

Abstract
Background
Cellular hypertrophy requires coordinated regulation of progrowth and antigrowth mechanisms. In cultured neonatal cardiomyocytes, Foxo transcription factors trigger an atrophy-related gene program that counters hypertrophic growth. However, downstream molecular events are not yet well defined.
Methods and Results
Here, we report that expression of either Foxo1 or Foxo3 in cardiomyocytes attenuates calcineurin phosphatase activity and inhibits agonist-induced hypertrophic growth. Consistent with these results, Foxo proteins decrease calcineurin phosphatase activity and repress both basal and hypertrophic agonist-induced expression of MCIP1.4, a direct downstream target of the calcineurin/NFAT pathway. Furthermore, hearts from Foxo3-null mice exhibit increased MCIP1.4 abundance and a hypertrophic phenotype with normal systolic function at baseline. Together, these results suggest that Foxo proteins repress cardiac growth at least in part through inhibition of the calcineurin/NFAT pathway. Given that hypertrophic growth of the heart occurs in multiple contexts, our findings also suggest that certain hypertrophic signals are capable of overriding the antigrowth program induced by Foxo. Consistent with this, multiple hypertrophic agonists triggered inactivation of Foxo proteins in cardiomyocytes through a mechanism requiring the PI3K/Akt pathway. In addition, both Foxo1 and Foxo3 are phosphorylated and consequently inactivated in hearts undergoing hypertrophic growth induced by hemodynamic stress.
Conclusions
This study suggests that inhibition of the calcineurin/NFAT signaling cascade by Foxo and release of this repressive action by the PI3K/Akt pathway are important mechanisms whereby Foxo factors govern cell growth in the heart.
Keywords: angiotensin, calcineurin, hypertrophy
In response to stress from neurohumoral activation, hypertension, or other myocardial injury, the heart initially compensates with an adaptive hypertrophic increase in mass. The resulting growth and remodeling response alters the balance between protein synthesis and protein degradation. In skeletal muscle, activation of progrowth signaling pathways is accompanied by deactivation of pathways that promote proteolysis. Prominent among the atrophy-inducing pathways are those governed by Forkhead box transcription factors, O subfamily (Foxo).
Foxo transcription factors regulate key physiological functions, including responses to stress, cell-cycle progression, protein degradation, and apoptosis.1,2 There are 4 mammalian Foxo genes: Foxo1 (FKHR), Foxo3 (FKHRL1), Foxo4 (AFX), and Foxo6. The transcriptional activities of Foxo proteins are governed by posttranslational modifications such as phosphorylation and acetylation. With respect to myocyte growth and remodeling, Foxo proteins induce ubiquitin ligases and promote proteolysis in skeletal muscle.3,4
In heart, a number of signaling cascades involving transcription factors, kinases, and G-protein–coupled receptors are implicated in the regulation of muscle growth (see reviews5–7). Among these, the calcineurin/nuclear factor of activated T cells (NFAT) pathway has been shown to be a key signaling cascade that promotes cardiac hypertrophy.8 It has been reported recently that calcineurin is a target for ubiquitin-mediated proteolysis via the E3 ubiquitin ligase atrogin-1.9 Atrogin-1 expression is under the control of Foxo transcription factors.3,4 Atrogin-1 expression suppresses cardiac hypertrophy9 and is upregulated in and required for skeletal muscle atrophy.10,11 Thus far, the direct effect of Foxo factors on calcineurin/NFAT signaling has not been explored.
Here, we demonstrate that overexpression of either Foxo1 or Foxo3 decreases calcineurin phosphatase activity and reduces the basal and agonist-induced expression of the modulatory calcineurin interacting protein, exon 4 isoform (MCIP1.4), a direct target of the calcineurin/NFAT pathway.12,13 In addition, Foxo proteins inhibit agonist-induced MCIP1.4 expression and hypertrophic growth in cardiomyocytes. This inhibition occurs at the transcriptional level and therefore is consistent with the finding that Foxo proteins downregulate calcineurin/NFAT signaling. Moreover, hearts from Foxo3-null mice have elevated levels of MCIP1.4 and manifest a hypertrophic phenotype, both of which are consistent with enhanced calcineurin activity. Given that hypertrophic growth of the heart occurs in multiple contexts, our findings also suggest that certain hypertrophic signals are capable of overriding the antigrowth program induced by Foxo. Consistent with this, we find that multiple progrowth triggers inactivate Foxo proteins in cardiac myocytes through a mechanism requiring the PI3K/Akt pathway. Together, these results suggest that inhibition of the calcineurin/NFAT signaling cascade by Foxo and release of this inhibition by the PI3K/Akt pathway are major mechanisms whereby Foxo factors govern cell growth in the heart.
Methods
Primary Cardiac Cell Preparation and Adenovirus Infection
Neonatal rat ventricular myocytes were isolated from the ventricles of Sprague-Dawley rats on postnatal day 1 to 2. Cells were preplated (2 hours) to enrich for cardiac myocytes, plated at a density of 1200 cells/mm2, and cultured for 24 hours in Dulbecco’s modified Eagle’s medium (DMEM)/M199 (3:1) containing 5% FBS, 10% horse serum, and 100 μmol/L bromodeoxyuridine (BrdU; Sigma). At 24 hours after plating, the culture media was replaced with serum-free DMEM/M199. As assessed by immunohistochemistry with a monoclonal antibody against sarcomeric α-actinin, ≥95% of the cells were cardiac myocytes at harvest.
Neonatal rat cardiac fibroblasts were prepared by 2 to 3 passages of cells adhering to the Petri dish during the preplating procedure. Cells were maintained in DMEM/M199 containing 10% FBS without BrdU.
For adenovirus-mediated protein overexpression, at 24 hours after plating, cells were incubated for 2 hours with adenovirus at a multiplicity of infection of 10 to 50 plaque-forming units per cell; then, the virus was removed, and cells were cultured for an additional 24 to 48 hours in fresh serum-free DMEM/M199. Under these conditions, infection efficiency routinely exceeded 90%.
