

Abstract
This phase 1b trial investigated several doses and schedules of midostaurin in combination with daunorubicin and cytarabine induction and high-dose cytarabine post-remission therapy in newly diagnosed patients with acute myeloid leukemia (AML). The discontinuation rate on the 50-mg twice-daily dose schedule was lower than 100 mg twice daily, and no grade 3/4 nausea or vomiting was seen. The complete remission rate for the midostaurin 50-mg twice-daily dose schedule was 80% (FLT3–wild-type: 20 of 27 [74%], FLT3-mutant: 12 of 13 [92%]). Overall survival (OS) probabilities of patients with FLT3-mutant AML at 1 and 2 years (0.85 and 0.62, respectively) were similar to the FLT3–wild-type population (0.78 and 0.52, respectively). Midostaurin in combination with standard chemotherapy demonstrated high complete response and OS rates in newly diagnosed younger adults with AML and was generally well-tolerated at 50 mg twice daily for 14 days. A phase III prospective trial is ongoing (CALGB 10603, NCT00651261).
Keywords: FMS-Like Tyrosine kinase 3 receptor (FLT3), acute myeloid leukemia (AML), midostaurin, PKC412, newly diagnosed
INTRODUCTION
Mutations in the FMS-Like Tyrosine kinase 3 receptor (FLT3) occur in approximately 25% of patients with acute myeloid leukemia (AML), cause constitutive activation, and are associated with poor prognosis.1-3 Cytogenetically normal patients with AML whose leukemia is characterized by the presence of internal tandem duplication (ITD) mutations in FLT3 (FLT3-ITD) have a significantly shorter disease-free survival (DFS) and overall survival (OS) than patients without the mutation.2,4
The prognostic significance of the FLT3 mutation has led to the pursuit of FLT3 inhibitors for the treatment of AML. Midostaurin (PKC412) is a potent kinase inhibitor of FLT3, c-KIT, PDGF-Rβ, VEGFR-2, and protein kinase C, with demonstrated activity against cell lines containing mutant FLT3 and against FLT3-induced myeloproliferative disease in a mouse model.5 Midostaurin’s two major metabolites, CGP62221 and CGP52421, are also capable of inhibiting FLT3 in vitro.6,7 In previous single-agent clinical studies with midostaurin at 75 mg three times daily8 or 50 mg or 100 mg twice daily,9 70% and 42% of patients with FLT3-mutant (n = 55) and FLT3–wild-type (n = 60) AML, respectively, had ≥ 50% peripheral blood blast reduction. Duration of response was short, and complete remissions (CRs) were not observed.
Midostaurin’s activity, although limited, prompted a search for optimization of outcomes with FLT3 inhibitors. Synergism between FLT3 inhibitors and standard chemotherapeutic agents, including daunorubicin and cytarabine, has been demonstrated in preclinical studies. These studies highlight the importance of the dosing schedule; pretreatment with the inhibitor is antagonistic with chemotherapy. FLT3 inhibition of cell cycle progression in AML cell lines renders such cells insensitive to S-phase-specific chemotherapeutic agents such as cytarabine.10
We conducted a phase 1b trial in newly diagnosed patients with wild-type and mutant FLT3 AML to examine the safety, efficacy, and pharmacokinetics of combining midostaurin with an induction regimen of daunorubicin and cytarabine followed by high-dose cytarabine consolidation. Several doses and schedules of midostaurin were investigated. After discontinuing dose schedules involving 100 mg twice daily, we found schedules of midostaurin (50 mg twice daily taken sequentially or concomitantly for 14 days per cycle) that could be combined tolerably with chemotherapy. This analysis describes 29 patients who received midostaurin 100 mg twice daily, and focuses on 40 patients who received midostaurin 50 mg twice daily.
PATIENTS AND METHODS
Patients and Objectives
Previously untreated patients, aged 18 to 60 years, diagnosed with AML according to the World Health Organization criteria and with Karnofsky performance status ≥ 70 were eligible. Exclusion criteria included known impaired gastrointestinal (GI) function or GI disease that could significantly alter the absorption of midostaurin; receipt of any investigational agent within 30 days of day 1; any surgical procedure within 14 days of day 1; an ejection fraction of < 50% as assessed by multigated acquisition scan or echocardiogram scan within 14 days of day 1; presence of pulmonary infiltrates; history of or newly diagnosed myelodysplastic syndrome; history of myeloproliferative disease or secondary AML; and prior chemotherapy (other than hydroxyurea) or radiation therapy.
The primary objectives were to evaluate the safety and tolerability of twice-daily midostaurin (50 mg and 100 mg) administered either concomitantly with standard chemotherapy or sequentially after completion of chemotherapy, and to determine the effect of midostaurin on the pharmacokinetics of daunorubicin and cytarabine. The secondary objectives were to evaluate the efficacy of midostaurin in combination with standard chemotherapy by measuring the response rate, DFS, and OS, and to investigate the effect of FLT3 mutational status on the rate of patient response.
Treatment Plan
Midostaurin 100 mg twice daily in combination with chemotherapy was administered on either a concomitant dose schedule starting on day 1 of a 28-day cycle or sequentially starting on day 8 (Fig 1, dose/schedule I). After the first 14 patients, prolonged exposure was deemed too toxic, and the study was amended to limit treatment to 14 days per chemotherapy course (days 1-7 and 15-21 of the concomitant schedule; days 8-21 of the sequential schedule) (Fig 1, dose/schedule II). Given intolerance of the 14-day-per-cycle exposure to midostaurin 100 mg twice daily, the study was again amended to reduce the dose of midostaurin to 50 mg twice daily in both the 14-day concomitant and sequential schedules (Fig 1, dose/schedule III).
Fig 1.
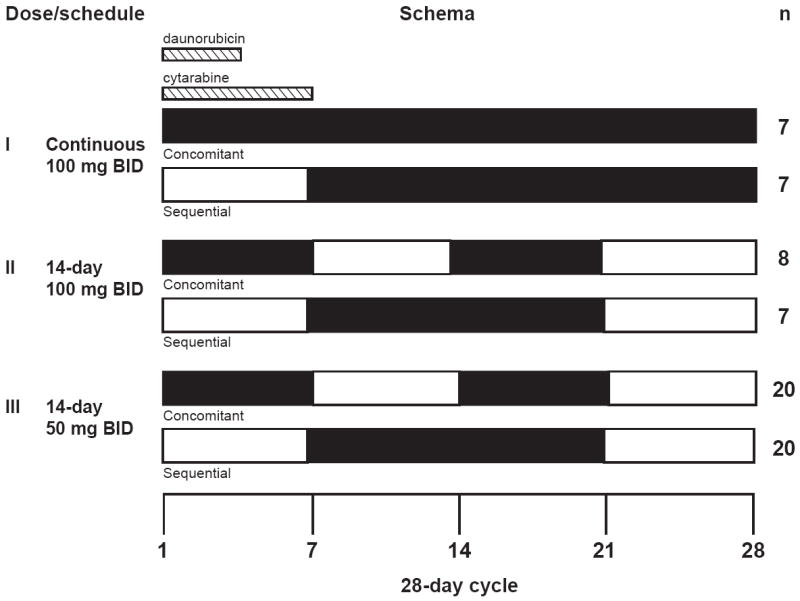
Schema of dose and schedule of midostaurin administration. Daunorubicin and cytarabine induction (3 + 7) and high-dose cytarabine post-remission therapy was administered on a standard schedule. In addition, patients received midostaurin (indicated by black bars) on one of three dose schedules: I. midostaurin 100 mg twice daily (BID) for 21 or 28 days; II. midostaurin 100 mg twice daily for 14 days; or III. midostaurin 50 mg twice daily for 14 days. Within each dose schedule, patients were assigned to receive midostaurin on day 1 (concomitant with chemotherapy, days 1-7; 14-21 in dose schedules II and III) or day 8 (sequential with chemotherapy; days 8-21 in dose schedules II and III).
