

Abstract
The islets of Langerhans normally contain resident antigen presenting cells (APCs), which in normal conditions are mostly represented by macrophages, with a few dendritic cells (DC). We present here the features of these islet APCs, making the point that they have a supportive function in islet homeostasis. Islet APCs express high levels of major histocompatibility complexes (MHC) molecules on their surfaces and are highly active in antigen presentation in the autoimmune diabetes of the NOD mouse: they do this by presenting peptides derived from molecules of the β-cells. These APCs also are instrumental in the localization of diabetogenic T cells into islets. The islet APC present exogenous peptides derived from secretory granules of the beta cell, giving rise to unique peptide-MHC complexes (pMHC) that activate those non-conventional T cells that bypass thymus selection.
Introduction
In autoimmune diabetes of the NOD mouse two tissues contain antigen presenting cells (APCs) responsible for presentation of diabetogenic antigens, i.e. the antigenic peptides that trigger the autoreactive T cells. These two tissues are the islets of Langerhans and the pancreatic lymph node (pLN). We reviewed the APCs of the islets recently (1). This paper is an update. Normal islets are made up of three sets of endocrine sets: the β-cells, which are the most abundant and generate the secretory granules containing insulin, the α-cells that produce glucagon, and the δ-cells, a minor component that produces pancreatic polypeptide. The islets have a rich vascular network. Within the islets normally resides a non-endocrine myeloid cell belonging to the phagocytic lineage, the center of our analysis.
The early studies on the islet APC and their functional relevance came from studies on allogeneic islet transplantation in mouse experiments. These were experiments first carried out in the laboratories of Paul E. Lacy and Kevin Lafferty, who identified a cell within the islets that expressed Major Histocompatibility Complex (MHC) molecules and that were a target of rejection of islet transplants (2, 3). Both laboratories observed that culturing allogeneic islets for a period of time resulted in a prolongation of graft rejection in some strain combinations. These were extensions of the passenger leukocyte observations initially made by George Snell as an element of allogeneic graft rejections (4), but extended by Lafferty’s group not only using islets but other tissues like thyroid (5). We now know now that the phagocytic cells of the islets are not passenger leukocytes but resident cells with characteristic features and properties; most important are their close adhesion to the vessels and their high content of products of the beta cell secretory granules (6).
Concomitant with the Lacy and Lafferty functional studies, examinations of either histological sections or highly purified islets established that islets contained a set of myeloid cells as a normal inhabitant (1). Hume, Gordon and associates had identified macrophages by the presence of the F4/80 molecule in all endocrine tissues including islets (7).
More recently, the features of the islet APCs were examined by a few laboratories (6,8,9). Some of the recent studies were done on islets of NOD mice, the main strain used to examine spontaneous diabetes (6). We, as well as others, initially classified the islet APC as part of the DC lineage, but our most current findings point to them as macrophages. Most of the islet APCs are characterized as positive for F4/80, MHCII, CD11b and CD11c. Most do not express Zbtb46, a protein that identifies the DC lineage (10, 11) (our unpublished findings), and depend on colony stimulating factor -1 (CSF-1), see below. Moreover, a transcriptome analysis of isolated islets of NOD and non-diabetic mouse strains of mice at different ages showed the appearance of a gene signature consistent with a tissue macrophages (12) (Fig 1, panel c). A small and variable number of islet APC are made by DC, but these are restricted to the CD103 DC subset. The CD103 DC are associated with the CD8 alpha DC, both of which are developmentally regulated by the batf3 transcription factor (13). Many of the present studies have not distinguished among subsets of islet APCs, so we best refer to them as “APC”.
Figure 1. Examination of islets for myeloid cells, T cells and myeloid transcriptional analysis.
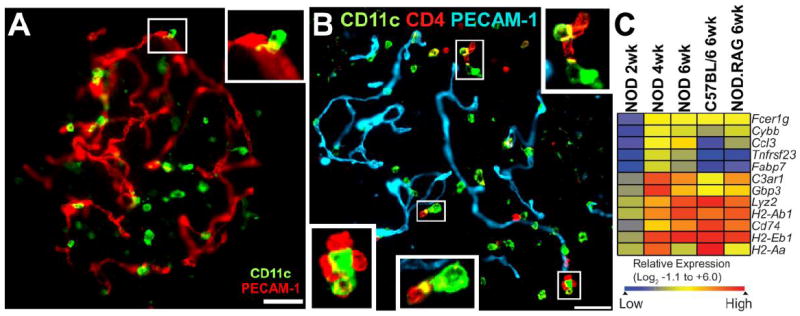
A) Islet of 8 week-old NOD.Rag1-/- mouse showing myeloid cells (CD11c+) and blood vessels (PECAM-1+). Note the close apposition of CD11c+ cells with vessels. Inset shows the apposition between islet myeloid cell and vessel. B) Islet from 6 week-old NOD female mice stained for blood vessels (PECAM-1+), islet myeloid cells (CD11c+), and T cells (CD4+). Insets show contacts between islet myeloid cells and CD4 T cells. White bars represent 50 μm. Panel B) and C) taken from Ref. 12 with permission. C) Microarray identification of myeloid gene signatures in islets of Langerhans. The heat map shows a hierarchically clustered gene subset that is enriched for macrophage/myeloid genes in NOD mice (2, 4 and 6 weeks), C57BL/6 and NOD.Rag-1-/- (6 weeks). Color intensity is based on the row normalized log2 scaled relative expression. The subset of genes represented in Panel C) is derived from the analysis detailed in Ref. 12.
There are important characteristics of the islet APCs. First, there is a high expression of both class I and class II MHC molecules as well as costimulatory molecules, making them suitable for presenting antigens (6). By all criteria these are activated APC. Second, most of them are associated with the intra-islet blood vessels, positioned next to them: by two photon microscopy on isolated islets, one can visualize them attached to the vessel wall emitting projections within the islet. Of interest is that some of these project into the islet lumen (14).
