

Clinical Vignette
A 74-year-old man presented with decreasing exercise tolerance and mild ankle edema. He was previously fit, but was now breathless on climbing 2 flights of stairs. He had no history of angina, orthopnea or paroxysmal nocturnal dyspnea. His past medical history included non-insulin dependent diabetes treated for 10 years and mild hypertension. Six years earlier he had been diagnosed with a monoclonal gammopathy of unknown significance (MGUS). At that time a bone marrow biopsy showed 30% overall cellularity with 5-10% plasmacytosis (normal <4%) and immunoglobulin light chain restriction. About 3 years ago he developed deep vein thrombosis, treated with low molecular weight heparin. A year later, leg swelling occurred and was attributed to venous insufficiency. The following year he developed progressive fatigue on exertion and an abnormal ECG (Figure 1) led to a treadmill test which was considered normal. An echocardiogram showed concentric wall thickening (Supplemental Video 1) and the possibility of cardiac amyloidosis was raised. A fat pad biopsy was negative for amyloid deposits. The bone marrow biopsy performed in 2005 (when his MGUS was diagnosed) was restained and was negative for amyloid. At that time serum free lambda light chains were 108.9 mg/L (normal range 5.7-26.3) with kappa light chains of 13 mg/L (normal 3.3-19.) and an abnormal ratio of 0.12 (normal 0.26-1.65). His B-natriuretic peptide (BNP) measured 275 pcg/mL. He was treated with oral diuretics which improved leg swelling, but, due to persistent symptoms he sought medical care at our institution. On review of symptoms, he denied jaw claudication, symptoms of postural hypotension, easy bruising or tongue swelling. He did give a history suggestive of neuropathy with a “leathery feeling” in his feet, but no numbness in his hands. Medications included metformin 500 mg twice a day, aspirin 80 mg daily, lisinopril 10 mg a day, glyburide 2.5 mg a day, atorvastatin 40 mg a day, furosemide 40 mg a day for 3-4 days. He had no family history of heart failure. His father had died of a heart attack at age 69 years and his mother had died at age 93 years. One sister had multiple myeloma and had died of pancreatic cancer. On physical examination, pulse was 80 beats per minute with a respiratory rate of 18 breaths per minute and blood pressure of 145/85 mm Hg seated and 130/60 mm Hg standing. The jugular venous pressure was elevated slightly with a prominent X and Y descent and a positive Kussmaul sign. Head and neck exam revealed submandibular swelling consistent with amyloid infiltration of the salivary glands. There was no macroglossia or periorbital bruising. Abdominal exam revealed a palpable smooth liver 2-3 fingerbreadths below the costal margin. Heart exam showed a non-displaced apex without murmurs or additional sounds. There was bilateral dullness at the lung bases suggesting effusions. He had mild right ankle edema with venous insufficiency. There was no hand wasting. Peripheral pulses were brisk.
Figure 1. ECG.
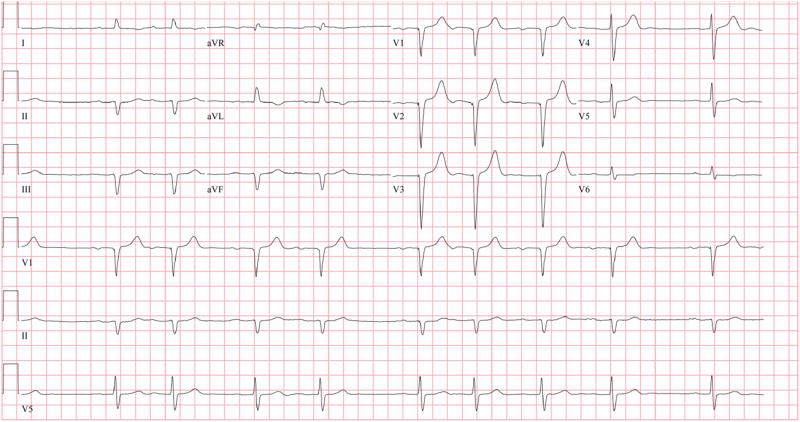
A 12 lead ECG showing first degree AV block with atrial premature beats and pseudo Q waves without typical low voltage complexes.
This case represents a patient with a plasma cell dyscrasia, presenting with symptoms of progressive congestive heart failure and an echocardiogram suggestive of cardiac amyloidosis. The task is to confirm the suspected diagnosis and, if confirmed, to determine the type of amyloid, as treatments and outcome differ among the different etiological forms. The most common types of amyloid fibrils that affect the heart are derived either from light chains produced by a plasma cell dyscrasia (AL amyloidosis) or from transthyretin (ATTR), a protein mainly synthesized by the liver, from either wild type or mutant ATTR-related amyloidosis. AL amyloidosis is in general characterized by a rapid and aggressive clinical course and systemic manifestations with cardiac involvement being associated with a worse prognosis. On the other hand, wild-type ATTR amyloidosis is a slowly progressive disease that tends to almost invariably affect the heart of elderly men (the disease is commonly referred to as senile systemic amyloidosis). Mutant ATTR amyloidosis is characterized by a high genotypic and phenotypic heterogeneity, with over 100 pathogenic mutations identified so far and with variable degrees of neurological, visceral and cardiac involvement. If this patient does have cardiac amyloidosis, the coexisting plasma cell dyscrasia with elevated free light chains suggests AL amyloidosis. However, his age, the slowly progressive symptoms and the disease limited to the heart are clinical features more consistent with wild-type ATTR amyloidosis.
Diagnosis of cardiac amyloidosis
Cardiac amyloidosis is often initially suspected in a patient presenting with unexplained heart failure symptoms and typical findings on ECG and 2D transthoracic echocardiography. Once suspected, advanced echocardiographic techniques such as strain imaging can be used, as can several other imaging modalities, although the diagnosis has to be eventually confirmed histologically, often requiring endomyocardial biopsy and special staining.
ECG findings in cardiac amyloidosis may include a low QRS voltage pattern (QRS amplitude < 0.1 mV in all the precordial leads or < 0.05 mV in all the standard limb leads) (56%), pseudo-infarction patterns (60%), supraventricular arrhythmias (mainly atrial fibrillation),1 atrio-ventricular or intra- and inter-ventricular conduction defects and unusual axis deviations.1 The presence of a low voltage ECG despite increased left ventricular wall thickening on echocardiography is highly suggestive of cardiac amyloidosis (particularly AL amyloidosis) (Supplemental Figure 1) and a typical voltage-mass ratio has been described.2, 3 Although the precise reasons for the low voltage ECG are not known, myocyte atrophy and cardiac toxicity exerted by circulating light chains are possible contributing factors.3 Of note, the ECG may not demonstrate low voltage complexes in individuals with ATTR amyloidosis, particularly in patients with underlying hypertensive heart disease.
Echocardiography is the cornerstone for the diagnosis and management of patients with known or suspected cardiac amyloidosis. Typical echocardiographic features (Table 1) may lead us to consider cardiac amyloidosis in patients with unexplained increased left ventricular wall thickness. Echocardiography also provides valuable information about amyloid burden, left ventricular filling pressures, and other hemodynamic features that are vital for the initial evaluation, management, and follow-up of these patients.
