

Abstract
Hypertrophic growth of the myocardium occurs in most forms of heart failure and may contribute to the pathogenesis of the failure state. Little is known about the regulatory mechanisms governing the often-coexisting phenotypes of hypertrophy, systolic failure, and diastolic stiffness that characterize clinical disease. We hypothesized that intracellular signaling pathways are differentially activated by graded degrees of hemodynamic stress. To test this, we developed models of graded pressure stress in mice and used them to directly compare compensated hypertrophy and pressure-overload heart failure. Surgical interventions were designed to be similar, on either side of a threshold separating compensated from decompensated responses. Our findings revealed two dramatically different hypertrophic phenotypes with only modest differences in the activation of relevant intracellular signaling pathways. Furthermore, we uncovered a functional requirement of calcineurin signaling in each model such that calcineurin suppression blunted hypertrophic growth. Remarkably, in each case, suppression of calcineurin signaling was not associated with clinical deterioration or increased mortality. Profiles of stress-response signaling and Ca2+ handling differ between the steady-state, maintenance phases of load-induced cardiac hypertrophy and failure. This information may be useful in identifying novel targets of therapy in chronic disease.
Keywords: signal transduction, myocardium
It is estimated that at least 5 million people in the United States suffer from chronic heart failure (HF), with nearly 1 million hospitalizations and over 500,000 new cases identified each year (1). The impact of HF on the healthcare system is expanding, as evidenced by the fact that hospitalizations for HF have increased 2.5-fold between 1979 and 1999 (1). Despite recent advances in therapy, mortality from HF remains high at ~50% at 5 yr. Development of new therapies awaits the elucidation of molecular events governing myocardial decompensation and the steady-state performance of the failing heart.
Hypertrophic growth of the myocardium occurs in most forms of HF and may contribute to the pathogenesis of the failure state (2, 26). Hypertrophic remodeling takes place as a response to chronic stress, such as that occuring in individuals with hypertension. Cardiac hypertrophy with concomitant ventricular dysfunction also develops as a consequence of acute-onset stress, such as after a loss of ventricular mass due to massive infarction. Little is known about the regulatory mechanisms governing the often-coexisting phenotypes of hypertrophy, systolic failure, and diastolic stiffness that characterize clinical disease.
Numerous signaling pathways have been implicated in hypertrophic transformation of the heart (for a review, see Ref. 18). Many studies have examined the association between activation of molecular signaling pathways and human HF, but, as yet, the reported findings have been incomplete and often contradictory. Thus it is not known whether activation of a distinct signaling pathway triggers a specific clinical phenotype or whether disparate degrees of activation of the same pathways are responsible for a clinical spectrum ranging from compensated hypertrophy (Comp Hyp) to decompensated HF.
The development of surgical models of cardiac pressure overload in mice has provided a platform for genetic and mechanistic studies of load-induced failure. An increase in left ventricular (LV) pressure is achieved by constriction of either the ascending, transverse or descending aorta. The magnitude of resulting afterload elevation is dependent on both the location and severity of the constriction. Unfortunately, similar techniques in different hands result in a wide spectrum of experimental outcomes ranging from Comp Hyp to decompensated HF. This has made it difficult to compare results from one group with those of another. It also suggests that relatively small differences in hemodynamic stress can result in vastly different clinical phenotypes.
We hypothesized that intracellular signaling pathways are differentially activated by graded degrees of hemodynamic stress. Furthermore, we postulated that unique profiles of stress-response signaling underlie unique phenotypes: ventricular hypertrophy with preserved systolic function (21) versus hypertrophy with ventricular dysfunction and clinical HF. To test this, we developed models of graded pressure stress in mice and used them to make a direct comparison between Comp Hyp and pressure-overload HF. Surgical interventions were designed to be similar, on either side of a threshold separating compensated from decompensated responses. Our findings revealed two dramatically different hypertrophic phenotypes with modest differences in the activation of relevant intracellular signaling pathways. Furthermore, we uncovered a functional requirement of calcineurin signaling in each model such that calcineurin suppression blunted hypertrophic growth. Remarkably, in each case, suppression of calcineurin signaling was not associated with clinical deterioration or increased mortality.
EXPERIMENTAL PROCEDURES
Pressure-overload hypertrophy models
Male C57BL6 mice (6–8 wk old) were subjected to pressure overload by thoracic aortic banding (TAB) (33), using protocols approved by the Univeristy of Texas Southwestern Animal Care and Use Committee. In this procedure, the constriction is placed in the transverse aorta between the innominate and left common carotid arteries. We (21) have shown previously that constriction to a 27-gauge stenosis induces moderate hypertrophy (≈40% increases in heart mass) without clinical signs of HF or malignant ventricular arrhythmia. Severe, decompensated hypertrophy was induced by banding the thoracic aorta to a 28-gauge diameter. At 3 wk, when the hypertrophic response had reached steady-state (21), integrity of aortic banding was confirmed by inspection of the surgical constriction and by visualization of marked differences in caliber of the right and left carotid arteries. Transstenotic pressures were measured in anesthetized mice as previously described (21).
Myocyte isolation and surface area determinations
The surface area of a two-dimensional silhouette was estimated by measuring the length and width of 20 randomly selected myocytes. Our reported two-dimensional surface area (length × width) is directly proportional to the surface area of a cylinder (2π radius × length).
RNA dot blots
Total RNA was isolated from the LV using TRIzol (Invitrogen) reagent. Expression of hypertrophic marker genes was assessed by hybridization of RNA dot blots as previously described (29).
Western blot analysis
Protein lysates were subjected to polyacrylamide gel electrophoresis, transferred to polyvinylidene difluoride membranes, and subjected to immunoblot analysis. The ratio of antigen to antibody, detergent concentration, and duration and temperature of the reaction was optimized for each antibody. Data were quantified by densitometric analysis and normalized to GAPDH protein levels.
Echocardiography
Transthoracic echocardiograms were recorded in conscious-sedated mice as described (21, 22). Briefly, views were taken in planes that approximated the parasternal short-axis view (chordal level) and the apical long-axis view in humans. LV internal diameters and wall thicknesses were measured (at least 3 cardiac cycles) at end systole and end diastole. LV mass and volumes were calculated using the area-length method (32).
Reagents
Antibodies were purchased from Cell Signaling [ERK no. 9102, phosphorylated (p)ERK no. 9106, JNK no. 9252, pJNK no. 9251, p38 no. 9212, pp38 no. 9211, Akt no. 9272, and pAkt no. 9271], Affinity Bioreagents [calsequestrin no. PA1-913, phospholamban (PLB) MA3–922], BD Biosciences [calcineuring A (CaN) no. 610259], Upstate (Ser16-pPLB 07-052), or Research Diagnostics (GAPDH no. TRK5G-6C5). Modulatory calcineuring-interacting protein 1 (MCIP1) antiserum (7) was a kind gift of Erik Bush (Myogen).