(The islet purification process becomes an important technical aspect when evaluating intra-islet leukocytes. Contamination of non-islet leukocytes is a major hurdle for islet examination due to the lack of rigorous islet purification. The routine protocols uses two purification steps, one being the separation of islets from the acinar and surrounding tissue component by Ficoll gradient centrifugation, following the collagenase treatment; the second step is handpicking the Ficoll purified islets to ensure a purify islet preparation. Omitting the hand-picking step consistently results in leukocyte contaminations from non-islet structures. Moreover, frequently small lymphoid aggregates are found in the intra-lobular fat tissue and contaminate the islets preparation. For 100% purity, isolated islets need to be distinguished from these aggregates by selecting them under a microscope using Dithizone which selectively stains islets in red.)
The trophic role of the islet APC
In the normal steady state, islet APCs have a supportive role, maintaining the health, the homeostasis of the islet endocrine cells. [Homeostasis is the famous word first enunciated by Walter B. Cannon, based on Claude Bernard’s famous concept of the milieu interieur: At the cellular level it can be defined as those cell activities that keep the tissue under a balanced steady state (15).] The trophic function of islet APC was first demostrated when examining the Csf op/Csfop mice (16). These mice have a spontaneous mutation in the Csf1 gene that results in a non-functional molecule, and therefore in a defect in macrophage differentiation. The mice are osteopetrotic as a result of the absence of osteoclasts required for the removal of bone matrix. Banel-Boucharap et al showed an absence of pancreatic macrophages in such mice and a number of functional abnormalities: reduced β-cell proliferation in late fetal life and a reduction in β-cell mass of about 30% in the adult (16). We confirmed these findings, proving that the isolated islets had a major loss of the macrophages: about 40% of islets did not have any, and the rest had one to two compared to about 10 in islets from normal mice. Islet mass was likewise reduced (6). These findings pointed to an important trophic support role that macrophages have in maintaining tissue homeostasis (17-19), the nature of which in regards to islet biology has not been determined. The islet macrophage may be a contributor to the local production of the vascular endothelial cell growth factor (VEGF) required for islets angiogenesis (20). VEGF and its receptors (VEGFRs) are highly expressed in several adult organs, including endocrine glands (pancreatic islets, thyroid, adrenal cortex, and pituitary), choroid plexus, intestine villi, lung and kidney. VEGF-A is responsible for the dense islet vascularization, being more highly expressed in the islet endocrine cells than the exocrine pancreas (20). Islet capillaries are known to express high levels of two VEGF receptors, VEGFR-2 and VEGFR-3. Endothelial fenestrations are induced by VEGF [21-23]. VEGF-A deficient islets are poorly vascularized and have few endothelial fenestrations (20, 24). As a result, islet capillaries are atrophic, not fenestrated and contain an unusual number of caveolae.
In brief, macrophages in all tissues have a major homeostatic function, which is to ensure the normal tissue physiology and to modulate its development (17-19). Macrophages release all sorts of modulatory molecules, including cytokines, angiogenic factors, chemokines and lipid mediators. These influence how the tissue cells react, how traffic is controlled and how vascular function is regulated; in turn, macrophages are highly plastic, as are the cells of the macrophage-DC lineage, which in turn adapt and are influenced by the tissue anatomy and cells.
The function of the islet APC in antigen presentation
Islet β-cells do not express class II-MHC molecules so they do not directly participate in antigen presentation, a point that established in early investigations. Incubation of CD4 T cells with β-cell and APCs results in the presentation of antigenic material, including that from allogeneic β-cells, a finding first reported by Haskins et al (25). Thus the β-cells need to interact with the APC and donate their antigens to them in order for presentation to take place (25, 26). We strongly argue that it is the uptake by APC of antigen from β-cells and their presentation to be central in the development of the diabetic autoimmunity (1,27).
APC isolated from purified islets were shown to present antigen highly efficiently (6, 8, 26, 28-32), indicating that they were highly charged with peptide-MHC (pMHC) complexes from products of the β-cell (Table I). Importantly, this presentation neither required the death of β-cells nor an inflammatory response. It was a constitutive function, which was apparent when testing islets from NOD.SCID or RAG.1-/- mice (6). Microscopy studies documented that islet APC were found to take up β-cell secretory granules. Using intravital microscopy examination, Melli et al showed uptake of GFP bearing particles from mice expressing this protein under the insulin promoter (8). We found by electron microscopy that islet APC contained granules (6). In one of our studies we compared the immunogenicity of beta cells expressing the small protein hen-egg white lysozyme (HEL) to free HEL and found the former to be about 20 times more potent (32), i.e. the cell-associated antigen appeared to be handled more efficiently. In a more recent study, all islet APCs were found to contain vesicles bearing the peptide (28). The number of vesicles per APC was about ten, explaining the high level of peptide-MHC from β-cell granule protein.
TABLE I. Antigen Presentation by Class II MHC Molecules.
| Antigen | Peptide | Islet APC | T Cell Localization | pLN | Refs |
|---|---|---|---|---|---|
| Insulin | B chain 12-20 (VEALYLVCG) | + | + | weak or negative | 28, 35 6 |
| Insulin | B chain 13-21 (EALYLVCGE) | + | not tested | not tested | 28 |
| C-peptide | INS-1 47-64 | + | not tested | not tested | 29 |
| BDC 2.5 | Chromogranin A (?) | + | + | + | 71, 72 6 |
| HEL | 48-62 | + | + | + | 31,32 6 |
| HEL | 31-47 | + | not tested | not tested | 32 |
Table indicates selected antigen epitopes studied for islet presentation. Islet APC refers to experiments in which islet or purified islet APC were used to present to T cells directed to the indicated peptides: it indicates that the APC are charged with the peptide-MHC complex. T cell localization refers to two sets of findings: either transferring the T cells and examining their localization in islets; or examining the localization of T cells from a transgenic TCR mouse. pLN refers to experiments showing presentation in pancreatic lymph nodes.
How the antigenic material is transferred from β-cell to the APC is not known, and needs to be established. The APC are in close proximity to the beta cells, with intense areas of membrane ruffling in the contact area (6). Electron microscopy studies have shown structures inside the APC structures identical to secretory granule with the dense core and the halo surrounding it. This image suggests a direct transfer of granules which is a puzzling issue from what is known of the biology of granule in which it opens as it traffics to plasma membrane to then release its cargo. Regardless, there appears to be a close interaction that needs to be defined functionally and molecularly. In other situations, purified secretory granules have been shown to be taken up by APC and their antigenic peptides presented to CD4 T cells (28).
