerotic plaque formation
After 8 weeks of a high-fat, cholesterol-rich diet, immunohistochemical stainings were carried out to assess atherosclerotic plaque burden. Interestingly, oil-red-O staining revealed significantly enhanced plaque formation in mice that received mono-MP compared with mice that received vehicle (0.29 ± 0.03 mm2 versus 0.19 ± 0.03 mm2, P < 0.05) (Fig. 2A).
Fig. 2.
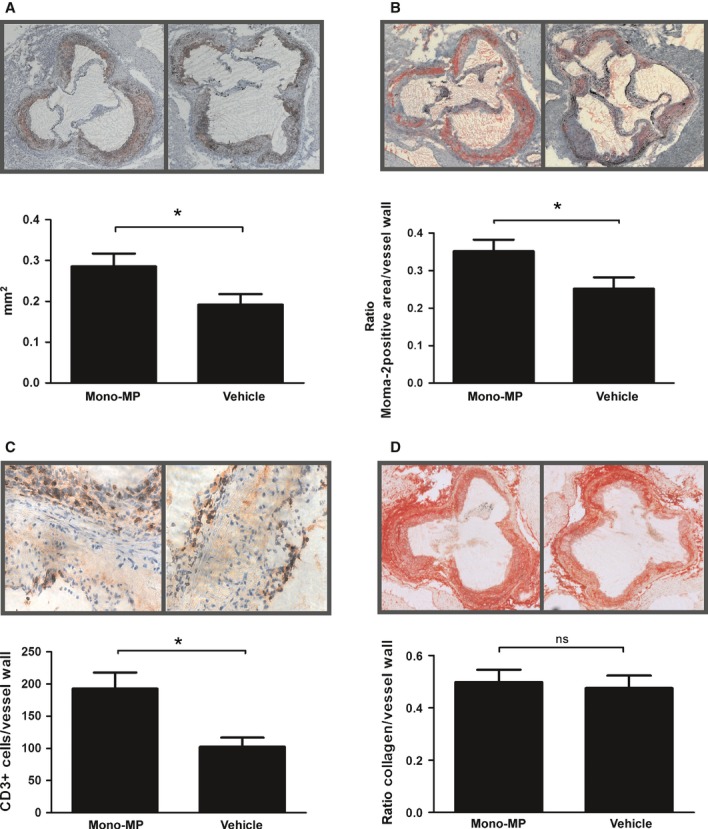
Atherosclerotic lesion formation and leucocyte infiltration. After 8 weeks of a high-fat, cholesterol-rich diet atherosclerotic burden was assessed in the aortic sinus by means of oil-red-O staining. Atherosclerotic lesion formation was increased in ApoE−/− mice treated with 1 × 106 mono-MP twice a week (n = 7) compared with ApoE−/− mice treated with vehicle (n = 5) (0.29 ± 0.03 mm2 versus 0.19 ± 0.03 mm2, * indicates P < 0.05) (A). Monocyte/macrophage accumulation in the vascular wall was assessed by means of Moma-2 staining. ApoE−/− mice treated with mono-MP showed significantly increased vascular accumulation of macrophages compared with control (0.35 ± 0.03 versus 0.25 ± 0.03, ratio Moma-2 positive area/vessel wall, * denotes P < 0.05) (B). The accumulation of T cells was determined using anti-CD3 staining. Mono-MP treatment significantly increased CD3+ T-cell accumulation (193 ± 25 versus 103 ± 14 CD3+ cells/vessel wall, *P < 0.05). T cells are depicted as little red spots (C). Mono-MP application did not influence plaque composition such as collagen content, as assessed by picrosirius red staining (0.49 ± 0.05 versus 0.48 ± 0.05, ratio collagen/vessel wall, ns indicates P > 0.05) (D).
Mono-MP treatment fosters accumulation of inflammatory cells in the vascular wall, but does not modulate plaque collagen content
Vascular accumulation of monocytes and macrophages was assessed using Moma-2 staining. Mono-MP treatment significantly increased the accumulation of monocytes/macrophages in the vascular wall of ApoE−/− mice compared with control (0.35 ± 0.03 versus 0.25 ± 0.03, ratio Moma-2 positive area/vessel wall, P < 0.05) (Fig. 2B). In addition, we have assessed the accumulation of T cells using anti-CD3 staining (Fig. 2C). Mono-MP treatment significantly increased CD3+ T-cell accumulation as well (193 ± 25 versus 103 ± 14 CD3+ cells/vessel wall, P < 0.05). Mono-MP application did not influence plaque composition such as collagen content, as assessed by picrosirius red staining (0.49 ± 0.05 versus 0.48 ± 0.05, ratio collagen/vessel wall, P > 0.05) (Fig. 2D).
