

Abstract
Purpose of Review
Novel medical approaches and personalized medicine seek to use genetic information to “individualize” and improve diagnosis, prevention, and therapy. The personalized management of cardiovascular disease involves a large spectrum of potential applications, from diagnostics of monogenic disorders, to prevention and management strategies based on modifier genes, to pharmacogenetics in which individual genetic information is used to optimize pharmacological treatments.
Recent Findings
Evidence suggests that common polymorphic variants of modifier genes could influence drug response in cardiovascular disease in a variety of areas including heart failure, arrhythmias, dyslipidemia and hypertension. In heart failure, common genetic variants of beta-adrenergic receptors, alpha-adrenergic receptors, and endothelin receptors (among others) have been associated with variable response to heart failure therapies. The challenge remains to develop strategies to leverage this information in ways that personalize and optimize cardiovascular therapy based on a patient's genetic profile.
Summary
While advances in technologies will continue to transition personalized medicine from the research to the clinical setting, health care providers will need to reshape clinical diagnostic paradigms. Ultimately, pharmacogenetics will give providers options for improving patient management on the basis of pharmacogenetic data.
Keywords: Pharmacogenomics, genetic variation, cardiovascular genetics, drug response, heart failure
Introduction
The International Human Genome Project was completed in 2003 after 13 years of extensive work by a network of laboratories and an approximately $3 billion investment. The 3 billion base pairs of the human genome became publicly available shortly after sequencing. This event and the extraordinary rapid change in technology have changed the way we perceive and practice medicine.
Today, with novel sequencing technologies like massively parallel sequencing or next-generation sequencing, (NGS) the cost of sequencing a human genome is less than $5,000 and can be completed in about a week. Furthermore, it is possible to sequence only the coding region of the genome, the exome, for less than $1,000. It is expected that continued improvements of technology and bioinformatics analysis will further reduce the cost of whole genome sequencing below $1,000 (The Road to the $1000 Genome — A Roundup of Sequencing Technology Developments, http://www.genome.gov). Meanwhile, the “1000 Genomes Project” has currently sequenced the genomes of 2,500 people of various ethnic origins and represents one of a growing number of sequenced human cohorts that provides data on genetic variation in populations (1).
The advances in genomics and high throughput technologies are projected to profoundly impact cardiovascular medicine (2). Out of 3 billion base pairs in the human genome, there are likely over 10 million common genetic variations that occur naturally, meaning that even two closely related individuals will possess thousands of genetic differences. A mutation rate of 2×10-8 per base pair in any given generation can thus lead to the accumulation of approximately 60 new mutations every generation (3). Within a genome there are common variants, termed polymorphisms that typically have no clinical consequences. However, some genetic variations in genes are functionally significant and account for differences in susceptibility or severity of diseases, or responses to drugs (pharmacogenetics). Drug response variation can be observed in large population studies and even within families. Genes that can aid in pharmacogenetic differences in the individual's response to disease or therapy are called modifier genes. Clinicians historically have included behavioral and environmental factors in personalized care, but it is only recently that genetic information has the opportunity to be used in medicine as well (4).
Pharmacogenetics
It has been estimated that more than 100,000 deaths in the United States occur each year are due to adverse drug reactions, and pharmacogenetics differences in drug response may be one factor that contributes to these deaths (5).
Genes associated with variation in drug response have been identified using three major approaches. The first is the candidate gene approach based on the identification of “candidate variants” in well-defined pharmacokinetic pathways. The second and most recent approach is based on genome-wide association studies (GWAS). Through 12/2013, the Catalog of Genome-Wide Association Studies of the National Human Genome Research Institute listed approximately 2,050 published GWAS with significant findings (P≤5×10-8) in 17 trait categories (www.genome.gov/gwastudies). In GWAS, the approach is based on whole genome screening of hundreds of thousands of single nucleotide polymorphisms (SNPs), rather than by candidate genes. Once an association is discovered between a polymorphism and the disease, the modifier gene in proximity to the SNP is identified. The approach, developed thanks to technological advances in high throughput sequencing methods and bioinformatic approaches, relies on the screening of large populations of patients and controls, and has already been successfully utilized in cardiovascular medicine in complex common disorders such as hypertension and coronary artery disease (3,6). The third tool to search for variants in the protein-coding regions of the genome, known as whole exome sequencing (WES), analyzes the region of the genome most likely to contain pathogenic mutations and has been recently used in pharmacogenetics of drug metabolism (7).