Thoracic Aortic Banding
Male C57BL6 mice (6 to 8 weeks old) were subjected to thoracic aortic constriction. At 3 weeks, when the hypertrophic response reaches steady state,14 the integrity of aortic banding was confirmed by inspection of the surgical constriction and by visualization of marked differences in caliber of the right and left carotid arteries. In some animals, transstenotic pressures were measured as described.14
Foxo3-Null Mice
Foxo3-null mice were generated as previously described.15 Foxo3+/− animals were backcrossed 6 times to FVB/n females, and the progeny of these matings were intercrossed with littermates to generate the experimental cohort.
Statistical Analysis
Data are expressed as mean±SEM. Unless otherwise specified, triplicate or quadruplicate samples were used, and each experiment was repeated at least twice. Statistical significance was analyzed with a Student unpaired t test or 1-way ANOVA, followed by Bonferroni correction for post hoc pairwise multiple comparisons. Values of P<0.05 were considered significant.
An expanded Methods section is provided in the online Data Supplement.
The authors had full access to the data and take full responsibility for their integrity. All authors have read and agree to the manuscript as written.
Results
Overexpression of Foxo1 Decreases Calcineurin Activity and MCIP1.4 Expression
To investigate the effects of Foxo transcription factors on the calcineurin/NFAT signaling cascade, we infected neonatal cardiomyocytes with adenovirus expressing Foxo1 or Foxo3. Relative to green fluorescent protein (GFP)–infected control cells, cells expressing constitutively active Foxo1 (caFoxo1) or caFoxo3 showed a significant decease in endogenous calcineurin phosphatase activity (Figure 1A). A decrease in calcineurin activity should diminish expression of downstream targets of the calcineurin/NFAT pathway. Therefore, we examined expression of the exon 4 isoform of MCIP1.4, a direct target of NFAT. In cells overexpressing either Foxo1 or Foxo3, there was a marked decrease in the abundance of MCIP1.4 protein (Figure 1B and 1C). MCIP1.4 mRNA levels, quantified by real-time reverse-transcription polymerase chain reaction (RT-PCR), also were reduced significantly (Figure 1D). In contrast, overexpression of either Foxo1 or Foxo3 did not affect the abundance of MCIP1.1, a splice variant that is not regulated by calcineurin/NFAT.
Figure 1.

Activation of Foxo1 inhibits endogenous calcineurin activity. Myocytes were infected with adenovirus as indicated. A, At 48 hours after infection, cells were harvested and calcineurin phosphatase activity was measured. B and C, Western blots and densitometric analysis of calcineurin, MCIP1.1, and MCIP1.4 in whole-cell lysates harvested 48 hours after infection. D, mRNA levels of atrogin-1 and GAPDH were detected by RT-PCR 24 hours after infection. E, Schematic of the MCIP1.4-luciferase construct. F, Myocytes were coinfected with adenovirus encoding MCIP1.4-luc plus GFP or caFoxo1-GFP and processed for luciferase activity. G, Cells were infected with adenovirus (48 hours), and MCIP1.4 and GAPDH mRNA levels were determined by real-time PCR. Data are mean±SEM (n=6 to 8). *P≤0.05; **P≤0.01; ***P≤0.001.
The proximal MCIP1.4 promoter region contains multiple NFAT-responsive elements (Figure 1E).13 Further studies explored the effect of Foxo proteins on the activity of the MCIP1.4 promoter, revealing that overexpression of caFoxo1 markedly decreased MCIP1.4 promoter activity (Figure 1F). Together, these findings suggest that Foxo factors decrease calcineurin/NFAT signaling in cardiac myocytes.
It has been reported that the E3-ubiquitin ligase atrogin-1, the expression of which is driven by Foxo factors, directly interacts with calcineurin and promotes its proteasomal degradation.9 Semiquantitative RT-PCR showed that overexpression of caFoxo1 led to elevated levels of atrogin-1 in cardiomyocytes (Figure 1G), demonstrating that Foxo1 induces the “atrogene program” in cardiomyocytes, as shown recently for Foxo3.16
Overexpression of Foxo Inhibits Angiotensin II–Induced Hypertrophic Growth
Activation of the calcineurin/NFAT pathway is sufficient to induce hypertrophic growth of the heart,17 and inhibition of calcineurin prevents hypertrophic growth in response to multiple stimuli.5 Given that Foxo1 inhibits calcineurin activity, we hypothesized that Foxo1 overexpression would blunt hypertrophic growth of cardiomyocytes. Indeed, in myocytes overexpressing GFP, angiotensin (Ang) II (100 nmol/L) provoked significant increases in both protein synthesis and cell size, but not in cells overexpressing Foxo1 or caFoxo1 (Figure 2A through 2C). Thus, activated Foxo1 opposes cell growth. We also examined the effect of increased Foxo1 activity on the expression of the β-myosin heavy chain (β-MHC) gene, the expression of which is often induced in pathological hypertrophy and requires calcineurin activity.18 PCR analysis revealed that overexpression of caFoxo1 blocked both basal and Ang II–induced β-MHC expression (Figure 2D). The marked decreases in the levels of β-MHC and MCIP1.4 transcript in Foxo1-expressing cells were not due to potential proapoptotic effects of Foxo1 because Western blot analysis revealed no increase in cleavage of caspase 3 by Foxo1 (Figure 2E) and there was no loss of total cell number.
Figure 2.

Foxo1 activity inhibits Ang-II–induced hypertrophic growth in cardiomyocytes. A, Myocytes were cultured and infected with adenovirus and treated with Ang II (48 hours). Cells were then assayed for [3H]leucine incorporation normalized to cellular DNA content. B, Representative fields of cardiomyocytes stained with an α-actinin antibody. C, Quantification of cell cross-sectional area from experiments shown in B. Sixty-five to 75 randomly selected cells from each group were measured. D, β-MHC mRNA levels were measured by real-time PCR. E, Western blots of caspase 3 in whole-cell lysates harvested from myocytes 48 hours after adenovirus infection (A, D, n=8; C, n=65 to 75). *P≤0.05, ***P≤0.001 vs untreated control; ††P≤0.01, †††P≤0.001 vs respective GFP control group.
