

Abstract
Understanding the mechanisms that direct mesenchymal stem cell (MSC) self-renewal fate decisions is a key to most tissue regenerative approaches. The aim of this study here was to investigate the mechanisms of action of platelet-derived growth factor receptor β (PDGFRβ) signalling on MSC proliferation and differentiation. MSC were cultured and stimulated with PDGF-BB together with inhibitors of second messenger pathways. Cell proliferation was assessed using ethynyl-2′-deoxyuridine and phosphorylation status of signalling molecules assessed by Western Blots. To assess differentiation potentials, cells were transferred to adipogenic or osteogenic media, and differentiation assessed by expression of differentiation association genes by qRT-PCR, and by long-term culture assays. Our results showed that distinct pathways with opposing actions were activated by PDGF. PI3K/Akt signalling was the main contributor to MSC proliferation in response to activation of PDGFRβ. We also demonstrate a negative feedback mechanism between PI3K/Akt and PDGFR-β expression. In addition, PI3K/Akt downstream signal cascades, mTOR and its associated proteins p70S6K and 4E-BP1 were involved. These pathways induced the expression of cyclin D1, cyclin D3 and CDK6 to promote cell cycle progression and MSC proliferation. In contrast, activation of Erk by PDGFRβ signalling potently inhibited the adipocytic differentiation of MSCs by blocking PPARγ and CEBPα expression. The data suggest that PDGFRβ-induced Akt and Erk pathways regulate opposing fate decisions of proliferation and differentiation to promote MSC self-renewal. Thus, activation of multiple intracellular cascades is required for successful and sustainable MSC self-renewal strategies.
Keywords: PDGFRβ, Akt, Erk, mesenchymal stem cell, cell cycle, self-renewal, differentiation
Introduction
Mesenchymal stem cells (MSC) are self-renewing cells with multipotent differentiation potentials. They usually reside in a niche with no tissue specific characteristics and remain quiescent until activated by appropriate signals to either self-renew to maintain the stem cell pool or differentiate to generate specialized cells. A precise regulatory mechanism should therefore be in place to control fate decisions of MSCs, in order to maintain the MSC population, prevent excessive proliferation and to induce specific lineage differentiation when required. Extensive studies have been carried out on a wide range of extracellular/intracellular mediators which may regulate commitment and differentiation of MSCs, including growth factors and hormones, such as Bone Morphogenetic Proteins, Wnt proteins, hedgehog proteins and G-protein coupled receptors [1–4]. These factors are thought to regulate the changes in the expression and activation of lineage specific transcription factors that act as master switches to drive the differentiation of uncommitted precursors down a specific lineage. On the other hand, cytokines and growth factors, such as fibroblast growth factor (FGF)-2, platelet-derived growth factor (PDGF)-BB and epidermal growth factor (EGF) have been suggested as regulators of self-renewal of MSCs and used for their in vitro expansion [5–9]. Although a number of studies have been conducted on signal pathways downstream of mitogenic inducers of proliferation or upstream to transcriptional regulators of differentiation, it is still unknown exactly how MSCs process similar signals elicited by different growth factors to either undergo proliferation or commit to differentiation. Such information could be gained by studying factors with dual actions on MSC fate decisions of proliferation versus differentiation. This knowledge may have a number of important applications of practical use in the future by regulating self-renewal capacity, such as managing diseases associated with insufficient MSC numbers or differentiation such as age related osteoporosis [10,11]. Other potential uses include prevention of cancer stem cell growth [12] and to enhance the ability of in vitro expansion of MSCs for use in tissue engineering and regenerative medicine [13].
Members of the PDGF signalling family have received considerable attention in recent years for their roles in MSC homeostasis, migration and recruitment [9,14–16]. The PDGF family of growth factors consists of four ligands (AA, BB, CC and DD) that regulate many physiological and pathophysiological conditions by interacting with two PDGF receptors (PDGFRα and PDGFRβ) [17]. PDGF binding leads to the autophosphorylation of the receptors on multiple tyrosine residues and subsequently activates several downstream cascades, such as mitogen-activated protein kinase (MAPK), extracellular signal-regulated kinase (Erk), the phosphatidylinositol 3-kinase (PI3K)/Akt, Janus-activated kinase (JAK) and signal transducers and activators of transcription (STAT) pathways [17,18]. Stimulation of these pathways by various stimuli has been implicated in various physiological functions including cell proliferation [19], differentiation [20–23] and migration [24], and in pathological conditions such as cancer and musculoskeletal diseases [25–27]. Although PDGFs have been implicated in the regulation of many aspects of MSC physiology, the underlying signal transduction pathways are poorly studied and remain largely unknown.
Here, we have investigated in detail the mechanisms involving PDGF-BB/PDGFRβ-induced MSC proliferation and differentiation. We demonstrate that MEK/Erk, PI3K/Akt and JAK/STAT signal pathways are all partially involved in MSC proliferation induced by serum, but the PI3K/Akt is the main contributor to MSC proliferation in response to PDGF-BB. Subsequently we studied the regulatory role of PI3K/Akt downstream signal cascades; mTOR and proteins associated with the cell cycle. We also investigated possible feedback mechanisms between protein kinases and PDGF-BB and PDGFR-β expression. Finally, we demonstrate that PDGF signalling inhibits differentiation of MSCs into adipocytes through activation of Erk. Based on these findings we propose that PDGFRβ signalling promotes self-renewal of MSCs through two distinct pathways, Akt to induce proliferation and Erk to maintain the cell in an unspecialized status by preventing differentiation.
Materials and methods
Antibodies and reagents
The antibodies used were as follow: rabbit antibody against phospho-PDGFRβ (Tyr751), phospho-SHP-2 (Tyr542), phospho-Akt (Ser473), phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204), phospho-mTOR (Ser2448), phospho-p70 S6 Kinase (Thr389), phospho-p70 S6 Kinase (Ser371), phospho-4E-BP1 (Thr37/46) eIF4E, p15 INK4B, p16 INK4A, p27 Kip1 (Cell Signalling, New England Biolabs, Hitchin, UK), cyclin B, cyclin E (BioLegend, Cambridge Bioscience, Cambridge, UK). Mouse antibody against p21 Waf1/Cip1, cyclin D1, cyclin D3, CDK4, CDK6 (Cell Signalling), CDK2, cyclin A (BioLegend). Secondary HRP-linked antibody against mouse and rabbit IgG were from Cell Signalling and Dako (Ely, UK) respectively. The inhibitors used were as follow: PDGFRβ inhibitor, SU 16f (Tocris Bioscience, Bristol, UK); MAPKK/MEK inhibitor, PD98059; JAK2, JAK3/STAT inhibitor, AG490; mTOR inhibitor, rapamycin (Cayman chemical, Cambridge Bioscience) and PI3K inhibitor, LY294002 (Sigma–Aldrich, Poole, Dorset, UK). Recombinant human PDGF-BB was purchased from R&D Systems (Abingdon, UK).
Cell culture
Human MSCs from healthy adults were obtained from Lonza (Slough, UK), characterized (Supplementary Figure 1) and used at passages 2–5. Cell were maintained in normal growth medium consisting of α-Minimal Essential Medium (MEM), penicillin (50 U/ml), streptomycin (50 μg/ml) (Sigma–Aldrich), Glutamax (2 mM) (Invitrogen, Paisley, UK) and 10% foetal bovine serum (FBS) (Sigma–Aldrich) at 37°C in a humidified 5% CO2:95% air atmosphere.