Pharmacogenetics of heart failure
Heart failure (HF) is one of the most serious and expensive conditions in health care worldwide due to its high prevalence (1-1.5% of adult population) and high morbidity (frequent hospitalization). In the United States, HF affects approximately 4 million people and causes about 200,000 deaths per year, with a generally rapid course with a mean survival of only 1.7 years for males and 3.2 for females after diagnosis (8). In Europe, data are substantially similar, which suggests that in spite of the improvement of HF therapy, disease progression has not changed and HF still remains one of the most important health issues in the world.
HF is a syndrome characterized by primary pathophysiological processes, which interact with a wide number of complex secondary interrelated pathophysiological mechanisms. HF is mediated by rare mutations in single Mendelian genes, in addition to common genetic polymorphisms in modifier genes that can modify the natural history of the cardiac disease. Known HF modifiers include genes of the renin-angiotensin-aldosterone (RAAS) and adrenergic systems (9,10). Furthermore, genetic polymorphisms can modify the response to therapy (9,10,11) by changing gene-gene interactions, such as β1 and α2 adrenergic receptors (12).
Several studies have provided evidence of the existence of modifier genes in HF that can modulate the severity and progression of the disease independently from the primary cause of the disease or monogenic disorder. Lee and coworkers in the Framingham Offspring Study have shown that the risk of HF is significantly increased in offspring of patients with HF compared to controls (13). Furthermore, in monogenic cardiomyopathies there is frequently high intra-familial variability of the phenotype, consistent with the presence of genetic variation contributing to phenotypic variation (9). Finally, studies on the effect of candidate gene polymorphisms have shown that genetic variations can influence the HF phenotype and the mutant protein function (9,10). Examples of modifier variants include the DD genotype of the angiotensin converting enzyme (ACE), where subjects homozygous for the deletion (D) have increase circulating and myocardial ACE levels. These patients are at risk of early heart remodeling after myocardial infarction, as well as risk of severe systolic dysfunction in dilated cardiomyopathy (DCM), and ischemic cardiomyopathy (9,10). Other polymorphisms that can modify the natural history of DCM are the AT1 receptor, β1- and β2-adrenergic receptors, and the α2C-adrenergic receptor (9-13).
A more comprehensive approach to identifying modifier genes in HF is expected to come from GWAS, such as in the Framingham Heart study. Two recent papers have reported a large meta-analysis of the risk of heart failure and mortality in the CHARGE Consortium (14,15). The study population included 20,926 European ancestry participants, and 2,895 African ancestry participants previously enrolled in four smaller studies from the US and the Netherlands: the Atherosclerosis Risk in Communities (ARIC) study, Cardiovascular Health Study (CHS), the Framingham Heart Study (FHS) and the Rotterdam Study (RS). The first analysis of Smith and coworkers on the risk of developing HF identified two loci, USP3 in subjects of European ancestry and LRIG3 in subjects of African ancestry, with genome-wide significance (P<10-8)(14). The USP3 gene encodes an ubiquitin-specific protease: ubiquitin is a highly conserved protein involved in important cellular processes, such as protein degradation, cell-cycle regulation and stress response, and is activated in cardiomyopathies and pathogenic cardiac hypertrophy. The LRIG3 gene encodes a member of the LRIG family, integral membrane proteins widely expressed and involved in tissue development (14). In the second study, Morrison and co-investigators analyzed the subgroup of CHARGE subjects who developed HF (2,526 individuals of European ancestry and 466 of African ancestry) and estimated the risk of mortality (15). One locus was identified in the European subgroup, with genome-wide significance (P<10-7) in the CKLF-like MARVEL transmembrane domain containing 7 (CMTM7) gene, one of the chemokine-like factor genes clustered on chromosome 3p22. Although its function is still unknown, CMTM7 appears to be expressed in leukocytes and in the heart, may be upregulated among in HF and may act as a chemoattractant to guide migration of cells in the heart (15). For this information to lead to a clinical impact, the functional significance of the modifier genes identified needs to be further elucidated and the studies replicated in independent prospective HF cohorts.
In HF, pharmacogenetics has a promising role: indeed, in spite of the improvement in the natural history of HF thanks to the therapeutic advancement in the last 20 years and the development of practice guidelines, large clinical trials such as BEST (Beta Blocker Evaluation Survival) and AHeFT (African American Heart Failure Trial) have suggested that some patients have a different response to treatment (responders versus nonresponders) due to underlying genetic differences (16). The most important genetic variations associated with these different pharmacological responses are listed in the Table.