Open boxes represent values from patients on the concomitant arm (day 1-7 and day 15-22 dosing); closed circles represent values from patients on the sequential arm (day 8-22 dosing).
The chemotherapy regimen consisted of a cycle of induction with daunorubicin 60 mg/m2 intravenously (IV) on days 1 to 3 and cytarabine 200 mg/m2 by continuous intravenous infusion (CIV) on days 1 to 7. A bone marrow biopsy performed between days 21 and 28 was used to determine if a second cycle of induction (daunorubicin 60 mg/m2 IV on days 1 to 2; cytarabine 200 mg/m2 CIV on days 1 to 5; and midostaurin given in the same schedule used in the initial induction course) should be administered. At the investigator’s discretion, the biopsy was delayed to insure that re-induction did not occur during administration of midostaurin in patients on the sequential arm. Patients who did not achieve CR at the end of a second cycle of induction (cycle 2) were discontinued from the study. Patients who achieved a CR at the end of cycle 1 or 2 received consolidation therapy for three cycles with high-dose cytarabine 3 g/m2 IV over 3 hours every 12 hours given every other day (days 1, 3, and 5) for six doses in addition to midostaurin administered according to the schedule assigned during induction.
Maintenance therapy with midostaurin alone was allowed after completion of the planned chemotherapy (because of the potential benefits of continuous inhibition of FLT3) and was administered for 14 days in each 28-day cycle according to the patient’s original assignment. A 50% dose reduction in midostaurin was allowed for grade 3/4 non-hematologic toxicity attributed to the drug. Midostaurin could be given at a 50% dose until toxicity was resolved, and then the drug could be re-escalated.
Study Assessments
The patient’s FLT3 mutation status was determined at baseline from bone marrow and/or blood samples. To determine the presence or absence of FLT3-ITD, FLT3 sequencing was carried out at a central laboratory (Transgenomic Labs, New Haven, CT, USA). If the central laboratory data were not available (e.g. sample was not sent or was insufficient), results from the local laboratory were used. The allelic ratio was not routinely determined. Cytogenetic abnormalities were characterized on the basis of the Cancer and Leukemia Group B criteria.11 All adverse events (AEs) were recorded, regardless of causality. Because the primary objective of the study was to determine safety and tolerability of the novel regimens, DFS duration (time from remission to relapse or death) was analyzed using investigator-reported data. However, all patients were followed for survival without censoring for alternative therapies such as stem cell transplant. Follow-up occurred every 3 months after treatment discontinuation until death.
Pharmacokinetic Analysis
Samples were collected to determine the concentration of midostaurin and metabolites CGP52421 (the monohydroxy metabolite) and CGP62221 (the desmethyl metabolite) during cycles 1 to 5. Concentrations were determined by high-performance liquid chromatography/mass spectrometry with a limit of quantification of 10 ng/mL. Plasma concentrations of daunorubicin and cytarabine (cycle 1, day 1) were analyzed separately by high-performance liquid chromatography with ultraviolet detection. The limit of quantification was 5 ng/mL for daunorubicin and 10 ng/mL for cytarabine.
Statistical Analysis
Enrolled patients (n = 69) who received at least one dose of midostaurin and/or standard chemotherapy were analyzed for both safety and efficacy. Overall survival was considered as the time from first dose of any study drug to death; otherwise, patients were censored at the date last known to be alive. Disease-free survival was considered as the time from first CR to relapse or death and was not censored for transplant.
Study Conduct
This trial was registered with ClinicalTrials.gov as Novartis-CPKC412A2106. All patients signed informed consent forms approved by the relevant Institutional Review Boards. The study was performed at four centers in the United States and two centers in Germany.
RESULTS
Dose Schedules I and II: 100 mg Twice Daily
Twenty-nine patients received midostaurin 100 mg orally twice daily, with 14 patients receiving dose schedule I (continuous dosing beginning on day 1 [n = 7] or day 8 [n = 7]) and 15 patients receiving dose schedule II (days 1-7 and 15-21 [n = 7] or days 8-21 [n = 8]) (Fig 1). The discontinuation rate was high, with 23 of 29 patients (79%) failing to complete all planned therapy (Fig 2). Gastrointestinal grade 3/4 AEs occurred at this dose: seven (24.1%) nausea, seven (24.1%) vomiting, and four (13.8%) diarrhea. Intolerable GI AEs led to discontinuation in two patients (both grade 2). The frequency and grade of other AEs occurring on dose schedules I and II were similar to that seen for patients treated with midostaurin 50 mg twice daily on dose schedule III (data not shown).
Fig 2.
