

Abstract
Dendritic cell (DC) immunotherapy is capable of generating tumour-specific immune responses. Different maturation strategies were previously tested to obtain DC capable of anti-cancer responses in vitro, usually with limited clinical benefit. Mutual comparison of currently used maturation strategies and subsequent complex evaluation of DC functions and their stimulatory capacity on T cells was performed in this study to optimize the DC vaccination strategy for further clinical application. DC were generated from monocytes using granulocyte–macrophage colony-stimulating factor (GM-CSF) and interleukin (IL)-4, pulsed with whole tumour cell lysate and then matured with one of five selected maturation strategies or cultured without additional maturation stimulus. DC were characterized with regard to their surface marker expression, cytokine profiles, migratory capacity, allogeneic and autologous T cell stimulatory capacity as well as their specific cytotoxicity against tumour antigens. We were able to demonstrate extensive variability among different maturation strategies currently used in DC immunotherapeutic protocols that may at least partially explain limited clinical benefit of some clinical trials with such DC. We identified DC matured with interferon-γ and lipopolysaccharide as the most attractive candidate for future clinical trials in cancer immunotherapy.
Keywords: cancer immunotherapy, cytotoxicity, dendritic cell, interleukin-12, maturation, migration
Introduction
Dendritic cells were discovered to be the most efficient antigen-presenting cells controlling B- and T cell-mediated immune reaction and linking innate and adaptive immunity [1,2]. Recently, they are studied and optimized for their possible use in cancer immunotherapy [reviewed in [3,4]]. DC can be isolated directly from circulating blood in limited amounts [5,6] or generated from various precursors in a larger extent, with the majority of studies using DC derived from peripheral blood monocytes [7–9]. Many strategies for DC antigen loading and maturation have been developed to improve their characteristics and amplify the immune response they induce [reviewed in [9–11]]. However, the transition to a powerful clinical tool has been limited [12].
The protocol commonly used today in DC production for cancer immunotherapy typically involves two steps: differentiation of immature DC from peripheral blood monocytes using granulocyte-macrophage colony-stimulating factor (GM-CSF) and interleukin (IL)-4, then stimulation with a cytokine cocktail to promote maturation [7]. Common strategies used at present for DC maturation include the following: (i) combination of tumour-necrosis factor (TNF)-α, IL-1β, IL-6 and prostaglandin E2 (PGE2) or its modifications [13–16]; (ii) sole use of TNF-α [17–19]; (iii) cocktails containing TNF-α, IL-1β, interferons (IFN) and Toll-like receptor (TLR) ligands [20–22]; (iv) cocktails containing TLR ligands and IFN-γ [23–25] and (v) cocktails containing CD40 ligand [26,27]. Newly designed maturation cocktails are usually compared only with the combination of TNF-α, IL-1β, IL-6 and PGE2, despite current knowledge that this combination is not optimal for inducing strong anti-tumour immune reaction [21,28,29]. Only few studies compared mutually more than two different maturation strategies [21,28,30]. Therefore, we performed a thorough in vitro comparison of six maturation strategies to obtain powerful DC for cancer immunotherapy. We tested complex functions of DC in a broad panel of parameters, including DC surface marker expression, cytokine production and migration, as well as T cell proliferation, activation and antigen-specific cytotoxicity upon DC stimulation. Here, we demonstrate that commonly used DC maturation strategies produce phenotypically and functionally different types of DC with different ability to induce specific T cell responses.
Materials and methods
Generation of immature DC
The study was approved by the Ethical Committee of the University Hospital Brno. Buffy coats of human peripheral blood were obtained from healthy donors at the Transfusion Department, University Hospital Brno. Peripheral blood mononuclear cells (PBMC) were separated by density gradient centrifugation over Histopaque (Sigma-Aldrich, Irvine, UK) and separated into six equal parts for later maturation. Monocytes were isolated as the adherent fraction of PBMC after 2 hrs incubation at 37°C in CellGro DC medium (CellGenix, Freiburg, Germany) with 50 μg/ml DNase I (Roche, Mannheim, Germany). After washing, adherent monocytes were cultured for 6 days in CellGro DC medium supplemented with 400 U/ml of rhIL-4 and 1000 U/ml of rhGM-CSF (both from CellGenix). No serum or antibiotics were added.
DC maturation
On the sixth day, 10 μg/ml of tumour lysate (prepared as described below) was added. Two hours later, different combinations of rhTNF-α, rhIL-1β (CellGenix), rhIL-1α, rhIL-6, rhIFN-γ (ProSpec, Rehovot, Israel), PGE2 (Sigma-Aldrich), R848 (InvivoGen, San Diego, CA, USA) and lipopolysaccharide (LPS) from E. coli strain O111:B4 (EMD Chemicals, Darmstadt, Germany) were added as specified in Table 1 to obtain DC1–DC6.
Table 1.
Maturation cocktails used
| Compounds | Concentrations | |
|---|---|---|
| DC1 | TNF-α | 40 ng/ml |
| DC2 | TNF-α, IL-1α, IL-6, PGE2 | 10 ng/ml, 10 ng/ml, 1000 U/ml, 10 ng/ml |
| DC3 | TNF-α, IL-1β, IFN-γ, R848, PGE2 | 10 ng/ml, 10 ng/ml, 5000 IU/ml, 1 µg/ml, 250 ng/ml |
| DC4 | IFN-γ, LPS | 50 ng/ml, 200 ng/ml |
| DC5 | IFN-γ, R848 | 50 ng/ml, 1 µg/ml |
| DC6 | Culture medium only | — |
TNF—tumour-necrosis factor; IL—interleukin; PGE2—prostaglandin E2; IFN—interferon; LPS—lipopolysaccharide.
Tumour lysate preparation
The human T98G glioblastoma cell line was obtained from the European Collection of Cell Cultures and cultured in DMEM containing 10% FCS, 100 μg/ml each of penicillin and streptomycin, 2 mM L-glutamine, 0.1 mM non-essential amino acids and 1 mM sodium pyruvate (all from Gibco Invitrogen, Paisley, UK). Cells were resuspended in sterile water and lysed by five repeated freeze–thaw cycles [23] at −196 and 37°C. Total protein concentration was quantified on a Beckman DU 530 spectrophotometer using the Bio-Rad protein assay (Bio-Rad, Hercules, CA, USA).