Table 1. Pharmacogenetics of Cardiovascular Disease.
| Disease | Gene | Polymorphism | Function | Therapeutic Implications |
|---|---|---|---|---|
| Heart Failure | ||||
| RAAS | ACE | D/I | ACE D: higher ACE activity and A II levels | ACE inhibitors |
| Beta-blockers | ||||
| Aldosterone synthase | Promoter -344 T/C | -344 C: increased transcriptional activity and aldosterone production | ACE inhibitors | |
| Aldosterone receptor antagonists | ||||
| β-adrenergic receptors | β1-adrenergic receptors | Arg389Gly | Arg: increased adrenergic signal | Beta-blockers |
| ACE inhibitors | ||||
| β2-adrenergic receptors | Gly49Ser | Gly: enhanced down regulation | Beta-blockers | |
| Gly16Arg | Receptor down regulation | Beta-blockers | ||
| Gln27Gly | ||||
| α-adrenergic signaling | α-2C receptor | α-2C Deletion | Deletion: decreased uptake of norepinephrine | Beta-blockers |
| G protein β3 subunit | C825T | C825T: increased α-adrenergic signaling, lower plasma renin | ACE inhibitors | |
| Nitric oxide | NOS3 | Asp298Glu | Asp: associated with lower NOS3 activity | ACE inhibitors |
| Endothelin system | EDN1 | IVS-4 G/A Lys198Asn | Unknown | Beta-blockers |
|
| ||||
| Arrhythmia | ||||
| QT interval | SCN5A, KCNH2, KCNQ1, KCNJ2, KCNE1 NOS1AP | Ion channel function | QT-prolonging antiarrhythmic drugs, antibiotics, antipsychotics | |
| NO synthase pathway | QT prolonging agents | |||
|
| ||||
| Anticoagulant therapy | ||||
| Cytochrome P450 | CYP2C9 | *2 and *3 | Clearance of warfarin, risk of bleeding | Warfarin |
| Vitamin K oxidase | VKORC1 | 1173C/T | Reduced metabolism of vit. K, higher warfarin dose requirement | Warfarin |
|
| ||||
| Antiplatelet Agents | ||||
| Cytochrome P450 | CYP2C19 | *2 | Decreased conversion of active metabolite, loss-of-function | Clopidogrel |
In the BEST trial, the initial evaluation of bucindolol, a β-blocker/sympatholytic agent, on patients with HF in class 3 and 4 was disappointing and did not reach statistical significance. However, when the investigators analyzed the response to treatment based on the β1 adrenergic receptor (AR) genotype encoded by ADBR1, they found a strong association with the amino acid at position 389 (17). Wild-type Arg389 homozygotes responded significantly better than Arg389Gly polymorphism carriers to the drug treatment with a 38% reduction in mortality. The wild-type Arg389 response was even better than that previously reported for carvedilol (10,17). The different behavior of the two allelic variants is explained by the fact that the wild-type allele is more responsive to the agonist stimulation (Isoproterenol) than the Arg389Gly variant allele (10, 17), a response confirmed by other studies involving different β-blockers including metoprolol and carvedilol (16) (Figure 1).
Figure 1. Cardiac adrenergic neuro-effector junction.
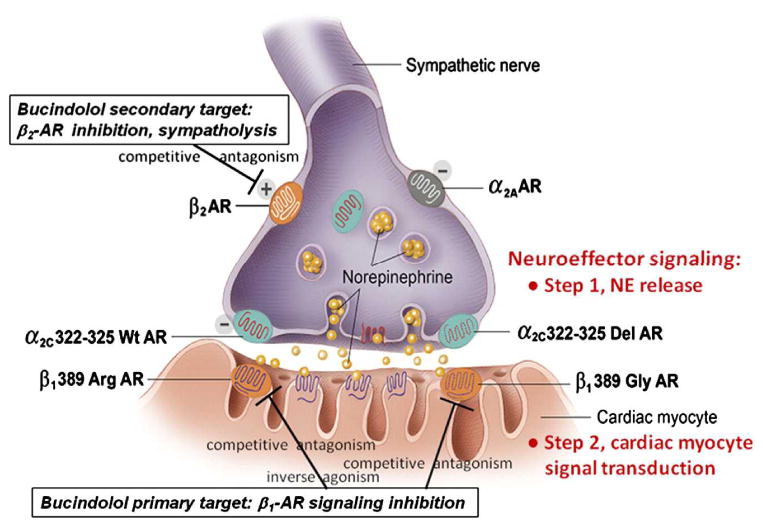
Prejunctional adrenergic receptors (ARs) that regulate norepinephrine (NE) release, and postjunctional β1-ARs that are the primary signal transduction target of NE are shown (from Bristow 2012, with permission). α2C Receptors regulate NE release; a loss-of-function deletion polymorphism (322–325 Del) produces dysregulation and predisposes to a much greater degree of NE lowering by the sympatholytic β-blocker bucindolol (19) which lowers NE via blockade of prejunctional β2-ARs. Postjunctional β1-ARs are also polymorphic, with the 389Arg variant having much greater signal transduction capacity, constitutive activity and NE affinity than the loss-of-function 389Gly variant.