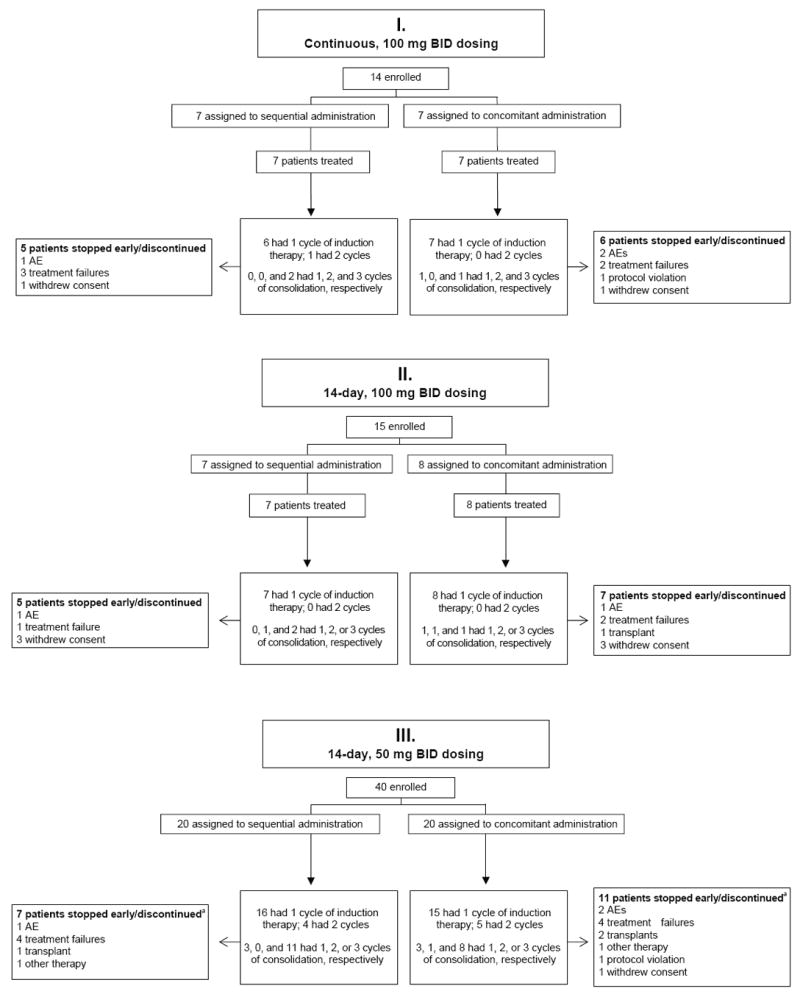
CONSORT flow diagram. Depicts patient numbers for enrollment, intervention allocation, and follow-up. Abbreviations: AE, adverse event; BID, twice daily.
a Three patients (n= 2 from the sequential arm; n = 1 from the concomitant arm) discontinued in complete remission before completing consolidation and were considered as completers by investigators.
Open boxes represent values from patients on the concomitant arm (day 1-7 and day 15-22 dosing); closed circles represent values from patients on the sequential arm (day 8-22 dosing).
Complete remission was achieved by 13 of 29 patients (45%), including 8 of 23 patients (35%) with FLT3–wild-type blasts and five of six patients (83%) with FLT3-mutant blasts (three ITD and two tyrosine kinase domain [TKD] mutations). One patient from dose schedule I with FLT3–wild-type blasts received two cycles of induction and did not respond. Two patients (33%) with FLT3-mutant blasts and nine patients (39%) with FLT3–wild-type blasts survived more than 4 years.
Dose Schedule III: 50 mg Twice Daily
Patients on dose schedule III received midostaurin 50 mg twice daily either concomitantly (days 1-7 and 15-21; n = 20) with chemotherapy or sequentially (days 8-21; n = 20) after chemotherapy (Fig 1). FLT3 mutations were noted in 13 of 40 patients: nine with FLT3-ITD mutations and four with TKD mutations. As expected, most patients (77%) with FLT3-mutated blasts displayed normal cytogenetics (Table 1).11 The percentage of patients with the FLT3 mutation in the sequential and concomitant arms was similar. The discontinuation rate in the 14-day 50-mg twice-daily arm (45%) was lower than the rate in the 14-day 100-mg twice-daily arm (80%).
Table 1. Characteristics of Patients Treated on Dose Schedule III (n = 40) by FLT3 Mutation Status.
| Characteristic,n (%) | FLT3–Wild-Type n = 27 |
FLT3-Mutant n= 13 |
Total N = 40 |
|---|---|---|---|
| Sex, male | 17 (63) | 7 (54) | 24 (60) |
| Age ≤ 60 years [median] | 27 (100) [50] | 12* (92) [46] | 39 (98) [48.5] |
| Karnofsky performance score | |||
| 100 | 7 (26) | 1 (8) | 8 (20) |
| 90 | 11 (41) | 3 (23) | 14 (35) |
| 80 | 4 (15) | 3 (23) | 7 (18) |
| 70 | 3 (11) | 3 (23) | 6 (15) |
| Unknown | 2 (7) | 3 (23) | 5 (13) |
| Cytogenetics11 | |||
| Adverse | 7 (26) | 2 (15) | 9 (23) |
| Intermediate (excluding normal) | 7 (26) | 1 (8) | 8 (20) |
| Favorable | 5 (19) | 0 | 5 (13) |
| Normal | 5 (19) | 10 (77) | 15 (38) |
| Unknown | 3 (11) | 0 | 3 (8) |
| White blood cell count | |||
| ≤ 50,000 × 109/L [median] | 21 (78) [9000] | 12 (92) [16,000] | 33 (83) [13,000] |
| Dosage schedule | |||
| Sequential | 13 (48) | 7 (54) | 20 (50) |
| Concomitant | 14 (52) | 6 (46) | 20 (50) |
| FLT3 mutation type | |||
| ITD | NA | 9 (69) | NA |
| Point mutation | NA | 4 (31) | NA |
Patient aged 65 years at enrollment was a protocol violation, but was included in all analyses.
Abbreviations: NA, not applicable; ITD, internal tandem duplication.
Efficacy: 50 mg Twice Daily
The CR rate (80% overall) was higher among patients with FLT3-mutant AML (12 of 13 patients [92%] compared with 20 of 27 patients [74%] with wild-type FLT3). The single patient with FLT3-mutant AML who did not respond had an ITD mutation consisting of a 43 base-pair insertion. The dosage schedule did not affect the rate of CR; 16 of 20 patients (80%) in both the sequential and concomitant groups achieved a CR. Of the patients who achieved CR, nine of 12 patients (75%) with FLT3-mutant AML and 15 of 20 patients (75%) with FLT3–wild-type disease achieved CR after the first cycle of induction.
Kaplan-Meier OS probabilities at 1 and 2 years, respectively, were 0.85 (95% CI, 0.65 to 1.0) and 0.62 (95% CI, 0.35 to 0.88) in patients with FLT3-mutant AML, and 0.78 (95% CI, 0.62 to 0.93) and 0.52 (96% CI, 0.33 to 0.71) in patients with FLT3–wild-type AML (Fig 3a). Kaplan-Meier DFS probabilities at 1 year for FLT3-mutant and FLT3–wild-type patients were 0.50 (95% CI, 0.22 to 0.78) and 0.60 (95% CI, 0.39 to 0.81), respectively (Fig 3b).
Fig 3.