DC flow cytometric analysis and cytokine production analysis
DC were harvested after 48 hrs of maturation and stained with fluorescently labelled monoclonal antibodies against CD80, CD86, HLA-DR (Coulter Immunotech, Marseille, France), CD83 and CD14 (BD Biosciences, San Jose, CA, USA). PI (Sigma-Aldrich) was added immediately before analysis to assess cell viability. For CCR7 expression analysis, DC were collected after 24 hrs of maturation and stained for CCR7 (R&D Systems, Minneapolis, MN, USA). Cells were analysed on a FACSCanto II cytometer using BD FACSDiva Software (both BD Biosciences). As IL-12 secretion by DC is limited to the first 24 hrs after maturation [31], the IL-12p70 and IL-10 release from DC was analysed in DC culture supernatant collected after 24 hrs of maturation. The cytokine release was measured using a Cytometric Bead Array kit on a FACSArray Bioanalyzer (both BD Biosciences) according to the manufacturer's instructions.
Scratch assay
DC migratory ability was tested in a test called ‘scratch assay’ after 24 hrs of maturation. Briefly, scratch was performed in a culture plate with DC monolayer by a pipette tip. The cells were then incubated in a humidified incubator at 37°C and observed using an inverted microscope after 3, 6 and 24 hrs. Differences in filling in the scratch were observed to establish the DC capability of migration and adherence to plastic.
Migration assay
We used semi-mature DC collected after 6 hrs of maturation for migration assay. Chemotaxis of DC in response to CCL21 chemokine was tested in 24-well plate transwell permeable supports with 5 μm pore size polycarbonate membrane (Corning Inc., Corning, NY, USA), according to [32] with a few modifications. Briefly, we placed 600 μl of CellGro DC medium, alone or supplemented with 1–100 ng/ml CCL21 (R&D Systems), into the lower compartment, and 105 DC in 100 μl of culture medium into the upper insert. Plates were incubated for 18 hrs in a humidified incubator in 5% CO2 at 37°C. Then we harvested cells from the lower compartment using accutase (PAA, Pasching, Austria) for 10 min. at 37°C to release adherent cells. Cells were centrifuged and resuspended in 300 μl of culture medium. The relative count of migrated DC was determined as events counted in a fixed time period of 60 sec. by FACSCanto II cytometer. Data were analysed with BD FACSDiva Software.
T cell proliferation assay
Mixed leucocyte reaction was performed with autologous or allogeneic T cells labelled with 2.5 μM of carboxyfluorescein succinimidyl ester (CFSE; Invitrogen, Eugene, OR, USA) for 10 min. at 37°C to track their proliferation. DC were harvested after 24 hrs of maturation and mixed with 105 CFSE-labelled T cells in DC:T ratios of 1:5, 1:10 and 1:20. As a negative control, spontaneous T cell proliferation with no DC was measured. As a positive control, 20 μg/ml of phytohemagglutinin (Sigma-Aldrich) was added to set maximal T cell proliferation. Cells were cultured in X-VIVO 10 (Lonza, Verviers, Belgium) supplemented with 2% heat inactivated human AB serum (Sigma-Aldrich) for 6 days. Then, cells were collected and analysed for expression of CD3 (Beckman Coulter, Fullerton, CA, USA), CD4, CD8 (Exbio, Prague, Czech Republic) and CFSE. The analysis was performed on a FACSCanto II cytometer using BD FACSDiva Software. CFSE-low cells were considered as proliferating. Results were calculated as: % proliferation = (proliferating experimental−proliferating spontaneous)/(proliferating maximal−proliferating spontaneous) × 100.
T cell activation assay
For cytokine production analysis, we cultured DC with 106 unstained autologous or allogeneic T cells at a ratio of DC:T cells 1:5. Same culture medium and conditions were used as in the proliferation assay. The IFN-γ and TNF-α release from activated T cells was measured in the cell culture supernatant using a Cytometric Bead Array kit on a FACSArray Bioanalyzer. As TNF-α is produced during early T cell activation by DC while IFN-γ is produced later during this process [33], we quantified IFN-γ production after 6 days of co-culture, whereas TNF-α production was quantified after 24 hrs. The exact time points were selected based on method optimization (data not shown). After 6 days of co-culture, activated T cells were further used for cytotoxicity assay.
Cytotoxicity assay
Autologous or allogeneic T cells (as effector cells), previously exposed to DC pulsed with T98G-tumour lysate, were cultured with 105 CFSE-labelled fresh tumour cells (as target cells). Those were either T98G cells, or cells of control tumour cell lines HCT116 (American Type Culture Collection) or RPMI-8226 (European Collection of Cell Cultures). An effector/target ratio of 10:1 was used. In all cases, extra wells were set up for determining spontaneous death of target cells (no T cells were added). Cells were incubated in X-VIVO 10 and 2% heat inactivated human AB serum for 24 hrs, then collected and analysed on a FACSCanto II cytometer. PI was added immediately before measurement to detect dead cells. CFSE+PI+ cells were evaluated as dead tumour cells. Data were analysed with BD FACSDiva Software. Results were calculated as: %cytotoxic lysis = %dead experimental−%dead spontaneous.
Statistical analysis
Results of individual techniques were obtained from at least six (maximum 17) different donors tested repeatedly in pair designed experiments. Data were analysed using Friedman anova followed by the non-parametric Wilcoxon matched pair test without correction, using commercial software (Statistica 9.1; StatSoft Inc., Tulsa, OK, USA). All possible pairs of DC (DC1 versus DC2, DC1 versus DC3, and etc.) were compared statistically. Significance was accepted at the level of P < 0.05.