Table 1. Typical echocardiographic features of cardiac amyloidosis.
| Parameters | Comments |
|---|---|
|
| |
| Characteristic granular / sparkling appearance of the left ventricular (LV) myocardium | Not specific. Need to differentiate from other infiltrative diseases* |
|
| |
| Increased LV wall thickness | Results from amyloid infiltration of interstitial space and may relate to amyloid burden |
|
| |
| Decreased LV end-diastolic volumes | Leads to reduced stroke volume despite near normal LVEF |
|
| |
| Typically preserved or mildly reduced LV ejection fraction | LV ejection fraction may decrease in end stage disease |
|
| |
| High E/A ratio | Is seen due to restrictive pathophysiology. But, a reduced amplitude A wave may suggest poor atrial function and higher risk of thrombus formation* |
|
| |
| Shortened mitral E deceleration time (restrictive filling pattern), High E/e’ ratio | High E/e’ suggests increased left atrial pressures |
|
| |
| Increased left and right atrial volumes and reduced atrial function | A common feature. Also imaged on CMR |
| Atrial strain can be significantly reduced | |
|
| |
| Longitudinal strain (LS) in the left ventricle is impaired and worse at the base and mid ventricular regions compared to the apex4 | Specific patterns of left ventricular LS may differentiate amyloid from aortic stenosis and hypertrophic cardiomyopathy7, 8 |
|
| |
| Right ventricular (RV) thickening, reduced RV myocardial velocities on tissue Doppler imaging and reduced RV LS.5, 6 | TAPSE and RV LS are early indicators of cardiac involvement in patients with systemic AL amyloidosis5, 6 |
| Reduced tricuspid annular plane excursion despite normal RV end-diastolic dimension5, 6 | RV LS may be an independent predictor of cardiac death5 |
|
| |
| Valve thickening | Nonspecific |
|
| |
| Pericardial effusion | Nonspecific |
|
| |
| Atrial septal thickening | A characteristic feature of cardiac amyloidosis |
|
| |
| Dynamic LV outflow tract obstruction | A less common feature, but, need to distinguish from hypertrophic cardiomyopathy |
Increased left ventricular wall thickness on echocardiography, often with a restrictive filling pattern, is a classic feature but it indicates advanced cardiac amyloidosis. The clinical manifestations of cardiac amyloidosis are manifold and misdiagnosis as true left ventricular hypertrophy is frequent; hence, it is important to be vigilant for the typical echocardiographic findings that suggest amyloidosis. Coexisting right ventricular free wall or atrial septal thickening strongly suggest an infiltrative cardiomyopathy as these are rarely seen in association with true left ventricular hypertrophy. Also, transthyretin amyloidosis due to mutant ATTR (variant V122I, valine to isoleucine substitution at position 122,) is common among African-Americans (3-4%) and usually manifests with heart failure symptoms in the 6th or 7th decades; unexplained left ventricular thickening in these individuals should be carefully evaluated, including genetic testing, before labeling as hypertensive heart disease. Patients with ATTR amyloidosis tend to demonstrate greater degrees of left ventricular thickening compared to those with AL amyloidosis. (Supplemental Figure 2) However, there is overlap and the echocardiographic features AL and ATTR amyloidosis are indistinguishable, despite a much better prognosis in the latter.
Also, compared to conventional mitral inflow spectral Doppler velocities (E wave and A wave), left4 and right 5ventricular tissue Doppler imaging (early diastolic parameters of e’ and a’) (Figure 2), tricuspid annular plane excursion6 (TAPSE) (Figure 3), and strain imaging of the right5, 6 and left ventricles (longitudinal 2D-strain) (Figure 4) are more insightful and sensitive for the early identification of cardiac amyloidosis. A specific pattern of longitudinal strain characterized by worse longitudinal strain in the mid and basal ventricle with relative sparing of the apex4 may help distinguish left ventricular infiltration due to amyloid from true ventricular hypertrophy of hypertensive heart disease or hypertrophic cardiomyopathy.7, 8 In one recent study,9 although there was overlap, an apex to base ratio of longitudinal systolic strain of >2.1 distinguished cardiac amyloidosis from other causes of left ventricular hypertrophy such as hypertension, Fabry’s disease, Friedreich’s ataxia with a sensitivity - 88%, specificity-85%, positive predictive value- 67% and negative predictive value - 96%). Furthermore, abnormal longitudinal strain predicted worse survival in patients with AL amyloidosis.10-12 Koyama et al demonstrated, in 119 patients with systemic AL amyloidosis (70 with cardiac amyloidosis), that abnormal basal segment longitudinal strain11 was an independent predictor of survival. In a subsequent study of 206 patients with systemic AL amyloidosis,10 a 2D LV global longitudinal strain of less than − 11.78% was shown to be an independent predictor of survival.
Figure 2. The value of tissue Doppler imaging in early diagnosis of cardiac amyloidosis.
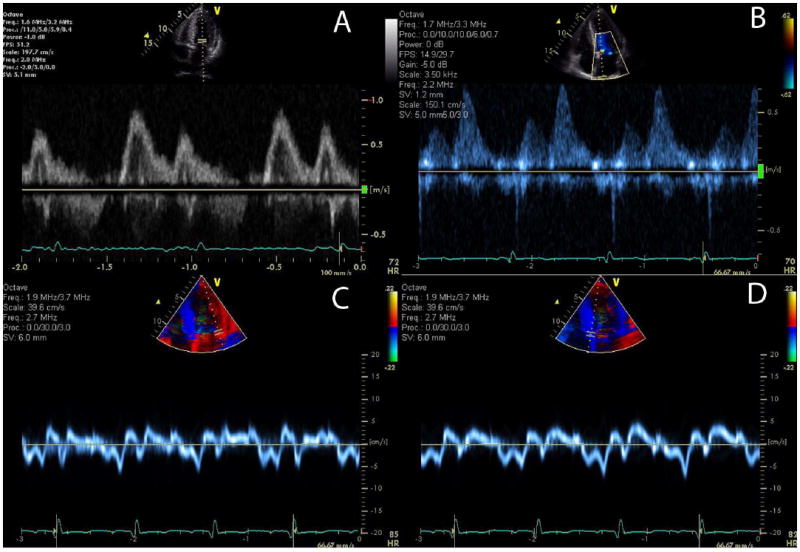
Images from a patient with AL amyloidosis demonstrate relatively normal mitral inflow parameters (E and A waves with normal deceleration time), but, abnormal pulmonary venous Doppler flow (diastolic predominance) and abnormal tissue Doppler images showing a reversed e’ and a’ ratio suggesting pseudonormalization of transmitral flow. E/e’ is elevated consistent with elevated left ventricular filling pressure.
Figure 3. The right ventricle in cardiac amyloidosis.
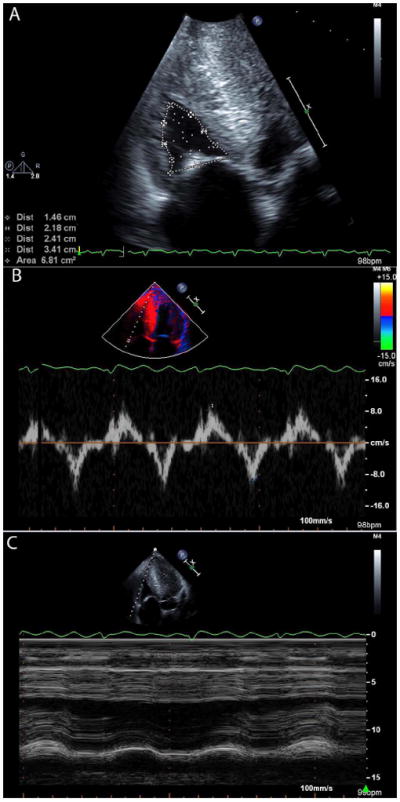
Images from a patient with wild-type ATTR (senile systemic) amyloidosis, showing increased right ventricular wall thickness with a small cavity size (A). Myocardial relaxation velocities on tissue Doppler imaging are reduced (B) and tricuspid annular plane excursion is reduced (B).
Figure 4. Left ventricular strain imaging in cardiac amyloidosis.