To induce osteogenesis, MSCs were seeded at a density of 1.2 × 104 cell/cm2 in 12-well plates (Nunc, Fisher Scientific, Loughborough, UK) and after 24 hrs treated with osteogenic medium consisting of growth medium supplemented with 0.1 μM dexamethasone, 0.05 mM ascorbic acid (AA) and 10 mM ß-glycerophosphate (Sigma–Aldrich). Osteogenic differentiation was assessed by quantitative (q)RT-PCR analysis of mRNA expression of markers of differentiation, i.e. Runx-2, alkaline phosphatase (ALP) and osteocalcin and accumulation of calcium deposits by staining with alizarin red dye. Briefly, cells were fixed (15 min. with 4% formaldehyde in PBS), stained for 10 min. with alizarin red S (1:100 dilution in H2O) and washed (five times) in 50% ethanol and air-dried. To induce adipogenic differentiation cells were seeded at a density of 4 × 104 cell/cm2 in 12-well plates (Nunc) and incubated for 24 hrs before switching to adipogenic medium (growth medium supplemented with 1 μM dexamethasone, 0.25 mM isobutylmethylxanthine, 50 μM indomethacin and 10 μg/ml insulin (Sigma–Aldrich)). Adipogenesis was assessed by qRT-PCR analysis of markers of differentiation, i.e. PPARγ, CEBPα and LPL and visualized by light microscopy alone or following staining with Oil-red-O dye as described previously [28]. Briefly, cells were fixed (15 min. with 4% formaldehyde in PBS), stained for 15 min. and washed with 60% isopropanol and with PBS.
EdU incorporation assay
MSCs were seeded at a density of 1.5 × 104 cell/cm2 in 6-well plates and incubated for 24 hrs in normal growth medium. Medium was changed with reduced (1%) FBS culture medium and cells were incubated for an additional 12 hrs prior to stimulation with PDGF-BB in the presence or absence of inhibitors as described in the results. Inhibitors were given 1 hr prior to stimulation with PDGF-BB. Cell proliferation was assessed by measuring 5-ethynyl-2′ -deoxyuridine (EdU) DNA incorporation using the Click-iT EdU Alexa Fluor 647 cell proliferation assay kit (Invitrogen). Cells were treated with EdU at 10 μg/ml at the start of stimulation. After 48 hrs of incubation, cells were harvested by trypsinization, washed in PBS/1% BSA and fixed with Click-iT fixative for 15 min. at room temperature. The cells were then permeabilized using saponin-based permeabilization reagent, treated with Click-iT EdU reaction cocktail for 30 min. at room temperature in the dark and washed with saponin-based permeabilization reagent. The number of EdU-positive cells was determined using FACS-Canto II flow cytometer, and data analysis was performed using DIVA software (Becton Dickinson Biosciences, San Jose, CA).
Western blotting analysis
MSCs were seeded at a density of 2 × 104 cell/cm2 in 6-well plates and incubated for 24 hrs in normal growth medium. The culture was then incubated with serum free α-MEM for 12 hrs prior to treatment with inhibitors (1 hr) and/or stimulation with PDGF-BB as indicated in the results section. Following incubation for the indicated period with PDGF-BB the reaction was terminated by two quick washes in ice-cold PBS containing 1 mM sodium orthovanadate and cells were lysed in ice-cold radioimmunoprecipitation assay buffer [50 mM tris(hydroxymethyl)aminomethane (Tris)-hydrochloric acid (HCl), pH 7.5, 150 mM sodium chloride (NaCl), 1% Nonidet P-40, 0.1% Sodium dodecyl sulphate (SDS), 0.5% sodium deoxycholate] containing a protease inhibitor cocktail (Sigma–Aldrich), 1 mM sodium orthovanadate and 0.1 mg/ml phenylmethylsulfonyl fluoride. Cell lysates (10–15 μg of protein) and biotinylated protein ladder (Cell Signalling) were mixed with Laemmli buffer (Bio-Rad, Hempstead, UK) and subjected to SDS-PAGE. Proteins were transferred onto PVDF membranes and incubated overnight at 4°C with primary antibody. Secondary antibodies (1:2000) conjugated to horseradish peroxidase were then applied for 1 hr at room temperature, and proteins visualized and photographed using ECL Plus detection reagent (GE Healthcare, Bucks, UK) and Molecular Imager Gel Doc XR+ documentation and analysis System with Image Lab Software (Bio-Rad). Equal sample loading was confirmed by probing with antibody to the housekeeping protein GAPDH.
qRT-PCR analysis
Total RNA was extracted using TRI reagent (Ambion, Warrington, UK) and Phase Lock Gel Heavy tubes (5 prime, VWR, Leicestershire, UK) according to the manufacturer's instructions. RNA purity and quantity was assessed by Nano Drop 1000 (Fisher Scientific) (A260/A280 1.8–2 was considered suitable for further analysis). Possible contaminating DNA was removed and cDNA prepared from 1 μg RNA using QuantiTect Reverse Transcription Kit (Qiagen, West Sussex, UK) according to the manufacturer's instructions. qRT-PCR was performed with a Rotor-Gene 6000 thermal cycler (Qiagen) using Brilliant III Ultra-Fast SYBR Green qPCR Master mix (Stratagene, Agilent Technologies, Cheshire, UK) and primer pairs as listed in Supplementary Table 1. PCR conditions consisted of 1 cycle of 95°C for 3 min. and 40 cycles of 95°C for 10 sec. and 60°C for 10 sec. followed by melting analysis of 1 cycle with gradual increase from 65°C to 95°C. RPL13a was used as an invariant housekeeping gene.
Data analysis
Statistical comparisons between means were made using one-way anova (SPSS 17, SPSS, Chicago, IL, USA), and post hoc analyses using the Tukey test to evaluate the differences among the mean values between groups. If comparisons were made only between two groups Student's t-test was used. A P-value of less than 0.05 was considered statistically significant.
Results
PDGF-BB induces MSC proliferation via PDGFRβ through PI3K signalling
To determine the mechanism involved in MSC proliferation we studied the effect of inhibitors of major signal pathways on MSC growth in the presence of normal growth medium containing 10% FBS, using EdU-DNA incorporation. As shown in Figure 1A, inhibition of MEK/Erk by PD98059, JAK/STAT by AG490 and PI3K/Akt by LY294002 all resulted in reduction (P < 0.01) in the number of cells entering cell cycle. In addition, inhibition of PDGFR-β by a specific inhibitor (SU 16f) reduced the EdU-positive cells by 60% (P < 0.001), suggesting that PDGF signalling is involved in normal MSC growth, possibly via MEK/Erk and/or JAK/STAT and/or PI3K/Akt signal mechanisms.
Fig. 1.
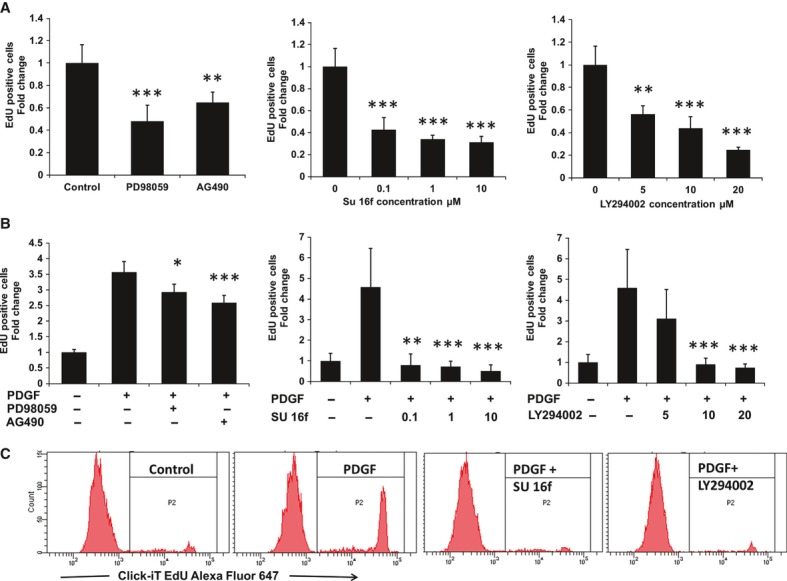
Effect of inhibition of signal transduction pathways on serum or PDGF-BB induced cell proliferation. MSCs were serum deprived (1%) for 12 hrs and subsequently cultured in (A) 10% serum or (B) in 10 ng/ml PDGF-BB in the presence or absence of PD98059, AG490, SU 16f or LY294002 at concentrations of 40 μM, 10 μM (0.1, 1 and 10 μM) and (5, 10 and 20 μM) respectively. Inhibitors were given 1 hr prior to stimulation with PDGF-BB. The proliferation rate was determined by analysing the proportion of cells that incorporated EdU following 48 hrs of incubation using flow cytometry. Samples were normalized to the mean fold change in untreated control which is set as 1 and expressed as mean ± S.D. [3 experiments; **P < 0.01 and ***P < 0.001 versus untreated control (A) or PDGF-BB treated (B)]. (C) Representative histograms showing EdU-Alexa Fluor® 647 fluorescence emission in unstimulated cells, PDGF-BB stimulated alone or in presence of 1 μM SU 16f or 10 μM LY294002.