Mono-MP are incorporated into target cells in vitro
To investigate whether mono-MP were incorporated into target cells, calcein-labelled mono-MP were co-cultured with murine macrophages. Calcein-labelled mono-MP were incorporated in a time-dependent manner with a peak at 4 hrs of co-cultivation. Heat-denaturation of mono-MP significantly diminished the uptake into murine macrophages (4 hrs mono-MP: 277 ± 26% versus heat-inactivated mono-MP: 157 ± 6%, n = 4, P < 0.05) (Fig. 3A). Uptake into murine macrophages was visualized using calcein-labelling (Fig. 3B) and PKH-26-labelling (Fig. 3C). Calcein-labelled mono-MP were incorporated into endothelial cells as well (Fig. 3D).
Mono-MP increase migration of murine macrophages to MCP-1 by upregulating CCR2
As we detected enhanced accumulation of inflammatory cells in the vascular wall of mice treated with mono-MP, we analysed the underlying cellular mechanisms in vitro. Murine macrophages were generated as described above and modified Boyden chamber experiments were carried out. First, we determined whether mono-MP possess chemoattractant functions. Either mono-MP or heat-inactivated mono-MP were filled into the lower compartment of the chambers in a 10:1 relation (2 × 105 mono-MP in the lower compartment and 2 × 104 macrophages in the upper compartment). Murine macrophages pre-treated with mono-MP or inactivated mono-MP in a 1:1-relation (1 × 106 mono-MP/heat inactivated mono-MP: 1 × 106 cells) for 24 hrs did not reveal an altered migratory behaviour to mono-MP (mono-MP to mono-MP: 111.2 ± 3.3% versus mono-MP to heat inactivated mono-MP: 108.8 ± 5.8% versus mono-MP to vehicle (control): 100.0 ± 3.2%, n = 5, P > 0.05) (Fig. 4A). Next, we assessed whether mono-MP stimulation influences migration of murine macrophages to well-described chemoattractant molecules such as MCP-1. Mono-MP treatment of murine macrophages for 24 hrs (1 × 106 mono-MP: 1 × 106 cells) significantly enhanced migration to MCP-1 (10 ng/ml, 2 × 104 macrophages/chamber), whereas heat inactivation of mono-MP molecules or vehicle treatment markedly attenuated this effect (mono-MP: 156 ± 7% versus hi mono-MP: 115 ± 5%, n = 5, P < 0.05) (Fig. 4B). As mono-MP pre-treatment increased migration of macrophages to MCP-1, CCR2 (receptor for MCP-1) levels were analysed using quantitative RT-PCR. Macrophages were stimulated on day 6 for 24 hrs with monocytic MP or heat-inactivated mono-MP. Mono-MP significantly enhanced the expression of CCR2 on murine macrophages, compared with control (macrophages + mono-MP: 293.5 ± 50.21% versus macrophages + hi mono-MP: 106.5 ± 49.35%, n = 14, P < 0.05) (Fig. 4C).
Fig. 4.