More recently, the BEST investigators reported the results of a substudy on the pharmacogenetic effect of the α2C-adrenergic receptor, whose role is to inhibit norepinephrine release in the prejunctional adrenergic nerve terminals. The polymorphism α2C Del322-325 had previously been associated with a worse prognosis in HF, with evidence for a synergistic effect with the β1 Arg389 allele in Black patients (18). In another study, Bristow et al. demonstrated that the norepinephrine-lowering and clinical therapeutic responses to bucindolol were strongly influenced by the α2C receptor genotype. The α2C Del322-325 carriers had an excessive sympatholytic effect and no evidence of any therapeutic benefit from bucindolol, whereas wild-type α2C carriers had a 30% reduction in mortality (19).
Many polymorphisms, such as the β1 extracellular adrenergic receptors with polymorphisms Ser49Gly and Arg389Gly can aid in modification of HF. Also, polymorphisms of the β2 adrenergic receptor can also modify HF (Table). In the RAAS system, patients with the ACE DD genotype had a worse prognosis, but at the same time were the best responders to β blocker therapy compared to the other genotypes (9). It is interesting to note that the evidence of a genetically driven response to therapy in HF dates back to the AHeFT study. In this trial, the investigators found that African-American patients had a much better response to the therapy with hydralazine and isosorbide dinitrate compared to Caucasian patients. Indeed, this is the first FDA approved therapy for HF based on racial differences and consequently on genetic background (20). Data of the GRAHF substudy (Genetic Risk Assessment of Heart Failure in AHeFT) suggests that at least one of the genetic causes lies in the -344C/T polymorphism located in the promoter of the alderosterone synthase gene, which is associated with a worse prognosis but a better response to the hydralazine/isosorbide dinitrate therapy in carriers: the same polymorphisms had previously been associated with higher enzymatic activity, hypertension and myocardial remodeling (16).
The studies on β1 – α2C receptors indicate the existence of complex gene-gene interactions in the genetic determinants of HF. In particular, the gene-gene interaction and the functional effect in the case of the adrenergic receptors are of particular interest. The β1 Arg389 receptor is more responsive to the adrenergic stimulation: patients homozygous for the Arg389 allele carrying also the α2C Del322-325 receptor characterized by decreased uptake of norepinephrine seem to have an enhanced adrenergic response, but a worst prognosis. However they have the greatest improvement in ejection fraction with β blocker therapy (18).
Finally, we have studied the association of polymorphisms of the endothelin system with HF in the BEST cohort (11). Two genetic variations (IVS-4 G/A and Lys198Asn) on a common haplotype in the endothelin-1 gene were associated with differential response to bucindolol in terms of a combined endpoint of HF hospitalization and cause of death (Figure 2). The effect of the endothelin-1 haplotype was only evident in the treatment group, supporting a pharmacogenetic interaction between bucindolol and the haplotype. Ultimately, these types of data collecting could be used to tailor beta-blocker therapy for individuals based on their underlying endothelin-1 haplotype (11).
Figure 2. Effect of endothelin-1 polymorphisms on survival and beta-blocker therapy.