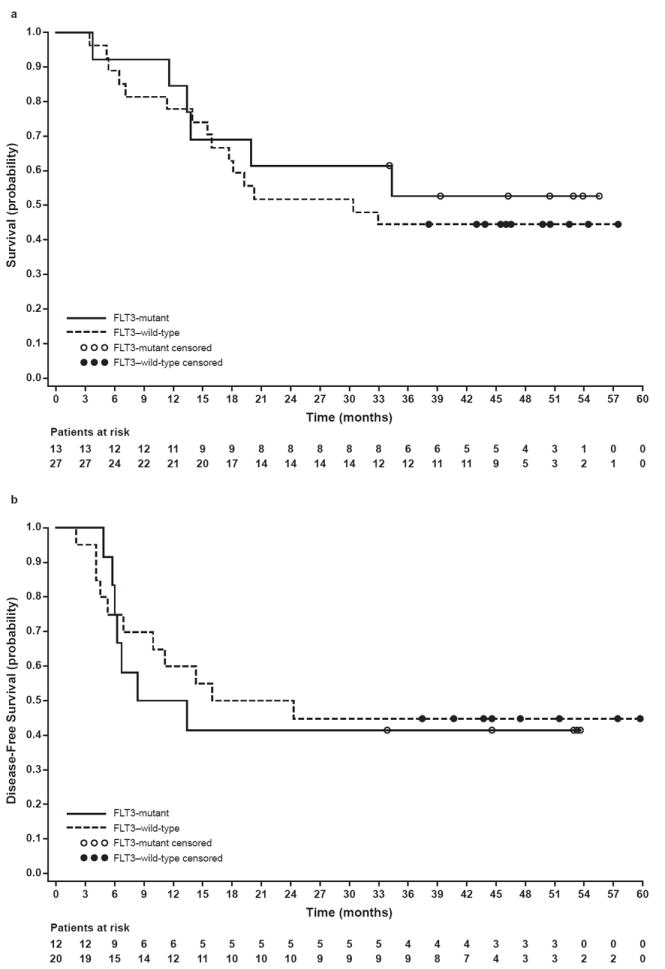
a. Overall survival probability in patients with FLT3–wild-type and FLT3-mutant AML treated on dose schedule III. Overall survival was assessed without censoring for alternative therapies such as stem cell transplant.
b. Disease-free survival probability in patients with FLT3–wild-type and FLT3-mutant AML treated on dose schedule III.
Open boxes represent values from patients on the concomitant arm (day 1-7 and day 15-22 dosing); closed circles represent values from patients on the sequential arm (day 8-22 dosing).
Of the eight patients with FLT3-ITD mutations assessed for DFS, six relapsed, all within 1 year. Six of the nine patients with FLT3-ITD mutations assessed for OS have died. Of the four patients with TKD mutations assessed for DFS and OS, one relapsed after 1 year and none have died.
Overall, three patients in the study received more than two cycles of maintenance therapy (between 23 and 29 cycles). All three were in the midostaurin 50–mg twice–daily cohort. Of these three patients, two had FLT3-mutant AML and one had FLT3–wild-type AML. These patients tolerated the drug well (two patients with no midostaurin dose adjustments). All discontinued treatment while in CR and remained in CR at last follow-up. Two other patients received less than 2 months of post-consolidation maintenance therapy. Both patients discontinued treatment in CR; one (FLT3–wild-type) relapsed 1 month after discontinuation. By investigator report, post-treatment transplant was received by five of 27 patients (19%) with FLT3–wild-type disease and four of 13 patients (31%) with FLT3-mutant disease.
Safety and Tolerability: 50 mg Twice Daily
Midostaurin was generally well tolerated in combination with chemotherapy at the 50-mg twice-daily dose. The amount of midostaurin and chemotherapy received during induction was similar on the two arms, with 14 of 20 patients (70%) in both the sequential and concomitant arms completing all planned induction therapy. All three cycles of consolidation treatment were received by 12 of 16 patients (75%) in the sequential arm and eight of 16 patients (50%) in the concomitant arm. A higher rate of discontinuation was noted in the concomitant (55%) compared with the sequential (35%) schedule (Fig 2). Patients on the sequential arm were exposed to midostaurin for a median of 130 days (range, 7 to 975 days) and patients on the concomitant arm were exposed for a median of 89 days (range, 8 to 1016 days). Exposure to midostaurin was similar between the FLT3-mutated group (median, 133 days; range, 21 to 975 days) and FLT3–wild-type group (median, 90 days; range, 7 to 1016 days).
Nausea, diarrhea, and vomiting were the most common nonhematologic events in the induction and consolidation periods (Table 2). Overall, one episode (3%) of grade 3 diarrhea occurred, lasted 1 day, and resolved without treatment. No grade 3/4 nausea or vomiting occurred. Grade 3/4 hepatic toxicity was infrequent. No grade 3/4 peripheral edema was observed. Overall, the toxicity reported was similar in the sequential and concomitant schedules. No deaths were recorded in either arm on treatment or within 28 days of the last dose of study drug.
Table 2. Adverse Events Occurring During the Induction/Consolidation Periods in Greater Than 20% of Patients Regardless of Attribution for Patients Treated on Dose Schedule III.
| Event, n (%) | Concomitant n = 20 |
Sequential n = 20 |
Total N = 40 |
|||
|---|---|---|---|---|---|---|
| Grade 1/2 | Grade 3/4 | Grade 1/2 | Grade 3/4 | Grade 1/2 | Grade 3/4 | |
| Nausea | 16 (80) | 0 | 17 (85) | 0 | 33 (83) | 0 |
| Neutropenia (including febrile) | 2 (10) | 16 (80) | 0 | 13 (65) | 2 (5) | 29 (73) |
| Thrombocytopenia | 1 (5) | 13 (65) | 0 | 16 (80) | 1 (3) | 29 (73) |
| Diarrhea | 13 (65) | 0 | 14 (70) | 1 (5) | 27 (68) | 1 (3) |
| Vomiting | 12 (60) | 0 | 14 (70) | 0 | 26 (65) | 0 |
| Hypokalemia | 10 (50) | 3 (15) | 8 (40) | 4 (20) | 18 (45) | 7 (18) |
| Pyrexia | 8 (40) | 0 | 9 (45) | 6 (30) | 17 (43) | 6 (15) |
| Headache | 9 (45) | 1 (5) | 11 (55) | 0 | 20 (50) | 1 (3) |
| Anemia | 1 (5) | 6 (30) | 1 (5) | 11 (55) | 2 (5) | 17 (43) |
| Insomnia | 10 (50) | 0 | 9 (45) | 0 | 19 (48) | 0 |
| Constipation | 9 (45) | 0 | 9 (45) | 0 | 18 (45) | 0 |
| Chills | 8 (40) | 0 | 9 (45) | 0 | 17 (43) | 0 |
| Petechiae | 5 (25) | 2 (10) | 10 (50) | 0 | 15 (38) | 2 (5) |
| Cough | 6 (30) | 0 | 9 (45) | 0 | 15 (38) | 0 |
| Hypomagnesemia | 9 (45) | 0 | 6 (30) | 0 | 15 (38) | 0 |
| Rash | 8 (40) | 1 (5) | 6 (30) | 0 | 14 (35) | 1 (3) |
| Abdominal pain | 3 (15) | 0 | 10 (50) | 1 (5) | 13 (33) | 1 (3) |
| Peripheral edema | 9 (45) | 0 | 5 (25) | 0 | 14 (35) | 0 |
| Epistaxis | 5 (25) | 0 | 4 (20) | 3 (15) | 9 (23) | 3 (8) |
| Hypotension | 8 (40) | 0 | 4 (20) | 0 | 12 (30) | 0 |
| ALT increased | 4 (20) | 0 | 4 (20) | 3 (15) | 8 (20) | 3 (8) |
| Alopecia | 7 (35) | 0 | 4 (20) | 0 | 11 (28) | 0 |
| Decreased appetite | 5 (25) | 0 | 5 (25) | 1 (5) | 10 (25) | 1 (3) |
| Hypocalcemia | 7 (35) | 1 (5) | 2 (10) | 1 (5) | 9 (23) | 2 (5) |
| Pruritus | 6 (30) | 1 (5) | 4 (20) | 0 | 10 (25) | 1 (3) |
| Anorexia | 4 (20) | 0 | 5 (25) | 1 (5) | 9 (23) | 1 (3) |
| Anxiety | 4 (20) | 0 | 5 (25) | 1 (5) | 9 (23) | 1 (3) |
| AST increased | 3 (15) | 1 (5) | 3 (15) | 3 (15) | 6 (15) | 4 (10) |
| Depression | 2 (10) | 0 | 8 (40) | 0 | 10 (25) | 0 |
| Fatigue | 5 (25) | 0 | 5 (25) | 0 | 10 (25) | 0 |
| Mucosal inflammation | 4 (20) | 1 (5) | 5 (25) | 0 | 9 (23) | 1 (3) |
| Blood bilirubin increased | 4 (20) | 1 (5) | 3 (15) | 1 (5) | 7 (18) | 2 (5) |
| Transfusion reaction | 4 (20) | 1 (5) | 2 (10) | 2 (10) | 6 (15) | 3 (8) |
Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase.