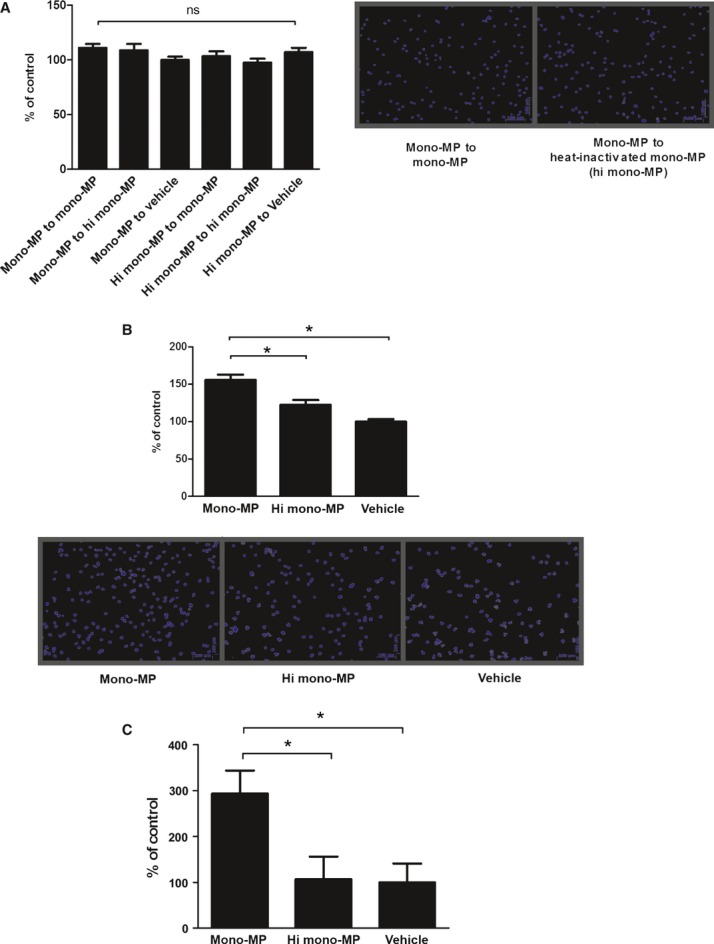
Migration of macrophages and CCR2. Chemotactic and migratory effects of mono-MP on murine macrophages were measured via modified Boyden chamber experiments. Murine macrophages pre-treated with mono-MP or control were allowed to migrate to mono-MP or control (A). No differences were detected between both groups [mono-MP to mono-MP: 111.2 ± 3.3%; mono-MP to heat-inactivated mono-MP: 108.8 ± 5.8%; mono-MP to vehicle (control): 100.0 ± 3.2%, n = 5, P > 0.05]. Next, migration to MCP-1 of murine macrophages pre-treated with mono-MP or control was determined (B). Migration of murine macrophages to MCP-1 was significantly enhanced after 24 hrs co-incubation with mono-MP compared with control (mono-MP: 156 ± 7% versus hi mono-MP: 115 ± 5%, n = 5, * denotes P < 0.05). CCR2 levels were analysed using quantitative RT-PCR. Macrophages, stimulated with mono-MP for 24 hrs, showed significantly increased CCR2 levels, compared with control (macrophages + mono-MP: 293.5 ± 50.21% versus macrophages+hi mono-MP: 106.5 ± 49.35%, n = 14, *P < 0.05) (C).
Mono-MP do not influence apoptosis or proliferation of murine macrophages
To determine whether the enhanced accumulation of inflammatory cells in the atherosclerotic vessel wall is rather because of a modified apoptotic or proliferatory behaviour than because of an intensified chemoattractance, apoptosis and proliferation of murine macrophages treated with mono-MP were assessed in vitro. To analyse apoptosis, 2 × 105 murine bone marrow-derived macrophages were cultured in 24-well plates and stimulated with 2 × 105 mono-MP or vehicle for 24 hrs on day 6 (1:1-relation, mono-MP:cells). Caspase-3 assay experiments were carried out as described above. Mono-MP application did not evoke significant apoptosis of murine macrophages after stimulation. Apoptosis of monocytes induced by either 1μmol or 5μmol camptothecin was not influenced by concomitant mono-MP stimulation as well (mono-MP: 1484 ± 183 versus heat-inactivated mono-MP: 1346 ± 127, DEVD-Cleavage in AU, n = 4, P > 0.05) (Fig. 5A). Beyond that, proliferation of murine macrophages after treatment with mono-MP was assessed using BrdU-FACS analysis. Macrophages were incubated with BrdU and either mono-MP or heat-inactivated mono-MP for 24 hrs on day 6 of differentiation (1:1-relation, mono-MP: cells). Cells were collected and proliferation was assessed by means of flow cytometry. Mono-MP did not influence proliferation of murine macrophages (BrdU + mono-MP: 97.63 ± 21.76% versus BrdU + heat-inactivated mono-MP: 74.65 ± 7.29%, n = 3, P > 0.05) (Fig. 5B).
Fig. 5.