Statistical methods
Data are reported as means ± SE. Statistical significance was analyzed using a Student’s unpaired t-test or one-way ANOVA followed by Bonferroni’s method for post hoc pair-wise multiple comparisons.
RESULTS
To study stress-induced ventricular dysfunction, we developed a model of pressure-overload HF by calibrated constriction of the thoracic aorta. The surgical intervention was designed to be qualitatively similar to an established model of TAB (33) but imposed slightly tighter (1/20th mm) constriction on the aorta (severe TAB). All other aspects of the TAB and severe TAB surgeries were identical, rendering the resulting disease phenotypes directly comparable.
Animals subjected to standard TAB were clinically indistinguishable from sham-operated controls. Measurements of heart mass (Fig. 1 and Table 1) revealed significant hypertrophic growth. In contrast, mice subjected to severe TAB gradually developed signs of circulatory failure, including lethargy, impaired mobility, and diminished appetite. Hearts dissected from these mice were markedly enlarged and dilated, and morphometric analysis revealed significant increases in heart mass (Fig. 1).
Fig. 1.

A: 4-chamber images of sham-operated, thoracic aortic banded (TAB), and severe TAB hearts. B: heart weights (HW) normalized to body weight (BW) or tibia length (T) are increased in mice with compensated hypertrophy (TAB) and heart failure (HF; severe TAB). C: hypertrophy of cardiac myocytes, measured as two-dimensional (2D) surface area, is similar in cells isolated from compensated hypertrophy (Comp Hyp) and HF myocardium.
Table 1.
Indexes of hypertrophy and failure in animals subjected to graded degrees of pressure stress
| Sham Operated | Compensated Hypertrophy | Heart Failure | |
|---|---|---|---|
| TNF, pg/ml | 5.1 ± 2.3 | 4.8 ± 2.4 | 16.9 ± 5.8* |
| n | 4 | 3 | 4 |
| Proximal aortic pressure, mmHg | NA | 166/69 ± 12/5 | 120/70 ± 28/20 |
| n | 4 | 3 | |
| HW/BW, mg/g | 5.0 ± 0.5 | 7.2 ± 0.08* | 10.4 ± 0.5† |
| n | 10 | 73 | 39 |
| HW/T, mg/mm | 7.1 ± 0.1 | 10.6 ± 0.2* | 13.6 ± 0.4† |
| n | 10 | 73 | 39 |
| Cell surface area, μm2 | 2,979 ± 749 | 4,735 ± 1,433* | 4,507 ± 1,195* |
| n | 540 | 200 | 480 |
Values are means ± SE; n, no. of animals. HW/BW and HW/T, heart weight (HW) normalized to body weight (BW) and tibia length (T).
P < 0.05 vs. sham;
P < 0.01 vs. sham.
Increases in heart size and mass may result from changes in myocyte and nonmyocyte fractions of myocardium. To investigate this, individual ventricular myocytes were enzymatically dissociated, and randomly selected cells were examined microscopically. Two-dimensional surface areas of dissociated ventricular myocytes were increased 59% (P < 0.05) in TAB hearts and 51% (P < 0.05) in severe TAB hearts (Fig. 1C and Table 1), demonstrating that significant hypertrophy of individual cardiomyocytes occurred in both models. Interestingly, despite significant hypertrophic cell growth in myocytes isolated from severe TAB hearts, the ratio of myocyte length to width (5.90 ± 0.12, n = 480) was unchanged compared with sham-operated controls [5.80 ± 0.06, n = 540, P = not significant (NS)]. Myocytes isolated from TAB hearts, however, manifested disproportionate growth in the longitudinal dimension (6.44 ± 0.18, n = 200, P < 0.01).
Circulating levels of tumor necrosis factor (TNF) and other cytokines are frequently increased in patients with HF, a response that has been implicated in the pathophysiology of the disease (13). Consistent with a clinical diagnosis of HF in severe TAB mice, levels of circulating TNF (17) were markedly increased (P < 0.05) relative to sham-operated controls (Fig. 2 and Table 1). In contrast, TNF levels were not elevated in TAB mice (P = NS vs. sham).
Fig. 2.
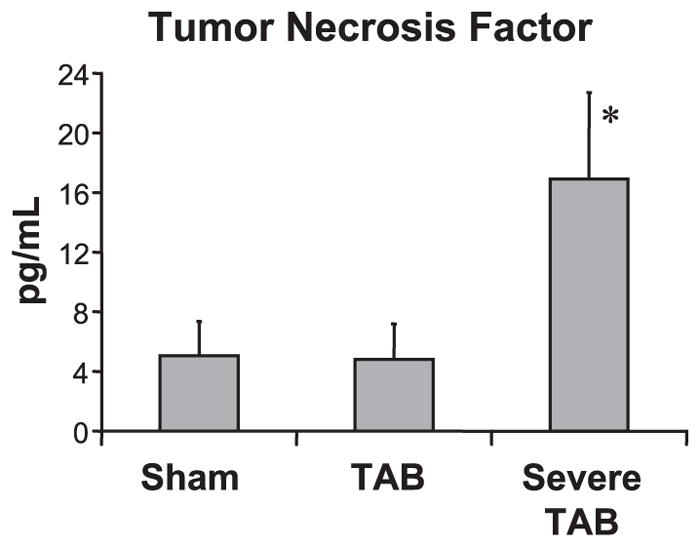
Tumor necrosis factor (TNF) levels are significantly elevated (*P < 0.05 vs. sham) in HF mice.
Whereas perioperative mortalities were similar (<10%) in the two models, survival was diminished (P < 0.01) in severe TAB mice with a 1-wk mortality of 52% (n = 202/388 mice) compared with 17% (n = 47/272) in TAB mice (see Supplemental Fig. S1; available at the Physiological Genomics web site).1 Necropsy revealed signs of cardiovascular compromise in severe TAB mice, including effusions and elevated lung-to-body weight ratios (increased 72%, P < 0.01), consistent with venous congestion from LV dysfunction. These features were absent in TAB mice or in sham-operated controls.
Development of ventricular dysfunction and clinical HF
At death, the integrity of aortic banding was confirmed by inspection of the surgical constriction and by visualization of marked differences in caliber of the right and left carotid arteries. Proximal aortic pressures (Fig. 3) were decreased (P < 0.05) in severe TAB mice relative to TAB mice, consistent with decreased cardiac output from diminished pump function (Table 1).
Fig. 3.