Pharmacokinetics: 50 mg Twice Daily
Plasma levels for midostaurin and its metabolites, on both the sequential and concomitant dosing schedules, reached levels similar to those previously reported for patients with AML treated with midostaurin monotherapy (Fig 4; Table 3).9 During off-treatment periods, trough concentrations of midostaurin and CGP62221 were similar, decreasing to low or undetectable levels. Differences were observed in plasma concentrations of midostaurin and CGP62221 between the two arms, which can be explained by the different treatment schedules. For example, on day 15, the plasma concentrations of drug in the concomitant arm are lower compared with the sequential arm because patients in the concomitant arm had not received midostaurin for 7 days whereas those in the sequential arm were in the middle of the 14-day treatment block. In contrast, the active metabolite CGP52421 displayed a long half-life and was maintained at stable levels between dosing treatment phases.
Fig 4.
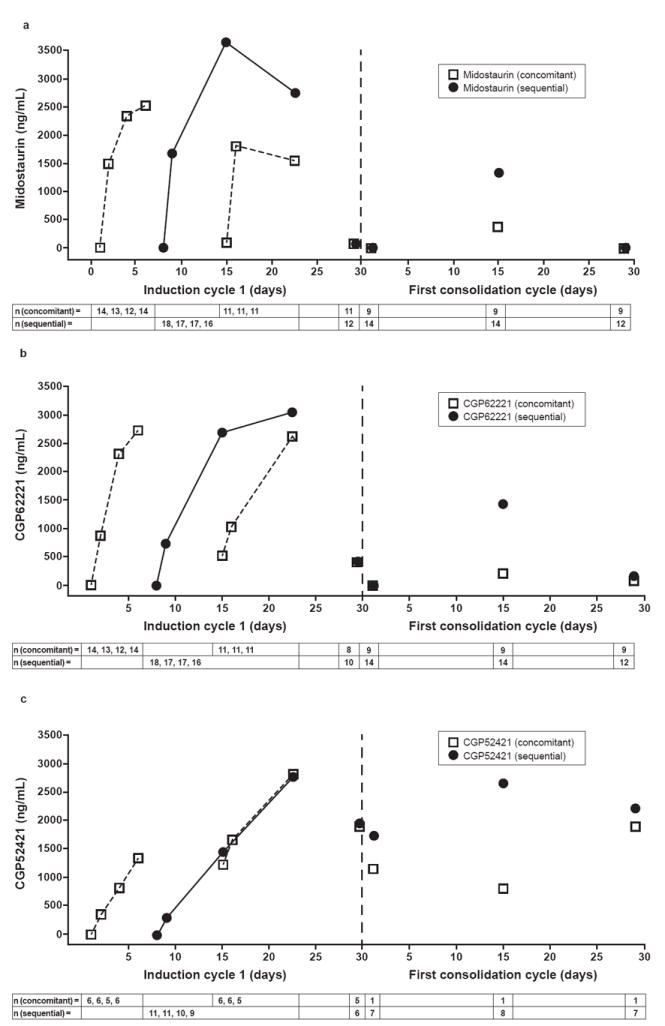
a. Median trough concentration-time profile of midostaurin during the first cycle of induction therapy (midostaurin 50-mg twice-daily cohort).
b. Median trough concentration-time profile of CGP62221 during the first cycle of induction therapy (midostaurin 50-mg twice-daily cohort).
c. Median trough concentration-time profile of CGP52421 during the first cycle of induction therapy (midostaurin 50-mg twice-daily cohort).
Open boxes represent values from patients on the concomitant arm (day 1-7 and day 15-22 dosing); closed circles represent values from patients on the sequential arm (day 8-22 dosing).
Table 3. Concentration of Midostaurin and Its Two Metabolites With 50-mg Twice-Daily Midostaurin Administration During Daunorubicin/Cytarabine Induction.