To assess this hypothesis we examined the effect of inhibitors on PDGF-BB stimulated MSC proliferation in reduced serum (1%) (Fig. 1B). The number of EdU-positive cells were enhanced by 3–4-fold (P < 0.001) in the presence of PDGF-BB after 48 hrs of culture. This stimulatory effect was completely abolished when the PDGFR-β antagonist, SU 16f, was included in cultures at concentrations between 0.1 and 10 μM (P < 0.001). LY294002 also blocked the increased proliferation in a dose-dependent manner by 30, 95 and 100% at 5, 10 and 20 μM (P < 0.001) concentrations, whereas the JAK/STAT inhibitor (AG490) only partially inhibited the effect by 30%. In contrast, inhibition of MAPK by PD98059 had a relatively modest effect on PDGF-BB induced MSC proliferation when compared with the other inhibitors. Addition of inhibitors (SU 16f, LY294002 or AG490) had only a slight effect on MSC proliferation in the absence of PDGF-BB in low serum culture conditions, indicating selectivity of these compounds on PDGF-BB signal pathways (data not shown).
PDGF-BB activation of PDGFRβ causes sustained Akt and transient Erk phosphorylation
To investigate further the downstream pathways involved in PDGF-BB stimulation of MSC proliferation, we assessed the alteration in phosphorylation of PDGFRβ, SHP2, Akt and P44/P42 MAPK in response to stimulation with PDGF-BB. Serum-starved MSCs were incubated for 5–60 min. with PDGF-BB and protein phosphorylation was determined using phospho-specific antibodies against PDGFRβ (Tyr751), SHP-2 (Tyr542), Akt (Ser473), p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (Fig. 2). PDGFRβ was rapidly and transiently phosphorylated, peaking at 5 min. and returning to near basal levels after 120 min. A similar transient stimulation was observed for SHP2 and Erk1/2 phosphorylation. The Akt was also rapidly phosphorylated after 5 min., but unlike PDGFRβ or Erk the level remained consistently high for the duration of 2 hrs stimulation (Fig. 2B). Maximal stimulation was seen with PDGF-BB concentrations of 5 ng/ml or greater (Fig. 2A).
Fig. 2.

Signal pathways regulated by PDGF-BB in MSCs. MSCs were serum starved for 12 hrs and subsequently (A) treated for 15 min. with various concentration of PDGF-BB (1–30 ng/ml) or (B) different time course with 10 ng/ml of PDGF-BB. (C) Serum-starved (12 hrs) MSCs were pre-treated with inhibitors of pathways involved in PDGF-BB signalling SU 16f (1 μM), LY294002 (10 μM), PD98059 (40 μM), AG490 (10 μM) or (D) various concentrations of LY294002 (5, 10 and 20 μM) for 1 hr prior to 15 min. stimulation with PDGF-BB at 10 ng/ml. Cells were lysed in RIPA buffer and analysed for expression of phospho-PDGFRβ, phospho-SHP-2, phospho-Akt, phospho-p44/42 MAPK (Erk1/2) by western blotting. NS – no stimulation. Samples were also probed for eIF4E in the same blot as a loading control. Representative blots from three separate experiments are shown.
To determine whether the stimulation of PDGFRβ was the sole activator of downstream pathways, we analysed the effect of simultaneous blockage of the receptor. As shown in Figure 2C, PDGF-BB induced stimulation of PDGFRβ, SHP2, Akt and P44/P42 MAPK was completely abolished by pre-treatment with a selective PDGFRβ inhibitor (SU 16f). We also conducted a similar assay using Erk, PI3K or JAK/STAT inhibitors. Pre-treatment with PD98059 resulted in complete loss of PDGF-BB-induced Erk1/2 phosphorylation. Pre-treatment with LY294002 also significantly inhibited Akt phosphorylation induced by PDGF in a dose-dependent manner (Fig. 2D). The effect of PD98059 and LY294002 was specific for Erk and Akt, respectively, and did not disrupt phosphorylation of each other by PDGF-BB.
PDGF-BB/PDGFRβ promote expression of cyclin D1/D3 and CDK6 via PI3K/Akt pathway to initiate G1 cell cycle progression
To verify the means by which PDGF drives cell cycle progression and thus MSC proliferation, we assessed the changes in major regulators of different stages of the cell cycle. As shown in Figure 3, stimulation of serum-starved MSCs by PDGF-BB for 6, 12 and 24 hrs resulted in a dramatic increase in the level of protein of activators of G1/S phase; cyclin D1, cyclin D3 and CDK6. However, the level of cyclin E, CDK4 and CDK2 remained unchanged during the duration of treatment. Cyclin A and cyclin B proteins were undetectable even after 48 hrs of PDGF-BB treatment, although high expression was detected in human umbilical vein endothelial cells (HUVEC) used as a positive control (Fig. 3). Treatment with PDGF-BB also had no effect on expression of cell cycle inhibitors, i.e. P15 INK4B, P16 INK4A, P21 Waf1/Cip1 and p27 Kip1 (Fig. 3).
Fig. 3.

Regulation of cell cycle proteins by PDGF-BB. MSCs were serum starved for 12 hrs and then treated with 10 ng/ml PDGF-BB for 6, 12 and 24 hrs. Cells were lysed and analysed by western blotting using antibodies against p15 INK4B, p16 INK4A, p27 Kip1, p21 Waf1/Cip1, CDK2, CDK4, CDK6, cyclin A, cyclin B, cyclin E, cyclin D1 and cyclin D3. Blots were then stripped and re-probed for GAPDH to confirm equal loading. PC – human umbilical vein endothelial cells used as positive control. Representative blots from at least two separate experiments are shown.
To confirm these findings and determine the likely upstream mechanisms involved, we assessed the effect of inhibitors on the PDGF-BB-induced cyclin D1, cyclin D3 and CDK6 expression (Fig. 4). In correlation with our EdU incorporation and our signal transduction data, pre-treatment with SU 16f completely abolished PDGF-BB-induced cyclin D1, cyclin D3 and CDK6 expression at every time-point examined. The effect was also reduced in a dose-dependent manner in the presence of LY294002 with complete inhibition at concentrations over 10 μM (Fig. 4B). Pre-treatment with PD98059 and AG490 had little or no effect on cyclin D1- and cyclin D3-induced protein expression. PDGF-BB-induced CDK6 expression was, however, significantly reduced in the presence of AG490, a potential route by which the partial reduction in PDGF-BB-induced MSC proliferation by AG490 (Fig. 1) might be explained.
Fig. 4.