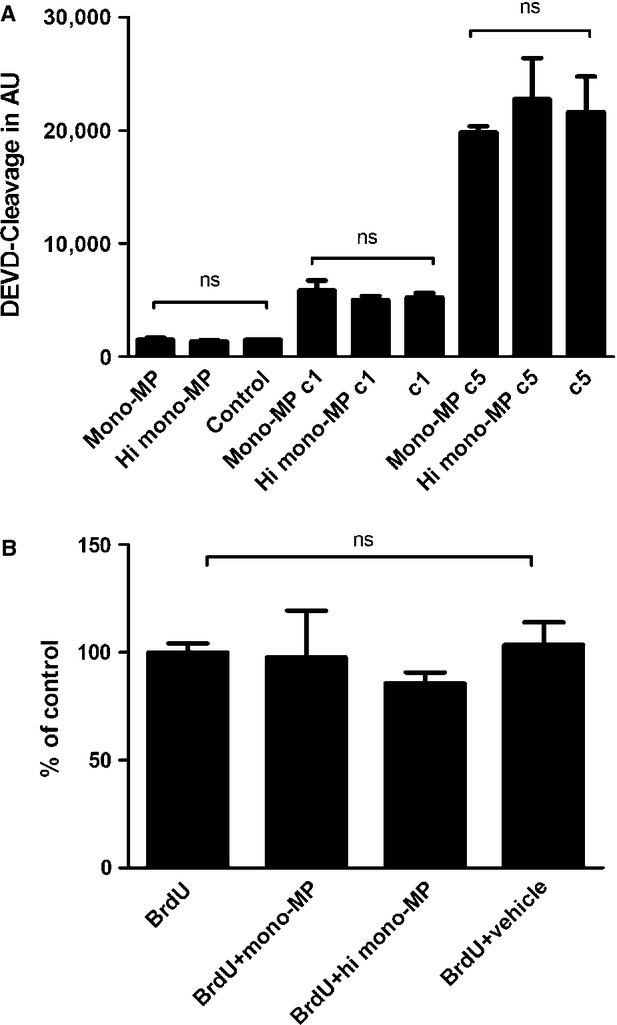
Apoptosis and proliferation of murine macrophages. Apoptotic effects of mono-MP on murine macrophages were assessed and analysed using a caspase-3 assay. Murine bone marrow-derived macrophages were cultured and stimulated with mono-MP or vehicle for 24 hrs on day 6 (1:1-relation mono-MP: cells). Mono-MP application did not influence apoptosis of target cells. Apoptosis of macrophages induced by either 1 μmol or 5 μmol camptothecin was not further influenced by mono-MP pre-stimulation as well (mono-MP: 1484 ± 183 versus heat inactivated mono-MP: 1346 ± 127, DEVD-Cleavage in AU, n = 4, P > 0.05) (A). The influence of mono-MP on the proliferation of murine macrophages was analysed using BrdU-FACS experiments. Macrophages were incubated with BrdU on day 6 of differentiation and concomitant stimulated with mono-MP or heat-inactivated mono-MP (1:1-relation mono-MP: cells). Mono-MP stimulation did not influence proliferation of murine macrophages: BrdU (control): 100.0 ± 4.2%; BrdU + mono-MP: 97.63 ± 21.76%; BrdU + heat-inactivated mono-MP: 74.65 ± 7.29%; BrdU + vehicle: 103.5 ± 10.42%, n = 3, P > 0.05 (B).
Mono-MP induce the release of IL-6 in vitro
To assess general pro-inflammatory responses provoked by mono-MP, IL-6 liberation of murine macrophages was determined after mono-MP application. A total of 1 × 105 murine macrophages per 24-well were stimulated for 24 hrs with 1 × 105 mono-MP or vehicle on day 6 of differentiation. Mono-MP activation significantly increased the release of IL-6 compared with control, as assessed by ELISA (mono-MP: 238.2 ± 17.64% versus vehicle: 100.0 ± 3.7%, n = 3, P < 0.05) (Fig. 6).
Fig. 6.
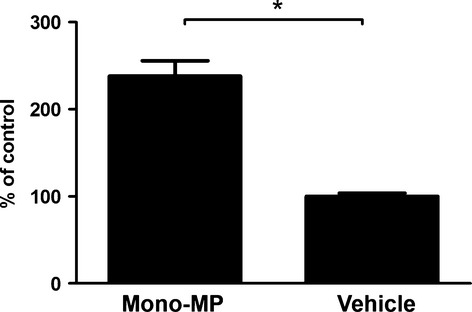
IL-6 release of murine macrophages. Mono-MP treatment of murine macrophages for 24 hrs induced a significantly increased liberation of IL-6 compared with control, as assessed by ELISA [mono-MP: 238.2 ± 17.64% versus vehicle (control): 100.0 ± 3.7%, n = 3, *P < 0.05].