Because an increase in calcineurin activity has been observed with agonist-induced hypertrophy in cardiomyocytes,19 we next examined the effects of Foxo1 on calcineurin activation triggered by insulin-like growth factor (IGF)-1 or Ang II. Western blot analysis showed that in cells overex-pressing GFP, IGF-1 (10 nmol/L) induced a slight increase in calcineurin protein abundance and a robust increase in the level of MCIP1.4 protein, suggesting that calcineurin activity was elevated (Figure 3A and 3B). However, in cells overex-pressing caFoxo1, agonist-triggered increases in calcineurin and MCIP1.4 abundance were both abolished (Figure 3C). Consistent with our previous experiments (Figure 1), forced expression of caFoxo1 also diminished the basal expression of MCIP1.4, indicating diminished tonic activation of calcineurin.
Figure 3.

Activation of Foxo1 blunts agonist-induced MCIP1.4 expression. Serum-starved myocytes were infected and treated with IGF-1 (A, B) or Ang II (C, D), and the protein levels of calcineurin and MCIP1 were analyzed (n=6). *P≤0.05 vs untreated control; ††P≤0.01 vs respective GFP control group.
Cardiofibroblasts contribute through paracrine mechanisms to the effects of Ang II on myocyte growth; hence, they may respond to changes in Foxo activity. To test this, we examined the effects of Foxo1 overexpression on Ang II–induced Erk activation in cultured neonatal cardiofibroblasts because the Erk signaling pathway is important for Ang II paracrine effects.20,21 Western blot analyses showed that overexpression of Foxo1 did not alter Ang II–induced Erk activation (Figure I in the online Data Supplement), indicating that Foxo does not antagonize this step in Ang II activation of fibroblasts. This finding suggests that inhibition of cardiac hypertrophy by Foxo is a direct effect on intracellular myocyte signaling rather than an alteration of paracrine mechanisms.
Foxo3-Null Hearts Have Elevated MCIP1.4 Levels and Are Hypertrophic
These findings demonstrate that an increase in either Foxo1 or Foxo3 activity inhibits the growth of cardiomyocytes in culture by suppressing the calcineurin/NFAT signaling cascade. To investigate whether a decrease in Foxo function increases calcineurin/NFAT signaling and enhances myocardial growth in vivo, we studied mice with targeted deletion of the Foxo3 gene (targeted deletion of Foxo1 is embryonic lethal15). Western blot analysis demonstrated that MCIP1.4 protein abundance was elevated in hearts from Foxo3-null mice at 12 weeks of age (Figure 4A and 4B), suggesting increased calcineurin activation, presumably from removal of tonic repression by Foxo. Furthermore, relative to wild-type littermates, Foxo3-null mice displayed a highly reproducible ≈10% increase in heart mass (P<0.01), measured as heart weight normalized to either body weight or tibia length (Figure 4C). These increases could not be attributed to differences in body size because body weight (wild type, 29.5±0.4 g; knockout, 30.90.6 g) and tibia length (wild type, 17.4±0.1 mm; knockout, 17.5±0.1 mm) were similar (P>0.05) between wild-type and Foxo3-null littermates. RNA analyses revealed no discernable changes in atrial natriuretic factor and β-MHC expression between wild-type and Foxo3-null hearts (data not shown), and systolic function in Foxo3-null mice was normal as assessed by echocardiography22 (Table I in the online Data Supplement). Together, these results support the notion that loss of Foxo function enhances calcineurin signaling and cardiac growth.
Figure 4.

Foxo3-null mice are hypertrophic. A and B, Western blot analysis of MCIP1.4 protein levels in extracts from sham-operated or TAB LVs from Foxo3–knockout (ko) or wild-type (wt) mice (n=6 to 8). **P≤0.01 vs sham-operated; †P≤0.05 vs wt sham group. C, Heart weight/body weight (HW/BW; left) and heart weight/tibia length (HW/T; right) ratios of control, sham-operated, or TAB mice 3 weeks after surgery. Male Foxo3-null mice and wild-type littermates were studied at 12 weeks.
To examine the function of Foxo proteins during hypertrophic growth in vivo, we assessed the growth response of Foxo3-null mice to thoracic aortic banding (TAB). Interestingly, pressure stress induced by TAB triggered similar degrees of hypertrophic growth in Foxo3-null mice relative to wild-type littermates (Figure 4C), and TAB-induced increases in MCIP1.4 abundance were not altered by loss of Foxo3 (Figure 4A and 4B). As expected, TAB induced robust increases in cardiac mass without evidence of left- or right-heart failure manifested as edematous congestion of lung or liver (Table II in the online Data Supplement). These findings—that wild-type and Foxo3-null mice manifest similar hypertrophic growth relative to their starting weight—are consistent with Foxo proteins being inactivated in the setting of pressure stress, resulting in the release of calcineurin from their inhibitory action.
Foxo Proteins Are Inactivated During Load-Induced Cardiac Hypertrophy
Robust hypertrophic growth in pressure-stressed heart suggests that the antigrowth effects of Foxo are antagonized. To investigate this, we assessed the phosphorylation level of Foxo proteins in TAB animals. Analysis of left ventricular (LV) extracts from TAB mice revealed marked increases in phosphorylated Foxo1 and Foxo3 relative to sham-operated controls (Figure 5A and 5B) detected as early as 3 days after aortic constriction (Figure 5C). No appreciable changes in the levels of total Foxo3 protein were detected. (In our hands, commercially available antibodies do not detect endogenous Foxo1 in cardiomyocytes or mouse LV.) These findings are consistent with Foxo protein inactivation by pressure stress.
Figure 5.