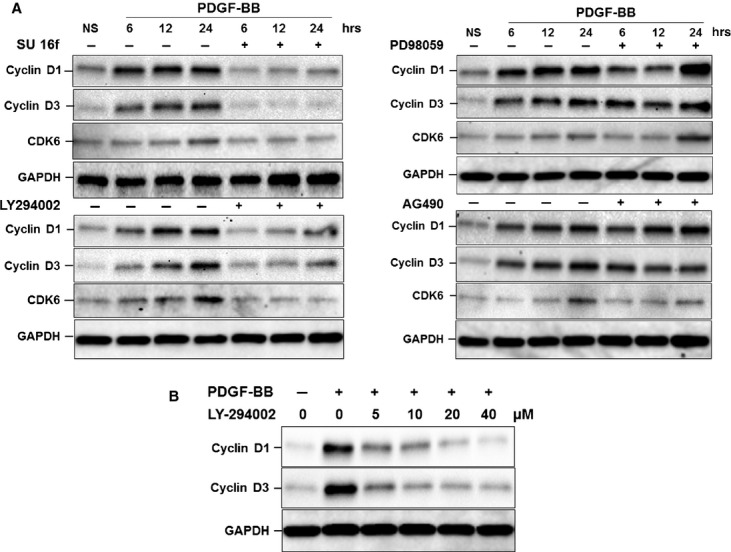
Effect of inhibition of signal transduction pathways on PDGF-BB-induced cell cycle protein expression. Serum-starved MSCs were pre-treated with (A) SU 16f (1 μM), LY294002 (10 μM), PD98059 (40 μM), AG490 (10 μM) for 1 hr and subsequently stimulated with PDGF-BB for 6, 12 and 24 hrs or (B) increasing concentrations of LY294002 (5, 10 and 20 μM) for 1 hr, and stimulated with PDGF-BB for 24 hrs. Cells were then lysed and analysed by Western blotting for changes in cyclin D1, cyclin D3 and CDK6 protein expression. Blots were then stripped and re-probed for GAPDH to confirm equal loading. Representative blots from three experiments are shown.
mTOR/p70S6K/4E-BP1 is a mediator of PDGFRβ/PI3K/Akt-induced G1 cell cycle progression and MSC proliferation
mTOR/p70S6K is one of the major signal pathways downstream of PI3K/Akt that has been implicated in mitogenic responses and in a number of cancer cell types. Thus, we examined whether the mTOR/p70S6K pathway was also a mediator of PDGF-BB induced MSC proliferation. Serum-starved MSCs were treated for 15 min. with PDGF-BB and phosphorylation of mTOR substrates was assessed using phospho-specific antibodies recognizing the phosphorylated forms of mTOR (Ser2448), 4E-BP1 (Thr37/46) and p70S6K (Thr389 and Ser371) (Fig. 5C). Although PDGF-BB elicited an up-regulation of mTOR phosphorylation, the effect was partly masked by the already high background. However, a high level of phosphorylation was observed for 4E-BP1 at Thr37/46 and for p70S6K at Thr389 but not at Ser371. The PDGF-BB induced 4E-BP1 and p70S6K phosphorylation was completely blocked when cells were pre-treated with LY294002 at 5–20 μM or rapamycin (an inhibitor of mTOR signalling) at 1–50 nM. In line with this, we also observed that pre-treatment with rapamycin led to a significant reduction in the PDGF-BB induced MSC proliferation (50%, P < 0.001), as assessed by EdU-DNA incorporation (Fig. 5A). Furthermore, rapamaycin pre-treatment reduced the expression of cyclin D1 and cyclin D3 induced by 24 hrs of treatment with PDGF-BB, however, no effect was detected on CDK6 protein level (Fig. 5D).
Fig. 5.
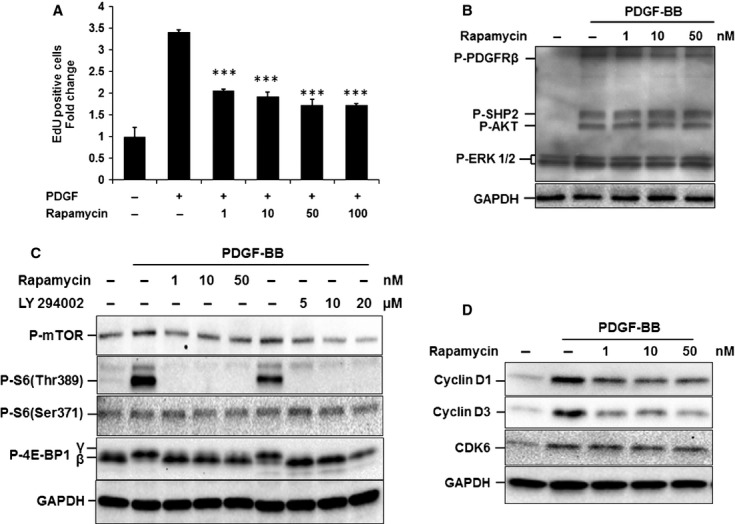
Role of mTOR signal pathway in PDGFRβ/PI3K/Akt-induced cell cycle progression and MSC proliferation. (A) Effect of mTOR inhibition on PDGF-BB induced proliferation. Serum-starved MSCs were pre-treated with indicated concentrations of rapamycin (1–100 nM) for 1 hr and then treated with 10 ng/ml PDGF-BB for 48 hrs. The change in MSC proliferation was determined by analysing the proportion of cells that incorporated EdU using flow cytometry. Samples were normalized to the mean fold change in untreated control which is set as 1 and expressed as mean ± S.D. (3 experiments; ***P < 0.001 versus untreated control). (B–D) The role of mTOR on signal transduction pathway and cell cycle protein expression. MSCs were serum starved for 12 hrs and treated for 1 hr with the indicated concentrations of rapamycin before stimulation with PDGF-BB (10 ng/ml) for 15 min. (B and C) or 24 hrs (D). Cells were then lysed and analysed by Western blotting for changes in (B) PDGF signal pathway proteins, (C) mTOR signalling proteins or (D) cell cycle proteins. Equal loading was determined by re-probing stripped blots with the housekeeping protein GAPDH. Representative blots from three experiments are presented.
Inhibition of PI3K/Akt induces the expression of PDGFRβ
Negative feedback from PI3K/Akt to inhibit upstream signal cascades through inhibition of the PDGFRβ expression has been reported previously [29]. To test whether a similar feedback loop is present in our setting, we analysed the effect of inhibition of downstream signal cascades on expression of PDGF-BB and PDGFRβ. MSCs in normal medium were treated for 24 hrs and the changes in mRNA expression were determined. Expression of PDGF-BB was barely detectable in MSCs and was not affected by inhibition of any of the signal pathways (data not shown). PDGFRβ on the other hand was highly expressed and significantly further induced in cells treated with LY294002 for 24 hrs (P < 0.001, Fig. 6). Other inhibitors had no significant effect on PDGFRβ expression.
Fig. 6.

Effect of inhibition of signal transduction pathways on expression of PDGFRβ. Cells were treated with SU 16f (1 μM), LY294002 (10 μM), PD98059 (40 μM), AG490 (10 μM), LY294002 (10 μM) or rapamycin (10 nM) in normal growth medium for 24 hrs. PDGFRβ mRNA expression determined by qRT-PCR and compared with untreated cells after normalization to RPL13a (3 experiments; total of 6 replicates; **P < 0.01 and ***P < 0.001 versus untreated).
PDGF-BB/PDGFRβ regulation of MSC differentiation is via Erk signalling
Activation or inhibition of Akt/mTOR and Erk signal pathways has previously been shown to regulate osteogenic and adipogenic differentiation of pre-osteoblast and pre-adipocyte cell lines [20–23]. The role of PDGF in adipogenesis of MSCs and the mechanisms involved is, however, not known. Our previous data showed that MSCs grown in the presence of PDGF-BB in normal growth medium express higher levels of the adipogenic transcription factor PPARγ (Supplementary Figure 2A). Subsequently, the same cultures showed a lower level of adipogenesis when compared with cells grown in the absence of PDGF in prior culture (Supplementary Figure 2B).