Secretory granules contain uniquely immunogenic insulin peptides
An important finding is that among the products of the secretory granules are free peptides mostly derived from insulin. Using an antibody specific for the peptide 9-23 of the insulin β chain, we identified in beta cells granules only containing the peptide but not the mature insulin molecule (28). These peptides were also identified by mass spectrometry analysis of the purified granules and likely represent by-products of insulin handling by the beta cells. Currently we are examining whether they are the consequences of endoplasmic reticulum stress to which the beta cell is submitted (33). The important consequence is that such peptides are presented to unique non-conventional CD4 T cells reactive to insulin (28, reviewed in 27).
The free peptides in the secretory granules give rise to unique insulin pMHC complexes that select for non-conventional T cells (28, 34, 35). These are T cells that recognize segments of the insulin molecules not found after processing of insulin, i.e. they are unique to the interactions of exogenous insulin peptides with I-Ag7, we define them as type B pMHC (27). The explanation follows.
The most important autoreactivity in NOD mice is against insulin (selected references include 28, 34-41) presented by the unique class II MHC molecule I-Ag7. (NOD diabetes and human Type 1 diabetes are highly influenced by class II MHC alleles, HLADQ2 and HLADQ8 in the human and I-Ag7 in the NOD mouse.) Many of the CD4 T cells recognize a peptide segment in the B chain from residues 9 to 23 (38). Cellular and biochemical analysis indicate that uptake of insulin results in the presentation of peptides covering the nine amino acid stretch from residues 13-21 (28). As segment differing by one single amino acid shift, i.e. 12-20 is not presented by insulin processing. The 12-20 peptide is rapidly eliminated by insulin processing in the late vesicular compartment of APC where the peptide-MHC complex is assembled. The 12-20 segment binds weakly and is rapidly eliminated by the H2-DM molecule in the assembly vesicle. This is very much in contrast when the APC takes up the free peptide from the beta cells. In APCs, free peptides bind by peptide exchange with class II MHC molecules found in plasma membrane or recycling vesicles, sites where H2-DM is not present. In such situations the 12-20 segment survives and can be presented to T cells with complementary CD4 T cell receptors (TCR) to it. In sum, processing of insulin restricts the repertoire of peptides that generate peptide-MHC complexes, while exogenous peptides give rise to a more diverse set of them.
Importantly, the insulin expression induced by Aire in thymus gives rise mostly to peptide-MHC complexes bearing the 13-21 segment, resulting in the purging of most T cells to it. In contrast, these T cells that recognize the weak 12-20 segment survive. A majority of the insulin reactive T cells in NOD are directed to this unstable segment. A recent study using a TCR expressed as a transgene has confirmed these findings (35).
Role of APC in the localization of CD4 T cells to islets
A key function of the islet APC is in fostering the localization of diabetogenic T cells into the islets. Both CD4 and CD8 T cells need to migrate and localize in islets in order to initiate the active attack on β-cells. Many laboratories have identified T cells in islets at the early stages of the process. In a recent detailed examination of islets during the early stages of diabetes, T cells were found inside the islet at a very early age at a time when no conspicuous lesions were evident (12). Most of them were in intimate contact with the islet APCs. Thus this initial entry precedes the peri-insulitic lesion that is found at a later stage preceding beta cell death. At the time of T cell localization a conspicuous interferon signature genes became evident (12). This interferon signature was not found in non diabetic strains of mice nor in NOD.RAG.1-/- mice.
Few studies have examined the nature by which diabetogenic T cells localize in islets. Diabetogenic T cells could enter at random, non-specifically, but retained only if there is effective cell interaction with the islet APCs. Alternatively, T cells could enter only through a specific immunological recognition. Lennon et al first reported on the specific localization of diabetogenic T cells into islets (42). They used a “retrogenic” approach, a retroviral vector containing genes encoding the α and β chains of the TCR to reconstitute stem cells of the NOD.SCID mice. By this approach they generated a number of TCR transgenic mice, which were then evaluated for the localization of T cells.
We followed a different approach by transferring known diabetogenic TCR transgenic cells into non-diabetic mice and following their localization into the islets several hours after injection (14, 43). Using this approach, the mechanisms of entry could be followed and manipulated in a short interval of time. In NOD mice, the pre-activated CD4 BDC2.5 T cell clone entered the islets in a process that was refractory to pertussis toxin treatment of the cell indicating that chemokine receptor signaling was most likely not taking place. In contrast, non-specifically activated CD4 T cells by themselves did not enter islets. The entry of the diabetogenic T cells only took place if the islets expressed the specific class II MHC molecules. Moreover, the localization was inhibited by a previous administration of monoclonal antibodies to the I-Ag7 molecules as well as to ICAM-1. This finding pointed to these two key molecules involved in the process of entry and localization and in a chemokine independent process.
The specificity of the localization also was shown in a non-NOD mouse system, that of B10.BR mice expressing the small protein HEL in β-cells by transgenesis. T cells specific for the HEL dominant epitope injected into such mice localized to their islets expressing HEL. Most of the CD4 T cells were in contact with the islet APC. By live microscopy of the isolated islets, the time of contact of the T cells was typical of specific interaction as it takes place in lymph nodes (6). However, no such localization took place when injected into control B10.BR mice not expressing HEL.
Two other experimental systems tested the specificity of migration. Santamaria’s laboratory examined the migration of CD8 T cells specific to a peptide 206-214 from the islet-specific glucose-6-phosphatase catalalytic subunit-related protein (IGRP) (44) and found their localization into islets. No such migration occurred when the cells were injected into mice with a knock of the IGRP gene having mutations on key amino acids required for its recognition. Chervonsky’s group also examined migration of another CD8 clone directed to an insulin peptide and found migration only into islets having the correct MHC class I allele (Kd) (45).