Results
DC surface marker expression
DC were pulsed with tumour lysate to initiate the maturation process and then further matured with different agents (DC1–DC5) or kept without further maturation stimulus (DC6) as shown in Table 1. Significant differences were observed in DC viability, CD80, CD83, CD86 and CD14 expression after 48 hrs of maturation (Fig. 1). DC3 and DC4 expressed higher levels of CD83 and also CD86 than did the four other DC alternatives (P < 0.05). Higher expression of CD80 was observed in DC3, DC4 and DC5 (P < 0.05). No significant differences were observed in HLA-DR expression. As expected, the monocyte marker CD14 was expressed in DC6 (85.3%), but partially also in DC2 (19.9%). Cells treated with other maturation cocktails contained less than 6.0% of CD14+ monocytes (P < 0.05). In summary, the fully mature phenotype (CD80+, CD83+, CD86+, HLA-DR+ and CD14-) was achieved only in DC3 and DC4. Viability over 93% was reached in all DC except of DC5 (87.9% living cells, P < 0.05).
Fig. 1.

DC surface markers and viability. Immature DC were differentiated from monocytes by 6-day culture in the presence of GM-CSF and IL-4. Maturation was induced by TNF-α (DC1); TNF-α, IL-1α, IL-6, PGE2 (DC2); TNF-α, IL-1β, IFN-γ, PGE2, R848 (DC3); IFN-γ, LPS (DC4) or IFN-γ, R848 (DC5). DC6 is a control using no maturation cocktail. Expression of CD80, CD83, CD86, HLA-DR, CD14 and cell viability were evaluated by flow cytometry after 48 hrs in the gate of cells with high FSC/SSC. (A) Data are presented as the median (▪), 25–75% quantiles (box), and non-outlier range (whiskers) of 17 donors. Marker ▲ indicates significant difference from all groups not indicated by this marker, P < 0.05, Wilcoxon matched pair test. (B) Representative gating strategy and representative histograms for particular tested parameters are shown.
IL-12p70 and IL-10 cytokine production by DC
IL-12p70 has been considered an important cytokine with Th1 polarizing and cytotoxic T cell stimulatory effect. We were able to detect broad variability in IL-12p70 production after treatment with the different maturation stimuli. The ratio of IL-12p70 and its antagonist IL-10 was calculated for each experiment. DC4 produced the highest amounts of IL-12p70 (median 3710 pg/ml, P < 0.05), followed by DC3 and DC5 (Fig. 2A). As expected, IL-12p70 production was followed by IL-10 immunosuppressive feedback. Calculating the IL-12p70/IL-10 ratio revealed the highest pro-stimulatory potential for DC4 (median 13.2, P < 0.05), again followed by DC3 and DC5 (median over 6.0, P < 0.05). In the cases of DC1, DC2 and DC6, the IL-12p70 production was negligible (<10 pg/ml) and the IL-12p70/IL-10 ratio was <1 (Fig. 2B).
Fig. 2.
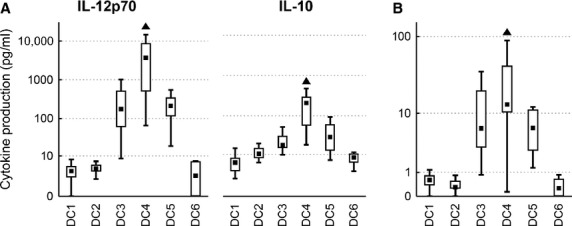
DC cytokine profiles. DC were matured with TNF-α (DC1); TNF-α, IL-1α, IL-6, PGE2 (DC2); TNF-α, IL-1β, IFN-γ, PGE2, R848 (DC3); IFN-γ, LPS (DC4); IFN-γ, R848 (DC5) or were cultured without maturation (DC6). Box plots represent (A) production of IL-12p70 and IL-10 after 24 hrs of maturation, and (B) the ratio of IL-12p70/IL-10 production. Data are presented as the median (▪), 25–75% quantiles (box), and non-outlier range (whiskers) of 17 donors. Marker ▲ indicates significant difference from all groups not indicated by this marker, P < 0.05, Wilcoxon matched pair test.
DC migratory ability
We observed differences in DC adherence to plastic and migratory capacity. We documented high adherence and minimal migration for DC4 and DC5 in a scratch assay. In these DC types, the scratch was almost empty even after 24 hrs (not shown). On the other hand, DC1, DC2 and DC6 exhibited higher migratory ability, as they filled in the scratch in less than 6 hrs (Fig. 3). As the scratch assay provides only semi-quantitative results, we proceeded with a transwell migration assay to establish the DC migratory ability more precisely. Spontaneous as well as CCL21 chemokine-induced migration was assessed in the transwell migration assay. DC3 exhibited the highest migratory capacity in both spontaneous (median 6719 events) and CCL21-induced migration (4175 events, P < 0.05) compared with all other alternatives. Higher migratory potential was also observed in DC2 and DC6 in both spontaneous and CCL21-induced migration. The lowest migration ability was documented in DC4 and DC5 (medians below 600 events) in both spontaneous and CCL21-induced migration (Fig. 4A and B). Interestingly, we did not observe a positive effect of CCL21 on DC migration in most cases (Fig. 4C). This finding corresponds to relatively low expression of CCR7 on all types of DC (Fig. 4D).
Fig. 3.
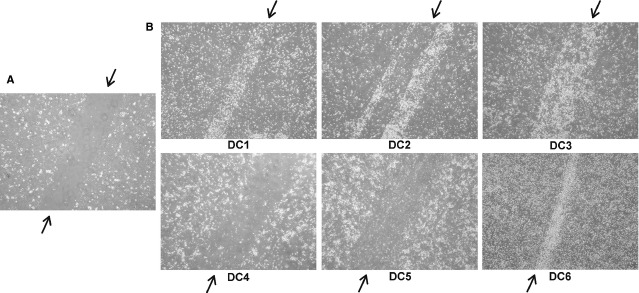
DC adherence to plastic tested in a ‘scratch assay’. DC were matured with TNF-α (DC1); TNF-α, IL-1α, IL-6, PGE2 (DC2); TNF-α, IL-1β, IFN-γ, PGE2, R848 (DC3); IFN-γ, LPS (DC4); IFN-γ, R848 (DC5) or were cultured without maturation (DC6). Scratch assay was performed after 24 hrs of maturation. The contrast of all pictures has been increased artificially and equally to facilitate the observation of the differences among DC types. (A) Scratch assay, 0 hr: a scratch without cells created in a DC monolayer by a pipette tip. The scratch is marked with an arrow on its both ends. (B) Scratch assay, after 6 hrs: adherent cells do not fill in the scratch, whereas migrating cells do. Each scratch is marked with an arrow. Pictures represent results obtained from four different donors, magnification of 400.