Here, we assessed the effect of inhibition of PDGFRβ, Erk, Akt, JAK-STAT and mTOR signal pathways on MSC differentiation in the absence or presence of PDGF-BB. As shown in Figure 7 A and B, treatment with SU 16f, PD98059 and AG490 had little effect on adipogenesic differentiation of MSCs. However, an increase in expression of CEBPα and LPL was seen in the presence of SU 16f and PD98059 respectively. Addition of LY294002 and rapamycin to the differentiation medium on the other hand significantly blocked the expression of PPARγ, CEBPα and LPL (P < 0.01) and the number of lipid containing adipocyte cells in culture. Interestingly, a potent inhibitory effect was also observed when PDGF-BB was included in adipogenic culture. PDGF-BB significantly inhibited the expression of adipogenic transcription factors PPARγ and CEBPα and subsequently blocked the adipogenesis process. As both inhibition of Akt/mTOR or activation of PDGF resulted in loss of adipogenic differentiation of MSCs, it can be concluded that other downstream pathways rather than Akt/mTOR are involved. Pre-treatment of PDGF-treated adipogenic cultures with inhibitors of signal pathways subsequently revealed that Erk is the mediator of the inhibitory effect of PDGF on adipogenesis. The effect of PDGF-BB on PPARγ and CEBPα was reversed by inhibition of either PDGFRβ or Erk and the level of adipogenesis returned to that of control. Figure 7A and B shows that SU 16f and PD98059 were both able to block the inhibition of adipogenic differentiation of MSCs in the presence of PDGF-BB.
Fig. 7.

Role of signal transduction pathways on adipogenic differentiation of MSCs in absence or presence of PDGF-BB. MSCs were incubated with adipogenic medium with or without SU 16f (1 μM), LY294002 (10 μM), PD98059 (40 μM), AG490 (10 μM), LY294002 (10 μM) or rapamycin (10 nM) in presence or absence of 10 ng/ml PDGF-BB for 7 days. (A) Expression of adipogenic markers PPARγ, CEBPα and LPL mRNA was analysed by qRT-PCR. (Mean ± SEM of 3 experiments of duplicates). *P < 0.05, **P < 0.01 and ***P < 0.001 when compared with cell in differentiation medium alone. (B) To assess phenotypic changes, cells were differentiated for a further 7 days. Lipid accumulation in treated and untreated populations was visualized by staining with Oil Red O (×200).
In contrast with adipogenesis, PDGF-BB had no significant effect on expression of osteogenic genes under normal growth condition (data not shown). Expression of Runx-2, osteopontin and osteocalcin (Osc) also remained unchanged in the presence of PDGF during the differentiation (Fig. 8A). There were, however, significant reductions in both expression of genes involved in osteoblast function, i.e. ALP, collagen type I and BMP-2 in the presence of PDGF, as well as mineralization of cultures that underwent differentiation (Fig. 8 A and B).
Fig. 8.

Effect of PDGFRβ activation on MSC osteogenic differentiation and function. MSCs were incubated with osteogenic medium with or without PDGF-BB (10 ng/ml) for 7 days. (A) mRNA expression of the osteogenic markers Runx-2, osteopontin, osteocalcin, alkaline phosphatase (ALP), collagen type I and BMP-2 was analysed by qRT-PCR. (Mean ± SEM of 3 experiments of duplicates). *P < 0.05, **P < 0.01 and ***P < 0.001 when compared with cells in differentiation medium alone. (B) To assess phenotypic changes, cells were differentiated for a further 14 days. The ability of the cultures to undergo matrix mineralization was assessed by staining with Alizarin red for calcium deposition.
Discussion
MSCs have received considerable attention in recent years for a variety of applications in regenerative medicine. Understanding the pathways that control MSC fate (proliferation versus differentiation) could be beneficial for directing tissue engineering approaches both in vitro and in vivo. PDGF has been shown to enhance the expansion of MSCs in vitro. In long-term culture MSCs lose their proliferative response to PDGF, and prolonged cultivation with PDGF-BB is associated with changes in their multipotency (Gharibi and Hughes, submitted for publication). MSCs from aged animals and individuals have been also shown to have reduced proliferation and differentiation capacity [30]. In addition, the rate of malignancies is higher in aged stem cells and the molecular pathways involved in tumorigenesis and in proliferation of stem cells have been shown to be the same [31–33]. Alterations in PDGF signalling such as overexpression, autocrine activation or hyper-stimulation of their receptors and their downstream cascades have also been implicated in tumour development and progression in many cancer cells [34–36]. Establishing the PDGF signal pathways involved in MSC proliferation and differentiation could therefore be useful for developing novel strategies for in vivo and in vitro MSC expansion, and differentiation and determining possible association with tumorigenesis.
In this study, we demonstrated that PDGFRβ is crucial for MSC proliferation in normal culture and that major signal pathways including MEK/Erk, PI3K/Akt or JAK/STAT are partly involved in growth MSCs. Although PDGFRβ activation by PDGF-BB was able to elicit a response from both MEK/Erk and PI3K/Akt pathways, only the inhibition PI3K/Akt could reverse the stimulatory effects of PDGF signalling on MSC proliferation. It is interesting to note that only the Akt activity was sustained during the duration of experiment, while Erk phosphorylation was rapid and transient, peaking at 5 min. and returning back to near a basal level after 30 min. It is well known that the duration of signal by these pathways is a determinant of the subsequent physiological response by the cells. Sustained Erk activation by FGF-2 has been shown to induce proliferation in both MSCs and dermal fibroblasts [37,38], whereas transient activation of Erk or Akt has been associated with cell proliferation, migration and differentiation [39–42].
The mechanisms by which activation of Akt or Erk leads to cell cycle progression and cell proliferation also differ depending on the upstream activator and with different cell types. For instance Erk regulates c-Fos to sustain cyclin B1 expression in keratinocytes but not in fibroblasts, and PDGF activates cell cycle G1 progression in beta pancreatic cells through Erk but not in smooth muscle via Akt [43–45]. Here, we have demonstrated that PDGFRβ exerts its proliferative effect on MSCs by promoting cyclin D1, cyclin D3 and CDK6 protein expression and G1/S progression. In correlation with our cell proliferation data, the stimulation of regulators of G1/S by PDGF was completely blocked by inhibitors of PDGFRβ and Akt pathways. The PI3K/Akt pathway role in proliferation and tumorigenesis is often coupled with its downstream target, the mTOR signalling molecules [46]. Our data show that the action of PDGF/Akt on proliferation of MSCs was at least partly mediated through mTOR, specifically via phosphorylation of 4E-BP1 at Thr37/46 and for p70S6K at Thr389. It is interesting to note that inhibition of mTOR by rapamycin only affected the expression of cyclin D1 and D3 and had no inhibitory action on CDK6 expression. In contrast, inhibition of Jak/Stat abolished PDGF-induced CDK6 expression without affecting cyclins D1 and D3, which could account for the partial reduction in PDGF-induced MSC proliferation seen in the presence of both rapamycin and AG490. Our finding is particularly important as the cyclin D family has been widely implicated in tumorigenesis as well as being a well-known regulator of G1/S phase transition. Overexpression of cyclin D1, for example, has been associated with early cancerogenesis, tumour progression and metastasis [47]. Changes in levels of regulators of cyclin D1 such as Akt and mTOR are also a common occurrence in cancer and are emerging as important therapeutic targets [27,48]. Therefore, the use of PDGF for in vitro MSC expansion and in vivo tissue regeneration should be carried out with caution and may require further investigation. It is noteworthy that PDGF-BB expression in MSCs was barely detectable and was not affected by inhibition of any of the signal cascades studied, which suggests requirement of paracrine induction. Pathways other than soluble PDGF-BB for PDGFRβ activation such as direct cell–cell interactions have been reported in fibroblasts and should also be considered [49]. In contrast, high levels of PDGFRβ were detectable in MSCs and were further induced in the absence of Akt signalling. Similar feedback mechanisms have been recently reported in mouse embryonic fibroblasts, wherein activation of Akt by several mechanisms (loss of PTEN, activation of PI3K or Akt) suppressed the expression of PDGFRs [29]. A better understanding of the interactions between these signal pathways could explain their role in cell proliferation and tumorigenesis and help in optimum targeting of these pathways for anticancer therapeutics.