A: measurements of distal and proximal carotid artery pressures demonstrate diminished pressure gradients in HF mice, consistent with decreased systolic performance. B: systolic function is preserved in mice subjected to TAB, whereas HF mice (severe TAB) manifest progressive declines in systolic performance. C: left ventricular (LV) size is preserved in all 3 models at 1 wk but progressively increases in HF mice.
Echocardiography was used to directly examine LV size and function. Systolic performance remained normal throughout the study period in TAB mice (Fig. 3). In severe TAB mice, however, there was a progressive deterioration in systolic performance over time. Ventricular volumes remained normal in TAB mice for at least 3 wk (Fig. 3) and initially in severe TAB mice. After the first postoperative week, however, end-diastolic volumes in severe TAB mice increased steadily.
Thus, despite the similarity in surgical intervention, the clinical phenotypes differed markedly in these models of pressure-induced ventricular remodeling, ranging from Comp Hyp in TAB mice to pressure-overload HF in severe TAB mice. Whereas markers of cardiac size and performance demonstrated hypertrophic growth in each case, echocardiographic and clinical markers of progressive failure were manifest exclusively in HF mice.
Activation of signaling cascades
Hypertrophic growth of the myocardium is accompanied by a pattern of gene expression mimicking that seen during embryonic development. To investigate reactivation of the “fetal gene program” in load-induced HF, we measured transcript levels of natriuretic peptides after each phenotype had attained steady state (3 wk postoperative). Increases in atrial natriuretic factor (ANF) and B-type natriuretic peptide (BNP) transcript were observed in both Comp Hyp and HF hearts (Fig. 4). Whereas the extent of hypertrophy, measured as heart weight normalized to body weight, was greater in HF hearts compared with Comp Hyp hearts, the increases in ANF and BNP were significantly greater in Comp Hyp hearts than in HF hearts (P < 0.05). In contrast, β-myosin heavy chain (MHC) transcript abundance was increased to a similar extent in both Comp Hyp and HF tissues. Together, these findings are suggestive of differential activation of transcriptional programs in compensated versus failing hypertrophic phenotypes.
Fig. 4.

Dot blots of steady-state transcript levels (representative of 3 independent measurements) reveal differential reactivation of the fetal gene program in Comp Hyp and HF hearts. Total RNA was isolated from the LV, and expression of hypertrophic marker genes was assessed by specific hybridization as previously described (29). ANF, atrial natriuretic factor; BNP, B-type natriuretic peptide; β-MHC, β-myosin heavy chain. *P < 0.05 vs. sham.
To investigate molecular signals that could underlie the striking differences in phenotype, we quantified the activation profiles of specific signaling cascades. First, we measured the activation of the three major arms of MAPKs (19). Steady-state levels of protein expression for ERK, JNK, and p38 were unchanged (P = NS) in Comp Hyp and HF hearts compared with sham-operated controls (Fig. 5). To study signaling kinase activation, we probed lysates prepared with phosphatase inhibitors using antibodies specific for the phosphorylated, activated isoform of each enzyme. These studies revealed that the activated form of ERK (pERK) was increased in severe TAB mice with HF (146 ± 32%, n = 3, P < 0.05 compared with sham normalized to 100%) but not in TAB mice with Comp Hyp (117 ± 8%, n = 3, P = NS). Similarly, activated JNK (pJNK) was selectively increased (149 ± 8%, n = 3, P < 0.05) in HF only. Steady-state activation of the p38 pathway was not different (P = NS) from control in either HF or Comp Hyp.
Fig. 5.

Western blot analysis of total protein expression (top blots and left bars in histogram) and phosphorylated (p) isoforms (bottom blots and right bars in histogram) reveal selective activation of ERK (A) and JNK (B) in HF. Evidence of significant activation of p38 (C) and Akt (D) was not obtained. Representative data from 3 independent measurements are illustrated. *P < 0.05 vs. sham.
Phosphatidylinositides are key regulators of membrane trafficking and ion channel/transporter function relevant to the hypertrophic process (8, 10). The intracellular kinase Akt is a downstream target of phosphatidylinositol 3-kinase (PI3K). Prior work from our group has shown that intracellular phosphatidylinositides accumulate in Comp Hyp and HF myocardium (S. Feng, J. A. Hill, and D. W. Hilgemann, unpublished observations). Among these lipids, phosphatidylinositol-4,5-biphosphate, the major substrate for PI3K (42), was increased 16% in Comp Hyp hearts and 28% in HF hearts. Therefore, we examined whether Akt, a major downstream target of PI3K, was activated. By Western blot analysis, we found no evidence of increased expression or activation of Akt in either Comp Hyp or HF hearts compared with controls (Fig. 5).
Alterations in calcium handling
Depletion of Ca2+ from sarcoplasmic reticulum (SR) stores contributes to the disordered excitation-contraction coupling of systolic HF. Calsequestrin is a high-capacity Ca2+-binding protein that regulates sarco(endo)plasmic reticulum Ca2+ release by the ryanodine receptor (28). In a mouse model of hypertrophic cardiomyopathy, calsequestrin levels are diminished in advance of changes in cardiac histology or morphology (37). Consistent with the notion that dysregulation of Ca2+-responsive signaling is a proximal event in the transition from hypertrophy to failure, we observed a trend toward diminished calsequestrin levels in TAB mice with Comp Hyp (Fig. 6). In failing severe TAB hearts, calsequestrin expression was significantly (P < 0.05) diminished, reflecting graded changes in calcium handling in response to hemodynamic stress.
Fig. 6.

Representative Western blot analysis of calsequestrin (CSQ; A), calcineurin A subunit (CnA; B), phospholamban (PLB) and Ser16-pPLB (Phos-S16 PLB; C), and the exon 4 isoform of modulatory calcineurin-interacting protein (MCIP; D) in Comp Hyp and HF hearts. Data illustrated are representative of 3 independent measurements. *P < 0.05 vs. sham.
After each contraction, uptake of calcium back into the SR occurs via sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA). Activity of the SERCA pump is regulated through an interaction with the SR membrane protein PLB. The inhibitory action of PLB on SERCA is tightly regulated; phosphorylation of PLB releases SERCA from inhibition, thereby increasing calcium uptake and improving contractility. To address this, we quantified levels of PLB and its phosphorylation at serine 16 (Ser16). There were no significant changes in the total abundance of PLB protein in both Comp Hyp and HF hearts compared with sham-operated controls (Fig. 6). Interestingly, we observed significant increases in Ser16 phosphorylation in HF hearts relative to control and variable increases in Comp Hyp hearts.
Calcineurin signaling
Calcineurin links cytoplasmic Ca2+ to transcriptional regulation of multiple genes involved in the cardiac hypertrophic program. We observed a slight but reproducible increase in the level of calcineurin catalytic subunit CnA protein levels in Comp Hyp hearts compared with sham-operated controls (P < 0.05; Fig. 6). Interestingly, there was no significant change in CnA levels in severe TAB failing hearts (P = NS) compared with sham-operated controls.