| Concomitant Administration of Midostaurin With Chemotherapy (Days 1-7 and 15-21 per Cycle) | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cycle | Days | Midostaurin (ng/mL) | CGP62221 (ng/mL) | CGP52421 (ng/mL) | |||||||||||||
| n | Median | Min | Max | n | Median | Min | Max | n | Median | Min | Max | ||||||
| Induction cycle 1 | 1 | 14 | LLOQ | LLOQ | LLOQ | 14 | LLOQ | LLOQ | LLOQ | 6 | LLOQ | LLOQ | LLOQ | ||||
| Day 1 (+ 2 h) | 13 | 1170 | 355 | 2300 | 13 | 189 | 64.8 | 593 | 5 | 170 | 52.9 | 812 | |||||
| Day 1 (+ 4 h) | 13 | 1010 | 630 | 3380 | 13 | 385 | 219 | 626 | 5 | 137 | 115 | 209 | |||||
| 2 | 13 | 1490 | LLOQ | 4980 | 13 | 871 | LLOQ | 1710 | 6 | 361 | 228 | 460 | |||||
| 4 | 12 | 2355 | 1680 | 8710 | 12 | 2315 | 919 | 3310 | 5 | 832 | 570 | 1090 | |||||
| 6 | 14 | 2525 | 1200 | 11300 | 14 | 2730 | 1970 | 4850 | 6 | 1360 | 770 | 1550 | |||||
| 15 | 11 | 87.6 | 11.7 | 7100 | 11 | 528 | 184 | 3950 | 6 | 1235 | 882 | 1680 | |||||
| 15 (+ 2 h) | 11 | 1870 | 87.1 | 7300 | 11 | 630 | 195 | 3400 | 6 | 1170 | 814 | 1920 | |||||
| 15 (+ 4 h) | 11 | 1420 | 96.3 | 7120 | 11 | 845 | 297 | 3510 | 6 | 1465 | 893 | 1860 | |||||
| 16 | 11 | 1820 | 60 | 7520 | 11 | 1030 | 283 | 4460 | 6 | 1665 | 880 | 2240 | |||||
| 22.5 | 11 | 1550 | LLOQ | 9470 | 11 | 2630 | 66.7 | 4870 | 5 | 2850 | 2390 | 3840 | |||||
| 29.5 | 11 | 63.05 | LLOQ | 1810 | 8 | 425.9 | LLOQ | 2980 | 5 | 1900 | 1570 | 2930 | |||||
| First consolidation cycle | 1.5 | 9 | LLOQ | LLOQ | LLOQ | 9 | LLOQ | LLOQ | 16.4 | 1 | 1170 | ||||||
| 15.5 | 9 | 387 | LLOQ | 1040 | 9 | 210 | 14.1 | 986 | 1 | 810 | |||||||
| 29.5 | 9 | LLOQ | LLOQ | 83.8 | 9 | 65.8 | LLOQ | 1090 | 2 | 809; 2990 | |||||||
| Sequential Administration of Midostaurin With Chemotherapy (Days 8-21 per Cycle) | |||||||||||||||||
| Cycle | Day | Midostaurin (ng/mL) | CGP62221 (ng/mL) | CGP52421 (ng/mL) | |||||||||||||
| n | Median | Min | Max | n | Median | Min | Max | n | Median | Min | Max | ||||||
| Induction cycle 1 | 8 | 18 | LLOQ | LLOQ | LLOQ | 18 | LLOQ | LLOQ | LLOQ | 11 | LLOQ | LLOQ | LLOQ | ||||
| 8 (+ 2 h) | 18 | 1495 | 460 | 2880 | 18 | 130 | LLOQ | 612 | 11 | 70.8 | 35.9 | 194 | |||||
| 8 (+ 4 h) | 18 | 1585 | 635 | 3660 | 18 | 244.5 | 42.2 | 810 | 11 | 113 | 58.2 | 207 | |||||
| 9 | 17 | 1680 | 922 | 3290 | 17 | 732 | 136 | 1420 | 11 | 293 | 145 | 414 | |||||
| 15 | 17 | 3660 | 1290 | 17100 | 17 | 2690 | 1280 | 5500 | 10 | 1460 | 903 | 1670 | |||||
| 22.5 | 16 | 2755 | 904 | 19200 | 16 | 3050 | 1570 | 8200 | 9 | 2800 | 1630 | 3540 | |||||
| 29.5 | 12 | 57.2 | 0 | 4970 | 10 | 409.5 | 38.9 | 2590 | 6 | 1955 | 1350 | 3330 | |||||
| First consolidation cycle | 1.5 | 14 | LLOQ | LLOQ | 91.2 | 14 | LLOQ | LLOQ | 217 | 7 | 1740 | 835 | 3110 | ||||
| 15.5 | 14 | 1330 | 635 | 3790 | 14 | 1435 | 513 | 3560 | 8 | 2685 | 1490 | 4210 | |||||
| 29.5 | 12 | 6.75 | LLOQ | 4000 | 12 | 157.5 | LLOQ | 1980 | 7 | 2230 | 1440 | 5320 | |||||
Shaded rows indicate the active dosing period. Induction cycle 1 refers to the daunorubicin/cytarabine induction. First consolidation cycle refers to the first of three high-dose cytarabine consolidation cycles. The lower limit of quantification (LLOQ) is 10 ng/mL.
Although the clearance of daunorubicin does not seem to be affected by midostaurin (Table 4), the mean concentration of daunorubicin observed at 24 hours (C24 h) after the first dose was 17.4 ng/mL and undetectable (< 5 ng/mL) with and without concomitant administration of midostaurin, respectively. Therefore, a pharmacokinetic interaction between daunorubicin and midostaurin could not be excluded. No interaction between cytarabine and midostaurin was observed (data not shown).
Table 4. Effect of 50-mg Twice-Daily Midostaurin Administration on Daunorubicin Pharmacokinetic Parameters, Cycle 1, Day 1.
| Daunorubicin Parameter, Median (Mean ± SD) | Concomitant midostaurin n = 13 | Sequential midostaurin n = 13 | Concomitant/sequential midostaurin |
|---|---|---|---|
| Cmax (ng/mL) | 89 (98 ± 38) | 76 (229 ± 267) | 1.17 |
| Tmax (h) | 0.5 (0.5 ± 0.2) | 0.5 (0.6 ± 0.4) | 1.0 |
| C24 h (ng/mL) | 17.4 (26 ± 32) | LLOQ (0 ± 0) | 17.4/LLOQ |
| AUC (0–24 h) (h•ng/mL) | 189 (373 ± 327) | 159 (267 ± 254) | 1.19 |
| CL (L/h/m2) | 283 (269 ± 130) | 270 (330 ± 226) | 1.05 |
| Vz (L/m2) | 694 (717 ± 266) | 775 (946 ± 696) | 0.90 |
Abbreviations: AUC0-24 h, area under the plasma concentration-time curve from time zero to 24 hours; CL, apparent oral clearance; Cmax, maximum plasma concentration; C24 h, plasma concentration at 24 hours; Tmax, time to Cmax; Vz, apparent volume of distribution. The lower limit of quantification (LLOQ) is 5 ng/mL.
DISCUSSION
It was originally hoped that single-agent FLT3 inhibitor therapy would have a profound effect on AML, similar to the benefit of tyrosine kinase inhibitors in chronic myeloid leukemia (CML). However, AML is genetically more similar to blast-phase CML, with many more mutations required for the development of the full disease phenotype. FLT3 may occur as a secondary mutation rather than an initiator of the leukemic clone.12 The absence of primacy of FLT3 mutations, lack of adequate pharmacokinetics, and protection of the leukemic stem cells in the marrow niche have each been invoked to explain the disappointing clinical efficacy seen when potent FLT3 inhibitors were used as single agents in early-stage clinical trials in advanced FLT3-mutant AML.8,9,13-15 However, the biologic activity (frequent reduction in peripheral blood blasts) coupled with preclinical studies showing synergy between FLT3 inhibitors and chemotherapy prompted an effort to address the feasibility of combination therapy.