Fig. 4.
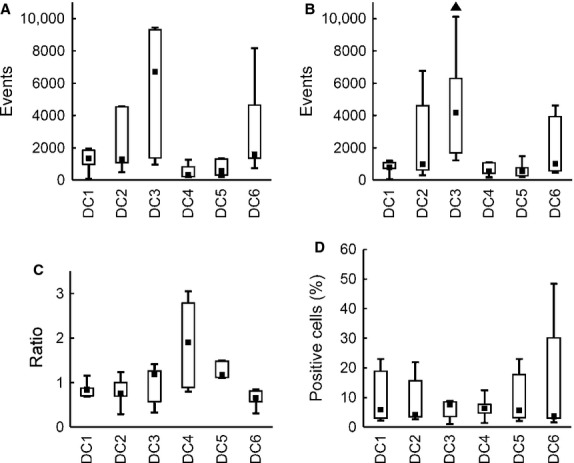
DC in vitro migratory capacity. DC were matured with TNF-α (DC1); TNF-α, IL-1α, IL-6, PGE2 (DC2); TNF-α, IL-1β, IFN-γ, PGE2, R848 (DC3); IFN-γ, LPS (DC4); IFN-γ, R848 (DC5) or were cultured without maturation (DC6). Box plots represent (A) spontaneous DC migration (no CCL21 added), (B) migration towards 100 ng/ml CCL21, (C) the ratio of CCL21-induced migration/spontaneous migration and (D) CCR7 expression by DC. Data are presented as the median (▪), 25–75% quantiles (box), and non-outlier range (whiskers) of three (A, B, C) or five (D) independent experiments, two donors per group. Marker ▲ indicates significant difference from all groups not indicated by this marker, P < 0.05, Wilcoxon matched pair test.
T cell stimulatory capacity
To evaluate the T cell stimulatory capacity of tested DC, mixed leucocyte reaction with autologous and allogeneic T cells was employed. Proliferation of total (CD3+), helper (CD3+CD4+) and cytotoxic (CD3+CD8+) T cells was analysed, as well as the release of IFN-γ and TNF-α. The results for T cell proliferation are shown for DC:T cell ratio 1:5, representing the trend observed in all three tested ratios. Using autologous T cells, the highest stimulatory capacity was observed for MC4 in all tested parameters. DC4 induced proliferation in 27.9% of autologous CD3+ T cells (median value, P < 0.05), whereas median T cell proliferation did not exceed 9% in the other five alternatives. Both helper CD4+ (median 29.7%) and cytotoxic CD8+ T cells (median 12.3%) were stimulated by DC4. Using the other five maturation strategies, autologous stimulation led to less than 11.0 and 2.1% proliferation of helper and cytotoxic T cells respectively (P < 0.05, Fig. 5A). DC4 was also superior for IFN-γ and TNF-α production (medians 2697 and 208 pg/ml, respectively, P < 0.05, Fig. 6A). The results were similar for allogeneic T cells. Same as in the autologous setting, the highest IFN-γ and TNF-α production by activated T cells was observed after DC4 stimulation (P < 0.05, Fig. 6B). The only difference was that the highest T cell proliferation was observed both upon DC3 and DC4 stimulation (P < 0.05, Fig. 5B).
Fig. 5.

Autologous and allogeneic T cell proliferation in a mixed leucocyte reaction. DC matured with TNF-α (DC1); TNF-α, IL-1α, IL-6, PGE2 (DC2); TNF-α, IL-1β, IFN-γ, PGE2, R848 (DC3); IFN-γ, LPS (DC4); IFN-γ, R848 (DC5) or without maturation (DC6) were mixed with CFSE-labelled (A) autologous or (B) allogeneic T cells. Proliferation of total (CD3+), helper (CD3+CD4+) and cytotoxic (CD3+CD8+) T cells was analysed by flow cytometry after 6 days of co-cultivation. CFSE-negative cells were considered as proliferating. The results are shown for DC:T cell ratio of 1:5. Data are presented as the median (▪), 25–75% quantiles (box) and non-outlier range (whiskers) of (A) 17 or (B) 6 donors. Marker ▲ indicates significant difference from all groups not indicated by this marker, P < 0.05, Wilcoxon matched pair test. (C) Representative gating strategies and representative histograms for particular tested parameters are shown.
Fig. 6.
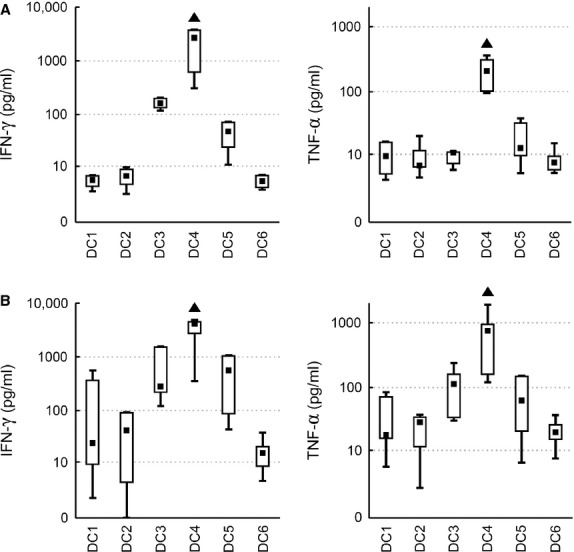
Cytokine production of autologous and allogeneic T cells activated by DC. DC matured with TNF-α (DC1); TNF-α, IL-1α, IL-6, PGE2 (DC2); TNF-α, IL-1β, IFN-γ, PGE2, R848 (DC3); IFN-γ, LPS (DC4); IFN-γ, R848 (DC5) or without maturation (DC6) were mixed with non-stained (A) autologous or (B) allogeneic T cells in a DC:T cell ratio of 1:5. Production of IFN-γ and TNF-α by activated T cells was measured with cytometric bead array. Data are presented as the median (▪), 25–75% quantiles (box), and non-outlier range (whiskers) of at least six donors. Marker ▲ indicates significant difference from all groups not indicated by this marker, P < 0.05, Wilcoxon matched pair test.