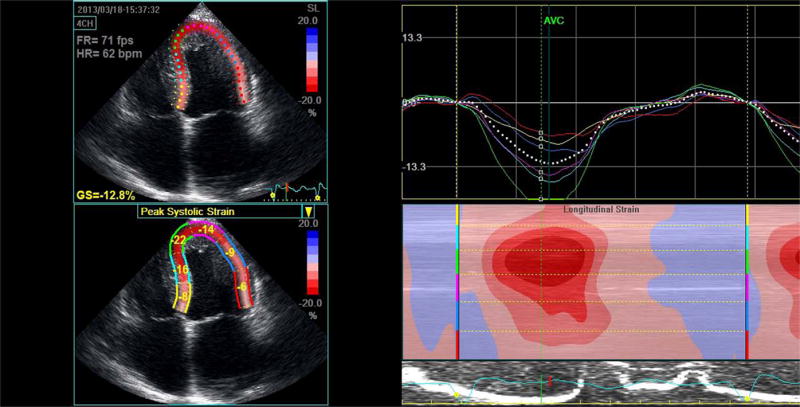
Apical 4-chamber peak systolic strain image illustrating a classic strain pattern of relatively well preserved apical strain (green and blue lines) with significant basal impairment (red and yellow lines). This is seen in the series of curves as well as the “bulls-eye” color coded strain image.
Of note, echocardiography can reveal the presence of left atrial dysfunction despite sinus rhythm, increasing the risk of stasis and thrombus formation; anticoagulation should be considered in individuals with amyloidosis who are in sinus rhythm but demonstrate impaired atrial function. Impairment of left atrial function may be identified by small late diastolic mitral inflow velocity (small “A”wave) (Figure 5, and Supplemental Video 2), reduced atrial ejection fraction, or reduced atrial strain (Figure 6). Modesto et al13 demonstrated abnormal septal left atrial strain in 60% of subjects with cardiac amyloidosis. Left atrial systolic strain was significantly lower in cardiac amyloid subjects compared to control subjects (peak lateral strain: 5.5±4% vs. 19±4%, P <0.0001).13
Figure 5. Loss of atrial function in cardiac amyloidosis.
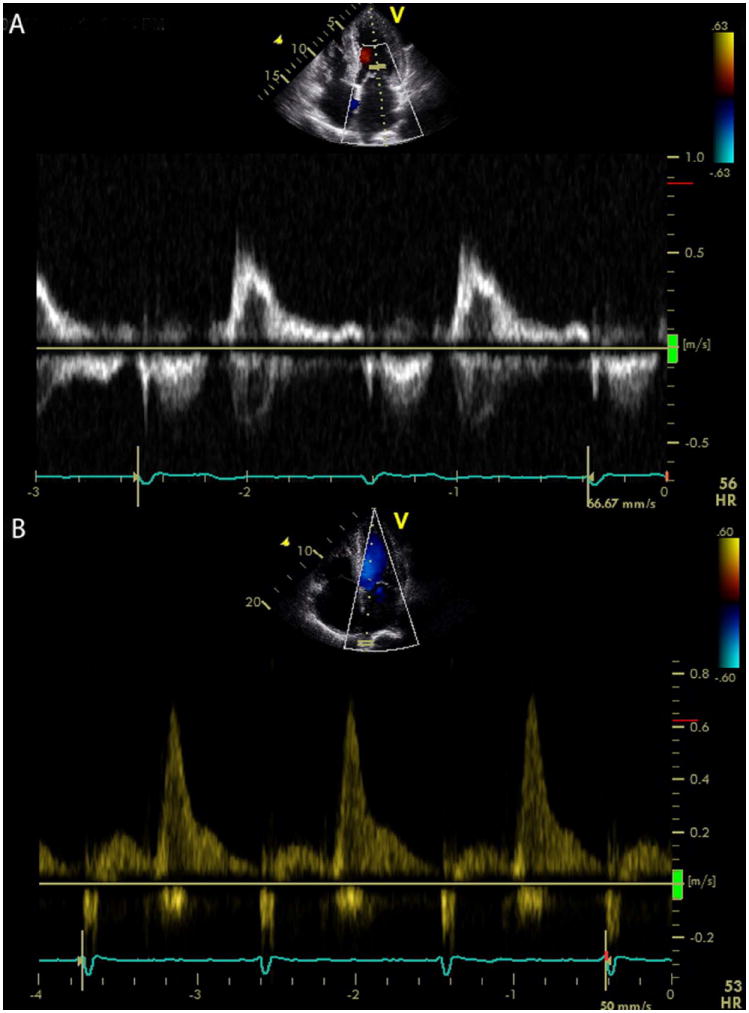
A transmitral Doppler image (A) showing a normal deceleration time but a very small A wave in an ATTR patient in sinus rhythm who presented with stroke, due to thrombus formation related to left atrial dysfunction. The pulmonary venous flow in a TTR amyloidosis patient (B) demonstrating almost exclusive diastolic flow due to restrictive filling-the atrium functions as a conduit with loss of contractile and reservoir function. This patient also had spontaneous echo contrast in the left ventricle (see Supplemental Video 2).
Figure 6. Atrial strain cardiac amyloidosis.
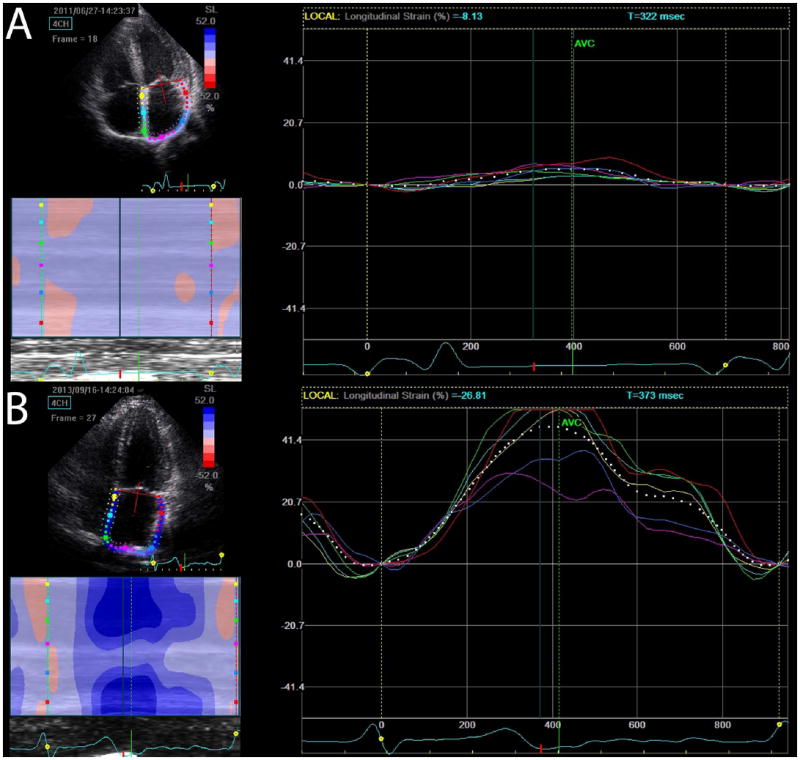
Atrial strain is significantly impaired globally in a subject with ATTR cardiac amyloidosis (A) contrasted with normal atrial strain in a gene positive phenotype negative subject (B).
Cardiac magnetic resonance imaging (CMR) provides high spatial resolution and high signal to noise ratio images without the limitation of poor echocardiographic windows. Increased myocardial mass, atrial structure, as well as atrial and ventricular function and other typical morphological features of restrictive cardiomyopathy can be imaged (Table 1, Figure 7 and Supplemental Video 3). Additional findings of amyloidosis on CMR imaging rely on tissue characterization: late gadolinium enhancement (LGE), abnormally prolonged T1 times (pre or post contrast) and an expansion of the extracellular volume.