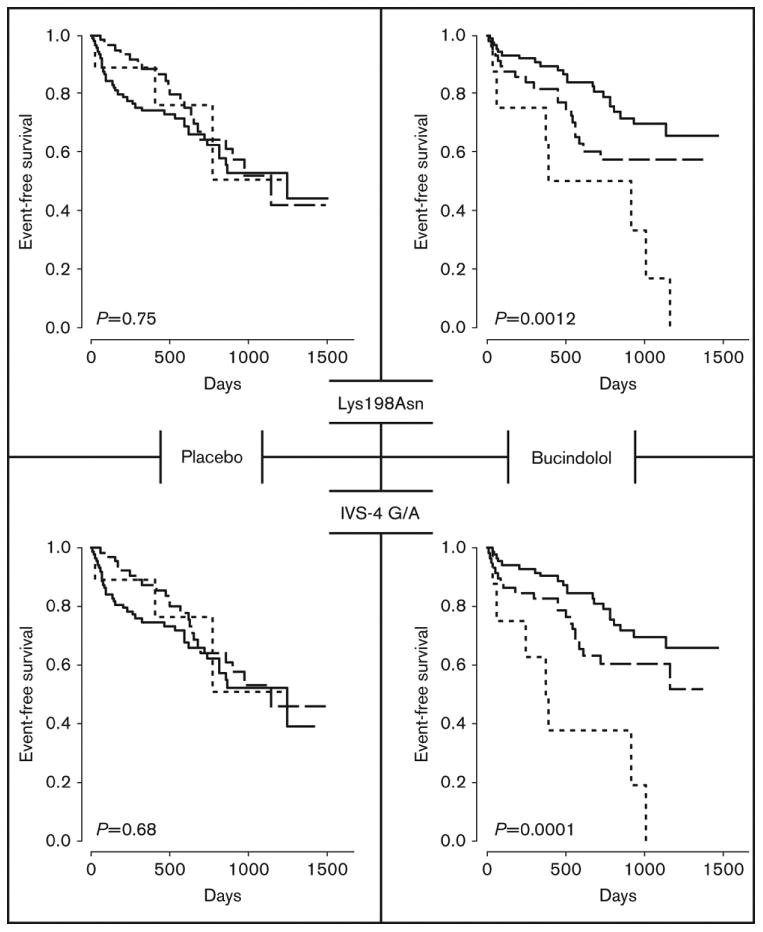
Time to the combined event of first heart failure hospitalization or death for endothelin-1 polymorphisms Lys198Asn (top) and IVS-4 G/A by genotype (bottom). Common homozygotes, heterozygotes, and rare homozygotes are depicted by the solid, dashed, and dotted lines respectively. Treatment groups separate the data with placebo treated and bucindolol treated subjects on the left and right, respectively. Reproduced with permission from Taylor et al. (11).
Other areas of pharmacogenetic investigations in HF associated therapies
Arrhythmias are frequent comorbidities in HF and have recently been associated with pharmacogenetic markers in the context of antiarrhythmic drugs. First of all, several antiarrhythmic drugs can induce arrhythmias by inducing a prolongation of the QT interval (drug-induced long QT syndrome), such as sotalol, dofetilide, and quinidine. Subclinical LQT syndrome appears to be the cause of a large proportion of these cases and seems to be associated with genetic variations of LQT genes (2). There is a critical need for a mechanism to identify patients at risk for potentially life-threatening drug induced arrhythmias. Pharmacogenetics of the G-protein beta3 subunit (GNB3) c825t polymorphism may allow a better identification of patient who will benefit from implantable cardiac defibrillators and biventricular pacing in HF (21).
A major field of interest has been the study of pharmacogenomics of warfarin therapy. The genes that appear to play the most important role are CYP2C9 and VKORC1 (40-50% drug variability) (22). The gene VKORC1 has been found to be the main predictor as well as variants in its promoter region (23). Furthermore, CYP2C9*2 and CYP2C9*3 are associated with lower warfarin dose requirements and increased risk of bleeding which can result in a longer hospital stay (4). In spite of several studies on warfarin pharmacogenomics, the clinical utility of pharmacogenetic testing for anticoagulation control is still not completely well established. Some studies have been conducted, but suffer from small sample sizes (3). A pharmacogenomic algorithm has been developed giving an estimate of the dosage requirement for patients taking warfarin (24) depending on polymorphisms in the warfarin candidate genes (CYP2C9, VKORC1) (http://www.warfarindosing.org)(22). The FDA has created a black box warning on the efficacy of this drug based on genetic testing conclusions (24,25). However, the real clinical utility of pharmacogenetic screening for anticoagulant therapy is still unclear, and the just published European EU-PACT (26) and US COAG studies (27) have provided mixed and apparently contradictory results.