Cytotoxicity induction
To detect antigen-controlled specific cytotoxic effect, we stimulated autologous and allogeneic T cells with T98G-tumour lysate-loaded and differently matured DC. Then we mixed the stimulated T cells with T98G cells as target cells. As a control, other tumour cell lines HCT116 and RPMI-8226 were used the same way as T98G cells. Autologous T cells demonstrated the highest cytotoxic potential after DC4 stimulation (median of 41% dead T98G target cells, P < 0.05) compared with other DC (median of less than 13% dead T98G target cells). Similarly, allogeneic T cells exhibited the highest cytotoxicity when stimulated with DC4 (median of 59% dead T98G target cells, P < 0.05) followed by DC3 (median 46%, P < 0.05). Reaction against control cell lines HCT116 and RPMI-8226 showed partial cytotoxic effect in both autologous and allogeneic T cells (Fig. 7A and B).
Fig. 7.
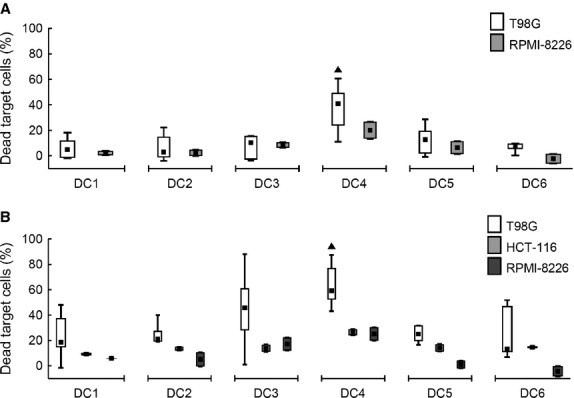
Cytotoxic effect of autologous and allogeneic T cells induced by DC. DC were loaded with a tumour lysate of a T98G tumour cell line and matured with TNF-α (DC1); TNF-α, IL-1α, IL-6, PGE2 (DC2); TNF-α, IL-1β, IFN-γ, PGE2, R848 (DC3); IFN-γ, LPS (DC4); IFN-γ, R848 (DC5) or cultured without maturation (DC6). Autologous or allogeneic T cells, co-cultured with those DC for 6 days, were mixed in a ratio 10:1 with CFSE-labelled fresh tumour cells. Those were target cells T98G, or control cells HCT116 and RPMI-8226 (as indicated in the figure legend). After 24 hrs of co-incubation, dead tumour cells were quantified on a flow cytometer. Results for cytotoxicity induced by (A) autologous and (B) allogeneic T cells are shown. Data are presented as the median (▪), 25–75% quantiles (box), and non-outlier range (whiskers) of at least 6 donors for T98G and two donors for both HCT116 and RPMI-8226 cell lines. Marker ▲ indicates significant difference from all T98G groups not indicated by this marker, P < 0.05, Wilcoxon matched pair test.
Discussion
In the last decade, various strategies to generate DC suitable for clinical application were described by many research groups. It can be difficult to decide which maturation strategy to choose for DC use for clinical application to obtain optimal tumour-antigen-specific T cell activation. Surprisingly, the broader comparison of more than two or three DC maturation strategies is missing. Thus, we aim to study 6 recently described approaches for a clinical-grade DC preparation and compare them using unified panel of functional tests. These tests were chosen to describe the most critical aspects and functions of DC suitable for cancer immunotherapy. Our data demonstrate that among others, the combination of IFN-γ and LPS generates DC with superior characteristics. These DC exhibit fully mature phenotype, the highest IL-12p70 production and stimulate T cell proliferation as well as their specific cytotoxic activity. These characteristics were previously identified as critical for cancer immunotherapy [9]. Partially beneficial characteristics were obtained using the combination of TNF-α, IL-1β, IFN-γ, R848 and PGE2 for DC maturation.
As IFN-γ reduces migratory ability of DC [24], DC matured with IFN-γ and LPS do not migrate well. Also high-IL-12p70 production is associated with decreased migratory capacity of DC [24,32,34]. This limitation can be easily overcome by direct and safe administration of DC directly into the lymph nodes [35–40].
Donor-dependent differences were observed in DC migratory capacity. That can indicate the possibility that DC migration does not depend only on the maturation strategy used, but also on individual characteristics of the donor. We can expect similar DC behaviour in vivo, resulting in personal variability in DC migration. Intranodal application of the DC could eliminate the effect of such personal-dependent variability, while leading to direct stimulation of lymph node T cells.
DC ability to migrate towards lymph node plays an important role when the vaccine is applied subcutaneously or intradermally. When injected intranodally, even week DC migration could be satisfactory to mediate DC interaction with T cells. In this study, the in vivo migratory ability of DC was not tested. However, it was demonstrated recently that weekly migrating DC can raise effective tumour-specific T cell responses in vivo when administrated intranodally [36,40].
The use of LPS was avoided by some authors for its possible direct toxicity [28]. However, as free LPS is not present after careful washing out before DC are administered, the risk of direct LPS toxicity is negligible. The safety of LPS-matured DC administration was already tested in previously published clinical trials with none or only minor adverse effects [36,40,41]. Clinical-grade LPS was used in these trials and is available up to date. It can be obtained from, e.g. US Pharmacopeia, Rockville, MD, USA [36].
Other TLR4 ligands have been tested recently to obtain DC with favourable characteristics [25,28,42]. Further exploration of the possible interchangeability of LPS and these LPS analogues (e.g. monophosphoryl lipid A and Angelan) within the advantageous DC maturation cocktails could bring another interesting insight into this issue.