Foxo1 and Foxo3 are inactivated in load-induced hypertrophy. A, Protein extracts from sham-operated or TAB LVs 3 weeks after surgery were separated by SDS-PAGE and probed with antibodies against phosphorylated or total Foxo1 and Foxo3. B, Mean values of phosphorylated Foxo/total Foxo ratios from sham- or TAB-treated groups were normalized to the mean value of the phosphorylated Foxo/total Foxo from the sham group (n=4 to 5). C, Time course of Foxo phosphorylation in hypertrophic hearts. LV protein extracts from sham-operated (S) or TAB (T) mice were probed with antibodies against phosphorylated or total Foxo protein. *P≤0.05, **P≤0.01 vs sham group.
Multiple Prohypertrophic Agonists Inactivate Foxo
To test whether Foxo factors are inactivated by other prohypertrophic factors, we first examined the effect of Ang II treatment on endogenous Foxo proteins. In serum-starved cardiomyocytes, Ang II treatment induced a time-dependent increase in the levels of phosphorylated Foxo1 and Foxo3 without affecting total protein levels (Figure 6A and 6B). Phosphorylation of Foxo occurred at an Akt consensus site (Thr24/32) and reached maximal levels by 15 to 30 minutes after Ang II treatment. Several other hypertrophic agonists, including phenylephrine (PE), iso-proterenol (ISO) (Figure 6C and 6D), and IGF-1 (data not shown), showed similar effects.
Figure 6.

Multiple hypertrophic ligands inactivate Foxo in cardiomyocytes. A–D, Western blots and densitometric analysis of levels of phosphorylated Foxo1 and phosphorylated Foxo3. Serum-starved cardiomyocytes were treated with Ang II, PE (10 μmol/L), or ISO (10 nmol/L) for indicated durations. E, Ang II induces Foxo1-GFP nuclear export. Myocytes were infected, serum starved overnight, and then treated with vehicle or Ang II for 30 minutes. Hoechst 33334 was used for nuclear staining. F, Hypertrophic ligands induce endogenous Foxo3 translocation. Serum-starved myocytes were treated with Ang II or PE. Nuclear and cytoplasmic extracts were analyzed for total Foxo3, GAPDH, or lamin A/C. G, Ang II decreases atrogin-1 expression. Serum-starved myocytes were treated with Ang II and assayed for atrogin-1 and GAPDH mRNA levels by RT-PCR (n=6). Con indicates vehicle-treated control; N, nuclear fraction; and C, cytoplasmic fraction. *P≤0.05 vs untreated control group.
Ang II treatment also triggered nuclear export of GFP-tagged Foxo1 in cardiomyocytes (Figure 6E). Foxo1 migrated to the cytoplasm as early as 5 minutes after Ang II exposure, with most cells showing cytoplasmic enrichment by 30 minutes. Other hypertrophic agonists, including IGF-1, PE, ISO, and endothelin-1, stimulated comparable degrees of Foxo1 nuclear export (data not shown). Phosphorylation of the Akt consensus sites Thr-24 and Ser-319 is required for agonist-induced Foxo translocation because the constitutively active Foxo1 mutant (T24A/S319A) did not undergo nuclear export in response to Ang II treatment (Figure 6E). Western blot analysis of nuclear and cytoplasmic fractions confirmed that most endogenous Foxo proteins were cytoplasmic in the presence of Ang II or PE (Figure 6F).
Finally, we have investigated the effects of Ang II treatment on the expression of atrogin-1. In serum-starved uninfected cells, Ang II induced a rapid, time-dependent decrease in atrogin-1 expression (Figure 6G). Together, these results provide strong evidence that Foxo transcription factors are inactivated by cardiac hypertrophic agonists.
Ang II Inactivates Foxo Proteins Through the PI3K/Akt Pathway
Foxo proteins were phosphorylated at Akt consensus sites, suggesting involvement of the PI3K/Akt pathway. To explore this, we examined Akt phosphorylation by Western blot, finding that Ang II promoted phosphorylation of Akt at Thr-308 and Ser-473 (Figure 7A and 7B), consistent with Akt activation. Similar results were obtained from PE or ISO treatment (Figure 7C and 7D). Interestingly, with all 3 ligands, phosphorylation of Thr308 consistently preceded that of Ser-473 (Figure 7A and 7C). Consistent with these results, the PI3K-selective inhibitor LY294002 abolished Ang II–induced increases in Foxo phosphorylation (Figure 7E and 7F) and nuclear export (Figure 7G).
Figure 7.

Ang II induces Foxo phosphorylation through the PI3K/Akt pathway. A–D, Western blots and densitometric analysis of levels of phosphorylated Foxo1 and phosphorylated Foxo3. Serum-starved cardiomyocytes were treated with Ang II, PE (10 μmol/L), or ISO (10 nmol/L) for indicated durations. E–G, LY294002 blocks Ang II–induced increases in Foxo phosphorylation (E, F) and Foxo1-GFP nuclear export (G). Serum-starved neonatal rat ventricular myocytes were preincubated with LY294002 (1 hour) before treatment with Ang II (30 minutes). H, I, Ang II–induced phosphorylation of both Foxo and Akt was blocked by LY294002 but not by inhibitors of other kinases. Western blots (H) and densitometric analysis (I) of levels of phosphorylated Foxo1 and phosphorylated Foxo3 (n=4). *P≤0.05, ***P≤0.001 vs vehicle-treated or Ang II–alone controls.
To query the role of other signaling pathways, a panel of selective inhibitors was used. In contrast to LY294002, inhibitors of calcineurin (CsA), cAMP-dependent protein kinase (H-89), protein kinase C (Gö6976), and p38 MAP kinase (SB202190) had no significant effect on Ang II–induced phosphorylation of Akt, Foxo1, or Foxo3 (Figure 7H and 7I). LY294002 also reduced basal levels of phosphorylated Foxo1 and Foxo3, presumably by inhibiting basal Akt activity; none of the other inhibitors had detectable effects on basal Foxo phosphorylation when tested alone (data not shown). Taken together, these results strongly suggest that Ang II inactivates Foxo proteins mainly through the PI3K/Akt pathway.