The specificity of the T cell entry was just documented for an insulin-reactive CD4 T cell. We examined the localization of an insulin reactive CD4 TCR transgenic NOD mouse specific to the insulin β chain 12-20 segment (8F10 mouse): their islets contained T cells in contact with the islet APCs already by the third week of life (35). The transfer of activated T cells from this TCR transgenic mouse into irradiated NOD resulted in their localization into islets with ensuing diabetes. However, no localization nor diabetes developed if the cells where injected into mice that contained a mutation in Tyr 16 (B16A) of the β chain of the insulin-2 gene (46,47). Experiments from Nakayama in Eisenbarth’s laboratory had shown that the tyrosine 16 residue was critical for the reactivity of insulin-specific T cells (46). Such mice did not develop diabetes, pointing to the importance of insulin as a major diabetic autoantigen. Another conspicuous results came from the examination of the unmanipulated B16A mouse in that their islets had very few T cells in contrast to the regular NODs. We interpret this finding to mean that a great number of the T cells found in islets in a regular NOD are most likely represented by the anti-insulin T cells or require the recognition of insulin. In brief, the localization depended on the presence in islets of insulin (35).
In toto, the evidence is convincing that there is specific localization of T cells only into islets in which the APC present the beta cell antigenic determinant: to sum, four experimental settings have shown this as discussed above, the transfer of the CD4 clone BDC2.5, the CD8 T cell to IGRP, the CD4 T cell to islet planted HEL and the CD4 T cell to insulin peptide (op cit). The mode of entry of the T cells is not entirely resolved. As mentioned above, one scenario is that there is a random non-specific entry and exit of T cells into islets, and only those reacting specifically with antigen in APC remain. We have repeatedly probed for such non-specific T cells and very rarely find them in islets. Moreover, systemic activation of T cells does not result in islet localization. The alternative scenario is that of specific entry. We found dendrites from the APC in many of the vessel lumens, which raises the question whether these extensions, a “periscope function” as we have called it, are used to interact with, capture and retain the circulating T cells. Circulation in islets can be slow and intermittent factors with differences in flow velocity among islets and within an islet (48-50). These differences are accentuated in periods of hypoglycemia, while in the hyperglycemic state circulation is increased (50). These features could favor contacts between the specific T cells with the APC. It is noteworthy that injection of 0.5μm latex beads coated with antibodies against class II MHC molecules were found to localize into islets of mice bearing the correct class II allele: the important finding was that the beads localized at the vessel wall next to the islet APC (14). These findings suggest that the interface APC/vessel wall is exposed to circulating particles and cells.
What about the localization of non-specifically T cells in islets? Lymphocytes that are not reactive with islet antigens can enter islets, but only under two special situations so far identified, that involve a pre-existing diabetogenic process or the presence of a level of chemokine expression. Part of the exuberant peri-insulitic lesion of the NOD is represented by T cells as well as B cells, in fact forming an ectopic lymph node-like structure in which there is an important role for lymphotoxin beta receptor interactions (51-54). Other studies in which a number of chemokines or cytokines were expressed in islets of non-diabetic mice by transgenesis results in the attraction of non-diabetogenic T cells and B cells without an effector reaction that would result in diabetes (55-58) These include quite a range of chemokines such as CXCL13 (55), CCL12 (56), CCL19(56), CXCL12 (56), CCL21 (56), CXCL10 (57) and CCL2 (58). The entry of lymphocytes without ensuing diabetes was also shown in islets expressing TNF alpha (59). Migration of non-diabetogenic lymphocytes was also shown in islets of NOD mice in which there was expression of a number of adhesion molecules (60).
We identified a relationship between the arrival of activated diabetogenic T cells into islets and the recruitment of non-specific T cells. While activated non-diabetogenic T cells did not enter a quiet islet, as mentioned above, they did so following the entry of diabetogenic T cells (43). The entry of the non-specific T cells differed from that of the specific T cells in two ways: it was blocked by prior treatment with pertussis toxin, pointing to a chemokine dependent response and was inhibited by blocking antibodies to VCAM-1. The entry of diabetogenic T cells rapidly induced the expression of new genes with an interferon signature, an upregulation of a variety of chemokines and expression of VCAM-1, changes that we have described as a “receptive” islet (43). We defined a “receptive islet” as one that allows the entry of leukocytes, which it does by upregulation of vascular adhesion molecules as well as by the presence of chemoattractants.
Pancreatic lymph nodes
The question can be raised on how the transfer of β-cell antigens to pLN, most of which are components of the secretory granule, takes place. Islets do not possess a lymphatic drainage, so direct flow into the APC of the pancreatic lymph node (pLN) cannot take place. Lymphatic vessels are in the stroma, some near the islets. It is hard to posit a transfer of soluble material from islets to the stroma and to lymphatics; insulin and other secretory granule material normally reaches the blood circulation. A logical explanation is that the islet APC (loaded with β-cell-derived antigens under steady state) moves out of islets into the stroma, and from there enters the lymphatics and finally reach the pLN. Our studies evaluating islet APC by two photon microscopy on isolated islets showed two physical profiles of APCs within the islet: one having a fixed point of localization with highly dynamic dendrite activity, while the second showing high motility with reduced dendrite activity, and in some cases leaving the islets during the observed time frame (~20 minutes) (6). Whether the highly motile islet APC is the one reaching the pLN still has to be determined. In a histochemical and electron microscope analysis of NOD mice, phagocytic cells as well as lymphocytes were found inside the lymphatics, with images showing stages of entry (or exit) of them (61). These images suggest but do not establish that the cells are derived from the islets since they could derive also from the stroma, which is rich in phagocytic cells. Microscopical analysis by Tang et al showed APC in pLN containing beta cell antigens from islets expressing GFP (62).
Longstanding information indicates that the central feature of the pLN is the presentation of β-cell-derived antigen to diabetogenic T cells for their initial activation and expansion prior to their islet migration. We examined this point in our previous review (1) and will only be briefly considered now. Some points emerge. First and foremost is the evidence indicating that the absence of pLN results in protection from diabetes, with low degree of islet inflammation. Gagnerault et al did this in a significant report by surgical removal during a critical time period of three weeks (63). Our laboratory examined it in offspring from NOD pregnant mice treated with lymphotoxin beta receptor –Fc fusion protein (64). As in the previous study, the incidence of diabetes was very low. That the pLN is a site of presentation has been documented by testing proliferation of CD8 and CD4 TCR transgenic T cells for limited β-cell antigens in the pLN (68-70). Although the sensitization of the pLN has been correlated with beta cell death (67,70), it has been documented in adult mice from non-diabetogenic strains of mice and also in SCID mice indicating a basal presentation not linked to inflammation or cell death (6).