Figure 7. Typical rest cardiac magnetic resonance imaging features in a patient with familial ATTR cardiac amyloidosis.
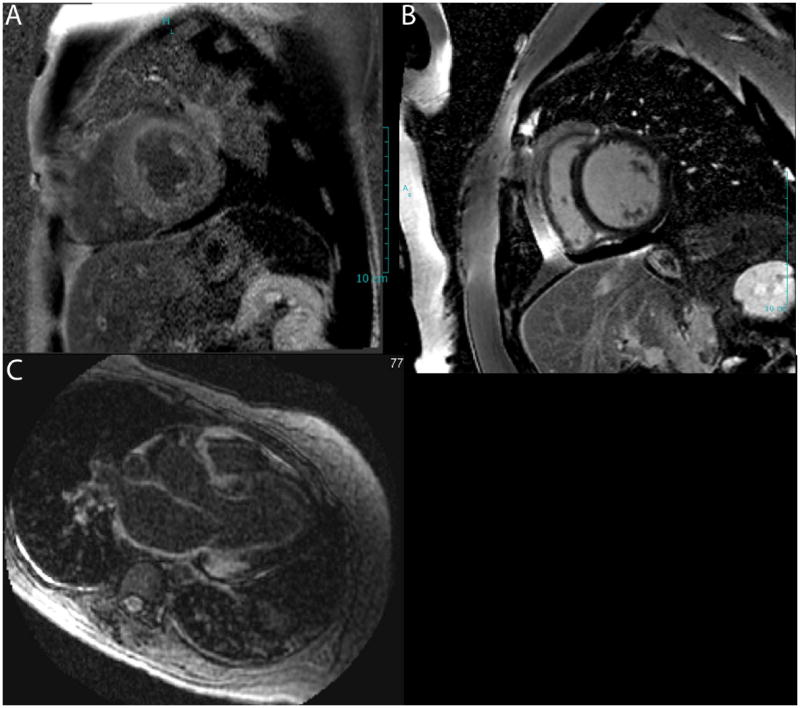
Late gadolinium enhanced images demonstrate diffuse LGE in the left ventricular myocardium (A) contrasted with dark myocardium a normal patient (B). The bottom panel (C) demonstrates LGE in the atrial wall, a characteristic feature of cardiac amyloidosis.
Gadolinium kinetics are abnormal in cardiac amyloidosis, with a faster washout of gadolinium from myocardium and blood pool compared to non-amyloid control subjects.14 Late gadolinium enhancement images are typically obtained about 5 minutes after infusion of 0.1 to 0.2 mmol/kg of gadolinium injection using a previously validated inversion recovery pulse sequence.14, 15 Characteristic LGE14, 16-22 patterns are observed in the left ventricle and the atria and permit an earlier diagnosis of cardiac amyloidosis (Table 2). Late gadolinium enhancement on CMR identified cardiac involvement in 47% of patients with known systemic amyloidosis and normal wall thickness on echocardiogram.20 In contrast, among patients with documented cardiac amyloidosis based on echocardiography or endomyocardial biopsy, left ventricular LGE was universal in both AL and ATTR patients; right ventricular LGE was present in all ATTR and in 72% of AL patients.23 Diffuse LGE in the atrial wall may be a characteristic feature of cardiac amyloidosis.
Table 2. Typical cardiac magnetic resonance (CMR) imaging features of cardiac amyloidosis.
| Parameters | Comments |
|---|---|
|
| |
| Characteristic morphological features of cardiac amyloidosis/restrictive cardiomyopathy as listed in Table 1 | Better resolution images than echocardiography |
| No limitation of difficult echo windows | |
|
| |
| Left ventricular LGE | Diffuse and subendocardial LGE of the LV myocardium is more common than patchy focal delayed enhancement |
| May be an early feature of cardiac involvement compared to increased wall thickness | |
|
| |
| Atrial LGE and function | A characteristic feature of cardiac amyloidosis |
| Atrial function can be studied well with CMR | |
|
| |
| T1 mapping | Subendocardial T1 relaxation time may be shortened in cardiac amyloidosis |
| This is an early feature of cardiac amyloid involvement | |
|
| |
| Extracellular volume estimation based on T1 mapping and hematocrit measures | Extracellular volume expansion may permit an early diagnosis of cardiac amyloid even before overt left ventricular LGE |
LGE = late gadolinium enhancement
Subendocardial T1 is shortened in cardiac amyloidosis18 and T1 mapping may identify cardiac involvement at an earlier stage compared to overt LGE images.18 Indeed, on T1 mapping, if the myocardium crosses the null point (becomes black) at a T1 time point prior to the blood pool, it indicates the presence of diffuse global hyperenhancement and is characteristic of cardiac amyloidosis. In a recent study,24 rapid visual T1 assessment was performed over 1-2 minutes about 5-10 minutes after gadolinium administration. In this study, among subjects with cardiac amyloidosis and LGE, a pattern of diffuse LGE was noted in 81%; both diffuse LGE [HR 6.0 (95% confidence interval 3.01-12.1), p < 0.0001] and any LGE [HR 6.5 (95% confidence interval 3.0 -14.2), p < 0.0001] were independently predictive of mortality.24
Native (pre-contrast) T1 mapping techniques have been studied in AL25 and ATTR 26cardiac amyloidosis, using a shortened modified look-locker inversion recovery (ShMOLLI) sequence. Although there was overlap, the native T1 times in patients with AL cardiac amyloidosis (1130±68 ms) and TTR cardiac amyloidosis (1097±43 ms) were significantly prolonged when compared to hypertrophic cardiomyopathy (1026±64 ms, p <0.0001) and normal subjects (967±34 ms, p <0.0001). Native T1 mapping techniques may be particularly helpful in patients with renal dysfunction (due to risk of gadolinium toxicity and nephrogenic systemic fibrosis).
Direct quantification of the volume of distribution of gadolinium (myocardial extracellular volume, ECV) by CMR is determined by pre and post contrast R1 (1/T1) for the blood and myocardium taking into account serum hematocrit (Hct). Serial R1 measurements after contrast injection can improve the accuracy of ECV measurement. Expansion of ECV is demonstrated even in myocardial segments without apparT1ent LGE, suggesting incremental diagnostic value.19 In AL amyloid patients, ECV correlated directly with LV mass, TDI “S” wave, brain natriuretic peptide (BNP) and troponin levels, suggesting that ECV may represent a marker of amyloid burden in the heart.17 T2 weighted sequences show hypointense signal in cardiac amyloidosis and a lower T2 ratio was independently associated with shortened survival.22
Late gadolinium enhancement, T1 prolongation, and extracellular volume expansion may be observed in amyloidosis, other infiltrative diseases,20 inflammation, and fibrosis. Therefore, although certain LGE patterns, T1 times (native or post contrast), and ECV values are sensitive to diagnose amyloidosis, they are not specific and cannot exclusively obviate the need for a definitive histological diagnosis. Notably, a difficulty in nulling of myocardial signal on the LGE images despite the use of a T1 scout may suggest underlying amyloid (especially if the blood pool images are unusually dark indicating rapid contrast sequestration) rather than suboptimal image quality. CMR is contraindicated in certain patients cardiac devices (pacemakers, implantable cardioverter defibrillators), and claustrophobia. Atrial fibrillation may limit image quality of gated sequences. Patient cooperation is required for brief periods of breath holds, and for approximately 45 minute scanning. Whether a contrast enhanced gated cardiac CT scan may provide at least some of the information as a CMR scan with short scan time, especially in patients with intracardiac devices, needs to be further evaluated.