Discussion
In this study, we demonstrate that Foxo1 and Foxo3 transcription factors inhibit cardiac hypertrophy as a result, at least in part, of suppression of calcineurin/NFAT signaling. Increased Foxo activity induces the expression of atrogin-1, an E3 ubiquitin ligase that promotes calcineurin proteolysis. This, in turn, leads to remarkable decreases in calcineurin phosphatase activity and reduced expression of its downstream target MCIP1.4. The decrease in MCIP1.4 expression occurs at the transcriptional level because Foxo suppresses MCIP1.4 promoter activity and reduces MCIP1.4 mRNA abundance. These results strongly suggest that Foxo activity downregulates calcineurin/NFAT signaling. Consistent with these results, Foxo3-null mice express more MCIP1.4 protein and manifest a cardiac hypertrophic phenotype under basal conditions. Our studies also show that Foxo proteins are inactivated by hemodynamic stress and by several hypertrophic ligands through a mechanism involving the PI3K/Akt pathway. These studies provide strong evidence that Foxo transcription factors attenuate calcineurin/NFAT signaling and are negative modulators of cardiac muscle growth. These repressive actions of Foxo are released in the presence of hypertrophic stimuli via mechanisms involving PI3K (Figure 8).
Figure 8.
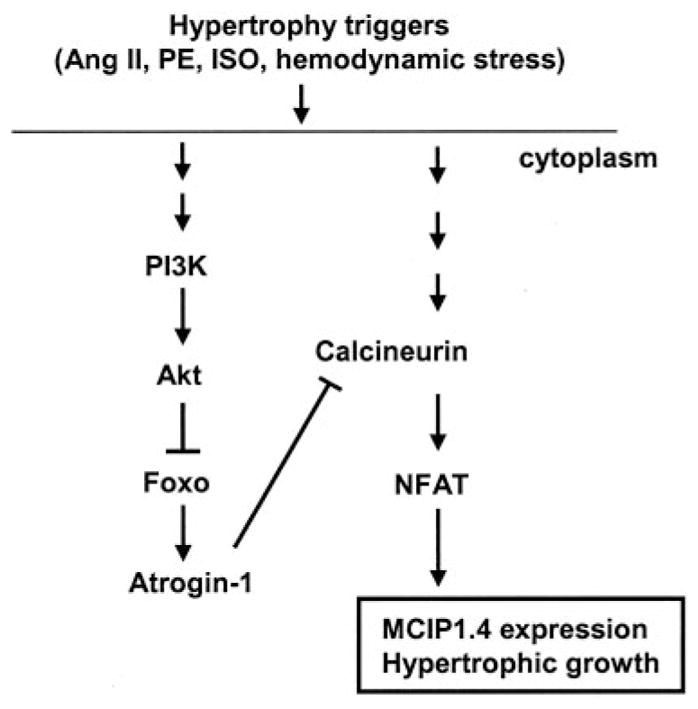
Role of Foxo proteins in cardiac hypertrophy. Neuro-hormonal activation or hemodynamic stress induces Foxo phosphorylation and consequent Foxo inhibition through the PI3K/Akt pathway. Activation of Foxo inhibits hypertrophic growth, at least in part, by blunting calcineurin-mediated pro-growth pathways.
Overexpression of Foxo1 in skeletal muscle resulted in both calcineurin-dependent slow muscle fiber loss and a decrease in total muscle mass.23 Therefore, Foxo1 activity in skeletal muscle may affect both calcineurin-dependent events and general protein degradation. Similarly, the antihypertrophic effects of Foxo in cardiac muscle may be mediated by mechanisms beyond targeting calcineurin signaling. For example, Foxo may counter cardiomyocyte growth through promotion of bulk sarcomeric protein degradation24 or through ubiquitin proteasome– dependent proteolysis.
It is important to note that Foxo proteins are not functionally interchangeable; Foxo1 knockout mice are embryonically nonviable,15,25 whereas Foxo3-null mice are viable. However, the present study, together with studies from others,16 demonstrates that both Foxo1 and Foxo3 have antigrowth functions in the heart and may work through similar mechanisms.
Regulation of Calcineurin Signaling
This study demonstrates for the first time that increased Foxo activity provokes a remarkable decrease in calcineurin/NFAT signaling. In Foxo1- or Foxo3 virus-infected myocytes, declines in calcineurin protein abundance are modest; however, there is a marked decrease in calcineurin activity and in both MCIP1.4 protein and transcript levels. MCIP1.4 is an endogenous modulator of calcineurin, the expression of which is controlled by calcineurin/NFAT.13 In cells, equilibrium is established between the abundance of MCIP1.4 protein and calcineurin activity. Therefore, MCIP1.4 protein abundance can serve as a functional surrogate for calcineurin activity.26 In fact, because calcineurin activation requires the binding of calcium and calmodulin, MCIP1.4 expression is a better measure of calcineurin activity than calcineurin protein levels because protein levels reflect both active and inactive calcineurin. This is supported by our results in cardiomyocytes, in which treatment with Ang II provoked marked increases in MCIP1.4 protein abundance, which paralleled increases in cell size. Previous studies have also shown that several other hypertrophic agonists, including PE, endothelin-1, and PAMH, promote a calcineurin-dependent increase in MCIP1.4.27,28 It is important to note that the expression of MCIP1.1, an isoform with an expression that is not calcineurin dependent,13 was not affected by forced Foxo expression.
Previous studies from our group have shown that the expression of β-MHC is dependent on calcineurin activity in vivo.18 Here, we observed that overexpression of Foxo1 in cardiomyocytes led to a significant loss of β-MHC expression and that of MCIP1.4 (Figure 2). These results agree with our finding of >50% reduction in calcineurin phosphatase activity in cells overexpressing Foxo1 (Figure 1A).