Our recent information testing an insulin reactive CD4 T cell to the insulin β chain 12-20 segment (8F10 CD4 T cell) showed that not all β-cell-derived antigens are presented in the pLN. 8F10 CD4 T cells recognized endogenous presentation of insulin only by the islet APCs and not by pLN APCs by in vitro studies. Recent in vivo evaluation using the 8F10 TCR transgenic mouse revealed three important findings: 1) naïve 8F10 CD4 T cells did not proliferate in the pLN and only proliferated in the islets of NOD mice upon adoptive transfer; 2) 8F10 transgenic nodeless mice (missing most lymph nodes, including the pLN) contained T cell infiltrated islets as early as 5 weeks of age; and 3) 8F10 Rag1-/- nodeless mice developed diabetes with similar kinetics to 8F10 Rag1-/- mice (35).
This new information brings a new perspective to the role of the pLN, which may be that of amplification of the diabetogenic T cell response upon the initial encounter with its cognate antigen inside the islet of Langerhans. The T cell encounter in the islet may trigger an initial insult that can augment the exit of β-cell-derived antigen. The flow of antigen to the pLN could result from egress of the islet-resident APC out of the islets, into the stroma and through the lymphatics; and/or the influx of circulating APCs into islets, to take up beta cell antigens and then exit into the stroma to move to the pLN. These are important issues that future studies will need to address.
Concluding remarks
Much more needs to be examined regarding the interactions among the three key cellular elements of the islets: beta cells, the APCs and the vascular endothelia. All indications point to a program of interactions that are key both in the integrity of islet function as well as for the development of autoreactivity. We describe here how these interactions are centered on the APCs of islets and pLN. Knowing that APC are normally charged with beta cell antigens, and have a basal level of activation; that the draining pLN is set up at an early stage to present antigens and to contribute to the autoimmune process; and that sets of diabetogenic T cells can bypass thymic negative selection and enter islets, sets a framework to decipher how each of these events are brought together, and to formulate a number of questions. What is the key initiating event? Are these initial events influenced by the early interferon gene signature in islets, and if so, how? Lastly, how does the islet, which appears well set for autoreactivity, defends itself to blood-borne pathogens and phlogogens while maintaining a lifelong homeostasis, without autoreactivity? These are seminal questions to answer in the future.
Highlights.
Islet APCs have a homeostatic supportive role under steady state.
Islet APC present beta-cell derived antigens.
Secretory granules of beta-cells contain immunogenic insulin peptides.
Diabetogenic CD4 T cells localize to islets by an antigen-dependent mechanism.
Acknowledgments
Our work cited in the text was funded by grants AI024742, DK058177 and DK20579 from the US National Institutes of Health and grants 1-2010-263 and 17-2012-141 from the Juvenile Diabetes Research Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Calderon B, Unanue ER. Antigen presentation events in autoimmune diabetes. Curr Opin Immunol. 2012;24:119–28. doi: 10.1016/j.coi.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2*.Lacy PE, Davie JM, Finke EH. Prolongation of islet allograft survival following in vitro culture (24 degrees C) and a single injection of ALS. Science. 1979;204:312–316. doi: 10.1126/science.107588. [DOI] [PubMed] [Google Scholar]
- 3*.Simeonovic CJ, Bowen KM, Kotlarski I, Lafferty KJ. Modulation of tissue immunogenicity by organ culture. Comparison of adult islets and fetal pancreas. Transplantation. 1980;30:174–179. doi: 10.1097/00007890-198009000-00004. [DOI] [PubMed] [Google Scholar]
- 4.Snell GD. The homograft reaction. Ann Rev Microbiol. 1957;11:439–458. doi: 10.1146/annurev.mi.11.100157.002255. [DOI] [PubMed] [Google Scholar]
- 5*.Lafferty KJ, Prowse SJ, Simeonovic CJ. Immunobiology of tissue transplantation: A return to the passenger leukocyte concept. Ann Rev Immunol. 1983;1:143–173. doi: 10.1146/annurev.iy.01.040183.001043. Ref. 2,3, and 5 are early papers reporting APC in islets of Langerhans. [DOI] [PubMed] [Google Scholar]
- 6**.Calderon B, Suri A, Miller M, Unanue ER. Dendritic cells in islets of Langerhans constitutively present beta cell-derived peptides bound to their class II MHC molecules. Proc Natl Acad Sci U S A. 2008;105:6121–6126. doi: 10.1073/pnas.0801973105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hume DA, Halpin D, Charlton H, Gordon S. The mononuclear phagocyte system of the mouse defined by immunohistochemical localization of antigen F4/80: macrophages of endocrine organs. Proc Natl Acad Sci USA. 1984;81:4174–4177. doi: 10.1073/pnas.81.13.4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8**.Melli K, Friedman RS, Martin AE, Finger EB, Miao G, Szot GL, Krummel MF, Tang Q. Amplification of autoimmune response through induction of dendritic cell maturation in inflamed tissues. J Immunol. 