Radionuclide imaging of cardiac amyloidosis can be performed using single photon emission computed tomography (SPECT) or positron emission tomography (PET) based radiotracers. Among SPECT tracers, direct amyloid imaging agents (I-123 labeled serum amyloid P component, SAP), bone imaging agents [Tc-99m pyrophosphate (PYP) or Tc-99m 3,3-diphosphono-1,2-propanodicarboxylic acid (DPD)], and agents to image cardiac sympathetic innervation [I-123 MIBG (Metaiodobenzylguanidine)] are available for use. MIBG is a norepinephrine analog which is actively taken up by sympathetic nerve terminals and not further metabolized, enabling us to image cardiac sympathetic innervation. I-123 labeled SAP scans have been used in systemic amyloidosis to image non-cardiac organs27 but the technique is not of proven value for imaging cardiac amyloidosis due to limited signal to noise ratio in the heart. Radionuclide scans (Figure 8A) may be helpful to diagnose cardiac amyloidosis in patients who cannot undergo a CMR study for any reason.
Figure 8. Tc-99m pyrophosphate (PYP) SPECT in cardiac amyloidosis.
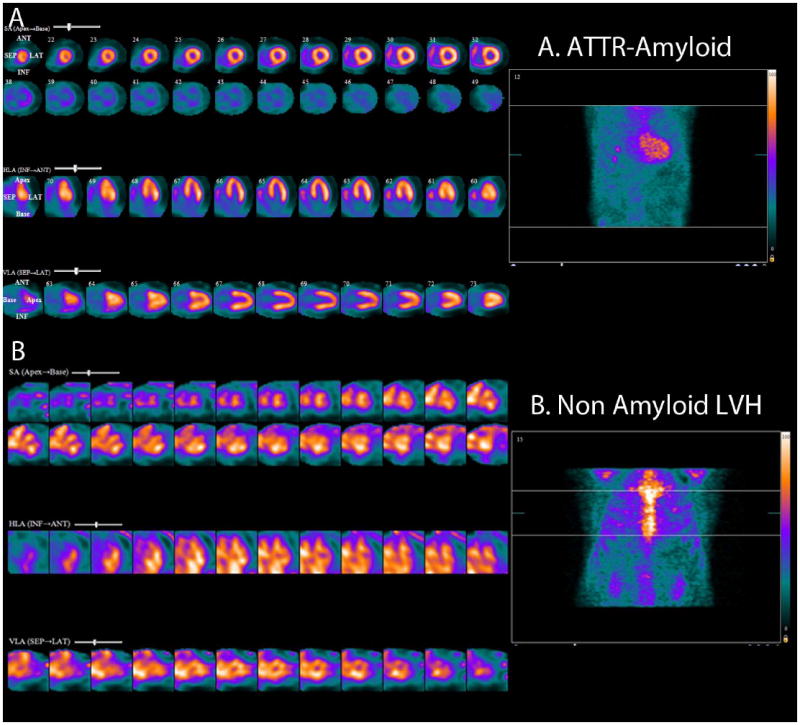
Note intense diffuse rest left and right ventricular myocardial uptake in a patient with ATTR cardiac amyloidosis (Top panel, standard cardiac imaging planes of short axis, horizontal long axis and vertical long axis projections). The bottom panel (standard cardiac imaging planes as above) shows blood pool activity and no myocardial uptake in a patient with non-amyloid LVH (left ventricular hypertrophy).
Bone imaging agents like Tc-99m pyrophosphate (PYP) or Tc-99m 3,3-diphosphono-1,2-propanodicarboxylic acid (DPD), are probably taken up via a calcium mediated mechanism, appears to be helpful to identify (both mutant and wild-type) ATTR amyloidosis.28, 29 In one study, Tc-99m DPD was able to noninvasively differentiate between AL and ATTR amyloidosis, with all ATTR patients showing DPD uptake and none of the AL patients showing DPD uptake (Supplemental Figure 3).28 It has been subsequently documented that DPD is taken up in the heart of some patients with AL amyloidosis (for reasons that remain unclear), but patients with ATTR amyloid undoubtedly show a more intense uptake than those with AL disease. DPD is not available in the USA but Tc-99m is, and is avidly taken up by the heart in ATTR amyloid cardiomyopathy. Bokhari et al30 showed that a heart to contralateral lung ratio of 1.5 on Tc-99m PYP imaging distinguished ATTR amyloidosis (≥ 1.5) from AL amyloidosis (< 1.5). The combination of increased wall thickness and high heart to whole body Tc-99m DPD uptake is also a significant independent predictor of major adverse cardiovascular events.29 Therefore, when ATTR amyloidosis of the heart is suspected based on the clinical presentation, Tc-99m PYP/DPD scan may be preferable to a CMR study; recognizing that, while a positive Tc-99m PYP/DPD scan suggests ATTR amyloidosis, a negative Tc-99m PYP/DPD scan does not rule out AL cardiac amyloid. Also, as bone imaging agents are not taken up by normal myocardium, blood pool activity maybe noted in negative scans and this must be distinguished from active myocardial uptake in amyloidosis (Figure 8B).
I 123 MIBG scintigraphy is used to image cardiac sympathetic denervation (not amyloidosis). Myocardial sympathetic denervation may be an early feature of cardiac involvement particularly in individuals with TTR familial amyloid polyneuropathy.31, 32 familial amyloid polyneuropathy is an autosomal dominant disease with single point mutations to the TTR gene presenting with neuropathy (sensorimotor and autonomic neuropathy, V30M) with or without cardiac involvement or isolated cardiac involvement (V122I). Also, in one recent study33 of 142 individuals with familial amyloid neuropathy V30M variant, a heart to mediastinal ratio of < 1.6 compared to ≥ 1.6 on I-123 MIBG scintigraphy identified individuals with a high mortality risk (HR 7.9, 95% CI, 3.38-15.2).
Direct imaging of amyloid fibrils, using PET tracers of C-11 Pittsburgh B compound34 (Supplemental Figure 4) and F-18 florbetapir (Figure 9) appears promising and is currently under investigation. These direct amyloid imaging agents offer the potential quantitation of amyloid burden and identification of early cardiac involvement before overt cardiac structural changes.
Figure 9. F-18 florbetapir imaging in cardiac amyloidosis.
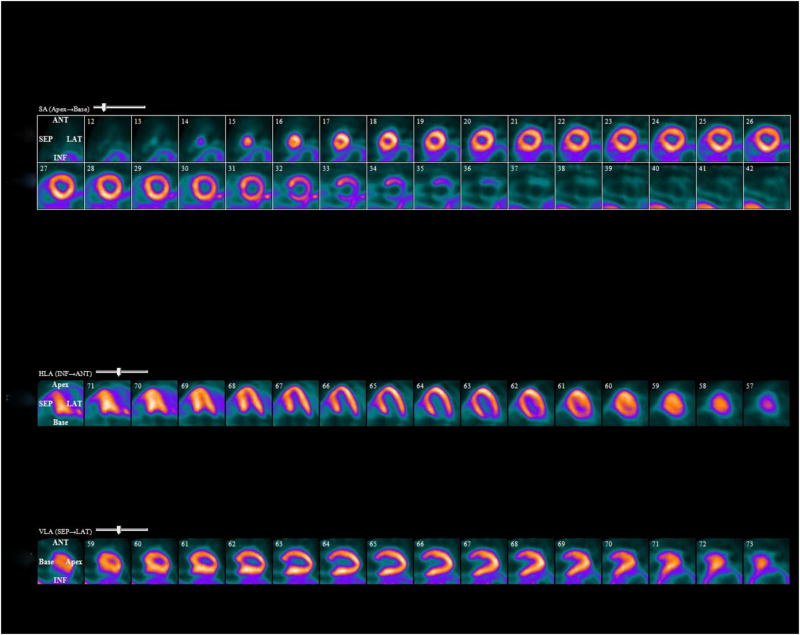
RestF-18 florbetapir PET images showing diffuse biventricular uptake of radiotracer in a patient with ATTR cardiac amyloidosis.