Foxo activity induces atrogin-1 expression, which likely contributes to the decreased calcineurin activity we observed in this study.9 The observed changes in calcineurin activity and MCIP1.4 levels are more pronounced than the changes in calcineurin protein levels (Figures 1 and 3). It is possible that under resting conditions the majority of calcineurin protein is inactive, and atrogin-1 preferentially targets the activated form of the enzyme. In addition, some evidence suggests that hypertrophic agonists mainly increase the calcineurin Aβ isoform.29 The antibody used in this study recognizes both calcineurin Aβ and Aβ isoforms, which may have masked the changes in the latter isoform. (Calcineurin isoform-specific antibodies from commercial sources did not work reliably in our hands.)
Suppression of MCIP1.4 Expression by Foxo
Our studies show that overexpression of Foxo1 or Foxo3 potently suppressed both basal and agonist-induced MCIP1.4 protein abundance and that these effects took place at the transcriptional level. We provide evidence for indirect inhibition of MCIP1.4 by Foxo through downregulation of calcineurin activity. However, it remains possible that Foxo affects MCIP1.4 protein abundance through additional mechanisms that do not involve calcineurin. For instance, the MCIP1.4 protein itself may be a target of atrogin-1. Alternatively, Foxo proteins may serve as transcriptional repressors of the MCIP1.4 gene. Indeed, we have identified a consensus Foxo binding site in the MCIP1.4 promoter and are currently investigating these alternative modes of Foxo function.
MCIP1.4 protein expression in Foxo3-null hearts is elevated compared with wild-type hearts, presumably because of reduced ubiquitin ligase activity. After TAB surgery, however, MCIP1.4 levels are increased to a similar extent in hearts from Foxo3-null and wild-type mice. In addition, pressure stress induced by TAB triggered similar degrees of cardiac hypertrophic growth in Foxo3-null mice relative to wild-type mice. These results are consistent with a model in which Foxo is active under resting conditions (leading to diminished calcineurin signaling) and inactivated by hypertrophic stress (so that targeted inactivation of the Foxo3 gene has no effect).
MCIP1.4 is involved in a variety of important physiological and pathological functions. It has been implicated in regulating cardiac hypertrophy, stress responses, angiogenesis, and neurological diseases, including Alzheimer’s disease and Down syndrome.30,31 The potent inhibition of MCIP1.4 by Foxo proteins and the abundant expression of Foxo1 in adult central nervous system suggest that Foxo factors may have unappreciated roles in these diseases.
Inactivation of Foxo During Hypertrophy
Results from this study and others show that Foxo proteins are inactivated during hemodynamic stress or agonist-induced muscle hypertrophy. In cultured cardiomyocytes, hypertrophic ligands such as Ang II, ISO, and PE decrease Foxo activity through activation of the PI3K/Akt pathway. It is worth noting that the suppression of Foxo functions by the PI3K/Akt pathway is typically triggered by growth factors.2 Here, we demonstrate that several G protein–coupled receptor ligands (Ang II, PE, ISO) also inactivate Foxo by activating Akt. These findings are consistent with previous reports of cross-talk between Gαq- or Gαs-mediated pathways and the PI3K pathway.6,32,33 Indeed, a recent report demonstrated that stimulation of β-adrenergic receptors induces phosphorylation of Akt at both the Thr-308 and Ser-473 sites in myocytes with kinetics similar to those described here.33
It has been reported that insulin-induced phosphorylation of Foxo1 results in its proteasomal degradation.34,35 These studies reported changes in Foxo protein abundance 4 hours after insulin exposure.35 In experiments reported here, cells were exposed to hypertrophic ligands for shorter duration, and changes in Foxo protein levels were not observed. We also did not observe changes in total Foxo protein in hypertrophic hearts after TAB surgery. It is possible that TAB induces multiple posttranslational modifications of Foxo proteins that protect them from degradation.
Progrowth and Antigrowth Mechanisms in Hypertrophy
A number of reports show that growth factors induce hypertrophic growth and net protein accumulation through the PI3K/Akt pathway. This progrowth function of PI3K/Akt pathway had been shown to be mediated by several downstream targets, including GSK3 and mTOR, which regulate protein translation. Recent studies of skeletal muscle atrophy, however, highlight the importance of another downstream target of the PI3K/Akt pathway, the Foxo transcription factor,3,4 and its role in proteolysis. Results from this study and others16 extend these findings to illustrate that Foxo proteins are also key regulators of catabolic processes in cardiac cells and that these Foxo-mediated antigrowth effects can be shut off during cardiac hypertrophy. The present study goes further to show that Foxo antagonizes cell growth by specifically targeting prohypertrophic signaling molecules.
Supplementary Material
CLINICAL PERSPECTIVE.
In response to stress, the heart initially compensates with an adaptive hypertrophic increase in mass. Under prolonged stress, the heart undergoes apparently irreversible decompensation, resulting in dilation of the failing heart. Thus, there is great interest in developing novel treatment strategies to block hypertrophy and to prevent heart failure. In this study, we report that Foxo transcription factors attenuate hypertrophic growth of the heart by suppressing the activity of calcineurin, an intracellular phosphatase important in pathological cardiac remodeling. Given that hypertrophic growth of the heart occurs in multiple contexts, our findings also suggest that certain hypertrophic signals are capable of overriding the antigrowth program induced by Foxo. Consistent with this, multiple hypertrophic agonists triggered the inactivation of Foxo proteins in cardiomyocytes through a mechanism requiring the PI3K/Akt pathway. This study suggests that inhibition of the calcineurin/NFAT signaling cascade by Foxo and release of this repressive action by the PI3K/Akt pathway are important mechanisms whereby Foxo factors govern cell growth in the heart. As such, Foxo proteins, and possibly other catabolic pathways, are novel targets of potential therapeutic interest in the prevention and treatment of load-induced heart disease.
Acknowledgments
We thank Drs Zhiping Liu and Stephen J. Gold for helpful discussions, Dr James Richardson and John Shelton for assistance with microscopy, and Erin Webster for technical assistance.