Although the PI3K/Akt/mTOR pathway was the principal mediator of PDGF-BB-induced MSC proliferation, it was not involved in regulation of MSC differentiation by PDGF. Indeed, treatment with PDGF-BB or inhibition of PI3K and mTOR had the same inhibitory effect on differentiation of MSCs to adipocytes. This is intriguing because activation of Akt/mTOR by factors such as insulin has been shown to have a stimulatory effect on adipogenesis of 3T3-L1 pre-adipocytes [20]. This effect is supported by our finding that LY294002 and rapamycin strongly inhibit adipogenesis by MSCs induced by differentiation medium in the absence or presence of PDGF. We have also shown that addition of PDGF-BB to normal medium induces the expression of the adipogenic ‘master switch’ PPARγ. Taken together these data suggest a role for PDGF-BB induced Akt in the proliferation of MSCs but not during their differentiation. Interestingly, Erk, which was also activated by PDGF signalling in MSCs but had only a negligible involvement in proliferation, was found to be responsible for the action of PDGF on adipogenic differentiation. The ability of PDGF-BB to inhibit adipogenesis through Erk appeared only when cells were induced to differentiate. In the undifferentiated state, PDGF-BB either upregulated or had no effect on genes involved in adipogenesis. Based on our data and others Akt/mTOR is required and sufficient for adipogenesis, suggesting that up-regulation of Akt could lead to differentiation as well as proliferation. However, simultaneous activation of Erk blocks the adipogenesis and maintains the cell in their unspecialized form. Erk signalling has been previously implicated in adipogenesis and shown to have a negative effect on adipocytes, although some have reported stimulation depending on the upstream signal [21,50,51]. Together these data imply that self-renewal of MSCs requires a complex interaction between various signal pathways. In this case, the Akt pathway promotes MSC proliferation and cell cycle progression, and Erk signalling is required to keep the cells in their undifferentiated state, countering the possibility of adipogenic differentiation through activation of Akt. This may explain why in pre-adipocytic 3T3L1 cells Akt induces adipogenic differentiation and even causes spontaneous differentiation when activated either by overexpression or by factors such as insulin [20] but not with PDGF in MSCs as we showed here. In the context of osteogenesis, we found little effect of PDGF-BB on differentiation process itself, but there were inhibitory effects on expression of genes involved in osteoblast function and mineralization. Although few studies have been previously carried out on the involvement of PDGF signalling on osteogenesis the findings are conflicting. Our results support the studies that report an effect on osteoblast function but not on MSC osteogenic differentiation. [14,52,53].
In summary our data demonstrate how a growth factor maintains self-renewal by inducing proliferation and preventing cell differentiation. In detail we show PDGFRβ signalling simultaneously activates two major transduction pathways with dual actions on proliferation versus differentiation. We propose that PDGFRβ activates PI3K/Akt/mTOR as the mediator of cell cycle progression and proliferation and Erk as the inhibitor of differentiation in MSCs (Fig 9). These data not only provide the signal mechanism of self-renewal, it may also explain the premature loss of differentiation with growth factor MSC cell expansion (unpublished data). It is clear that with PDGF there is a system in place to avoid the unwanted differentiation while MSCs undergo extensive proliferation. These findings contribute to developing novel strategies for regulation of stem cell proliferation and differentiation that may be applicable for future clinical applications.
Fig. 9.

Schematic diagram illustrating signal pathways involved in regulation of MSC self-renewal by PDGFRβ signalling. Binding of PDGF-BB to the PDGFRβ stimulates PI3K/Akt/mTOR phosphorylation which subsequently induces expression of the cell cycle proteins cyclin D1, cyclin D3 and CDK6 to initiate cell cycle progression and MSC proliferation. Phosphorylation of MEK/Erk by PDGF-BB/PDGFRβ on the other hand inhibits expression of PPARγ and CEBPα and blocks adipogenesis of MSCs.
Acknowledgments
This work was supported by research funding from the Guy's & St. Thomas Charity. The authors acknowledge financial support from the Department of Health via the National Institute for Health Research (NIHR) comprehensive Biomedical Research Centre award to Guy's & St Thomas' NHS Foundation Trust in partnership with King's College London and King's College Hospital NHS Foundation Trust. We thank Dr Susanne Heck and Mr P.J. Chana for assistance with flow cytometry analysis.
Conflict of interest
The authors confirm that there are no conflicts of interest.
Disclaimers: None.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Primer sequences. Product sizes were between 98 and 102 bp.
Characterization of MSCs. (A) Flow cytometry analysis of MSC positive (CD105, CD44, CD146 and CD90) and negative (CD14 and CD45) cell surface markers. (B) Alizarin red staining of calcium deposits and (C) accumulation of lipid droplets following differentiation to osteoblast and adipocyte lineages.