This study was designed to determine a safe and tolerable dose of midostaurin that could be administered with standard induction and post-remission chemotherapy. Achieving such a regimen was more challenging than initially expected. We had previously shown that 75 mg three times daily8 and 50 mg and 100 mg each twice daily9 were well tolerated and reasonably efficacious when given continuously as single agents to patients with AML. However, 100-mg twice-daily midostaurin given either concomitantly with induction chemotherapy (beginning on the first day of chemotherapy) or sequentially (on the eighth day after the start of chemotherapy) led to grade 3/4 nausea and vomiting and a high rate of discontinuation. Tolerability improved for patients who received midostaurin 50 mg twice daily for 14 days per cycle in both the concomitant and sequential arms. Given the slightly higher degree of tolerability in the sequential arm, the fact that a pharmacokinetic interaction between midostaurin and daunorubicin could not be excluded in our study, and concerns in other studies about possible antagonism if a FLT3 inhibitor was given prior to chemotherapy,10 the sequential schedule was chosen for further evaluation.
Although there was a 45% discontinuation rate in the cohort of 40 patients receiving midostaurin 50 mg twice daily in combination with chemotherapy, most of these patients stopped for reasons other than toxicity, such as relapse, failure to achieve CR, or stem cell transplant. Consequently, we supported moving ahead with this schedule, although further dose optimization may be possible.
This study demonstrated an encouragingly high CR rate of 92% in 13 patients with FLT3-mutant AML exposed to midostaurin 50 mg for 14 days of each 28-day cycle. Sorafenib plus induction chemotherapy also led to a high CR rate (93%) in 15 newly diagnosed patients with FLT3-mutant AML.16 While the poor prognosis noted in patients with FLT3-ITD mutations is thought to be due to a high relapse rate,1-4,17-21 it is possible that a higher likelihood of CR or “deeper” CR could be beneficial in reducing relapse rates. Although no firm conclusions can be drawn on the basis of 13 patients, four of whom had TKD mutations, it is interesting to note that Kaplan-Meier DFS and OS probabilities in this group of patients were similar to those in the 27 patients with FLT3–wild-type disease. The extent to which post-protocol therapy (eg, stem cell transplant) influenced this relatively favorable survival outcome in the FLT3-mutant cohort is unclear.
The results of our study, showing high CR and OS rates in patients with FLT3-mutant AML with acceptable tolerability, suggest that the addition of midostaurin to chemotherapy may improve outcomes for newly diagnosed younger patients with FLT3-mutant AML. However, AML is a heterogeneous disease and the current study was small and did not account for the influence of parameters such as stem cell transplantation or gene mutations other than FLT3. Nevertheless, these promising safety results enabled initiation of the ongoing, international, prospective, randomized double-blind phase III study (CALGB 10603, NCT00651261) of standard induction and post-remission chemotherapy with placebo or midostaurin at 50 mg twice daily on days 8 through 21 of each chemotherapy cycle and as maintenance therapy in newly diagnosed patients with AML younger than 60 years of age.
Acknowledgments
The authors acknowledge the study coordinators, nurses, and physicians who contributed to this study and the patients and their families for their participation. Susie Crowley provided secretarial assistance. Sandra Harris and Erinn Goldman of Articulate Science, LLC, provided medical writing assistance.
This study was sponsored by Novartis Pharmaceuticals Corporation.
Research Funding: Richard M. Stone, Novartis; Thomas Fischer, Novartis; Gary Schiller, Genzyme; Charles Schiffer, Pfizer, Ariad, Novartis, Celgene, Bristol-Myers Squibb, Ambit; Jorge Cortes, Novartis, Ariad, Ambit; Hagop M. Kantarjian, Novartis, Pfizer, Bristol-Myers Squibb; Francis Giles, Novartis
Footnotes
Author Contributions
Conception and design: Richard M. Stone, Thomas Fischer, Charles A. Schiffer, Gerhard Ehninger, Jorge Cortes, Hagop M. Kantarjian, Daniel J. DeAngelo, and Francis Giles
Collection and assembly of data: Richard M. Stone, Ronald Paquette, Charles A. Schiffer, Gerhard Ehninger, Jorge Cortes, Hagop M. Kantarjian, Daniel J. DeAngelo, Alice Huntsman-Labed, Catherine Dutreix, Adam del Corral, and Francis Giles
Data analysis and interpretation: Richard M. Stone, Thomas Fischer, Gary Schiller, Charles A. Schiffer, Jorge Cortes, Hagop M. Kantarjian, Daniel J. DeAngelo, Alice Huntsman-Labed, Catherine Dutreix, Adam del Corral, and Francis Giles
Manuscript writing: Richard M. Stone wrote the first draft of the manuscript and all authors edited and commented on subsequent drafts.
Final approval of manuscript: All authors gave final approval of the manuscript.
Provision of study materials or patients: Richard M. Stone, Thomas Fischer, Gary Schiller, Charles A. Schiffer, Jorge Cortes, Daniel J. DeAngelo, and Francis Giles
Previous presentations: Preliminary data from this study were presented at the 51st American Society of Hematology Annual Meeting in 2009: Stone RM, Fischer T, Paquette R, et al. A phase 1b study of midostaurin (PKC412) in combination with daunorubicin and cytarabine induction and high-dose cytarabine consolidation in patients under age 61 with newly diagnosed de novo acute myeloid leukemia: overall survival of patients whose blasts have FLT3 mutations is similar to those with wild-type FLT3. Blood. 2009;114:634.