Calcineurin is activated upon the binding of a calcium/calmodulin complex. Thus an increase in calcineurin protein levels per se (Fig. 6) does not necessarily indicate that calcineurin signaling is activated. Rather, the level of calcineurin activity depends both on the abundance of calcineurin enzyme and the availability of intracellular Ca2+ and calmodulin. To assess calcineurin activity, we measured the abundance of modulatory calcineurin-interacting protein-1 (MCIP1). MCIP1 is an endogenous feedback inhibitor of calcineurin, whose expression is under the control of calcineurin (47). Under steady-state conditions, equilibrium is established between MCIP1 protein levels and calcineurin activity, such that the level of MCIP1 protein is proportional to calcineurin activity (41). MCIP1 protein levels were elevated in both Comp Hyp and HF hearts (Fig. 6). MCIP1 protein was more abundant in HF hearts than in Comp Hyp hearts, a pattern consistent with that observed for PLB phosphorylation.
To determine the functional requirement for calcineurin activity in the pathogenesis of compensated and failing hypertrophy, we inhibited calcineurin with systemic administration of cyclosporin A (CsA). Consistent with previous reports (21, 44), CsA abolished the hypertrophic growth response in TAB mice (Fig. 7). Inhibition of calcineurin also blunted hypertrophy in the context of severe TAB failing hearts, measured in terms of heart mass normalized to body mass (decreased 62%, P < 0.01) or tibia length (decreased 58%, P < 0.01). As reported previously, CsA had no detrimental effect on the survival of TAB mice (21). However, CsA decreased the survival of mice subjected to severe TAB (Fig. 8).
Fig. 7.
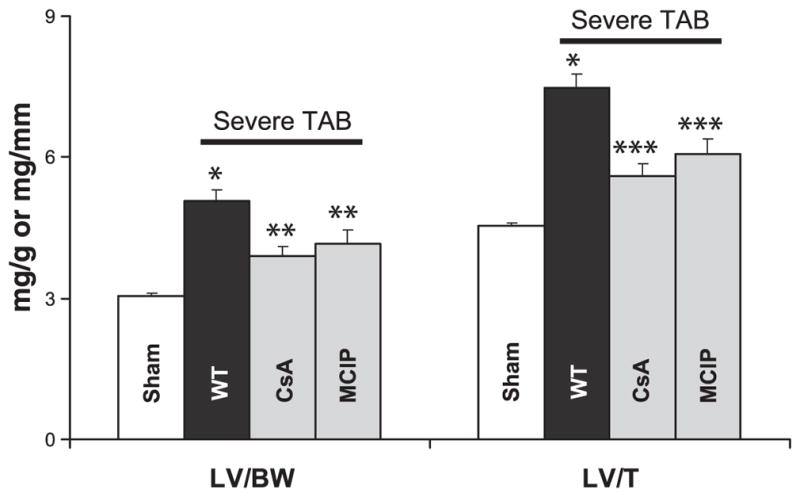
LV weight normalized to body weight or tibia length in wild-type (WT) animals subjected to sham surgery (n = 5) or severe TAB as listed [WT, n = 12; cyclosporin A (CsA), n = 8; MCIP, n = 12]. *P < 0.01 vs. sham; **P < 0.05 vs. vehicle-treated WT; ***P < 0.01 vs. vehicle-treated WT.
Fig. 8.
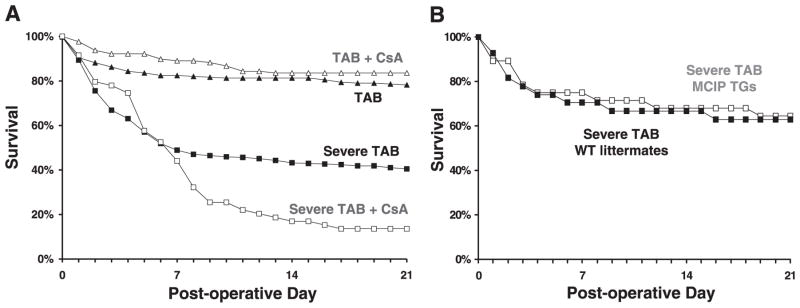
Kaplan-Meier survival analysis. A: Comp Hyp mice (▲, n = 262) and Comp Hyp mice treated with CsA (△, n = 128) manifested similar survival. In contrast, HF mice treated with CsA (□, n = 59) manifested worse survival compared with HF mice treated with vehicle (■, n = 268). B: MCIP1 transgenic mice subjected to severe TAB (□, n = 14) survived similarly to WT littermates subjected to similar surgery (■, n = 5).
Recent studies have called into question the notion that pressure-overload hypertrophy is necessarily adaptive and protective (for a review, see Ref. 18). To determine whether the increase in mortality in CsA-treated severe TAB mice was due to the loss of a beneficial calcineurin-dependent response or because of secondary CsA toxicity, we studied transgenic animals with cardiac-specific overexpression of MCIP1 (35). MCIP1 binds directly to the catalytic subunit (CnA) of the calcineurin holoenzyme, inhibiting activating effects on nuclear factor of activated T cells or myocyte enhancer factor-2 (35). Cardiomyocyte-autonomous suppression of calcineurin by the α-MHC-MCIP1 transgene blunted the hypertrophic response to TAB (data not shown), as reported previously (34). Similar findings were obtained in failing hearts (Fig. 7); forced expression of the α-MHC-MCIP1 transgene was sufficient to blunt hypertrophic growth triggered by severe TAB. Together, these findings are consistent with the notion that calcineurin-dependent signaling is required for hypertrophic growth under conditions of severe hemodynamic stress.
In contrast to CsA, the α-MHC-MCIP1 transgene did not cause in increase in mortality (P = NS) relative to wild-type littermate controls subjected to similar stress (Fig. 8). These results suggest that extracardiac effects of CsA may provoke toxicity in the setting of clinical HF. Our results with the α-MHC-MCIP1 transgenic mice, however, suggest that cardiomyocyte-specific suppression of calcineurin-dependent signaling pathways–and consequent blunting of heart growth–can be well tolerated, despite the persistence of elevated afterload stress.