Sources of Funding
This work was supported by grants from the Donald W. Reynolds Cardiovascular Clinical Research Center (Dr Hill), NIH (HL-075173 and HL-006296 to Dr Hill; HL-006296 to Dr Gerard; HL-072016 to Dr Rothermel), AHA (0640084N to Dr Hill; 0565100Y to Dr Ni), the Lance Armstrong Foundation (Dr Castrillon), and the Sidney Kimmel Foundation (Dr Castrillon).
Footnotes
The online-only Data Supplement is available with this article at http://circ.ahajournals.org/cgi/content/full/CIRCULATIONAHA.106.637124/DC1.
Disclosures
None.
References
- 1.Accili D, Arden KC. FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell. 2004;117:421–426. doi: 10.1016/s0092-8674(04)00452-0. [DOI] [PubMed] [Google Scholar]
- 2.Tran H, Brunet A, Griffith EC, Greenberg ME. The many forks in FOXO’s road. Sci STKE. 2003:RE5. doi: 10.1126/stke.2003.172.re5. [DOI] [PubMed] [Google Scholar]
- 3.Stitt TN, Drujan D, Clarke BA, Panaro F, Timofeyva Y, Kline WO, Gonzalez M, Yancopoulos GD, Glass DJ. The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol Cell. 2004;14:395–403. doi: 10.1016/s1097-2765(04)00211-4. [DOI] [PubMed] [Google Scholar]
- 4.Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, Walsh K, Schiaffino S, Lecker SH, Goldberg AL. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell. 2004;117:399–412. doi: 10.1016/s0092-8674(04)00400-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Frey N, Katus HA, Olson EN, Hill JA. Hypertrophy of the heart: a new therapeutic target? Circulation. 2004;109:1580–1589. doi: 10.1161/01.CIR.0000120390.68287.BB. [DOI] [PubMed] [Google Scholar]
- 6.Dorn GW, 2nd, Force T. Protein kinase cascades in the regulation of cardiac hypertrophy. J Clin Invest. 2005;115:527–537. doi: 10.1172/JCI24178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berenji K, Drazner MH, Rothermel BA, Hill JA. Does load-induced ventricular hypertrophy progress to systolic heart failure? Am J Physiol Heart Circ Physiol. 2005;289:H8–H16. doi: 10.1152/ajpheart.01303.2004. [DOI] [PubMed] [Google Scholar]
- 8.Vega RB, Bassel-Duby R, Olson EN. Control of cardiac growth and function by calcineurin signaling. J Biol Chem. 2003;278:36981–36984. doi: 10.1074/jbc.R300023200. [DOI] [PubMed] [Google Scholar]
- 9.Li HH, Kedar V, Zhang C, McDonough H, Arya R, Wang DZ, Patterson C. Atrogin-1/muscle atrophy F-box inhibits calcineurin-dependent cardiac hypertrophy by participating in an SCF ubiquitin ligase complex. J Clin Invest. 2004;114:1058–1071. doi: 10.1172/JCI22220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K, Pan ZQ, Valenzuela DM, DeChiara TM, Stitt TN, Yancopoulos GD, Glass DJ. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. 2001;294:1704–1708. doi: 10.1126/science.1065874. [DOI] [PubMed] [Google Scholar]
- 11.Gomes MD, Lecker SH, Jagoe RT, Navon A, Goldberg AL. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc Natl Acad Sci U S A. 2001;98:14440–14445. doi: 10.1073/pnas.251541198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rothermel BA, Vega RB, Williams RS. The role of modulatory calcineurin-interacting proteins in calcineurin signaling. Trends Cardiovasc Med. 2003;13:15–21. doi: 10.1016/s1050-1738(02)00188-3. [DOI] [PubMed] [Google Scholar]
- 13.Yang J, Rothermel B, Vega RB, Frey N, McKinsey TA, Olson EN, Bassel-Duby R, Williams RS. Independent signals control expression of the calcineurin inhibitory proteins MCIP1 and MCIP2 in striated muscles. Circ Res. 2000;87:E61–E68. doi: 10.1161/01.res.87.12.e61. [DOI] [PubMed] [Google Scholar]
- 14.Hill JA, Karimi M, Kutschke W, Davisson RL, Zimmerman K, Wang Z, Kerber RE, Weiss RM. Cardiac hypertrophy is not a required compensatory response to short-term pressure overload. Circulation. 2000;101:2863–2869. doi: 10.1161/01.cir.101.24.2863. [DOI] [PubMed] [Google Scholar]
- 15.Castrillon DH, Miao L, Kollipara R, Horner JW, DePinho RA. Suppression of ovarian follicle activation in mice by the transcription factor Foxo3a. Science. 2003;301:215–218. doi: 10.1126/science.1086336. [DOI] [PubMed] [Google Scholar]
- 16.Skurk C, Izumiya Y, Maatz H, Razeghi P, Shiojima I, Sandri M, Sato K, Zeng L, Schiekofer S, Pimentel D, Lecker S, Taegtmeyer H, Goldberg AL, Walsh K. The FOXO3a transcription factor regulates cardiac myocyte size downstream of AKT signaling. J Biol Chem. 2005;280:20814–20823. doi: 10.1074/jbc.M500528200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Molkentin JD, Lu JR, Antos CL, Markham B, Richardson J, Robbins J, Grant SR, Olson EN. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell. 1998;93:215–228. doi: 10.1016/s0092-8674(00)81573-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oh M, Rybkin II, Copeland V, Czubryt MP, Shelton JM, van Rooij E, Richardson JA, Hill JA, De Windt LJ, Bassel-Duby R, Olson EN, Rothermel BA. Calcineurin is necessary for the maintenance but not embryonic development of slow muscle fibers. Mol Cell Biol. 2005;25:6629–6638. doi: 10.1128/MCB.25.15.6629-6638.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taigen T, De Windt LJ, Lim HW, Molkentin JD. Targeted inhibition of calcineurin prevents agonist-induced cardiomyocyte hypertrophy. Proc Natl Acad Sci U S A. 2000;97:1196–1201. doi: 10.1073/pnas.97.3.1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zou Y, Komuro I, Yamazaki T, Kudoh S, Aikawa R, Zhu W, Shiojima I, Hirori Y, Tobe K, Kadowaki T, Yazaki Y. Cell type-specific angiotensin II-evoked signal transduction pathways: critical roles of Gbettagamma subunit, Src family, and Ras in cardiac fibroblasts. Circ Res. 1998;82:337–345. doi: 10.1161/01.res.82.3.337. [DOI] [PubMed] [Google Scholar]