Another field of interest in cardiovascular pharmacogenomics is the dose-response in antiplatelet agents, especially the dual antiplatelet therapy of aspirin with a P2Y12 inhibitor drug. Clopidogrel shows a wide range of dose-responses, and variability has been associated with loss-of-function mutations in CYP2C19*2 and CYP2C19*3, which cause a reduced conversion of clopidogrel (prodrug) into its active metabolite (2,28). Carriers have an increased risk of cardiovascular events and in-stent restenosis. The FDA approved a written warning alerting to the pharmacogenetic findings and the availability of alternative therapies for CYP2C19*2 carriers. Prasugrel utilizes CPY3A4 and CYP2B6 for the drug activation, and is therefore recommended when the CYP2C19 gene has a loss of function allele. The TRITON TIMI 38 trial found that CYP2C19 polymorphisms can cause major cardiovascular events, but dual antiplatelet therapy helps to relieve that issue (3).
Finally, pharmacogenetic associations with response to exercise have been examined by Wagoner et al. who found that beta(2)-adrenergic receptor polymorphisms may determine exercise capacity in patients with heart failure (29).
Conclusions
A new era of personalized medicine is poised to enter clinical practice, and it is fueled by the decrease in the cost of DNA sequencing. Ideally, tailoring therapy based on pharmacogenomic tests would save lives and improve patient care. Advances in technologies continue to facilitate this transition from the research to the clinical setting, reshaping clinical diagnostic paradigms and challenging the healthcare team to consider how new genomic information may be leveraged to influence management decisions and to realize the promise of personalized medical care. GWAS studies and other large population studies along with enhanced mechanisms to analyze genetic data are critical for the progression of pharmacogenomics. However, many challenges still remain in applying pharmacogenetics to the clinical practice in heart failure management, and the clinical utility of pharmacogenetic testing in cardiovascular patients still remains elusive (30). Physicians and cardiologists will need to understand, communicate, and manage this new genomic information to provide the patient with appropriate education and management recommendations.
Key Points (2-5 points).
Polymorphisms in many genes lead to variability in drug effectiveness and safety, as demonstrated by studies involving beta-blockers, warfarin, clopidogrel, and others.
As the cost of DNA sequencing drops, GWAS and WES will lead to the identification of new gene-drug interactions with clinical relevance.
Additional studies are needed to elucidate the mechanisms of gene-drug interactions leading to differences in patient responses.
Clinicians' understanding of pharmacogenetic interactions is key to helping patients get the best treatment with cardiovascular drugs that are produced today.
Acknowledgments
This work was supported by the NIH grants UL1 RR025780, UL1 TR001082 N01-HV-48194, R01 HL69071, R01 116906 to LM, and K23 JL067915 and R01HL109209 to MRGT.
Abbreviations
- GWAS
Genome Wide Association Study
- SNPs
single nucleotide polymorphisms
- HF
heart failure
- NGS
next generation sequencing
- RAAS
Renin-angiotensin-aldosterone system
- WES
whole exome sequencing
- DCM
dilated cardiomyopathy
- BEST
Beta Blocker Evaluation Survival
- CHARGE
The Cohorts for Heart and Aging Research in Genetics Epidemiology
- ARIC
Atherosclerosis Risk in Communities
- CYP2C9
cytochrome P450, family 2, subfamily C, polypeptide 9
- CHS
Cardiovascular Health Study
- FHS
Framingham Heart Study
- RS
Rotterdam Study
- AHeFT
African American Heart Failure Trial
- GRAHF
Genetic Risk Assessment of Heart Failure
- SEARCH
Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine
- LQT
long QT syndrome
- VKORC1
vitamin K epoxide reductase complex, subunit 1
Footnotes
Conflicts of Interest: The author declares that there are no conflicts of interest, grants or relationship with industry. The author owns a patent on “Endothelin Single Nucleotide Polymorphisms and Methods of Predicting B-Adrenergic Receptor Targeting Agent Efficacy” Sources and funding: there are no sources or funding for this paper.