Changes in mRNA expression of markers of adipogenesis in MSCs cultured in absence or presence of PDGF-BB. MSCs were cultured with 10 ng/ml PDGF-BB. Expression of adipogenic markers was assessed by qRT-PCR (A) before differentiation or (B) after differentiation with adipogenic media alone.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Noël D, Gazit D, Bouquet C, et al. Short-term BMP-2 expression is sufficient for in vivo osteochondral differentiation of mesenchymal stem cells. Stem Cells. 2004;22:74–85. doi: 10.1634/stemcells.22-1-74. doi: 10.1634/stemcells.22-1-74. [DOI] [PubMed] [Google Scholar]
- 2.Ling L, Nurcombe V, Cool SM. Wnt signaling controls the fate of mesenchymal stem cells. Gene. 2009;433:1–7. doi: 10.1016/j.gene.2008.12.008. doi: 10.1016/j.gene.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 3.Yu J, Carroll TJ, McMahon AP. Sonic hedgehog regulates proliferation and differentiation of mesenchymal cells in the mouse metanephric kidney. Development. 2002;129:5301–12. doi: 10.1242/dev.129.22.5301. [DOI] [PubMed] [Google Scholar]
- 4.Gharibi B, Abraham AA, Ham J, et al. Adenosine receptor subtype expression and activation influence the differentiation of mesenchymal stem cells to osteoblasts and adipocytes. J Bone Miner Res. 2011;26:2112–24. doi: 10.1002/jbmr.424. doi: 10.1002/jbmr.424. [DOI] [PubMed] [Google Scholar]
- 5.Tamama K, Fan VH, Griffith LG, et al. Epidermal growth factor as a candidate for ex vivo expansion of bone marrow–derived mesenchymal stem cells. Stem Cells. 2006;24:686–95. doi: 10.1634/stemcells.2005-0176. doi: 10.1634/stemcells.2005-0176. [DOI] [PubMed] [Google Scholar]
- 6.Tsutsumi S, Shimazu A, Miyazaki K, et al. Retention of multilineage differentiation potential of mesenchymal cells during proliferation in response to FGF. Bioch Bioph Res Co. 2001;288:413–9. doi: 10.1006/bbrc.2001.5777. doi: 10.1006/bbrc.2001.5777. [DOI] [PubMed] [Google Scholar]
- 7.Solchaga LA, Penick K, Porter JD, et al. FGF-2 enhances the mitotic and chondrogenic potentials of human adult bone marrow-derived mesenchymal stem cells. J Cell Physiol. 2005;203:398–409. doi: 10.1002/jcp.20238. doi: 10.1002/jcp.20238. [DOI] [PubMed] [Google Scholar]
- 8.Bianchi G, Banfi A, Mastrogiacomo M, et al. Ex vivo enrichment of mesenchymal cell progenitors by fibroblast growth factor 2. Exp Cell Res. 2003;287:98–105. doi: 10.1016/s0014-4827(03)00138-1. S0014482703001381 [pii] [DOI] [PubMed] [Google Scholar]
- 9.Kang YJ, Jeon ES, Song HY, et al. Role of c-Jun N-terminal kinase in the PDGF-induced proliferation and migration of human adipose tissue-derived mesenchymal stem cells. J Cell Biochem. 2005;95:1135–45. doi: 10.1002/jcb.20499. doi: 10.1002/jcb.20499. [DOI] [PubMed] [Google Scholar]
- 10.Stolzing A, Jones E, McGonagle D, et al. Age-related changes in human bone marrow-derived mesenchymal stem cells: consequences for cell therapies. Mech Ageing Dev. 2008;129:163–73. doi: 10.1016/j.mad.2007.12.002. doi: 10.1016/j.mad.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 11.Bonyadi M, Waldman SD, Liu D, et al. Mesenchymal progenitor self-renewal deficiency leads to age-dependent osteoporosis in Sca-1/Ly-6A null mice. Proc Natl Acad Sci U S A. 2003;100:5840–5. doi: 10.1073/pnas.1036475100. doi: 10.1073/pnas.1036475100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mishra PJ, Mishra PJ, Glod JW, et al. Mesenchymal stem cells: flip side of the coin. Cancer Res. 2009;69:1255–8. doi: 10.1158/0008-5472.CAN-08-3562. doi: 10.1158/0008-5472.CAN-08-3562. [DOI] [PubMed] [Google Scholar]
- 13.Satija NK, Singh VK, Verma YK, et al. Mesenchymal stem cell-based therapy: a new paradigm in regenerative medicine. J Cell Mol Med. 2009;13:4385–402. doi: 10.1111/j.1582-4934.2009.00857.x. doi: 10.1111/j.1582-4934.2009.00857.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tokunaga A, Oya T, Ishii Y, et al. PDGF receptor β is a potent regulator of mesenchymal stromal cell function. J Bone Miner Res. 2008;23:1519–28. doi: 10.1359/jbmr.080409. doi: 10.1359/jbmr.080409. [DOI] [PubMed] [Google Scholar]
- 15.Ding W, Knox TR, Tschumper RC, et al. Platelet-derived growth factor (PDGF)–PDGF receptor interaction activates bone marrow–derived mesenchymal stromal cells derived from chronic lymphocytic leukemia: implications for an angiogenic switch. Blood. 2010;116:2984–93. doi: 10.1182/blood-2010-02-269894. doi: 10.1182/blood-2010-02-269894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Veevers-Lowe J, Ball SG, Shuttleworth A, et al. Mesenchymal stem cell migration is regulated by fibronectin through α5β1-integrin-mediated activation of PDGFR-β and potentiation of growth factor signals. J Cell Sci. 2011;124:1288–300. doi: 10.1242/jcs.076935. doi: 10.1242/jcs.076935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andrae J, Gallini R, Betsholtz C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008;22:1276–312. doi: 10.1101/gad.1653708. doi: 10.1101/gad.1653708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu E, Palmer N, Tian Z, et al. Comprehensive dissection of PDGF-PDGFR signaling pathways in PDGFR genetically defined cells. PLoS ONE. 2008;3:e3794. doi: 10.1371/journal.pone.0003794. doi: 10.1371/journal.pone.0003794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elorza A, Hyde B, Mikkola HK, et al. UCP2 modulates cell proliferation through the MAPK/ERK pathway during erythropoiesis and has no effect on heme biosynthesis. J Biol Chem. 2008;283:30461–70. doi: 10.1074/jbc.M805400200. doi: 10.1074/jbc.M805400200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang HH, Huang J, Düvel K, et al. Insulin stimulates adipogenesis through the Akt-TSC2-mTORC1 pathway. PLoS ONE. 2009;4:e6189. doi: 10.1371/journal.pone.0006189. doi: 10.1371/journal.pone.0006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang T, Wang Y, Yamashita H. Evodiamine inhibits adipogenesis via the EGFR–PKCα–ERK signaling pathway. FEBS Lett. 2009;583:3655–9. doi: 10.1016/j.febslet.2009.10.046. doi: 10.1016/j.febslet.2009.10.046. [DOI] [PubMed] [Google Scholar]
- 22.Wang X, Harimoto K, Liu J, et al. Spata4 promotes osteoblast differentiation through Erk-activated Runx2 pathway. J Bone Miner Res. 2011;26:1964–73. doi: 10.1002/jbmr.394. doi: 10.1002/jbmr.394. [DOI] [PubMed] [Google Scholar]
- 23.Mukherjee A, Rotwein P. Akt promotes BMP2-mediated osteoblast differentiation and bone development. J Cell Sci. 2009;122:716–26. doi: 10.1242/jcs.042770. doi: 10.1242/jcs.042770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dasari VR, Kaur K, Velpula KK, et al. Upregulation of PTEN in glioma cells by cord blood mesenchymal stem cells inhibits migration via downregulation of the PI3K/Akt pathway. PLoS ONE. 2010;5:e10350. doi: 10.1371/journal.pone.0010350. doi: 10.1371/journal.pone.0010350. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 25.de Launay D, van de Sande MG, de Hair MJ, et al. Selective involvement of ERK and JNK mitogen-activated protein kinases in early rheumatoid arthritis (1987 ACR criteria compared to 2010 ACR/EULAR criteria): a prospective study aimed at identification of diagnostic and prognostic biomarkers as well as therapeutic targets. Ann Rheum Dis. 2012;71:415–23. doi: 10.1136/ard.2010.143529. doi: 10.1136/ard.2010.143529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hers I, Vincent EE, Tavaré JM. Akt signalling in health and disease. Cell Signal. 2011;23:1515–27. doi: 10.1016/j.cellsig.2011.05.004. doi: 10.1016/j.cellsig.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 27.Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9:550–62. doi: 10.1038/nrc2664. doi: 10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]