DISCUSSION
The mass of the LV increases in numerous, diverse forms of heart disease. Whereas great strides have been made in identifying signaling pathways that promote pressure-overload cardiac hypertrophy, the molecular events leading to decompensation and failure are poorly understood. In this study, we developed models of graded hemodynamic stress that induce dramatically different clinical phenotypes to compare Comp Hyp with pressure-overload HF. Using this platform, we demonstrated that 1) an incremental increase in the severity of aortic constriction is sufficient to cause decompensated, hypertrophic HF that recapitulates many aspects of clinical HF; 2) differences in stress-response signaling and calcium homeostasis are detectable, although modest, in compensated versus decompensated phenotypes under steady-state conditions; 3) calcineurin suppression is sufficient to blunt hypertrophic growth in the context of persistently severe hemodynamic stress; and 4) cardiomyocyte-autonomous suppression of calcineurin is well tolerated despite persistence of severe hemodynamic stress. In addition, we described a new model of pressure-overload HF in mice that can serve as a platform for analyzing mechanisms of pathological ventricular remodeling and for testing novel therapies [e.g., phosphodiesterase inhibition (39)].
In our hands, working with male C57BL6 mice, surgical stenosis of the thoracic aorta to the degree of a 27-gauge needle induces Comp Hyp without clinical HF or increased mortality [Hill et al. (21) and the present study]. Slightly tighter stenosis (differing by only 1/20th mm) induced ventricular dysfunction and clinical HF despite comparable degrees of cardiomyocyte hypertrophic growth. We found that the phenotypes resulting from these surgical interventions were highly reproducible, allowing us to make direct comparisons between compensated and decompensated forms of hypertrophy.
A wide range of parameters was used to assess the clinical outcome of our surgical procedures. Severe TAB animals manifested numerous features of HF not observed in TAB animals. These included lethargic behavior, increased levels of circulating TNF-α, and evidence of both left-sided and right-sided venous engorgement and edema. There was a decrease in systolic function and an increase in end-diastolic volume, consistent with HF. Simultaneous measurements of distal and proximal carotid artery pressures demonstrated that transstenotic pressure gradients were diminished in Severe TAB mice, consistent with impaired pump function of failing hearts. Animals subjected to standard TAB demonstrated none of these clinical features despite the fact that they manifested an increase in cardiac mass. The geometry of hearts with stable Comp Hyp was very different from that of the failing, severe TAB hearts.
Within 24 h after surgery, there were similar rates of mortality among the sham-operated, TAB, and severe TAB mice, presumably due to the initial surgical trauma. However, after this initial 24-h recovery period, TAB was well tolerated, whereas half of the severe TAB animals died over the next 7 days. Three weeks was chosen as an end point to assure sufficient animals for analysis and because we (21) have shown previously that the Comp Hyp resulting from standard TAB has reached steady state at this point. Survival experiments extending up to 18 mo have shown that TAB is well tolerated. In contrast, mortality in the severe TAB model continues with few surviving past 2 mo.
In this model, an incremental increase in the degree of aortic constriction is sufficient to trigger hypertrophic HF as opposed to Comp Hyp. It is important to note that, despite vastly different geometry and function, both the TAB and severe TAB hearts expressed elevated levels of ANF and BNP (molecular markers of HF), suggesting that both undergo pathological hypertrophic remodeling. Physiological hypertrophy, in contrast, occurs during development, in response to exercise, and during pregnancy, without an associated reactivation of the fetal gene program.
Although indicative that the heart is under stress, expression of ANF and BNP has antihypertrophic properties, as mice lacking the gene encoding guanylyl cyclase A, a common receptor for ANF and BNP, have marked hypertrophy and fibrosis (24, 31). Recombinant human BNP (nesiritide) is efficacious in the clinical treatment of decompensated HF (11). Thus the elevated expression of ANF and BNP in the heart may be a protective stress response. It is interesting to note that the severe TAB failing hearts had significantly lower levels of ANF and BNP expression than hearts with stable Comp Hyp. It is impossible to determine from these steady-state, end-point measurements whether this stems from inability of the failing heart to mount an effective stress response or because expression of these protective peptides decreases as HF progresses. Close examination of the temporal dynamics of this response could yield important clues relevant to the role of vasoactive peptides in the pathogenesis and therapeutics of HF.
Profiles of stress-responsive signaling and calcium handling
Evidence in the literature is conflicting regarding the role of intracellular signaling mechanisms in the pathogenesis of HF. Several studies have reported an association between HF and activation of MAPK (9, 20), protein kinase C (3, 4), Akt (20), or calcineurin (20, 25, 46) pathways. Others find no such association (40). A number of signaling cascades are activated within minutes of the imposition of hemodynamic load, triggering an array of downstream kinases during the acute phase response (16). Some evidence points to a second, later phase of activation, suggesting that certain pathways may have distinct roles at different stages of HF progression (23). By inference, this double peak of activation implicates feedback by negative regulators capable of arresting disease progression under certain circumstances. An attractive approach to the treatment and management of heart disease would be to capitalize on these endogenous negative regulators. It is for this reason that we assessed the activation profile of signaling cascades during the steady-state, maintenance phases of load-induced hypertrophy and failure, with a goal of identifying novel targets of therapy in chronic disease.
MAPK pathways provide a crucial link between extracellular stimuli and transcriptional regulation of numerous genes. Each of the three major MAPK arms has been implicated in hypertrophic signaling, and each has been shown to be activated during the acute response to TAB, although controversy persists regarding their relative importance and their contribution to the maladaptive features of hypertrophy (6, 19). TAB and severe TAB provoke acute-onset mechanical stress on the heart; the resulting, transient activation of ERK, JNK, and p38 kinases, however, likely has little bearing on chronic heart disease. Focusing on load-induced HF under steady-state conditions, we observed selective activation of ERK and JNK. We speculate that persistent activation of these stress-responsive signaling pathways may contribute to progressive myocardial decompensation, a hypothesis that we are currently testing.
Both Comp Hyp and HF hearts manifested changes in calsequestrin and PLB that would result in altered calcium handling. These changes were more pronounced in HF hearts. Decreased calsequestrin will reduce the Ca2+ buffering capacity of the SR, reducing both Ca2+ release and uptake. Protein kinase A-dependent phosphorylation of PLB at Ser16, however, would be expected to release SERCA from tonic inhibition, working to augment Ca2+ uptake (27). Indeed, PLB phosphorylation was highest in failing, severe TAB hearts. Taken as a whole, differential Ca2+ handling provokes changes in contractility and directly impacts the activity of Ca2+/calmodulin kinase and calcineurin, both of which have been linked to pathological remodeling of the heart. Changes in calsequestrin levels and PLB phosphorylation were greatest in HF hearts compared with Comp Hyp hearts, indicative of a larger perturbation in Ca2+ handling.
Calcineurin signaling
Calcineurin is a Ca2+/calmodulin-regulated cytoplasmic protein phosphatase whose activity is required for hypertrophic transformation of the heart in the setting of many, but not all, forms of stress (for a review, see Ref. 18). The importance of calcineurin signaling in HF has been the subject of debate, with some investigators reporting activation (20, 25) but others detecting none (40). Some evidence from infarction models suggests that calcineurin activation correlates with the severity of the pathological condition (46).