2009;182:2590–2600. doi: 10.4049/jimmunol.0803543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9**.Ginhoux F, Liu K, Heift J, Bogunovic M, Greter M, Hashimoto D, Price J, Yin N, Bromberg J, Lira SA, Stanley ER, Nussenzweig M, Merad M. The origin and development of nonlymphoid tissue CD103+ DCs. J Exp Med. 2009;206:3115–3130. doi: 10.1084/jem.20091756. Ref. 6, 8, and 9 are reports identifying the phenotype, localization and steady state of islet APC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Satpathy AT, KC W, Albring JC, Edelson BT, Kretzer NM, Bhattacharya D, Murphy TL, Murphy KM. Zbtb46 expression distinguishes classical dendritic cells and their committed progenitors from other immune lineages. J Exp Med. 2012;209:1135–1152. doi: 10.1084/jem.20120030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meredith MM, Liu K, Darrasse-Jeze G, Kamphorst AO, Schreiber HA, Guermonprez P, Idoyaga J, Cheong C, Yao KH, Niec RE, Nussenzweig MC. Expression of the zinc finger transcription factor zDC (Zbtb46, Btbd4) defines the classical dendritic cell lineage. J Exp Med. 2012;209:1153–1165. doi: 10.1084/jem.20112675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12**.Carrero JA, Calderon B, Towfic F, Artyomov MN, Unanue ER. Defining the transcriptional and cellular landscape of type 1 diabetes in the NOD mouse. PLoS One. 2013;8:e59701. doi: 10.1371/journal.pone.0059701. This is a detailed cellular and genetic analysis of the early stage of diabetes in NOD. It shows the early entrance of CD4 T cells and their relationship to the islet APC.And the finding of an interferon gene signature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS, et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science. 2008;322:1097–1100. doi: 10.1126/science.1164206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Calderon B, Carrero JA, Miller MJ, Unanue ER. Cellular and molecular events in the localization of diabetogenic T cells to islets of Langerhans. Proc Natl Acad Sci U S A. 2011;108:1561–1566. doi: 10.1073/pnas.1018973108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cannon WB. Organization for physiological homeostasis. Physiol Rev. 1929;9:399–431. [Google Scholar]
- 16**.Banaei-Bouchareb L, Gouon-Evans V, Samara-Boustani D, Castellotti MC, Czernichow P, Pollard JW, Polak M. Insulin cell mass is altered in Csf1op/Csf1 op macrophage-deficient mice. J Leukocyte Biol. 2004;76:359–367. doi: 10.1189/jlb.1103591. An excellent study of the changes in pancreatic islets in mice deficient in the CSF-1 mutant mice. It shows the trophic function of macrophages in islet development. [DOI] [PubMed] [Google Scholar]
- 17.Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. 2013;496:445–455. doi: 10.1038/nature12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18**.Pollard JW. Trophic macrophages in development and disease. Nature Rev Immunol. 2009;9:259–270. doi: 10.1038/nri2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19**.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nature Rev Immunol. 2011;11:723–737. doi: 10.1038/nri3073. Two excellent updates on the trophic function of macrophages. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lammert E, Gu G, McLaughlin M, Brown D, Brekken R, Murtaugh LC, Gerber HP, Ferrara N, Melton DA. Role of VEGF-A in vascularization of pancreatic islets. Curr Biol. 2013;13:1070–1074. doi: 10.1016/s0960-9822(03)00378-6. [DOI] [PubMed] [Google Scholar]
- 21.Roberts WG, Palade GE. Increased microvascular permeability and endothelial fenestration induced by vascular endothelial growth factor. J Cell Sci. 1995;108:2369–2379. doi: 10.1242/jcs.108.6.2369. [DOI] [PubMed] [Google Scholar]
- 22.Esser S, Wolburg K, Wolburg H, Breier G, Kurzchalia T, Risau W. Vascular endothelial growth factor induces endothelial fenestrations in vitro. J Cell Biol. 1998;140:947–959. doi: 10.1083/jcb.140.4.947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kamba T, Tam BY, Hashizume H, Haskell A, Sennino B, Mancuso MR, Norberg SM, O’Brien SM, Davis RB, Gowen LC, et al. VEGF-dependent plasticity of fenestrated capillaries in the normal adult microvasculature. Am J Physiol Heart Circ Physiol. 2006;290:H560–576. doi: 10.1152/ajpheart.00133.2005. [DOI] [PubMed] [Google Scholar]
- 24.Iwashita N, Uchida T, Choi JB, Azuma K, Ogihara T, Ferrara N, Gerber H, Kaawamori R, Inoue M, Watada H. Impaired insulin secretion in vivo but enhanced insulin secretion from isolated islets in pancreatic beta cell-specific vascular endothelial growth factor-A knock-out mice. Diabetologia. 2007;50:380–389. doi: 10.1007/s00125-006-0512-0. [DOI] [PubMed] [Google Scholar]
- 25**.Haskins K, Portas M, Bradley B, Wegmann D, Lafferty K. T-lymphocyte clone specific for pancreatic islet antigen. Diabetes. 1988;37:1444–1448. doi: 10.2337/diab.37.10.1444. An important first report identifying T cells directed to islet antigens. [DOI] [PubMed] [Google Scholar]
- 26.Shimizu J, Carrasco-Marin E, Kanagawa O, Unanue ER. Relationship between beta cell injury and antigen presentation in NOD mice. J Immunol. 1995;155:4095–4099. [PubMed] [Google Scholar]
- 27.Mohan JF, Unanue ER. Unconventional recognition of peptides by T cells and the implications for autoimmunity. Nat Rev Immunol. 2012;12:721–728. doi: 10.1038/nri3294. [DOI] [PubMed] [Google Scholar]
- 28**.Mohan JF, Levisetti MG, Calderon B, Herzog JW, Petzold SJ, Unanue ER. Unique autoreactive T cells recognize insulin peptides generated within the islets of Langerhans in autoimmune diabetes. Nat Immunol. 2010;11:350–354. doi: 10.1038/ni.1850. These is a report on the finding non conventional T cells in NOD diabetogenesis. Number 27 is a summary of the previous studies on type A and B T cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Levisetti MG, Lewis DM, Suri A, Unanue ER. Weak proinsulin peptide-MHC complexes are targeted in autoimmune diabetes in mice. Diabetes. 2008;57:1852–1860. doi: 10.2337/db08-0068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yin N, Xu J, Ginhoux F, Randolph GJ, Merad M, Ding Y, Bromberg JS. Functional specialization of islet dendritic cell subsets. J Immunol. 2012;188:4921–4930. doi: 10.4049/jimmunol.1103725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.DiPaolo RJ, Unanue ER. The level of peptide-MHC complex determines the susceptibility to autoimmune diabetes: Studies in HEL transgenic mice. Eur J Immunol. 2001;31:3453–3459. doi: 10.1002/1521-4141(200112)31:12<3453::aid-immu3453>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 32.Byersdorfer CA, Schweitzer GG, Unanue ER. Diabetes is predicted by the beta cell level of autoantigen. J Immunol. 2005;175:4347–4354. doi: 10.4049/jimmunol.175.7.4347. [DOI] [PubMed] [Google Scholar]