Mono-MP induce generation of intracellular reactive oxygen species
Enhanced levels of reactive oxygen species occur during atherogenesis and foster progression of vascular inflammation. Therefore, we have assessed whether mono-MP influence oxidative stress in murine macrophages. A total of 1 × 106 murine bone marrow-derived macrophages per 6 cm dish were pre-treated with 1 × 106 mono-MP or vehicle, on day 7 of differentiation, for 4 hrs at 37°C and intracellular ROS generation was determined by means of C2′,7′-dichlorodihydrofluorescin-diacetate. Mono-MP application markedly increased oxidative stress in murine macrophages compared with control (mono-MP: 120.2 ± 4.9% versus heat-inactivated mono-MP: 104.6 ± 3.9%, n = 4, P < 0.05) (Fig. 7).
Fig. 7.
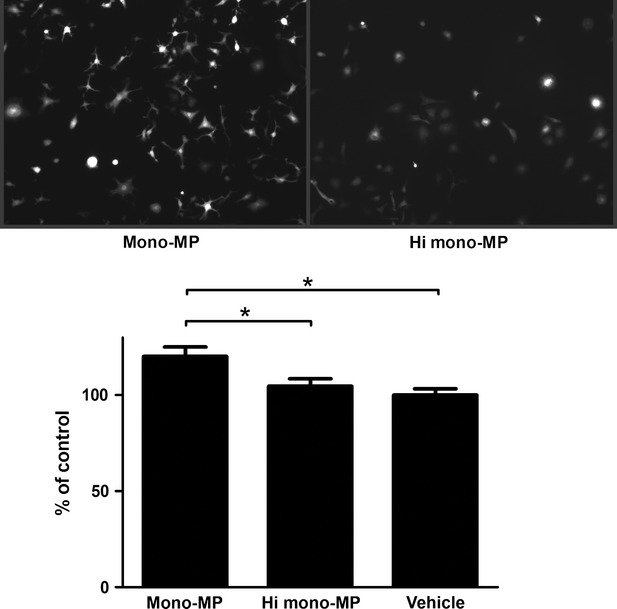
Generation of reactive oxygen species in murine macrophages.To assess the influence of mono-MP on the generation of intracellular oxidative stress in macrophages, C2′,7′-dichlorodihydrofluorescin-diacetate experiments were performed. Murine bone marrow-derived macrophages were stimulated with mono-MP or vehicle for 4 hrs on day 7 of differentiation. Cells pre-treated with mono-MP showed significantly increased intracellular ROS generation compared with control (mono-MP: 120.2 ± 4.9% versus heat inactivated mono-MP: 104.6 ± 3.9%, n = 4, * denotes P < 0.05).
Mono-MP increase expression of ICAM-1
To explore further underlying effects that can explain enhanced accumulation of macrophages in the vascular wall, the expression of endothelial ICAM-1 was assessed after mono-MP stimulation. HUVEC were incubated with mono-MP or heat-inactivated mono-MP for 24 hrs in a 10:1 relation (mono-MP: cells). RNA was extracted, reverse-transcribed into cDNA and quantitative RT-PCR experiments were performed. Mono-MP stimulation significantly increased ICAM-1 levels in HUVEC (HUVEC+ mono-MP: 313.5 ± 60.14% versus HUVEC+ heat-inactivated mono-MP: 130.8 ± 23.60%, n = 4, P < 0.05) (Fig. 8A).
Fig. 8.