We found evidence of increased calcineurin protein levels in Comp Hyp but not in pressure-overload HF. These data do not necessarily, however, point to increased calcineurin activity, as calcineurin is not active without a rise in cytoplasmic Ca2+ and binding of calmodulin. However, increased MCIP1 protein abundance is an indication that calcineurin is more active in both the hypertrophic and failing hearts compared with sham-operated control hearts. Once again, this is consistent with a greater shift in Ca2+ handling in the failing heart.
The calcineurin inhibitor CsA blunted hypertrophic growth in both the Comp Hyp and HF hearts, demonstrating functional involvement of calcineurin in both. Recent strides in deciphering mechanisms that underlie pathological growth of the heart suggest that calcineurin mediates a maladaptive form of hypertrophy. In fact, suppression of calcineurin-dependent hypertrophy is surprisingly well tolerated in the setting of moderate hemodynamic stress (for a review, see Ref. 18). Thus calcineurin inhibition has been proposed as a therapeutic approach to cardiac hypertrophy and failure. It was therefore of concern that CsA increased mortality in severe TAB hearts. Similarly, CsA has been shown to worsen outcomes in genetic models of hypertrophy (36). CsA can alter expression of the cardiac Na+/Ca2+ exchanger (45) and L-type Ca2+ channel (unpublished observations), perhaps contributing to the increased concentrations of intracellular Ca2+ observed in cells treated with CsA (14). CsA can also affect cellular targets other than calcineurin, such as the mitochondrial permeability transition pore (38, 43). Calcineurin is a ubiquitous protein, and systemic administration of CsA has effects on other, noncardiac organ systems.
Transgenic overexpression of MCIP1 was employed to inhibit calcineurin specifically in cardiac myocytes. MCIP1 overexpression blunts the hypertrophic response to activated calcineurin (35), hormonal or exercise stress (34), and pressure overload (22). Here, we report that MCIP1 blunted the hypertrophic growth response to severe pressure-overload stress, yet blunted hypertrophy was not associated with clinical deterioration or mortality detriment. Together, these findings point to calcineurin as a nodal point in the complex network of hypertrophic–and now failure–signaling. Furthermore, they suggest that the increased mortality in severe TAB mice treated with CsA may be a consequence of secondary toxicity.
Targeted calcineurin inhibition by MCIP1 has been shown to blunt hypertrophic remodeling in response to myocardial infarction and improve survival (41). In our model of HF, however, calcineurin inhibition by MCIP1 did not improve survival despite substantial blunting of hypertrophic growth. Certainly, much of the morbidity associated with HF stems from its negative impact on quality of life; in our animal models, however, it is impossible to assess improvement in symptoms. In any event, our results with cardiac-specific MCIP1 overexpression suggest that calcineurin inhibitors that target the myocardium with calcineurin specificity may provide clinical benefit. A major challenge for the future will be the development of such compounds. In addition, further work will be required to understand the compensatory mechanisms that regulate circulatory performance when “compensatory” hypertrophy is eliminated.
Limitations
Aortic banding induces abrupt elevations in ventricular wall stress, analogous to that seen in the spared regions of the infarcted LV. This model is less applicable to hypertension or aortic stenosis, however, where ventricular pressures typically rise gradually over time and myocyte hypertrophy precedes failure. Furthermore, these studies focus on rodents whose levels of physical exertion are limited by their being maintained in cages. To minimize the effects of gender, we studied only male mice. However, apparent strain differences were seen, as C57BL6 mice manifested a trend (P = NS) toward higher mortality from severe TAB (Fig. 8A) compared with wild-type littermates of the MCIP transgenic line (Fig. 8B). For all these reasons, it is difficult to extrapolate our findings to freely mobile large animals subjected to chronic, low-level afterload stress.
Summary and perspective
Recent studies have demonstrated that therapeutic targeting of hypertension-induced hypertrophy is a viable strategy that confers long-term mortality benefit (12, 30). The prospect of modulating hypertrophic growth of the myocardium in other contexts, without provoking cardiovascular compromise, requires that we identify commonalities and differences in the signaling systems that promote stable Comp Hyp versus HF. In this study, we found that signaling pathways and Ca2+ handling are differentially altered in pressure-overload hypertrophy and failure, suggesting that targets of therapeutic relevance may differ in these two important disease phenotypes. In both cases, however, calcineurin activation led to pathological remodeling, and suppression of calcineurin signaling was well tolerated.
Supplementary Material
Acknowledgments
We thank Dr. Joseph Francis for measurements of TNF.
GRANTS
This study was supported by grants from the Donald W. Reynolds Cardiovascular Clinical Research Center and American Heart Association and by National Heart, Lung, and Blood Institute Grants HL-03908, HL-072016, and HL-075173.
Footnotes
The Supplemental Material for this article (Supplemental Fig. S1) is available online at http://physiolgenomics.physiology.org/cgi/content/full/00061.2005/DC1.