- 28.Gharibi B, Abraham AA, Ham J, et al. Contrasting effects of A1 and A2b adenosine receptors on adipogenesis. Int J Obes. 2012;36:397–406. doi: 10.1038/ijo.2011.129. doi: 10.1038/ijo.2011.129. [DOI] [PubMed] [Google Scholar]
- 29.Zhang H, Bajraszewski N, Wu E, et al. PDGFRs are critical for PI3K/Akt activation and negatively regulated by mTOR. J Clin Invest. 2007;117:730–8. doi: 10.1172/JCI28984. doi: 10.1172/JCI28984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roobrouck VD, Ulloa-Montoya F, Verfaillie CM. Self-renewal and differentiation capacity of young and aged stem cells. Exp Cell Res. 2008;314:1937–44. doi: 10.1016/j.yexcr.2008.03.006. doi: 10.1016/j.yexcr.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 31.Zheng H, Ying H, Wiedemeyer R, et al. PLAGL2 regulates Wnt signaling to impede differentiation in neural stem cells and gliomas. Cancer Cell. 2010;17:497–509. doi: 10.1016/j.ccr.2010.03.020. doi: 10.1016/j.ccr.2010.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu HK, Wang Y, Belz T, et al. The nuclear receptor tailless induces long-term neural stem cell expansion and brain tumor initiation. Genes Dev. 2010;24:683–95. doi: 10.1101/gad.560310. doi: 10.1101/gad.560310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu L, Rando TA. Manifestations and mechanisms of stem cell aging. J Cell Biol. 2011;193:257–66. doi: 10.1083/jcb.201010131. doi: 10.1083/jcb.201010131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Suzuki S, Dobashi Y, Hatakeyama Y, et al. Clinicopathological significance of platelet-derived growth factor (PDGF)-B and vascular endothelial growth factor-A expression, PDGF receptor-beta phosphorylation, and microvessel density in gastric cancer. BMC Cancer. 2010;10:659. doi: 10.1186/1471-2407-10-659. doi: 10.1186/1471-2407-10-659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ustach CV, Huang W, Conley-LaComb MK, et al. A novel signaling axis of atriptase/PDGF-D/β-PDGFR in human prostate cancer. Cancer Res. 2010;70:9631–40. doi: 10.1158/0008-5472.CAN-10-0511. doi: 10.1158/0008-5472.CAN-10-0511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jones AV, Cross NC. Oncogenic protein tyrosine kinases: oncogenic derivatives of platelet-derived growth factor receptors. Cell Mol Life Sci. 2004;61:2912–23. doi: 10.1007/s00018-004-4272-z. doi: 10.1007/s00018-004-4272-z. [DOI] [PubMed] [Google Scholar]
- 37.Zaragosi LE, Ailhaud G, Dani C. Autocrine fibroblast rowth Factor 2 signaling is critical for self-Renewal of human multipotent adipose-derived stem cells. Stem Cells. 2006;24:2412–9. doi: 10.1634/stemcells.2006-0006. doi: 10.1634/stemcells.2006-0006. [DOI] [PubMed] [Google Scholar]
- 38.Makino T, Jinnin M, Muchemwa FC, et al. Basic fibroblast growth factor stimulates the proliferation of human dermal fibroblasts via the ERK1/2 and JNK pathways. Br J Dermatol. 2010;162:717–23. doi: 10.1111/j.1365-2133.2009.09581.x. doi: 10.1111/j.1365-2133.2009.09581.x. [DOI] [PubMed] [Google Scholar]
- 39.Liu S, Li Y, Lin T, et al. High dose human insulin and insulin glargine promote T24 bladder cancer cell proliferation via PI3K-independent activation of Akt. Diabetes Res Clin Pract. 2011;91:177–82. doi: 10.1016/j.diabres.2010.11.009. doi: 10.1016/j.diabres.2010.11.009. [DOI] [PubMed] [Google Scholar]
- 40.Tarcic G, Avraham R, Pines G, et al. EGR1 and the ERK-ERF axis drive mammary cell migration in response to EGF. FASEB J. 2012;26:1582–92. doi: 10.1096/fj.11-194654. doi: 10.1096/fj.11-194654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Choi SC, Kim SJ, Choi JH, et al. Fibroblast growth factor-2 and -4 promote the proliferation of bone marrow mesenchymal stem cells by the activation of the PI3K-Akt and ERK1/2 signaling pathways. Stem cells dev. 2008;17:725–36. doi: 10.1089/scd.2007.0230. doi: 10.1089/scd.2007.0230. [DOI] [PubMed] [Google Scholar]
- 42.Krysan K, Reckamp KL, Dalwadi H, et al. Prostaglandin E2 activates mitogen-activated protein kinase/Erk pathway signaling and cell proliferation in non–small cell lung cancer cells in an epidermal rowth factor receptor–independent manner. Cancer Res. 2005;65:6275–81. doi: 10.1158/0008-5472.CAN-05-0216. doi: 10.1158/0008-5472.CAN-05-0216. [DOI] [PubMed] [Google Scholar]
- 43.Dumesic PA, Scholl FA, Barragan DI, et al. Erk1/2 MAP kinases are required for epidermal G2/M progression. J Cell Biol. 2009;185:409–22. doi: 10.1083/jcb.200804038. doi: 10.1083/jcb.200804038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen H, Gu X, Liu Y, et al. PDGF signalling controls age-dependent proliferation in pancreatic β-cells. Nature. 2011;478:349–55. doi: 10.1038/nature10502. doi: 10.1038/nature10502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Perez J, Torres RA, Rocic P, et al. PYK2 signaling is required for PDGF-dependent vascular smooth muscle cell proliferation. Am J Physiol Cell Physiol. 2011;301:C242–51. doi: 10.1152/ajpcell.00315.2010. doi: 10.1152/ajpcell.00315.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cho D, Mier JW, Atkins MB. PI3K/Akt/mTOR pathway: a growth and proliferation pathway. In: Bukowski RM, Figlin RA, Motzer RJ, editors. Renal cell carcinoma. New York: Humana Press; 2009. pp. 267–85. [Google Scholar]
- 47.Fu M, Wang C, Li Z, et al. Minireview: cyclin D1: normal and abnormal functions. Endocrinology. 2004;145:5439–47. doi: 10.1210/en.2004-0959. doi: 10.1210/en.2004-0959. [DOI] [PubMed] [Google Scholar]
- 48.Meric-Bernstam F, Gonzalez-Angulo AM. Targeting the mTOR signaling network for cancer therapy. J Clin Oncol. 2009;27:2278–87. doi: 10.1200/JCO.2008.20.0766. doi: 10.1200/JCO.2008.20.0766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sundberg C, Branting M, Gerdin B, et al. Tumor cell and connective tissue cell interactions in human colorectal adenocarcinoma. Transfer of platelet-derived growth factor-AB/BB to stromal cells. Am J Pathol. 1997;151:479–92. [PMC free article] [PubMed] [Google Scholar]
- 50.Wu L, Cai X, Dong H, et al. Serum regulates adipogenesis of mesenchymal stem cells via MEK/ERK-dependent PPARγ expression and phosphorylation. J Cell Mol Med. 2010;14:922–32. doi: 10.1111/j.1582-4934.2009.00709.x. doi: 10.1111/j.1582-4934.2009.00709.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang M, Wang JJ, Li J, et al. Pigment epithelium-derived factor suppresses adipogenesis via inhibition of the MAPK/ERK pathway in 3T3-L1 preadipocytes. Am J Physiol Endocrinol Metab. 2009;297:E1378–87. doi: 10.1152/ajpendo.00252.2009. doi: 10.1152/ajpendo.00252.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kratchmarova I, Blagoev B, Haack-Sorensen M, et al. Mechanism of divergent growth factor effects in mesenchymal stem cell differentiation. Science. 2005;308:1472–7. doi: 10.1126/science.1107627. doi: 10.1126/science.1107627. [DOI] [PubMed] [Google Scholar]
- 53.Kumar A, Salimath BP, Stark GB, et al. Platelet-derived growth factor receptor signaling is not involved in osteogenic differentiation of human mesenchymal stem cells. Tissue Eng Part A. 2009;16:983–93. doi: 10.1089/ten.TEA.2009.0230. doi: 10.1089/ten.TEA.2009.0230. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Primer sequences. Product sizes were between 98 and 102 bp.
Characterization of MSCs. (A) Flow cytometry analysis of MSC positive (CD105, CD44, CD146 and CD90) and negative (CD14 and CD45) cell surface markers. (B) Alizarin red staining of calcium deposits and (C) accumulation of lipid droplets following differentiation to osteoblast and adipocyte lineages.
Changes in mRNA expression of markers of adipogenesis in MSCs cultured in absence or presence of PDGF-BB. MSCs were cultured with 10 ng/ml PDGF-BB. Expression of adipogenic markers was assessed by qRT-PCR (A) before differentiation or (B) after differentiation with adipogenic media alone.