Is there a gold standard for the noninvasive diagnosis of cardiac amyloidosis?
The definitive diagnosis of amyloidosis is generally accepted as requiring a biopsy with the typical appearance of amyloid deposits, staining with amyloid-specific stains (classically Congo red, with apple-green birefringence under polarized light.) A positive myocardial biopsy is indicative of amyloid in the heart, but with the caveat that very elderly patients have a high prevalence of cardiac amyloid deposits, which may be infrequent and patchy and of no clinical significance. Once there is enough amyloid in the heart to cause ventricular wall thickening, endomyocardial biopsy almost always contains a significant amount of amyloid. However, in unskilled hands, with inadequate staining, false negative reports may occur. Thus if noninvasive imaging is strongly suggestive of amyloidosis, particularly in the setting of a typical clinical picture, a “negative” cardiac biopsy should not be viewed as ruling out the diagnosis, and an expert second-opinion review should be sought, with consideration of rebiopsy in selected cases. A typical noninvasive abnormality on cardiac imaging, whether echocardiography, MRI or, possibly technetium pyrophosphate imaging in TTR amyloidosis is specific enough in the setting of a positive non-cardiac biopsy such as a fat pad biopsy or renal biopsy) to conclude that cardiac amyloid is definitely present in clinically significant amounts. Similarly, it is almost certain that a strongly positive technetium pyrophosphate scan in a patient who tests positive for a variant transthyretin known to be amyloidogenic will have ATTR cardiac amyloidosis.
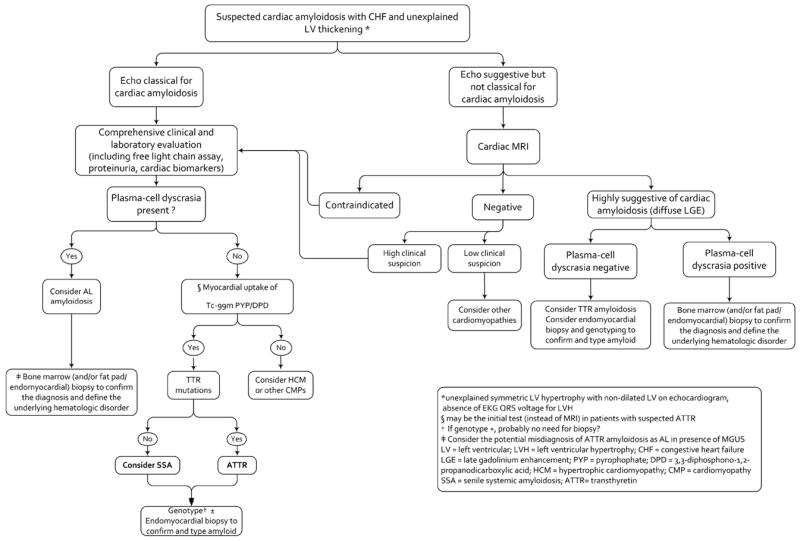
We do not, however, believe that there is one noninvasive test that can be considered either the ultimate gold standard for the diagnosis, nor even a gold standard when considered against other noninvasive testing modalities. Most commonly, the noninvasive diagnosis of cardiac involvement in patients with systemic amyloidosis is suspected initially by echocardiography based on increased wall thickness and a restrictive LV filling pattern. Typical left ventricular longitudinal strain abnormalities with preserved apical strain compared to basal strain may strengthen the likelihood that increased LV mass is due to amyloid infiltration, yet, there may be overlap with other left ventricular diseases associated with true hypertrophy. Characteristic patterns of LGE on CMR, increase in ECV fraction and prolongation of the T1 times may suggest cardiac amyloidosis even before overt left ventricular wall thickening, but, by themselves are also not pathognomonic for this diagnosis. Radionuclide techniques with specific binding to components of amyloid can identify systemic amyloid deposits. However, the amyloid specific radiotracer used (I123 SAP), is not available in North America, nor is it useful for imaging cardiac amyloidosis due to blood pool activity. The diagnostic accuracy of amyloid specific PET radiotracers, F-18 florbetapir and C-1 PiB is still currently under investigation. Although bone imaging radiotracers (Tc-99m DPD and Tc-99m PYP) are excellent for imaging ATTR cardiac amyloidosis, to the extent they may identify early cardiac involvement in asymptomatic familial ATTR disease when other imaging techniques are negative, they are poorly, or not at all, taken up by the myocardium in individuals with AL cardiac amyloidosis. Thus rather than seeking a gold standard for diagnosing this disease, the clinician and imaging specialist is better served by taking a broader, approach, utilizing where appropriate, multiple modalities while considering the clinical picture and the appropriate timing of cardiac biopsy. A diagnostic algorithm to evaluate patients with suspected cardiac amyloidosis is shown in Figure 10.
Figure 10. A Proposed diagnostic algorithm for the evaluation of patients with suspected cardiac amyloidosis.
Approach to the patient described above
The echocardiogram was repeated and showed an echogenic granular myocardium with moderate concentric left ventricular hypertrophy (14 mm), ejection fraction of 60%, a restrictive filling pattern, with an early diastolic mitral tissue Doppler velocity, (e’) of 0.06 cm/sec (Supplemental Video 1) . There was no aortic stenosis. Both atria were enlarged and there was moderate mitral valve regurgitation. Longitudinal strain imaging by speckle tracking revealed a typical pattern of apical sparing, with severe basal longitudinal dysfunction and depressed global longitudinal strain (Figure 4). A bone marrow biopsy was repeated and it showed a moderately hypocellular marrow (80% fat), with approximately 10% plasma cells. Immunoperoxidase staining revealed monotypic CD138 positive plasma cells positive for immunoglobulin lambda light chains and IgG heavy chain. There was a corresponding IgG lambda paraprotein in the serum, detected by immunofixation. Although this hematologic picture strongly suggested a diagnosis of AL amyloidosis, the clinical picture of isolated cardiac amyloidosis in an older male was more akin to the presentation of wild-type ATTR amyloidosis. Hence, a Tc-99m PYP scintigraphy was performed and strongly positive, (Figure 8A) suggesting a diagnosis of ATTR amyloidosis. Endomyocardial biopsy was then performed and documented an extensive amyloid deposition along with moderate myocyte hypertrophy. Immunofluorescence staining of the amyloid deposits, performed on a fresh sample of myocardium, was strongly positive for transthyretin and negative for IgG, IgM, IgA, kappa light chain, lambda light chain, and protein A. Because of the unusual clinical picture, proteomics analysis was performed on the biopsy samples which confirmed transthyretin-derived amyloidosis. ATTR genotyping showed no evidence of a mutant protein, thereby confirming a diagnosis of senile systemic amyloidosis.
In summary, the plasma cell dyscrasia in this patient, although usually highly suggestive of AL amyloidosis when associated with a positive biopsy for amyloid and an abnormal free light chain ratio, was an unrelated phenomenon and represents a MGUS, which may be found in up to 5% of the elderly population. The clinical impression that the disease was behaving more like senile systemic amyloidosis led to performance of the PYP scan, which by strengthening the clinical impression in the face of conflicting hematologic data, led to the ultimate performance of definitive molecular analysis of biopsy tissue, confirming ATTR and there by sparing the patient inappropriate chemotherapy.