References
- 1.American Heart Association. Heart Disease and Stroke Statistics–2004 Update. Dallas, TX: American Heart Association; 2003. [Google Scholar]
- 2.Berenji K, Drazner MH, Rothermel BA, Hill JA. Does load-induced ventricular hypertrophy progress to systolic heart failure? Am J Physiol Heart Circ Physiol. 2005;289:H8–H16. doi: 10.1152/ajpheart.01303.2004. [DOI] [PubMed] [Google Scholar]
- 3.Bowling N, Walsh RA, Song GJ, Estridge T, Sandusky GE, Fouts RL, Mintze K, Pickard T, Roden R, Bristow MR, Sabbah HN, Mizrahi JL, Gromo G, King GL, Vlahos CJ. Increased protein kinase C activity and expression of Ca2+-sensitive isoforms in the failing human heart. Circulation. 1999;99:384–391. doi: 10.1161/01.cir.99.3.384. [DOI] [PubMed] [Google Scholar]
- 4.Braz JC, Gregory K, Pathak A, Zhao W, Sahin B, Klevitsky R, Kimball TF, Lorenz JN, Nairn AC, Liggett SB, Bodi I, Wang S, Schwartz A, Lakatta EG, DePaoli-Roach AA, Robbins J, Hewett TE, Bibb JA, Westfall MV, Kranias EG, Molkentin JD. PKC-alpha regulates cardiac contractility and propensity toward heart failure. Nat Med. 2004;10:248–254. doi: 10.1038/nm1000. [DOI] [PubMed] [Google Scholar]
- 5.Brown DW, Giles WH, Croft JB. Left ventricular hypertrophy as a predictor of coronary heart disease mortality and the effect of hypertension. Am Heart J. 2000;140:848–856. doi: 10.1067/mhj.2000.111112. [DOI] [PubMed] [Google Scholar]
- 6.Bueno OF, Molkentin JD. Involvement of extracellular signal-regulated kinases 1/2 in cardiac hypertrophy and cell death. Circ Res. 2002;91:776–781. doi: 10.1161/01.res.0000038488.38975.1a. [DOI] [PubMed] [Google Scholar]
- 7.Bush E, Fielitz J, Melvin L, Martinez-Arnold M, McKinsey TA, Plichta R, Olson EN. A small molecular activator of cardiac hypertrophy uncovered in a chemical screen for modifiers of the calcineurin signaling pathway. Proc Natl Acad Sci USA. 2004;101:2870–2875. doi: 10.1073/pnas.0308723101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 9.Cook SA, Sugden PH, Clerk A. Activation of c-Jun N-terminal kinases and p38-mitogen-activated protein kinases in human heart failure secondary to ischaemic heart disease. J Mol Cell Cardiol. 1999;31:1429–1434. doi: 10.1006/jmcc.1999.0979. [DOI] [PubMed] [Google Scholar]
- 10.Czech MP. Dynamics of phosphoinositides in membrane retrieval and insertion. Annu Rev Physiol. 2003;65:791–815. doi: 10.1146/annurev.physiol.65.092101.142522. [DOI] [PubMed] [Google Scholar]
- 11.De Lemos JA, McGuire DK, Drazner MH. B-type natriuretic peptide in cardiovascular disease. Lancet. 2003;362:316–322. doi: 10.1016/S0140-6736(03)13976-1. [DOI] [PubMed] [Google Scholar]
- 12.Devereux RB, Wachtell K, Gerdts E, Boman K, Nieminen MS, Papademetriou V, Rokkedal J, Harris K, Aurup P, Dahlof B. Prognostic significance of left ventricular mass change during treatment of hypertension. JAMA. 2004;292:2350–2356. doi: 10.1001/jama.292.19.2350. [DOI] [PubMed] [Google Scholar]
- 13.Diwan A, Tran T, Misra A, Mann DL. Inflammatory mediators and the failing heart: a translational approach. Curr Mol Med. 2003;3:161–182. doi: 10.2174/1566524033361537. [DOI] [PubMed] [Google Scholar]
- 14.Fatkin D, McConnell BK, Mudd JO, Semsarian C, Moskowitz IG, Schoen FJ, Giewat M, Seidman CE, Seidman JG. An abnormal Ca2+ response in mutant sarcomere protein-mediated familial hypertrophic cardiomyopathy. J Clin Invest. 2000;106:1351–1359. doi: 10.1172/JCI11093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fischer TA, Ludwig S, Flory E, Gambaryan S, Singh K, Finn P, Pfeffer MA, Kelly RA, Pfeffer JM. Activation of cardiac c-Jun NH2-terminal kinases and p38-mitogen-activated protein kinases with abrupt changes in hemodynamic load. Hypertension. 2001;37:1222–1228. doi: 10.1161/01.hyp.37.5.1222. [DOI] [PubMed] [Google Scholar]
- 17.Francis J, Weiss RM, Johnson AK, Felder RB. Central mineralo-corticoid receptor blockade decreases plasma TNF-α after coronary artery ligation in rats. Am J Physiol Regul Integr Comp Physiol. 2003;284:R328–R335. doi: 10.1152/ajpregu.00376.2002. [DOI] [PubMed] [Google Scholar]
- 18.Frey N, Katus HA, Olson EN, Hill JA. Hypertrophy of the heart–a new therapeutic target? Circulation. 2004;109:1580–1589. doi: 10.1161/01.CIR.0000120390.68287.BB. [DOI] [PubMed] [Google Scholar]
- 19.Frey N, Olson EN. Cardiac hypertrophy: the good, the bad, and the ugly. Annu Rev Physiol. 2003;65:45–79. doi: 10.1146/annurev.physiol.65.092101.142243. [DOI] [PubMed] [Google Scholar]
- 20.Haq S, Choukroun G, Lim H, Tymitz KM, del Monte F, Gwathmey J, Grazette L, Michael A, Hajjar R, Force T, Molkentin JD. Differential activation of signal transduction pathways in human hearts with hypertrophy versus advanced heart failure. Circulation. 2001;103:670–677. doi: 10.1161/01.cir.103.5.670. [DOI] [PubMed] [Google Scholar]
- 21.Hill JA, Karimi M, Kutschke W, Davisson RL, Zimmerman K, Wang Z, Kerber RE, Weiss RM. Cardiac hypertrophy is not a required compensatory response to short-term pressure overload. Circulation. 2000;101:2863–2869. doi: 10.1161/01.cir.101.24.2863. [DOI] [PubMed] [Google Scholar]
- 22.Hill JA, Rothermel BA, Yoo KD, Cabuay B, Demetroulis E, Weiss RM, Kutschke W, Bassel-Duby R, Williams RS. Targeted inhibition of calcineurin in pressure-overload hypertrophy: Preservation of systolic function. J Biol Chem. 2002;277:10251–10255. doi: 10.1074/jbc.M110722200. [DOI] [PubMed] [Google Scholar]
- 23.Hoshijima M, Chien KR. Mixed signals in heart failure: cancer rules. J Clin Invest. 2002;109:849–855. doi: 10.1172/JCI15380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kishimoto I, Dubois SK, Garbers DL. The heart communicates with the kidney exclusively through the guanylyl cyclase-A receptor: acute handling of sodium and water in response to volume expansion. Proc Natl Acad Sci USA. 1996;93:6215–6219. doi: 10.1073/pnas.93.12.6215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lim HW, Molkentin JD. Calcineurin and human heart failure. Nat Med. 1999;5:246–247. doi: 10.1038/6430. [DOI] [PubMed] [Google Scholar]