- 33.Fonseca SG, Gromada J, Urano F. Endoplasmic reticulum stress and pancreatic β-cell death. Trends Endocrinol Metab. 2011;22:266–274. doi: 10.1016/j.tem.2011.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34**.Mohan JF, Petzold SJ, Unanue ER. Register shifting of an autoimmune insulin peptide-MHC II complex allows for the escape of diabetogenic T cells from negative selection. J Exp Med. 2011;208:2375–2383. doi: 10.1084/jem.20111502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35**.Mohan JF, Calderon B, Anderson MA, Unanue ER. Pathogenic CD4 T cells recognizing an unstable peptide of insulin are directly recruited into islets bypassing local lymph nodes. J Exp Med. 2013 doi: 10.1084/jem.20130582. in press. These two papers characterize the non-conventional CD4 T cells to insulin peptides. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wegmann DR, Eisenbarth GS. It’s insulin. J Autoimmun. 2000;15:286–291. doi: 10.1006/jaut.2000.0444. [DOI] [PubMed] [Google Scholar]
- 37**.Wegmann DR, Norbury-Glaser M, Daniel D. Insulin-specific T cells are a predominant component of islet infiltrates in pre-diabetic NOD mice. Eur J Immunol. 1994;24:1853–1857. doi: 10.1002/eji.1830240820. [DOI] [PubMed] [Google Scholar]
- 38**.Daniel D, Gill RG, Schloot N, Wegmann D. Epitope specificity, cytokine production profile and diabetogenic activity of insulin-specific T cell clones isolated from NOD mice. Eur J Immunol. 1995;25:1056–1062. doi: 10.1002/eji.1830250430. [DOI] [PubMed] [Google Scholar]
- 39**.Halbout P, Briand J-P, Becourt C, Muller S, Boitard C. T cell response to preproinsulin I and II in the nonobese diabetic mouse. J Immunol. 2004;169:2436–2443. doi: 10.4049/jimmunol.169.5.2436. Three key papers reporting the findings of T cells to insulin peptides. [DOI] [PubMed] [Google Scholar]
- 40.French MB, Allison J, Cram DS, Thomas HE, Dempsey-Collier M, Silva A, Georgiou HM, Kay TW, Harrison LC, Lew AM. Transgenic expression of mouse proinsulin II prevents diabetes in nonobese diabetic mice. Diabetes. 1997;46:34–39. doi: 10.2337/diab.46.1.34. [DOI] [PubMed] [Google Scholar]
- 41.Jaeckel E, Lipes MA, von Boehmer H. Recessive tolerance to preproinsulin 2 reduces but does not abolish type 1 diabetes. Nat Immunol. 2004;5:1028–1035. doi: 10.1038/ni1120. [DOI] [PubMed] [Google Scholar]
- 42**.Lennon GP, Bettini M, Burton AR, Vincent E, Arnold PY, Santamaria P, Vignai DA. T cell islet accumulation in type 1 diabetes is a tightly regulated, cell-autonomous event. Immunity. 2009;31:643–653. doi: 10.1016/j.immuni.2009.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43**.Calderon B, Carrero JA, Miller MJ, Unanue ER. Entry of diabetogenic T cells into islets induces changes that lead to amplification of the cellular response. Proc Natl Acad Sci U S A. 2011;108:567–572. doi: 10.1073/pnas.1018975108. These two papers report on the specific localization of CD4 T cells in pancreatic islets, wach following different technical approaches. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lieberman SM, Evans AM, Han B, Takaki T, Vinnitskaya Y, Caldwell JA, Serreze DV, Shabanowitz J, Hunt DF, Nathenson SG, et al. Identification of the β cell antigen targeted by a prevalent population of pathogenic CD8+ T cells in autoimmune diabetes. Proc Natl Acad Sci USA. 2003;100:8384–8388. doi: 10.1073/pnas.0932778100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Savinov AY, Wong FS, Stonebraker AC, Chervonsky AV. Presentation of antigen by endothelial cells and chemoattraction are required for homing of insulin-specific CD8+ T cells. J Exp Med. 2003;197:643–656. doi: 10.1084/jem.20021378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nakayama M, Abiru N, Moriyama H, Babaya N, Liu E, Miao D, Yu L, Wegmann DR, Hutton JC, Elliott JF, et al. Prime role for an insulin epitope in the development of type 1 diabetes in NOD mice. Nature. 2005;435:220–223. doi: 10.1038/nature03523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nakayama M, Beilke JN, Jasinski JM, Kobayashi M, Miao D, Li M, Coulombe MG, Liu E, Elliott JF, Gill RG, et al. Priming and effector dependence on insulin B:9-23 peptide in NOD islet autoimmunity. J Clin Invest. 2007;117:1835–1843. doi: 10.1172/JCI31368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wayland H. Microcirculation in pancreatic function. Microsc Res Tech. 1997;15:418–433. doi: 10.1002/(SICI)1097-0029(19970601)37:5/6<418::AID-JEMT6>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 49.Liu YM, Guth PH, Kaneko K, Livingston EH, Brunicardi FC. Dynamic in vivo observation of rat islet microcirculation. Pancreas. 1993;8:15–21. doi: 10.1097/00006676-199301000-00005. [DOI] [PubMed] [Google Scholar]
- 50.Nyman LR, Ford E, Powers AC, Piston DW. Glucose-dependent blood flow dynamics in murine pancreatic islets in vivo. Am J Physiol Endocrinol Metab. 2010;298:E807–814. doi: 10.1152/ajpendo.00715.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu Q, Salomon B, Chen M, Wang Y, Hoffman LM, Bluestone JA, Fu YX. Reversal of spontaneous autoimmune insulitis in nonobese diabetic mice by soluble lymphotoxin receptor. J Exp Med. 2001;193:1327–1332. doi: 10.1084/jem.193.11.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ettinger R, Munson SH, Chao CC, Vadeboncoeur M, Toma J, McDevitt HO. A critical role for lymphotoxin-beta receptor in the development of diabetes in nonobese diabetic mice. J Exp Med. 2001;4:1333–1340. doi: 10.1084/jem.193.11.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Luther SA, Bidgol A, Hargreaves DC, Schmidt A, Xi Y, Paniyadi J, Matloubian M, Cyster JG. Differing activities of homeostatic chemokines CCL19, CCL21, and CXCL12 in lymphocyte and dendritic cell recruitment and lymphoid neogenesis. J Immunol. 2002;169:424–433. doi: 10.4049/jimmunol.169.1.424. [DOI] [PubMed] [Google Scholar]