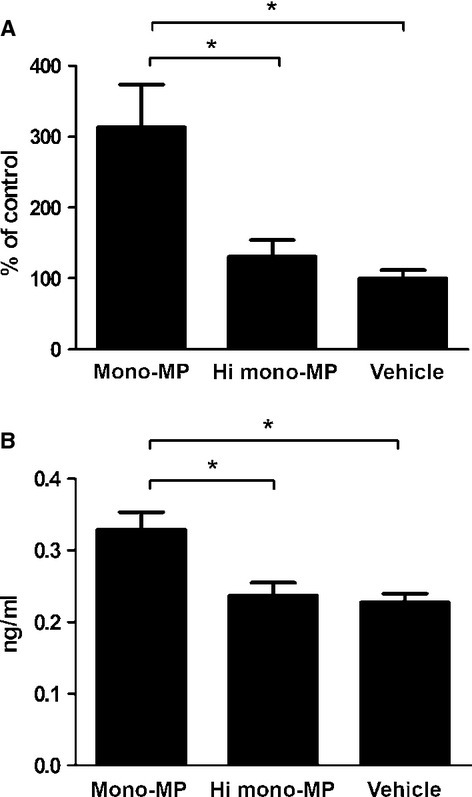
Mono-MP up-regulate ICAM-1. HUVEC pre-treated with mono-MP for 24 hrs showed significantly enhanced ICAM-1 level compared with control in quantitative RT-PCR (HUVEC+mono-MP: 313.5 ± 60.14% versus HUVEC+ heat-inactivated mono-MP: 130.8 ± 23.60%, n = 4, P < 0.05) (A). ELISA experiments confirmed the upregulation of ICAM-1, measuring soluble ICAM-1 in HUVEC supernatant after stimulation with mono-MP or control. ICAM-1 generation was significantly increased after mono-MP stimulation compared with control (mono-MP: 0.33 ± 0.024 ng/ml versus hi mono-MP: 0.24 ± 0.018 ng/ml versus vehicle: 0.32 ± 0.10 ng/ml, n = 4, * denotes P < 0.05) (B).
In addition, soluble ICAM-1 levels were analysed via ELISA. HUVEC were seeded in 24-well plates, stimulated with mono-MP or heat-inactivated mono-MP in a 1:1 relation for 3 days. Generation of sICAM-1 was significantly increased after mono-MP stimulation compared with control (mono-MP: 0.33 ± 0.024 ng/ml versus hi mono-MP: 0.24 ± 0.018 ng/ml, n = 4, P < 0.05) (Fig. 8B).
Circulating inflammatory cells
To investigate whether the increased vascular inflammation provoked by mono-MP treatment occurred because of an enhanced liberation of inflammatory cells into the circulation, additional flowcytometric experiments were performed. Wild-type mice were treated with 1 × 106 mono-MP or an equal amount of vehicle (RPMI 1640 medium without supplements, in which mono-MP were solved) for 3 days. Mono-MP treatment did not alter the level of circulating inflammatory cells such as CD3, CD4, CD8, CD11b and CD14 compared with control (Fig. 2).
Discussion
During atherogenesis, monocytes invade the atherosclerotic vessel wall and provoke and propagate vascular inflammation. These inflammatory processes are accompanied by the release of MP stemming from different cells including monocytic cells [15]. In this study, we analysed effects of monocytic microparticles (mono-MP) on vascular inflammation, demonstrating for the first time biological relevance in vivo. Mono-MP treatment resulted in enhanced atherosclerotic plaque formation and increased macrophage accumulation in the vessel wall of ApoE−/− mice. In vitro, mono-MP treatment provoked several pleiotropic pro-inflammatory effects. These results suggest a relevant interaction between mono-MP and inflammatory cells as well as vascular-bound cells during atherogenesis. To date, research mainly focussed on the role of endothelial microparticles (EMP) in vascular biology. In this context, it has been shown that endothelial apoptotic bodies induce proliferation and differentiation of endothelial progenitor cells [16] and act as activators of angiogenesis [17]. Therefore, EMP are considered a protective and replenishing element by supporting tissue repair, regeneration, anti-inflammation and vascular protection [3]. Possible mechanisms for message transduction are e.g., the transport of chemokine receptors, mRNA and micro-RNA [7,8,16,17].