- 26.Lorell BH, Carabello BA. Left ventricular hypertrophy–pathogenesis, detection, and prognosis. Circulation. 2000;102:470–479. doi: 10.1161/01.cir.102.4.470. [DOI] [PubMed] [Google Scholar]
- 27.MacLennan DH, Kranias EG. Phospholamban: a crucial regulator of cardiac contractility. Nat Rev Mol Cell Biol. 2003;4:566–577. doi: 10.1038/nrm1151. [DOI] [PubMed] [Google Scholar]
- 28.Muller FU, Kirchhefer E, Begrow F, Reinke U, Neumann J, Schmitz W. Junctional sarcoplasmic reticulum transmembrane proteins in the heart. Basic Res Cardiol. 2003;97:I52–I55. doi: 10.1007/s003950200030. [DOI] [PubMed] [Google Scholar]
- 29.Nicol RL, Frey N, Pearson G, Cobb M, Richardson J, Olson EN. Activated MEK5 induces serial assembly of sarcomeres and eccentric cardiac hypertrophy. EMBO J. 2001;20:2757–2767. doi: 10.1093/emboj/20.11.2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Okin PM, Devereux RB, Jern S, Kjeldsen SE, Julius S, Nieminen MS, Snapinn S, Harris KE, Aurup P, Edelman JM, Wedel H, Lindholm LH, Dahlof B. Regression of electrocardiographic left ventricular hypertrophy during antihypertensive treatment and the prediction of major cardiovascular events. JAMA. 2004;292:2343–2349. doi: 10.1001/jama.292.19.2343. [DOI] [PubMed] [Google Scholar]
- 31.Oliver PM, Fox JE, Kim R, Rockman HA, Kim HS, Reddick RL, Pandey KN, Milgram SL, Smithies O, Maeda N. Hypertension, cardiac hypertrophy, and sudden death in mice lacking natriuretic peptide receptor A. Proc Natl Acad Sci USA. 1997;94:14730–14735. doi: 10.1073/pnas.94.26.14730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ptacek LJ. Channelopathies: ion channel disorders of muscle as a paradigm for paroxysmal disorders of the nervous system. Neuromuscul Disord. 1997;7:250–255. doi: 10.1016/s0960-8966(97)00046-1. [DOI] [PubMed] [Google Scholar]
- 33.Rockman HA, Ross RS, Harris AN, Knowlton KU, Steinhelper ME, Field LJ, Ross J, Jr, Chien KR. Segregation of atrial-specific and inducible expression of an atrial natriuretic factor transgene in an in vivo murine model of cardiac hypertrophy. Proc Natl Acad Sci USA. 1991;88:8277–8281. doi: 10.1073/pnas.88.18.8277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rothermel BA, McKinsey TA, Vega RB, Nicol RL, Mammen P, Yang J, Antos CL, Shelton JM, Bassel-Duby R, Olson EN, Williams RS. Myocyte-enriched calcineurin-interacting protein, MCIP1, inhibits cardiac hypertrophy in vivo. Proc Natl Acad Sci USA. 2001;98:3328–3333. doi: 10.1073/pnas.041614798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rothermel BA, Vega RB, Yang J, Wu H, Bassel-Duby R, Williams RS. A protein encoded within the Down syndrome critical region is enriched in striated muscles and inhibits calcineurin signaling. J Biol Chem. 2000;275:8719–8725. doi: 10.1074/jbc.275.12.8719. [DOI] [PubMed] [Google Scholar]
- 36.Schmitt JP, Semsarian C, Arad M, Gannon J, Ahmad F, Duffy C, Lee RT, Seidman CE, Seidman JG. Consequences of pressure overload on sarcomere protein mutation-induced hypertrophic cardiomyopathy. Circulation. 2003;108:1133–1138. doi: 10.1161/01.CIR.0000086469.85750.48. [DOI] [PubMed] [Google Scholar]
- 37.Semsarian C, Ahmad I, Giewat M, Georgakopoulos D, Schmitt JP, McConnell BK, Reiken S, Mende U, Marks AR, Kass DA, Seidmann CE, Seidmann JG. The L-type calcium channel inhibitor diltiazem prevents cardiomyopathy in a mouse model. J Clin Invest. 2002;109:1013–1020. doi: 10.1172/JCI14677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Snyder SH, Sabatini DM, Lai MM, Steiner JP, Hamilton GS, Suzdak PD. Neural actions of immunophilin ligands. Trends Pharmacol Sci. 1998;19:21–26. doi: 10.1016/s0165-6147(97)01146-2. [DOI] [PubMed] [Google Scholar]
- 39.Takimoto E, Champion HC, Li M, Belardi D, Ren S, Rodriguez ER, Bedja D, Gabrielson KL, Wang Y, Kass DA. Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nat Med. 2005;11:214–222. doi: 10.1038/nm1175. [DOI] [PubMed] [Google Scholar]
- 40.Tsao L, Neville C, Musaro A, McCullagh KJ, Rosenthal N. Revisiting calcineurin and human heart failure. Nat Med. 2000;6:2–3. doi: 10.1038/71478. [DOI] [PubMed] [Google Scholar]
- 41.Van Rooij E, Doevendans PA, Crijns HJ, Heeneman S, Lips DJ, van Bilsen M, Williams RS, Olson EN, Bassel-Duby R, Rothermel BA, De Windt LJ. MCIP1 overexpression suppresses left ventricular remodeling and sustains cardiac function after myocardial infarction. Circ Res. 2004;94:e18–e26. doi: 10.1161/01.RES.0000118597.54416.00. [DOI] [PubMed] [Google Scholar]
- 42.Vanhaesebroeck B, Leevers SJ, Ahmadi K, Timms J, Katso R, Driscoll PC, Woscholski R, Parker PJ, Waterfield MD. Synthesis and function of 3-phosphorylated inositol lipids. Annu Rev Biochem. 2001;70:535–602. doi: 10.1146/annurev.biochem.70.1.535. [DOI] [PubMed] [Google Scholar]
- 43.Waldmeier PC, Zimmermann K, Qian T, Tintelnot-Blomley M, Lemasters JJ. Cyclophilin D as a drug target. Curr Med Chem. 2003;10:1485–1506. doi: 10.2174/0929867033457160. [DOI] [PubMed] [Google Scholar]
- 44.Wang Z, Kutschke W, Richardson KE, Karimi M, Hill JA. Electrical remodeling in pressure-overload cardiac hypertrophy: Role of calcineurin. Circulation. 2001;104:1657–1663. doi: 10.1161/hc3901.095766. [DOI] [PubMed] [Google Scholar]
- 45.Wang Z, Nolan B, Kutschke W, Hill JA. Na+/Ca2+ exchanger remodeling in pressure-overload cardiac hypertrophy. J Biol Chem. 2001;276:17706–17711. doi: 10.1074/jbc.M100544200. [DOI] [PubMed] [Google Scholar]
- 46.Wilkins BJ, Dai YS, Bueno OF, Parsons SA, Xu J, Plank DM, Jones F, Kimball TR, Molkentin JD. Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ Res. 2004;94:110–118. doi: 10.1161/01.RES.0000109415.17511.18. [DOI] [PubMed] [Google Scholar]
- 47.Yang J, Rothermel B, Vega RB, Frey N, McKinsey TA, Olson EN, Bassel-Duby R, Williams RS. Independent signals control expression of the calcineurin inhibitory proteins MCIP1 and MCIP2 in striated muscles. Circ Res. 2000;87:E61–E68. doi: 10.1161/01.res.87.12.e61. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.