All tests in our study were performed with healthy volunteers' cells and we can speculate that cancer patient immune cells can be compromised by the presence of cancer itself and/or by previous anti-cancer therapy. To what extent this may limit a therapeutic potential of such DC vaccination is not clear and further studies are urgently needed. At least prior to clinical vaccination, patient's cells can be tested in vitro for their individual reactivity against autologous tumour cells. Such testing, as proposed in this study, can identify patients who may benefit from DC vaccination and possibly lead to better clinical outcome of DC immune therapy.
We decided to perform all experiments with a tumour cell lysate as a source of tumour antigen. Despite there are some limitations of such approach, it is not HLA restricted, it is broadly available generally for each cancer patients undergoing surgery, and represents the broad spectrum of tumour antigens specific for the particular patient. Here, we also demonstrate that, in comparison to immature DC (DC6) that are usually associated with tolerance induction [4], the IL-12p70-producing DC matured with IFN-γ and LPS can easily overcome the tolerance and lead to strong cytotoxic T cell activation even in an autologous setting.
The cytotoxic reaction was performed in T cells activated previously by co-cultivation with tumour lysate-pulsed DC. For production of tumour lysate we have chosen a glioblastoma cell line T98G, which was also used as a target in the cytotoxicity assay. As controls for this assay we used tumour cell lines derived from different types of cancer, e.g. colon cancer (HCT116) and multiple myeloma (RPMI-8226), to ensure that the results will not be influenced by possible similarity in the presented tumour antigens. However, some universal tumour antigens (e.g. hTERT) can be expressed by all cell lines used. That could partially contribute to T cell cytotoxicity against non-target cell lines.
We documented that DC matured with TNF-α, IL-1β, IFN-γ, R848 and PGE2 are potent inducers of allogeneic but not autologous T cell proliferation. We also observed extensive variability in DC ability to induce allogeneic T cell proliferation among different donors. This was probably caused by differences in HLA antigens between donor-derived DC and allogeneic T cells. Allogeneic setting is often tested in vitro and frequently reported in pre-clinical studies. Unfortunately, such in vitro design is rather artificial because in clinical practice, dominantly autologous DC are used for vaccination. There are very limited pre-clinical data demonstrating DC stimulatory capacity to autologous T cells which may also explain some failures in establishing clinical responses after DC vaccinations that tested only allogeneic stimulatory capacity without any knowledge of autologous setting.
We also demonstrated that DC matured with TNF-α only, with combination of TNF-α, IL-1, IL-6 and PGE2 or DC generated without maturation stimulus are not ideal candidates for cancer immunotherapy. Their low capacity of IL-12p70 production and low expression of CD83 and co-stimulatory molecules in consequence leads to low immunostimulatory capacity [20,43–45].
It was described previously that PGE2 supports the yield, quality and migratory ability of DC [24,32,34,46]. However, later studies showed that PGE2 inhibits IL-12p70 secretion from DC in response to CD40 ligation after they come into the lymph nodes and directs the DC development into the Th2-stimulating DC [47]. Maturation strategies, which were testified to elicit sufficient anti-tumour cytotoxicity in vitro, often avoid using PGE2 [20,23,25].
In conclusion, the DC matured with IFN-γ and LPS exhibit the most favourable characteristics for cancer immunotherapy compared with the other maturation cocktails tested.
At least two clinical trials using DC matured with IFN-γ and LPS have been published [36,40,41]. In both of them, immune responses to the vaccination were observed. Significant increase in intracellular IFN-γ level in both helper and cytotoxic T cells was documented after DC vaccination and the delayed-type hypersensitivity skin test performed before and after vaccination documented a skin reaction in response to tumour antigen in three of six patients after vaccination [36]. In breast cancer patients, the level of tumour marker HER-2/neu decreased after DC vaccination [40,41]. However, phase II clinical trials proving clinical efficacy of such DC type have not yet been published. Further clinical trials are needed to demonstrate the in vivo effect of such immunotherapeutic approach.
Acknowledgments
We thank the Transfusion Department and Blood Bank of the University Hospital Brno for providing the healthy donors' blood samples and the Institute of Biostatistics and Analyses of the Masaryk University, Brno, for consulting the statistical analysis. This work was supported by grants from the Ministry of Health, Czech Republic (IGA NT 11137-5 and IGA NS9871–4 to J.M.), grants from the Ministry of Education, Youth and Sports, Czech Republic (NPVII 2B06058 and NPVII 2B08066 to J.M.) and via the International Consortium for Cell Therapy and Immunotherapy (CZ.1.07/2.3.00/20.0012).