- 54.Lee Y, Chin RK, Christiansen P, Sun Y, Tumanov AV, Wang J, Chervonsky AV, Fu YX. Recruitment and activation of naïve T cells in the islets by lymphotoxin beta receptor-dependent tertiary lymphoid structure. Immunity. 2006;25:499–509. doi: 10.1016/j.immuni.2006.06.016. [DOI] [PubMed] [Google Scholar]
- 55.Luther SA, Lopez T, Bai W, Hanahan D, Cyster JG. BLC expression in pancreatic islets causes B cell recruitment and lymphotoxin-dependent lymphoid neogenesis. Immunity. 2000;12:471–481. doi: 10.1016/s1074-7613(00)80199-5. [DOI] [PubMed] [Google Scholar]
- 56.Chen SC, Vassileva G, Kinsley D, Holzmann S, Manfra D, et al. Ectopic expression of the murine chemokines CCL21a and CCL21b induces the formation of lymph node-like structures in pancreas, but not skin, of transgenic mice. J Immunol. 168:1001–1008. doi: 10.4049/jimmunol.168.3.1001. [DOI] [PubMed] [Google Scholar]
- 57.Rhode A, Pauza ME, Barral AM, Rodrigo E, Oldstone MB, von Herrath MG, Christen U. Islet-specific expression of CXCL10 causes spontaneous islet infiltation and accelerates diabetes development. J Immunol. 2005;175:3516–3524. doi: 10.4049/jimmunol.175.6.3516. [DOI] [PubMed] [Google Scholar]
- 58.Kriegel MA, Rathinam C, Flavell RA. Pancreatic islet expression of chemokine CCL2 suppresses autoimmune diabetes via tolerogenic CD11c+ CD11b+ dendritic cells. Proc Natl Acad Sci USA. 2012;109:3457–3462. doi: 10.1073/pnas.1115308109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Picarella DE, Kratz A, Li CB, Ruddle NH, Flavell RA. Transgenic tumor necrosis factor (TNF)-α production in pancreatic islets leads to insulitis, not diabetic patterns of inflammation in TNF-α and TNF-β transgenic mice. J Immunol. 1993;150:4136–4150. [PubMed] [Google Scholar]
- 60.Faveeuw C, Gagnerault MC, Kraal G, Lepault F. Homing of lymphocytes into islets of Langerhans in prediabetic non-obese diabetic mice is not restricted to autoreactive T cells. Int Immunol. 1995;7:1905–1913. doi: 10.1093/intimm/7.12.1905. [DOI] [PubMed] [Google Scholar]
- 61.Qu P, Ji RC, Kato S. Histochemical analysis of lymphatic endothelial cells in the pancreas of non-obese diabetic mice. J Anat. 2003;203:523–530. doi: 10.1046/j.1469-7580.2003.00234.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tang Q, Adams JY, Tooley AJ, Bi M, Fife BT, Serra P, Santamaria P, Locksley RM, Krummel MF, Bluestone JA. Visualizing regulatory T cell control of autoimmune responses in nonobese diabetic mice. Nat Immunol. 2006;7:83–92. doi: 10.1038/ni1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63**.Gagnerault MC, Luan JJ, Lotton C, Lepault F. Pancreatic lymph nodes are required for priming of β cell reactive T cells in NOD mice. J Exp Med. 2001;196:369–377. doi: 10.1084/jem.20011353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64**.Levisetti MG, Suri A, Frederick K, Unanue ER. Absence of lymph nodes in NOD mice treated with lymphotoxin-β receptor immunoglobulin protects from diabetes. Diabetes. 2004;53:3115–3119. doi: 10.2337/diabetes.53.12.3115. These two papers examine the consequences of absent pancreatic lymph node in NOD diabetogenesis. [DOI] [PubMed] [Google Scholar]
- 65**.Kurts C, Kosaka H, Carbone FR, Miller JF, Heath WR. Class I-restricted cross-presentation of exogenous self-antigens leads to deletion of autoreactive CD8(+) T cells. J Exp Med. 1997;186:239–245. doi: 10.1084/jem.186.2.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66**.Kurts C, Miller JFAP, Subramaniam RM, Carbone FR, Heath WR. Major histocompatibility complex class I-restricted cross-presentation is biased towards high dose antigens and those released during cellular destruction. J Exp Med. 1998;188:409–414. doi: 10.1084/jem.188.2.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67**.Zhang Y, O’Brien B, Trudeau J, Tan R, Santamaria P, Dutz JP. In situ beta cell death promotes priming of diabetogenc CD8 T lymphocytes. J Immunol. 2002;168:1466–1472. doi: 10.4049/jimmunol.168.3.1466. References 65, 66 and 6 show the proliferation in the pancreatic lymph nodes of CD8 T cells to beta-cell antigens. [DOI] [PubMed] [Google Scholar]
- 68.Hoglund P, Mintern J, Waltzinger C, Heath W, Benoist C, Mathis D. Initiation of autoimmune diabetes by developmentally regulated presentation of islet cell antigens in the pancreatic lymph nodes. J Exp Med. 1999;189:331–339. doi: 10.1084/jem.189.2.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mintern JD, Sutherland RM, Lew AM, Shortman K, Carbone FR, Heath WR. Constitutive, but not inflammatory, cross-presentation is disabled in the pancreas of young mice. Eur J Immunol. 2002;32:1044–1051. doi: 10.1002/1521-4141(200204)32:4<1044::AID-IMMU1044>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 70.Turley S, Poirot L, Hattori M, Benoist C, Mathis D. Physiological β cell death triggers priming of self-reactive T cells by dendritic in a type-1 diabetes model. J Exp Med. 2003;198:1527–1537. doi: 10.1084/jem.20030966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Katz JD, Wang B, Haskins K, Benoist C, Mathis D. Following a diabetogenic T cell receptor from genesis through pathogenesis. Cell. 1993;74:1089–1100. doi: 10.1016/0092-8674(93)90730-e. [DOI] [PubMed] [Google Scholar]
- 72.Stadinski BD, Delong T, Reisdorph N, Reisdorph R, Powell RL, Armstrong M, Piganelli JD, Barbour G, Bradley B, Crawford F, Marrack P, Mahata SK, Kappler JW, Haskins K. Chromogranin A is an autoantigen in type 1 diabetes. Nat Immunol. 2010;11:225–231. doi: 10.1038/ni.1844. [DOI] [PMC free article] [PubMed] [Google Scholar]