- 21.Kodama H, Fukuda K, Pan J, Sano M, Takahashi T, Kato T, Makino S, Manabe T, Murata M, Ogawa S. Significance of ERK cascade compared with JAK/STAT and PI3-K pathway in gp130-mediated cardiac hypertrophy. Am J Physiol Heart Circ Physiol. 2000;279:H1635–H1644. doi: 10.1152/ajpheart.2000.279.4.H1635. [DOI] [PubMed] [Google Scholar]
- 22.Rothermel BA, Berenji K, Tannous P, Kutschke W, Dey A, Nolan B, Yoo KD, Demetroulis E, Gimbel M, Cabuay B, Karimi M, Hill JA. Differential activation of stress-response signaling in load-induced cardiac hypertrophy and failure. Physiol Genomics. 2005;23:18–27. doi: 10.1152/physiolgenomics.00061.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kamei Y, Miura S, Suzuki M, Kai Y, Mizukami J, Taniguchi T, Mochida K, Hata T, Matsuda J, Aburatani H, Nishino I, Ezaki O. Skeletal muscle FOXO1 (FKHR) transgenic mice have less skeletal muscle mass, down-regulated type I (slow twitch/red muscle) fiber genes, and impaired glycemic control. J Biol Chem. 2004;279:41114–41123. doi: 10.1074/jbc.M400674200. [DOI] [PubMed] [Google Scholar]
- 24.Kedar V, McDonough H, Arya R, Li HH, Rockman HA, Patterson C. Muscle-specific RING finger 1 is a bona fide ubiquitin ligase that degrades cardiac troponin I. Proc Natl Acad Sci U S A. 2004;101:18135–18140. doi: 10.1073/pnas.0404341102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hosaka T, Biggs WH, 3rd, Tieu D, Boyer AD, Varki NM, Cavenee WK, Arden KC. Disruption of forkhead transcription factor (FOXO) family members in mice reveals their functional diversification. Proc Natl Acad Sci U S A. 2004;101:2975–2980. doi: 10.1073/pnas.0400093101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Rooij E, Doevendans PA, Crijns HJ, Heeneman S, Lips DJ, van Bilsen M, Williams RS, Olson EN, Bassel-Duby R, Rothermel BA, De Windt LJ. MCIP1 overexpression suppresses left ventricular remodeling and sustains cardiac function after myocardial infarction. Circ Res. 2004;94:e18–e26. doi: 10.1161/01.RES.0000118597.54416.00. [DOI] [PubMed] [Google Scholar]
- 27.Wang Y, De Keulenaer GW, Weinberg EO, Muangman S, Gualberto A, Landschulz KT, Turi TG, Thompson JF, Lee RT. Direct biomechanical induction of endogenous calcineurin inhibitor Down syndrome critical region-1 in cardiac myocytes. Am J Physiol Heart Circ Physiol. 2002;283:H533–H539. doi: 10.1152/ajpheart.00002.2002. [DOI] [PubMed] [Google Scholar]
- 28.Bush E, Fielitz J, Melvin L, Martinez-Arnold M, McKinsey TA, Plichta R, Olson EN. A small molecular activator of cardiac hypertrophy uncovered in a chemical screen for modifiers of the calcineurin signaling pathway. Proc Natl Acad Sci U S A. 2004;101:2870–2875. doi: 10.1073/pnas.0308723101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oka T, Dai YS, Molkentin JD. Regulation of calcineurin through transcriptional induction of the calcineurin A beta promoter in vitro and in vivo. Mol Cell Biol. 2005;25:6649–6659. doi: 10.1128/MCB.25.15.6649-6659.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rothermel BA, McKinsey TA, Vega RB, Nicol RL, Mammen P, Yang J, Antos CL, Shelton JM, Bassel-Duby R, Olson EN, Williams RS. Myocyte-enriched calcineurin-interacting protein, MCIP1, inhibits cardiac hypertrophy in vivo. Proc Natl Acad Sci U S A. 2001;98:3328–4433. doi: 10.1073/pnas.041614798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harris CD, Ermak G, Davies KJ. Multiple roles of the DSCR1 (Adapt78 or RCAN1) gene and its protein product calcipressin 1 (or RCAN1) in disease. Cell Mol Life Sci. 2005;62:2477–2486. doi: 10.1007/s00018-005-5085-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dugourd C, Gervais M, Corvol P, Monnot C. Akt is a major downstream target of PI3-kinase involved in angiotensin II–induced proliferation. Hypertension. 2003;41:882–890. doi: 10.1161/01.HYP.0000060821.62417.35. [DOI] [PubMed] [Google Scholar]
- 33.Morisco C, Condorelli G, Trimarco V, Bellis A, Marrone C, Sadoshima J, Trimarco B. Akt mediates the cross-talk between beta-adrenergic and insulin receptors in neonatal cardiomyocytes. Circ Res. 2005;96:180–188. doi: 10.1161/01.RES.0000152968.71868.c3. [DOI] [PubMed] [Google Scholar]
- 34.Aoki M, Jiang H, Vogt PK. Proteasomal degradation of the FoxO1 transcriptional regulator in cells transformed by the P3k and Akt oncoproteins. Proc Natl Acad Sci U S A. 2004;101:13613–13617. doi: 10.1073/pnas.0405454101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matsuzaki H, Daitoku H, Hatta M, Tanaka K, Fukamizu A. Insulin-induced phosphorylation of FKHR (Foxo1) targets to proteasomal degradation. Proc Natl Acad Sci U S A. 2003;100:11285–11290. doi: 10.1073/pnas.1934283100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.