In contrast, and in accordance with our findings, the biological function of monocyte-/macrophage-derived MP is rather destructive by mediating pro-inflammatory and pro-apoptotic effects [18–21]. Mono-MP disrupt endothelial cell integrity, provoke membrane blebbing and increase endothelial thrombogenicity [18]. Mono-MP induce the expression of von-Willebrand factor and tissue factor on endothelial cells [19]. Furthermore, mono-MP induce synthesis of pro-inflammatory mediators in lung epithelial cells [21] and enhance nitrosative stress in human endothelial cells [12]. These studies emphasize that vascular cells, particularly endothelial cells, play an essential role in mediating inflammatory effects of monocytic and macrophagic MP. However, to date, it has remained unclear to what extent these inflammatory MP influence atherogenesis in vivo. To directly address this question, we have examined the role of mono-MP in chronic vascular inflammation. Mono-MP application during 8 weeks of a high-fat, cholesterol-rich diet accelerated atherosclerotic plaque formation and macrophage accumulation in the vessel wall, demonstrating, for the first time, important pro-inflammatory in vivo-effects as well. Enhanced expression of endothelial ICAM-1 after mono-MP stimulation in vitro might represent one important finding that can explain the increased vascular inflammation in mice treated with mono-MP. However, we have also observed effects in murine macrophages induced by mono-MP: While mono-MP alone did not act as chemoattractant for macrophages, co-incubation of macrophages with mono-MP resulted in an enhanced migration to MCP-1 by modulating the expression of CCR2. Therefore, the release of MP from monocytes during inflammatory processes might additionally augment and amplify activity of inflammatory cells at the place of inflammation and lead to a vicious circle. As mono-MP impact on the migratory behaviour of murine macrophages in vitro, it is conceivable that apoptotic macrophages maintain and spread inflammation in their vascular environment. However, the enhanced accumulation of monocytes in atherosclerotic plaques is rather because of an increased migration and attraction than because of a diminished apoptosis or enhanced proliferation of these cells, as we did not detect modified apoptosis or proliferation of macrophages after treatment with mono-MP in vitro. It has to be mentioned that we used vehicle instead of heat-inactivated mono-MP for our in vivo experiments as it is extremely difficult to generate sufficient amounts of MP. This can be a limiting factor of our study. However, in vitro studies indicated comparable biological effects of heat-inactivated MP and vehicle.
Taken together, the role of mono-MP might be oppositional to the role of EMP during atherosclerosis, underlining completely different functions of MP depending on their cellular origin.
In conclusion, we present evidence that mono-MP accelerate atherosclerosis by stimulating macrophages and endothelial cells in their vicinity. Mono-MP are pro-inflammatory molecules and aggravate vascular inflammation in vivo. The cross-talk between mono-MP and macrophages demonstrate a novel regulatory cardiovascular pathway and might be of interest for the understanding of atherosclerosis.
Acknowledgments
K.P. and H.S. provided outstanding technical assistance. This work was supported by Deutsche Forschungsgemeinschaft, Deutsche Gesellschaft für Kardiologie and by the European Vascular Genomics Network, a Network of Excellence granted by the European Commission.
Conflicts of interest
The authors declare that there are no conflicts of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article:
MMP quantification.
Circulating inflammatory cells.
Murine bone marrow-derived macrophages.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685–95. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 2.Hoyer FF, Nickenig G, Werner N. Microparticles—messengers of biological information. J Cell Mol Med. 2010;14:2250–6. doi: 10.1111/j.1582-4934.2010.01114.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dignat-George F, Boulanger CM. The many faces of endothelial microparticles. Arterioscler Thromb Vasc Biol. 2011;31:27–33. doi: 10.1161/ATVBAHA.110.218123. [DOI] [PubMed] [Google Scholar]
- 4.Horstman LL, Jy W, Jimenez JJ, et al. Endothelial microparticles as markers of endothelial dysfunction. Front Biosci. 2004;9:1118–35. doi: 10.2741/1270. [DOI] [PubMed] [Google Scholar]