References
- 1*.The 1000 Genomes Project Consortium. Abecasis GR, Auton A, Brooks LD, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. Update of the 1000 Genome Project: validated haplotype map of 38 million single nucleotide polymorphisms, 1.4 million short in/del, and over 14,000 larger deletions in 1,092 individuals from 14 populations generated by a combination of WGS and WES. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roden DM, Johnson JA, Kimmel SE, et al. Cardiovascular pharmacogenomics. Circ Res. 2011;109:807–20. doi: 10.1161/CIRCRESAHA.110.230995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3**.Roberts R, Marian AJ, Dandona S, Stewart AF. Genomics in cardiovascular disease. J Am CollCardiol. 2013;61:2029–37. doi: 10.1016/j.jacc.2012.12.054. Recent update of the impact of genomics in all areas of cardiovascular medicine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4**.Salari K, Watkins H, Ashley EA. Personalized medicine: hope or hype? Eur Heart J. 2012;33:1564–70. doi: 10.1093/eurheartj/ehs112. Critical review of the impact of genomic information in cardiology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guttmacher AE, McGuire AL, Ponder B, Stefansson K. Personalized genomic information: preparing for the future of genetic medicine. Nat Rev Genet. 2010;11:161–165. doi: 10.1038/nrg2735. [DOI] [PubMed] [Google Scholar]
- 6**.The CARDIoGRAMplusC4D Consortium. Coronary artery disease risk loci identified in over 190,000 individuals implicate lipid metabolism and inflammation as key causal pathways. Nat Genet. 2013;45:25–33. GWAS leading to the identification of four pathways linked to lipid metabolism and inflammation. [Google Scholar]
- 7**.Gordon AS, Tabor HK, Johnson AD, et al. On Behalf of the NHLBI GO Exome Sequencing Project. Quantifying rare, deleterious variation in 12 human cytochrome P450 drug-metabolism genes in a large-scale exome dataset. Hum Mol Genet. 2013 Nov; doi: 10.1093/hmg/ddt588. [Epub ahead of prin]. use of whole exome sequencing in pharmacogenetics of drug metabolism. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O'Connell JB, Bristow MR. Economic impact of heart failure in the United States: Time for a different approach. J Heart Lung Transplant. 1994;13:S107–S112. [PubMed] [Google Scholar]
- 9.Mestroni LJ, Gilbert EM, Lowes BL. In: Dilated Cardiomyopathies In Hurst's the Heart. 13th. Fuster V, Walsh RA, Harrington RA, editors. The McGraw-Hill Companies, Inc.; New York: 2011. pp. 821–836. Ch 32. [Google Scholar]
- 10.Bristow MR. Pharmacogenetic targeting of drugs for heart failure. Pharmacol Ther. 2012;134:107–15. doi: 10.1016/j.pharmthera.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 11.Taylor MR, Slavov D, Humphrey K, et al. Pharmacogenetic effect of an endothelin-1 haplotype on response to bucindolol therapy in chronic heart failure. Pharmacogenet Genomics. 2009;19:35–43. doi: 10.1097/FPC.0b013e328317cc57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Small KM, Wagoner LE, Levin AM, et al. Synergistic polymorphisms of beta1- and alpha2C-adrenergic receptors and the risk of congestive heart failure. N Engl J Med. 2002;347:1135–42. doi: 10.1056/NEJMoa020803. [DOI] [PubMed] [Google Scholar]
- 13.Lee DS, Pencina MJ, Benjamin EJ, et al. Association of parental heart failure with risk of heart failure in offspring. N Engl J Med. 2006;355:138–47. doi: 10.1056/NEJMoa052948. [DOI] [PubMed] [Google Scholar]
- 14.Smith NL, Felix JF, Morrison AC, et al. Association of genome-wide variation with the risk of incident heart failure in adults of European and African ancestry: a prospective meta-analysis from the cohorts for heart and aging research in genomic epidemiology (CHARGE) consortium. Circ Cardiovasc Genet. 2010;3:256–266. doi: 10.1161/CIRCGENETICS.109.895763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morrison AC, Felix JF, Cupples LA, et al. Genomic variation associated with mortality among adults of European and African ancestry with heart failure: the cohorts for heart and aging research in genomic epidemiology consortium. Circ Cardiovasc Genet. 2010;3:248–255. doi: 10.1161/CIRCGENETICS.109.895995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McNamara DM. Emerging role of pharmacogenomics in heart failure. Curr Opin Cardiol. 2008;23:261–268. doi: 10.1097/HCO.0b013e3282fcd662. [DOI] [PubMed] [Google Scholar]