Authors’ Disclosures of Potential Conflicts of Interest
Employment or Leadership Position: Alice Huntsman-Labed, Novartis Pharma AG (C); Catherine Dutreix, Novartis Pharma AG (C); Adam del Corral, Novartis Pharmaceuticals Corporation (C) Consultant or Advisory Role: Richard M. Stone, Genzyme (C), Celgene (C), Ariad (C); Ronald Paquette, Novartis (C); Gary Schiller, Genzme (C); Charles A. Schiffer, Pfizer (C), Micromet (C), Celgene (C), Ambit (C), Ariad (C); Jorge Cortes, Novartis (C), Ariad (C), Ambit (U); Hagop M. Kantarjian, Novartis (C); Daniel J. DeAngelo, Novartis (C); Francis Giles, Novartis (C) Stock Ownership: Gerhard Ehninger, Novartis; Alice Huntsman-Labed, Novartis Honoraria: Thomas Fischer, Novartis; Ronald Paquette, Novartis; Gerhard Ehninger, Novartis Expert Testimony: None Other Remuneration:None
References
- 1.Thiede C, Steudel C, Mohr B, Schaich M, Schäkel U, Platzbecker U, et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood. 2002;99:4326–4335. doi: 10.1182/blood.v99.12.4326. [DOI] [PubMed] [Google Scholar]
- 2.Whitman SP, Archer KJ, Feng L, Baldus C, Becknell B, Carlson BD, et al. Absence of the wild-type allele predicts poor prognosis in adult de novo acute myeloid leukemia with normal cytogenetics and the internal tandem duplication of FLT3: a cancer and leukemia group B study. Cancer Res. 2001;61:7233–7239. [PubMed] [Google Scholar]
- 3.Weisberg E, Barrett R, Liu Q, Stone R, Gray N, Griffin JD. FLT3 inhibition and mechanisms of drug resistance in mutant FLT3-positive AML. Drug Resist Updat. 2009;12:81–89. doi: 10.1016/j.drup.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schnittger S, Schoch C, Dugas M, Kern W, Staib P, Wuchter C, et al. Analysis of FLT3 length mutations in 1003 patients with acute myeloid leukemia: correlation to cytogenetics, FAB subtype, and prognosis in the AMLCG study and usefulness as a marker for the detection of minimal residual disease. Blood. 2002;100:59–66. doi: 10.1182/blood.v100.1.59. [DOI] [PubMed] [Google Scholar]
- 5.Weisberg E, Boulton C, Kelly LM, Manley P, Fabbro D, Meyer T, et al. Inhibition of mutant FLT3 receptors in leukemia cells by the small molecule tyrosine kinase inhibitor PKC412. Cancer Cell. 2002;1:433–443. doi: 10.1016/s1535-6108(02)00069-7. [DOI] [PubMed] [Google Scholar]
- 6.Manley PW, Boulton C, Caravatti G, Gilliland DG, Griffin J, Kung A, et al. Preclinical profile of PKC412 (Midostaurin) as an FLT3 inhibitor for the therapy of AML. AACR. 2003 Poster 1004. [Google Scholar]
- 7.Levis M, Brown P, Smith BD, Stine A, Pham R, Stone R, et al. Plasma inhibitory activity (PIA): a pharmacodynamic assay reveals insights into the basis for cytotoxic response to FLT3 inhibitors. Blood. 2006;108:3477–3483. doi: 10.1182/blood-2006-04-015743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stone RM, DeAngelo DJ, Klimek V, Galinsky I, Estey E, Nimer SD, et al. Patients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small-molecule FLT3 tyrosine kinase inhibitor, PKC412. Blood. 2005;105:54–60. doi: 10.1182/blood-2004-03-0891. [DOI] [PubMed] [Google Scholar]
- 9.Fischer T, Stone RM, DeAngelo DJ, Galinsky I, Estey E, Lanza C, et al. Phase IIB trial of oral Midostaurin (PKC412), the FMS-like tyrosine kinase 3 receptor (FLT3) and multi-targeted kinase inhibitor, in patients with acute myeloid leukemia and high-risk myelodysplastic syndrome with either wild-type or mutated FLT3. J Clin Oncol. 2010;28:4339–4345. doi: 10.1200/JCO.2010.28.9678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Levis M, Pham R, Smith BD, Small D. In vitro studies of a FLT3 inhibitor combined with chemotherapy: sequence of administration is important to achieve synergistic cytotoxic effects. Blood. 2004;104:1145–1150. doi: 10.1182/blood-2004-01-0388. [DOI] [PubMed] [Google Scholar]
- 11.Byrd JC, Mrozek K, Dodge RK, Carroll AJ, Edwards CG, Arthur DC, et al. Pretreatment cytogenetic abnormalities are predictive of induction success, cumulative incidence of relapse, and overall survival in adult patients with de novo acute myeloid leukemia: results from Cancer and Leukemia Group B (CALGB 8461) Blood. 2002;100:4325–4336. doi: 10.1182/blood-2002-03-0772. [DOI] [PubMed] [Google Scholar]
- 12.Ley TJ, Mardis ER, Ding L, Fulton B, McLellan MD, Chen K, et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature. 2008;456:66–72. doi: 10.1038/nature07485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith BD, Levis M, Beran M, Giles F, Kantarjian H, Berg K, et al. Single-agent CEP-701, a novel FLT3 inhibitor, shows biologic and clinical activity in patients with relapsed or refractory acute myeloid leukemia. Blood. 2004;103:3669–3676. doi: 10.1182/blood-2003-11-3775. [DOI] [PubMed] [Google Scholar]
- 14.DeAngelo DJ, Stone RM, Heaney ML, Nimer SD, Paquette RL, Klisovic RB, et al. Phase 1 clinical results with tandutinib (MLN518), a novel FLT3 antagonist, in patients with acute myelogenous leukemia or high-risk myelodysplastic syndrome: safety, pharmacokinetics, and pharmacodynamics. Blood. 2006;108:3674–3681. doi: 10.1182/blood-2006-02-005702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang W, Konopleva M, Shi YX, McQueen T, Harris D, Ling X, et al. Mutant FLT3: a direct target of sorafenib in acute myelogenous leukemia. J Natl Cancer Inst. 2008;100:184–198. doi: 10.1093/jnci/djm328. [DOI] [PubMed] [Google Scholar]
- 16.Ravandi F, Cortes JE, Jones D, Faderl S, Garcia-Manero G, Konopleva MY, et al. Phase I/II study of combination therapy with sorafenib, idarubicin, and cytarabine in younger patients with acute myeloid leukemia. J Clin Oncol. 2010;28:1856–1862. doi: 10.1200/JCO.2009.25.4888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kayser S, Schlenk RF, Londono MC, Breitenbuecher F, Wittke K, Du J, et al. Insertion of FLT3 internal tandem duplication in the tyrosine kinase domain-1 is associated with resistance to chemotherapy and inferior outcome. Blood. 2009;114:2386–2392. doi: 10.1182/blood-2009-03-209999. [DOI] [PubMed] [Google Scholar]
- 18.Breitenbuecher F, Markova B, Kasper S, Carius B, Stauder T, Böhmer FD, et al. A novel molecular mechanism of primary resistance to FLT3-kinase inhibitors in AML. Blood. 2009;113:4063–4073. doi: 10.1182/blood-2007-11-126664. [DOI] [PubMed] [Google Scholar]
- 19.Kottaridis PD, Gale RE, Frew ME, Harrison G, Langabeer SE, Belton AA, et al. The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: analysis of 854 patients from the United Kingdom Medical Research Council AML 10 and 12 trials. Blood. 2001;98:1752–1759. doi: 10.1182/blood.v98.6.1752. [DOI] [PubMed] [Google Scholar]
- 20.Whitman SP, Ruppert AS, Radmacher MD, Mrozek K, Paschka P, Langer C, et al. FLT3 D835/I836 mutations are associated with poor disease-free survival and a distinct gene-expression signature among younger adults with de novo cytogenetically normal acute myeloid leukemia lacking FLT3 internal tandem duplications. Blood. 2008;111:1552–1559. doi: 10.1182/blood-2007-08-107946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mead AJ, Linch DC, Hills RK, Wheatley K, Burnett AK, Gale RE. FLT3 tyrosine kinase domain mutations are biologically distinct from and have a significantly more favorable prognosis than FLT3 internal tandem duplications in patients with acute myeloid leukemia. Blood. 2007;110:1262–1270. doi: 10.1182/blood-2006-04-015826. [DOI] [PubMed] [Google Scholar]