- 5.Baj-Krzyworzeka M, Majka M, Pratico D, et al. Platelet-derived microparticles stimulate proliferation, survival, adhesion, and chemotaxis of hematopoietic cells. Exp Hematol. 2002;30:450–9. doi: 10.1016/s0301-472x(02)00791-9. [DOI] [PubMed] [Google Scholar]
- 6.Janowska-Wieczorek A, Majka M, Kijowski J, et al. Platelet-derived microparticles bind to hematopoietic stem/progenitor cells and enhance their engraftment. Blood. 2001;98:3143–9. doi: 10.1182/blood.v98.10.3143. [DOI] [PubMed] [Google Scholar]
- 7.Mack M, Kleinschmidt A, Brühl H, et al. Transfer of the chemokine receptor CCR5 between cells by membrane-derived microparticles: a mechanism for cellular human immunodeficiency virus 1 infection. Nat Med. 2000;6:769–75. doi: 10.1038/77498. [DOI] [PubMed] [Google Scholar]
- 8.Zernecke A, Bidzhekov K, Noels H, et al. Delivery of microRNA-126 by apoptotic bodies induces CXCL12-dependent vascular protection. Sci Signal. 2009;2:ra81. doi: 10.1126/scisignal.2000610. [DOI] [PubMed] [Google Scholar]
- 9.Rautou PE, Leroyer AS, Ramkhelawon B, et al. Microparticles from human atherosclerotic plaques promote endothelial ICAM-1-dependent monocyte adhesion and transendothelial migration. Circ Res. 2011;108:335–43. doi: 10.1161/CIRCRESAHA.110.237420. [DOI] [PubMed] [Google Scholar]
- 10.Li M, Yu D, Williams KJ, et al. Tobacco smoke induces the generation of procoagulant microvesicles from human monocytes/macrophages. Arterioscler Thromb Vasc Biol. 2010;30:1818–24. doi: 10.1161/ATVBAHA.110.209577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang JG, Williams JC, Davis BK, et al. Monocytic microparticles activate endothelial cells in an IL-1{beta}-dependent manner. Blood. 2011;118:2366–74. doi: 10.1182/blood-2011-01-330878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mastronardi ML, Mostefai HA, Soleti R, et al. Microparticles from apoptotic monocytes enhance nitrosative stress in human endothelial cells. Fundam Clin Pharmacol. 2011;25:653–60. doi: 10.1111/j.1472-8206.2010.00898.x. [DOI] [PubMed] [Google Scholar]
- 13.Aharon A, Tamari T, Brenner B. Monocyte-derived microparticles and exosomes induce procoagulant and apoptotic effects on endothelial cells. Thromb Haemost. 2008;100:878–85. doi: 10.1160/th07-11-0691. [DOI] [PubMed] [Google Scholar]
- 14.Teunissen CE, Mulder M, de Vente J, et al. Concentrations of different sterols in the striatum and serum of 3-nitropropionic acid-treated Wistar and Lewis rats. Neurochem Res. 2001;26:1237–44. doi: 10.1023/a:1013919407311. [DOI] [PubMed] [Google Scholar]
- 15.Martínez MC, Tesse A, Zobairi F, et al. Shed membrane microparticles from circulating and vascular cells in regulating vascular function. Am J Physiol Heart Circ Physiol. 2005;288:1004–9. doi: 10.1152/ajpheart.00842.2004. [DOI] [PubMed] [Google Scholar]
- 16.Hristov M, Erl W, Linder S, et al. Apoptotic bodies from endothelial cells enhance the number and initiate differentiation of human endothelial progenitor cells in vitro. Blood. 2004;104:2761–6. doi: 10.1182/blood-2003-10-3614. [DOI] [PubMed] [Google Scholar]
- 17.Deregibus MC, Cantaluppi V, Calogero R, et al. Endothelial progenitor cell derived microvesicles activate an angiogenic program in endothelial cells by a horizontal transfer of mRNA. Blood. 2007;110:2440–8. doi: 10.1182/blood-2007-03-078709. [DOI] [PubMed] [Google Scholar]
- 18.Piccin A, Murphy WG, Smith OP. Circulating microparticles: pathophysiology and clinical implications. Blood Rev. 2007;21:157–71. doi: 10.1016/j.blre.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 19.Essayagh S, Xuereb JM, Terrisse AD, et al. Microparticles from apoptotic monocytes induce transient platelet recruitment and tissue factor expression by cultured human vascular endothelial cells via a redox-sensitive mechanism. Thromb Haemost. 2007;98:831–7. [PubMed] [Google Scholar]
- 20.Mesri M, Altieri DC. Endothelial cell activation by leukocyte microparticles. J Immunol. 1998;161:4382–7. [PubMed] [Google Scholar]
- 21.Neri T, Armani C, Pegoli A, et al. Role of NF-kappaB and PPAR-gamma in lung inflammation induced by monocyte-derived microparticles. Eur Respir J. 2011;37:1494–502. doi: 10.1183/09031936.00023310. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
MMP quantification.
Circulating inflammatory cells.
Murine bone marrow-derived macrophages.