- 17.Pereira NL, Weinshilboum RM. Cardiovascular pharmacogenomics and individualized drug therapy. Nat Rev Cardiol. 2009;6:632–8. doi: 10.1038/nrcardio.2009.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lobmeyer MT, Gong Y, Terra SG, et al. Synergistic polymorphisms of beta1 and alpha2C-adrenergic receptors and the influence on left ventricular ejection fraction response to beta-blocker therapy in heart failure. Pharmacogenet Genomics. 2007;17:277–282. doi: 10.1097/FPC.0b013e3280105245. [DOI] [PubMed] [Google Scholar]
- 19.Bristow MR, Murphy GA, Krause-Steinrauf H, et al. An alpha2C-adrenergic receptor polymorphism alters the norepinephrine-lowering effects and therapeutic response of the beta-blocker bucindolol in chronic heart failure. Circ Heart Fail. 2010;3:21–28. doi: 10.1161/CIRCHEARTFAILURE.109.885962. [DOI] [PubMed] [Google Scholar]
- 20.Carson P, Ziesche S, Johnson G, Cohn JN. Racial differences in response to therapy for heart failure: analysis of the vasodilator-heart failure trials. Vasodilator-Heart Failure Trial Study Group. J Card Fail. 1999;5:178–187. doi: 10.1016/s1071-9164(99)90001-5. [DOI] [PubMed] [Google Scholar]
- 21.Wieneke H, Naber CN, Piaszek L, et al. Better identification of patients who benefit from implantable cardioverter defibrillators by genotyping the G-protein beta3 subunit (GNB3) c825t polymorphism. Basic Res Cardiol. 2006;101:447–e51. doi: 10.1007/s00395-006-0600-9. [DOI] [PubMed] [Google Scholar]
- 22.Verschuren JJ, Trompet S, Wessels JA, et al. A systematic review on pharmacogenetics in cardiovascular disease: is it ready for clinical application? Eur Heart J. 2012;33:165–75. doi: 10.1093/eurheartj/ehr239. [DOI] [PubMed] [Google Scholar]
- 23.Ashley EA, Hershberger RE, Caleshu C, et al. Genetics and cardiovascular disease: a policy statement from the American Heart Association. Circulation. 2012;126:142–57. doi: 10.1161/CIR.0b013e31825b07f8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson JA, Cavallari LH, Beitelshees AL, et al. Pharmacogenomics: application to the management of cardiovascular disease. Clin Pharmacol Ther. 2011;90:519–31. doi: 10.1038/clpt.2011.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25**.Ganesh SK, Arnett DK, Assimes TL, et al. Genetics and Genomics for the Prevention and Treatment of Cardiovascular Disease: Update: A Scientific Statement From the American Heart Association. Circulation. 2013;128(25):2813–51. doi: 10.1161/01.cir.0000437913.98912.1d. Recent guidelines for the management of genetic determinants of cardiovascular disease. [DOI] [PubMed] [Google Scholar]
- 26**.Pirmohamed M, Burnside G, Eriksson N, et al. EU-PACT Group. A randomized trial of genotype-guided dosing of warfarin. N Engl J Med. 2013;369:2294–303. doi: 10.1056/NEJMoa1311386. The European EU-PACT study has found utility in pharmacogenetic-based dosing showing a higher percentage of time in the therapeutic INR range than was standard dosing during the initiation of warfarin therapy. [DOI] [PubMed] [Google Scholar]
- 27*.Kimmel SE, French B, Kasner SE, et al. COAG Investigators. A pharmacogenetic versus a clinical algorithm for warfarin dosing. N Engl J Med. 2013;369:2283–93. doi: 10.1056/NEJMoa1310669. The US COAG found no significant utility in the genotype-guided dosing of warfarin compared to clinically-guided control. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davis EM, Knezevich JT, Teply RM. Advances in antiplatelet technologies to improve cardiovascular disease morbidity and mortality: a review of ticagrelor. Clin Pharmacol. 2013;5:67–83. doi: 10.2147/CPAA.S41859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wagoner LE, Craft LL, Singh B, et al. Polymorphisms of the beta(2)-adrenergic receptor determine exercise capacity in patients with heart failure. Circ Res. 2000;86:834–40. doi: 10.1161/01.res.86.8.834. [DOI] [PubMed] [Google Scholar]
- 30*.Fiuzat M, O'Connor CM, Gueyffier F, et al. Biomarker-guided therapies in heart failure: a forum for unified strategies. J Card Fail. 2013;19(8):592–9. doi: 10.1016/j.cardfail.2013.05.012. review of the current knowledge of biomarkers including pharmacogenetic factors in heart failure. [DOI] [PubMed] [Google Scholar]