Supplementary Material
Acknowledgments
We are extremely grateful to Dr. Raymond Y. Kwong for his review and comments on the CMR section of the paper.
Sources of Funding
Dr. Dorbala was supported by NIH-NHLBI K23HL092299, the Amyloidosis foundation and the American Society of Nuclear Cardiology Foundation.
Dr. Falk is supported in part by the Demarest Lloyd, Jr. Foundation.
Footnotes
Disclosures
Dr. Rodney H. Falk: None
Dr. Candida C. Quarta: None.
Dr. Dorbala: Research grant - Astellas Pharma
References
- 1.Rahman JE, Helou EF, Gelzer-Bell R, Thompson RE, Kuo C, Rodriguez ER, Hare JM, Baughman KL, Kasper EK. Noninvasive diagnosis of biopsy-proven cardiac amyloidosis. J Am Coll Cardiol. 2004;43:410–415. doi: 10.1016/j.jacc.2003.08.043. [DOI] [PubMed] [Google Scholar]
- 2.Murtagh B, Hammill SC, Gertz MA, Kyle RA, Tajik AJ, Grogan M. Electrocardiographic findings in primary systemic amyloidosis and biopsy-proven cardiac involvement. Am J Cardiol. 2005;95:535–537. doi: 10.1016/j.amjcard.2004.10.028. [DOI] [PubMed] [Google Scholar]
- 3.Rapezzi C, Merlini G, Quarta CC, Riva L, Longhi S, Leone O, Salvi F, Ciliberti P, Pastorelli F, Biagini E, Coccolo F, Cooke RM, Bacchi-Reggiani L, Sangiorgi D, Ferlini A, Cavo M, Zamagni E, Fonte ML, Palladini G, Salinaro F, Musca F, Obici L, Branzi A, Perlini S. Systemic cardiac amyloidoses: Disease profiles and clinical courses of the 3 main types. Circulation. 2009;120:1203–1212. doi: 10.1161/CIRCULATIONAHA.108.843334. [DOI] [PubMed] [Google Scholar]
- 4.Koyama J, Ray-Sequin PA, Falk RH. Longitudinal myocardial function assessed by tissue velocity, strain, and strain rate tissue doppler echocardiography in patients with al (primary) cardiac amyloidosis. Circulation. 2003;107:2446–2452. doi: 10.1161/01.CIR.0000068313.67758.4F. [DOI] [PubMed] [Google Scholar]
- 5.Cappelli F, Porciani MC, Bergesio F, Perlini S, Attana P, Moggi Pignone A, Salinaro F, Musca F, Padeletti L, Perfetto F. Right ventricular function in al amyloidosis: Characteristics and prognostic implication. Eur Heart J Cardiovasc Imaging. 2012;13:416–422. doi: 10.1093/ejechocard/jer289. [DOI] [PubMed] [Google Scholar]
- 6.Bellavia D, Pellikka PA, Dispenzieri A, Scott CG, Al-Zahrani GB, Grogan M, Pitrolo F, Oh JK, Miller FA., Jr Comparison of right ventricular longitudinal strain imaging, tricuspid annular plane systolic excursion, and cardiac biomarkers for early diagnosis of cardiac involvement and risk stratification in primary systematic (al) amyloidosis: A 5-year cohort study. Eur Heart J Cardiovasc Imaging. 2012;13:680–689. doi: 10.1093/ehjci/jes009. [DOI] [PubMed] [Google Scholar]
- 7.Phelan D, Collier P, Thavendiranathan P, Popovic ZB, Hanna M, Plana JC, Marwick TH, Thomas JD. Relative apical sparing of longitudinal strain using two-dimensional speckle-tracking echocardiography is both sensitive and specific for the diagnosis of cardiac amyloidosis. Heart. 2012;98:1442–1448. doi: 10.1136/heartjnl-2012-302353. [DOI] [PubMed] [Google Scholar]
- 8.Baccouche H, Maunz M, Beck T, Gaa E, Banzhaf M, Knayer U, Fogarassy P, Beyer M. Differentiating cardiac amyloidosis and hypertrophic cardiomyopathy by use of three-dimensional speckle tracking echocardiography. Echocardiography. 2012;29:668–677. doi: 10.1111/j.1540-8175.2012.01680.x. [DOI] [PubMed] [Google Scholar]
- 9.Liu D, Hu K, Niemann M, Herrmann S, Cikes M, Stork S, Gaudron PD, Knop S, Ertl G, Bijnens B, Weidemann F. Effect of combined systolic and diastolic functional parameter assessment for differentiation of cardiac amyloidosis from other causes of concentric left ventricular hypertrophy. Circ Cardiovasc Imaging. 2013;6:1066–1072. doi: 10.1161/CIRCIMAGING.113.000683. [DOI] [PubMed] [Google Scholar]
- 10.Buss SJ, Emami M, Mereles D, Korosoglou G, Kristen AV, Voss A, Schellberg D, Zugck C, Galuschky C, Giannitsis E, Hegenbart U, Ho AD, Katus HA, Schonland SO, Hardt SE. Longitudinal left ventricular function for prediction of survival in systemic light-chain amyloidosis: Incremental value compared with clinical and biochemical markers. J Am Coll Cardiol. 2012;60:1067–1076. doi: 10.1016/j.jacc.2012.04.043. [DOI] [PubMed] [Google Scholar]
- 11.Koyama J, Falk RH. Prognostic significance of strain doppler imaging in light-chain amyloidosis. JACC Cardiovasc Imaging. 2010;3:333–342. doi: 10.1016/j.jcmg.2009.11.013. [DOI] [PubMed] [Google Scholar]
- 12.Bellavia D, Pellikka PA, Al-Zahrani GB, Abraham TP, Dispenzieri A, Miyazaki C, Lacy M, Scott CG, Oh JK, Miller FA., Jr Independent predictors of survival in primary systemic (al) amyloidosis, including cardiac biomarkers and left ventricular strain imaging: An observational cohort study. J Am Soc Echocardiogr. 2010;23:643–652. doi: 10.1016/j.echo.2010.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Modesto KM, Dispenzieri A, Cauduro SA, Lacy M, Khandheria BK, Pellikka PA, Belohlavek M, Seward JB, Kyle R, Tajik AJ, Gertz M, Abraham TP. Left atrial myopathy in cardiac amyloidosis: Implications of novel echocardiographic techniques. Eur Heart J. 2005;26:173–179. doi: 10.1093/eurheartj/ehi040. [DOI] [PubMed] [Google Scholar]
- 14.Maceira AM, Joshi J, Prasad SK, Moon JC, Perugini E, Harding I, Sheppard MN, Poole-Wilson PA, Hawkins PN, Pennell DJ. Cardiovascular magnetic resonance in cardiac amyloidosis. Circulation. 2005;111:186–193. doi: 10.1161/01.CIR.0000152819.97857.9D. [DOI] [PubMed] [Google Scholar]
- 15.Simonetti OP, Kim RJ, Fieno DS, Hillenbrand HB, Wu E, Bundy JM, Finn JP, Judd RM. An improved mr imaging technique for the visualization of myocardial infarction. Radiology. 2001;218:215–223. doi: 10.1148/radiology.218.1.r01ja50215. [DOI] [PubMed] [Google Scholar]
- 16.Austin BA, Tang WH, Rodriguez ER, Tan C, Flamm SD, Taylor DO, Starling RC, Desai MY. Delayed hyper-enhancement magnetic resonance imaging provides incremental diagnostic and prognostic utility in suspected cardiac amyloidosis. JACC Cardiovasc Imaging. 2009;2:1369–1377. doi: 10.1016/j.jcmg.2009.08.008. [DOI] [PubMed] [Google Scholar]