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–52. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 2.Steinman RM, Banchereau J. Taking dendritic cells into medicine. Nature. 2007;449:419–26. doi: 10.1038/nature06175. [DOI] [PubMed] [Google Scholar]
- 3.Schuler G, Schuler-Thurner B, Steinman RM. The use of dendritic cells in cancer immunotherapy. Curr Opin Immunol. 2003;15:138–47. doi: 10.1016/s0952-7915(03)00015-3. [DOI] [PubMed] [Google Scholar]
- 4.Banchereau J, Palucka AK. Dendritic cells as therapeutic vaccines against cancer. Nat Rev Immunol. 2005;5:296–306. doi: 10.1038/nri1592. [DOI] [PubMed] [Google Scholar]
- 5.Figdor CG, de Vries IJ, Lesterhuis WJ, et al. Dendritic cell immunotherapy: mapping the way. Nat Med. 2004;10:475–80. doi: 10.1038/nm1039. [DOI] [PubMed] [Google Scholar]
- 6.Berger TG, Strasser E, Smith R, et al. Efficient elutriation of monocytes within a closed system (Elutra) for clinical-scale generation of dendritic cells. J Immunol Methods. 2005;298:61–72. doi: 10.1016/j.jim.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 7.Anguille S, Smits E, Cools N, et al. Short-term cultured, interleukin-15 differentiated dendritic cells have potent immunostimulatory properties. J Transl Med. 2009;7:109. doi: 10.1186/1479-5876-7-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schreibelt G, Tel J, Sliepen KH, et al. Toll-like receptor expression and function in human dendritic cell subsets: implications for dendritic cell-based anti-cancer immunotherapy. Cancer Immunol Immunother. 2010;59:1573–82. doi: 10.1007/s00262-010-0833-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schuler G. Dendritic cells in cancer immunotherapy. Eur J Immunol. 2010;40:2123–30. doi: 10.1002/eji.201040630. [DOI] [PubMed] [Google Scholar]
- 10.Tuyaerts S, Aerts JL, Corthals J, et al. Current approaches in dendritic cell generation and future implications for cancer immunotherapy. Cancer Immunol Immunother. 2007;56:1513–37. doi: 10.1007/s00262-007-0334-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Skalova K, Mollova K, Michalek J. Human myeloid dendritic cells for cancer therapy: does maturation matter? Vaccine. 2010;28:5153–60. doi: 10.1016/j.vaccine.2010.05.042. [DOI] [PubMed] [Google Scholar]
- 12.Higano CS, Schellhammer PF, Small EJ, et al. Integrated data from 2 randomized, double-blind, placebo-controlled, phase 3 trials of active cellular immunotherapy with sipuleucel-T in advanced prostate cancer. Cancer. 2009;115:3670–9. doi: 10.1002/cncr.24429. [DOI] [PubMed] [Google Scholar]
- 13.Bürdek M, Spranger S, Wilde S, et al. Three-day dendritic cells for vaccine development: antigen uptake, processing and presentation. J Transl Med. 2010;8:90. doi: 10.1186/1479-5876-8-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Curti A, Trabanelli S, Onofri C, et al. Indoleamine 2,3-dioxygenase-expressing leukemic dendritic cells impair a leukemia-specific immune response by inducing potent T regulatory cells. Haematologica. 2010;95:2022–30. doi: 10.3324/haematol.2010.025924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Soleimani A, Berntsen A, Svane IM, et al. Immune responses in patients with metastatic renal cell carcinoma treated with dendritic cells pulsed with tumor lysate. Scand J Immunol. 2009;70:481–9. doi: 10.1111/j.1365-3083.2009.02322.x. [DOI] [PubMed] [Google Scholar]
- 16.Jarnjak-Jankovic S, Hammerstad H, Saebøe-Larssen S, et al. A full scale comparative study of methods for generation of functional den- dritic cells for use as cancer vaccines. BMC Cancer. 2007;7:119. doi: 10.1186/1471-2407-7-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adamson L, Palma M, Choudhury A, et al. Generation of a dendritic cell-based vaccine in chronic lymphocytic leukaemia using CliniMACS platform for large-scale production. Scand J Immunol. 2009;69:529–36. doi: 10.1111/j.1365-3083.2009.02249.x. [DOI] [PubMed] [Google Scholar]
- 18.Ocadlíková D, Zahradová L, Kovárová L, et al. The preparation of anticancer vaccine for patients with multiple myeloma on the base of monoclonal immunoglobulin loaded dendritic cells. Klin Onkol. 2009;22:67–72. [PubMed] [Google Scholar]
- 19.Palmer DH, Midgley RS, Mirza N, et al. A phase II study of adoptive immunotherapy using dendritic cells pulsed with tumor lysate in patients with hepatocellular carcinoma. Hepatology. 2009;49:124–32. doi: 10.1002/hep.22626. [DOI] [PubMed] [Google Scholar]
- 20.Mailliard RB, Wankowicz-Kalinska A, Cai Q, et al. Alpha-type-1 polarized dendritic cells: a novel immunization tool with optimized CTL-inducing activity. Cancer Res. 2004;64:5934–7. doi: 10.1158/0008-5472.CAN-04-1261. [DOI] [PubMed] [Google Scholar]
- 21.Zobywalski A, Javorovic M, Frankenberger B, et al. Generation of clinical grade dendritic cells with capacity to produce biologically active IL-12p70. J Transl Med. 2007;5:18. doi: 10.1186/1479-5876-5-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang D, Kim M, Hong CY, et al. Alpha-type 1-polarized dendritic cells loaded with apoptotic allogeneic myeloma cell line induce strong CTL responses against autologous myeloma cells. Ann Hematol. 2010;89:795–801. doi: 10.1007/s00277-010-0931-3. [DOI] [PubMed] [Google Scholar]
- 23.Felzmann T, Hüttner KG, Breuer SK, et al. Semi-mature IL-12 secreting dendritic cells present exogenous antigen to trigger cytolytic immune responses. Cancer Immunol Immunother. 2005;54:769–80. doi: 10.1007/s00262-004-0637-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lehner M, Stilper A, Morhart P, et al. Plasticity of dendritic cell function in response to prostaglandin E2 (PGE2) and interferon-gamma (IFN-gamma) J Leukoc Biol. 2008;83:883–93. doi: 10.1189/jlb.0307153. [DOI] [PubMed] [Google Scholar]
- 25.Ten Brinke A, van Schijndel G, Visser R, et al. Monophosphoryl lipid A plus IFNgamma maturation of dendritic cells induces antigen-specific CD8 + cytotoxic T cells with high cytolytic potential. Cancer Immunol Immunother. 2010;59:1185–95. doi: 10.1007/s00262-010-0843-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim S, Kim HO, Kim HJ, et al. Generation of functionally mature dendritic cells from elutriated monocytes using polyinosinic: polycytidylic acid and soluble CD40 ligand for clinical application. Clin Exp Immunol. 2008;154:365–74. doi: 10.1111/j.1365-2249.2008.03757.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang X, Fan Z, Borowski L, et al. Dendritic cells reveal a broad range of MHC class I epitopes for HIV-1 in persons with suppressed viral load on antiretroviral therapy. PLoS ONE. 2010;5:e12936. doi: 10.1371/journal.pone.0012936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ten Brinke A, Karsten ML, Dieker MC, et al. The clinical grade maturation cocktail monophosphoryl lipid A plus IFNgamma generates monocyte-derived dendritic cells with the capacity to migrate and induce Th1 polarization. Vaccine. 2007;25:7145–52. doi: 10.1016/j.vaccine.2007.07.031. [DOI] [PubMed] [Google Scholar]
- 29.Hovden A, Karlsen M, Jonsson R, et al. Maturation of monocyte derived dendritic cells with OK432 boosts IL-12p70 secretion and conveys strong T-cell responses. BMC Immunol. 2011;12:2. doi: 10.1186/1471-2172-12-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaka AS, Foster AE, Weiss HL, et al. Using dendritic cell maturation and IL-12 producing capacity as markers of function: a cautionary tale. J Immunother. 2008;31:359–69. doi: 10.1097/CJI.0b013e318165f5d2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Langenkamp A, Messi M, Lanzavecchia A, et al. Kinetics of dendritic cell activation: impact on priming of TH1, TH2 and nonpolarized T cells. Nat Immunol. 2000;1:311–6. doi: 10.1038/79758. [DOI] [PubMed] [Google Scholar]
- 32.Boullart ACI, Aarntzen EH, Verdijk P, et al. Maturation of monocyte-derived dendritic cells with Toll-like receptor 3 and 7/8 ligands combined with prostaglandin E2 results in high interleukin-12 production and cell migration. Cancer Immunol Immunother. 2008;57:1589–97. doi: 10.1007/s00262-008-0489-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pacheco R, Martinez-Navio JM, Lejeune M, et al. CD26, adenosine deaminase, and adenosine receptors mediate costimulatory signals in the immunological synapse. Proc Natl Acad Sci USA. 2005;102:9583–8. doi: 10.1073/pnas.0501050102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luft T, Jefford M, Luetjens P, et al. Functionally distinct dendritic cell (DC) populations induced by physiologic stimuli: prostaglandin E2 regulates the migratory capacity of specific DC subsets. Blood. 2002;100:1362–72. doi: 10.1182/blood-2001-12-0360. [DOI] [PubMed] [Google Scholar]
- 35.Salcedo M, Bercovici N, Taylor R, et al. Vaccination of melanoma patients using dendritic cells loaded with an allogeneic tumor cell lysate. Cancer Immunol Immunother. 2006;55:819–29. doi: 10.1007/s00262-005-0078-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dohnal A, Witt V, Hügel H, et al. Phase I study of tumor Ag-loaded IL-12 secreting semi-mature DC for the treatment of pediatric cancer. Cytotherapy. 2007;9:755–70. doi: 10.1080/14653240701589221. [DOI] [PubMed] [Google Scholar]
- 37.Hersey P, Halliday GM, Farrelly ML, et al. Phase I/II study of treatment with matured dendritic cells with or without low dose IL-2 in patients with disseminated melanoma. Cancer Immunol Immunother. 2008;57:1039–51. doi: 10.1007/s00262-007-0435-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Verdijk P, Aarntzen EH, Lesterhuis WJ, et al. Limited amounts of dendritic cells migrate into the T-cell area of lymph nodes but have high immune activating potential in melanoma patients. Clin Cancer Res. 2009;15:2531–40. doi: 10.1158/1078-0432.CCR-08-2729. [DOI] [PubMed] [Google Scholar]
- 39.Yi Q, Szmania S, Freeman J, et al. Optimizing dendritic cell-based immunotherapy in multiple myeloma: intranodal injections of idiotype-pulsed CD40 ligand-matured vaccines led to induction of type-1 and cytotoxic T-cell immune responses in patients. Br J Haematol. 2010;150:554–64. doi: 10.1111/j.1365-2141.2010.08286.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sharma A, Koldovsky U, Xu S, et al. HER-2 pulsed dendritic cell vaccine can eliminate HER-2 expression and impact ductal carcinoma in situ. Cancer. 2012 doi: 10.1002/cncr.26734. doi: 10.1002/cncr.26734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Czerniecki BJ, Koski GK, Koldovsky U, et al. Targeting HER-2/neu in early breast cancer development using dendritic cells with staged interleukin-12 burst secretion. Cancer Res. 2007;67:1842–52. doi: 10.1158/0008-5472.CAN-06-4038. [DOI] [PubMed] [Google Scholar]
- 42.Kim JY, Kim YJ, Kim JS, et al. Adjuvant effect of a natural TLR4 ligand on dendritic cell-based cancer immunotherapy. Cancer Lett. 2011;313:226–34. doi: 10.1016/j.canlet.2011.09.009. [DOI] [PubMed] [Google Scholar]
- 43.Dilioglou S, Cruse JM, Lewis RE. Function of CD80 and CD86 on monocyte- and stem cell-derived dendritic cells. Exp Mol Pathol. 2003;75:217–27. doi: 10.1016/s0014-4800(03)00072-8. [DOI] [PubMed] [Google Scholar]
- 44.Aerts-Toegaert C, Heirman C, Tuyaerts S, et al. CD83 expression on dendritic cells and T cells: correlation with effective immune responses. Eur J Immunol. 2007;37:686–95. doi: 10.1002/eji.200636535. [DOI] [PubMed] [Google Scholar]
- 45.Prechtel AT, Turza NM, Theodoridis AA, et al. CD83 knockdown in monocyte-derived dendritic cells by small interfering RNA leads to a diminished T cell stimulation. J Immunol. 2007;178:5454–64. doi: 10.4049/jimmunol.178.9.5454. [DOI] [PubMed] [Google Scholar]
- 46.Jonuleit H, Kühn U, Müller G, et al. Pro-inflammatory cytokines and prostaglandins induce maturation of potent immunostimulatory dendritic cells under fetal calf serum-free conditions. Eur J Immunol. 1997;27:3135–42. doi: 10.1002/eji.1830271209. [DOI] [PubMed] [Google Scholar]
- 47.Kalinski P, Schuitemaker JH, Hilkens CM, et al. Prostaglandin E2 induces the final maturation of IL-12-deficient CD1a+CD83 + dendritic cells: the levels of IL-12 are determined during the final dendritic cell maturation and are resistant to further modulation. J Immunol. 1998;161:2804–9. [PubMed] [Google Scholar]