- 17.Banypersad SM, Sado DM, Flett AS, Gibbs SD, Pinney JH, Maestrini V, Cox AT, Fontana M, Whelan CJ, Wechalekar AD, Hawkins PN, Moon JC. Quantification of myocardial extracellular volume fraction in systemic al amyloidosis: An equilibrium contrast cardiovascular magnetic resonance study. Circ Cardiovasc Imaging. 2013;6:34–39. doi: 10.1161/CIRCIMAGING.112.978627. [DOI] [PubMed] [Google Scholar]
- 18.Maceira AM, Prasad SK, Hawkins PN, Roughton M, Pennell DJ. Cardiovascular magnetic resonance and prognosis in cardiac amyloidosis. J Cardiovasc Magn Reson. 2008;10:54. doi: 10.1186/1532-429X-10-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mongeon FP, Jerosch-Herold M, Coelho-Filho OR, Blankstein R, Falk RH, Kwong RY. Quantification of extracellular matrix expansion by cmr in infiltrative heart disease. JACC Cardiovasc Imaging. 2012;5:897–907. doi: 10.1016/j.jcmg.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Syed IS, Glockner JF, Feng D, Araoz PA, Martinez MW, Edwards WD, Gertz MA, Dispenzieri A, Oh JK, Bellavia D, Tajik AJ, Grogan M. Role of cardiac magnetic resonance imaging in the detection of cardiac amyloidosis. JACC Cardiovasc Imaging. 2010;3:155–164. doi: 10.1016/j.jcmg.2009.09.023. [DOI] [PubMed] [Google Scholar]
- 21.Vogelsberg H, Mahrholdt H, Deluigi CC, Yilmaz A, Kispert EM, Greulich S, Klingel K, Kandolf R, Sechtem U. Cardiovascular magnetic resonance in clinically suspected cardiac amyloidosis: Noninvasive imaging compared to endomyocardial biopsy. J Am Coll Cardiol. 2008;51:1022–1030. doi: 10.1016/j.jacc.2007.10.049. [DOI] [PubMed] [Google Scholar]
- 22.Wassmuth R, Abdel-Aty H, Bohl S, Schulz-Menger J. Prognostic impact of t2-weighted cmr imaging for cardiac amyloidosis. Eur Radiol. 2011;21:1643–1650. doi: 10.1007/s00330-011-2109-3. [DOI] [PubMed] [Google Scholar]
- 23.Dungu JN, Valencia O, Pinney JH, Gibbs SD, Rowczenio D, Gilbertson JA, Lachmann HJ, Wechalekar A, Gillmore JD, Whelan CJ, Hawkins PN, Anderson LJ. Cmr-based differentiation of al and attr cardiac amyloidosis. JACC Cardiovasc Imaging. 2014;7:133–142. doi: 10.1016/j.jcmg.2013.08.015. [DOI] [PubMed] [Google Scholar]
- 24.White JA, Kim HW, Shah D, Fine N, Kim KY, Wendell DC, Al-Jaroudi W, Parker M, Patel M, Gwadry-Sridhar F, Judd RM, Kim RJ. Cmr imaging with rapid visual t1 assessment predicts mortality in patients suspected of cardiac amyloidosis. JACC Cardiovasc Imaging. 2014;7:143–156. doi: 10.1016/j.jcmg.2013.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karamitsos TD, Piechnik SK, Banypersad SM, Fontana M, Ntusi NB, Ferreira VM, Whelan CJ, Myerson SG, Robson MD, Hawkins PN, Neubauer S, Moon JC. Noncontrast t1 mapping for the diagnosis of cardiac amyloidosis. JACC Cardiovasc Imaging. 2013;6:488–497. doi: 10.1016/j.jcmg.2012.11.013. [DOI] [PubMed] [Google Scholar]
- 26.Fontana M, Banypersad SM, Treibel TA, Maestrini V, Sado DM, White SK, Pica S, Castelletti S, Piechnik SK, Robson MD, Gilbertson JA, Rowczenio D, Hutt DF, Lachmann HJ, Wechalekar AD, Whelan CJ, Gillmore JD, Hawkins PN, Moon JC. Native t1 mapping in transthyretin amyloidosis. JACC Cardiovasc Imaging. 2014;7:157–165. doi: 10.1016/j.jcmg.2013.10.008. [DOI] [PubMed] [Google Scholar]
- 27.Hawkins PN, Lavender JP, Pepys MB. Evaluation of systemic amyloidosis by scintigraphy with 123i-labeled serum amyloid p component. N Engl J Med. 1990;323:508–513. doi: 10.1056/NEJM199008233230803. [DOI] [PubMed] [Google Scholar]
- 28.Perugini E, Guidalotti PL, Salvi F, Cooke RM, Pettinato C, Riva L, Leone O, Farsad M, Ciliberti P, Bacchi-Reggiani L, Fallani F, Branzi A, Rapezzi C. Noninvasive etiologic diagnosis of cardiac amyloidosis using 99mtc-3,3-diphosphono-1,2-propanodicarboxylic acid scintigraphy. J Am Coll Cardiol. 2005;46:1076–1084. doi: 10.1016/j.jacc.2005.05.073. [DOI] [PubMed] [Google Scholar]
- 29.Rapezzi C, Guidalotti P, Salvi F, Riva L, Perugini E. Usefulness of 99mtc-dpd scintigraphy in cardiac amyloidosis. J Am Coll Cardiol. 2008;51:1509–1510. doi: 10.1016/j.jacc.2007.12.038. [DOI] [PubMed] [Google Scholar]
- 30.Bokhari S, Castano A, Pozniakoff T, Deslisle S, Latif F, Maurer MS. (99m)tc-pyrophosphate scintigraphy for differentiating light-chain cardiac amyloidosis from the transthyretin-related familial and senile cardiac amyloidoses. Circ Cardiovasc Imaging. 2013;6:195–201. doi: 10.1161/CIRCIMAGING.112.000132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Delahaye N, Dinanian S, Slama MS, Mzabi H, Samuel D, Adams D, Merlet P, Le Guludec D. Cardiac sympathetic denervation in familial amyloid polyneuropathy assessed by iodine-123 metaiodobenzylguanidine scintigraphy and heart rate variability. Eur J Nucl Med. 1999;26:416–424. doi: 10.1007/s002590050406. [DOI] [PubMed] [Google Scholar]
- 32.Tanaka M, Hongo M, Kinoshita O, Takabayashi Y, Fujii T, Yazaki Y, Isobe M, Sekiguchi M. Iodine-123 metaiodobenzylguanidine scintigraphic assessment of myocardial sympathetic innervation in patients with familial amyloid polyneuropathy. J Am Coll Cardiol. 1997;29:168–174. doi: 10.1016/s0735-1097(96)00438-x. [DOI] [PubMed] [Google Scholar]
- 33.Coutinho MC, Cortez-Dias N, Cantinho G, Conceicao I, Oliveira A, Bordalo e Sa A, Goncalves S, Almeida AG, de Carvalho M, Diogo AN. Reduced myocardial 123-iodine metaiodobenzylguanidine uptake: A prognostic marker in familial amyloid polyneuropathy. Circ Cardiovasc Imaging. 2013;6:627–636. doi: 10.1161/CIRCIMAGING.112.000367. [DOI] [PubMed] [Google Scholar]
- 34.Antoni G, Lubberink M, Estrada S, Axelsson J, Carlson K, Lindsjo L, Kero T, Langstrom B, Granstam SO, Rosengren S, Vedin O, Wassberg C, Wikstrom G, Westermark P, Sorensen J. In vivo visualization of amyloid deposits in the heart with 11c-pib and pet. J Nucl Med. 2013;54:213–220. doi: 10.2967/jnumed.111.102053. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.