

Abstract
Because the kidney allograft has the potential to function as an in-vivo flowcytometer and facilitate the access of immune cells and kidney parenchymal cells in to the urinary space, we hypothesized that mRNA profiling of urinary cells offers a noninvasive means of assessing the kidney allograft status. We overcame the inherent challenges of urinary cell mRNA profiling by developing pre-amplification protocols to compensate for low RNA yield from urinary cells and by developing robust protocols for absolute quantification mRNAs using RT-PCR assays. Armed with these tools, we undertook first single-center studies urinary cell mRNA profiling and then embarked on the multicenter Clinical Trials in Organ Transplantation-04 study of kidney transplant recipients. We report here our discovery and validation of diagnostic and prognostic biomarkers of acute cellular rejection and of interstitial fibrosis and tubular atrophy (IF/TA). Our urinary cell mRNA profiling studies, in addition to demonstrating the feasibility of accurate diagnosis of acute cellular rejection and IF/TA in the kidney allograft, advance mechanistic and potentially targetable biomarkers. Interventional trials, guided by urinary cell mRNA profiles, may lead to personalized immunosuppression in recipients of kidney allografts.
Keywords: Kidney transplantation, Rejection, Gene expression, Polymerase chain reaction
Introduction
The number of individuals being treated for end stage kidney disease (ESRD) continues to grow worldwide [1–3]. For those afflicted with ESRD, kidney transplantation results in superior survival compared to maintenance dialysis [4, 5]. The adjusted first-year all-cause mortality rate for the year 2010 was 268.8 per 1000 patient years at risk for patients managed with hemodialysis, 121.4 for patients treated with peritoneal dialysis and only 54.4 for recipients of kidney allografts [1]. The salutary effect of kidney transplantation however is realized by very few since because of the shortage of organs for transplantation. In the US alone, each year, approximately 100,000 patients compete for the 11,000 deceased donor kidneys available for transplantation [6].
There are multiple causes for the organ shortage crisis and the growth of the transplant wait list [7]. It is well documented that acute rejection (AR) is a major contributor to allograft failure and a significant contributor to the escalating wait list is the return of patients with a failed graft to the list [8–12]. Reducing such graft failures would help mitigate the existing crisis in organ availability.
We have focused our research efforts on reducing the impact of allograft failure. We reasoned that better diagnosis of acute cellular rejection (ACR), the most common type of AR, and of interstitial fibrosis and tubular atrophy (IF/TA) is an essential first step. We review here our studies towards the development and validation of noninvasively ascertainable diagnostic and prognostic biomarkers of kidney allograft status. We provide first an overview of AR and chronic rejection, and follow this précis with our findings from interrogating AR and chronic rejection with the use of urinary cell mRNA profiling.
Biology of Immune Rejection
Kidney allograft rejection is defined as functional and structural deterioration due to an active immune response expressed by the recipient and directed at the transplanted organ [13]. Alloreactivity is primarily but not exclusively directed at the proteins encoded by genes located within the donor’s major histocompatibility complex region and involves a highly coordinated action of multiple cell types and mediators with donor antigen-reactive lymphocytes being the principal drivers of the immune repertory [14–16].
Two distinct but not mutually exclusive pathways, T-lymphocyte- based pathway and B-lymphocyte-based pathway contributes to immune rejection of the transplanted organ. T cell-mediated rejection is the commonest type of AR [15, 17–19]. In early 1970s, donor-specific and cytolytically active T lymphocytes were identified within the rejecting experimental or human allografts [20, 21]. These cytolytically active cells kill their target by inducing apoptosis/necrosis via the Fas-Fas ligand pathway and the granule exocytosis pathway in which perforin and granzyme B collaborate [22, 23]. Experiments in a number of laboratories including ours have shown intrarenal expression of mRNA encoding proteins involved in both major lytic pathways, and the differential expression of these molecular executors of cytotoxicity during AR or chronic rejection of the human kidney allograft [24–27].
Robust data exist that antibodies contribute to the three major types of immune rejection; hyperacute, acute or chronic [28–33]. The clinical introduction of highly sensitive assays to detect circulating antibodies have improved our appreciation of the role of antibodies in the pathogenesis of AR–a mechanism recognized as early as the 70s with the use of high-sensitive antibody-dependent cell-mediated cytotoxicity assays in which recipient’s sera are tested against 51 chromium labeled donor cells [34]. It is important to recognize however that dichotomization of rejection in to either cellular or antibody based might be simplistic and in reality both arms of immunity contribute to the rejection, albeit with one or the other arm appearing dominant based on the tools used to interrogate the biology of rejection. Indeed, we recently found that a third of the patients diagnosed as acute antibody mediated rejection by the current Banff criteria also fulfilled the criteria for ACR, and that the presence of concomitant ACR was an independent risk factor for kidney allograft failure following biopsy confirmed diagnosis of complement component 4d (C4d) positive acute antibody mediated rejection [35].
Although AR has garnered much attention, chronic rejection/chronic allograft nephropathy (CAN) is the commonest feature of chronically failing allografts [36, 37]. Interstitial fibrosis and tubular atrophy (IF/TA) are histologic features of chronic rejection and likely to be the final common pathway for allograft failure [38–41]. It is highly likely that alloantigen driven mechanisms as well as non-immune mechanisms contribute to the pathogenesis of IF/TA [39, 42–49]. Among the several non-immune mechanisms implicated in chronic allograft injury, the calcineurin inhibitors are prime suspects since cyclosporine as well as tacrolimus induces TGF-β1 hyperexpression in a number of cell types including T-cells and renal tubular cells [50–56]. As TGF-β1 is a potent fibrogenic cytokine and is involved in repair following tissue injury, a TGF-β1-centered hypothesis for integrating immune and non-immune events in the pathogenesis of chronic rejection/CAN (manifested by IF/TA) appears biologically plausible (Figure 1) [57]. Experimental support for this formulation includes: (i) increased intragraft expression of TGF-β1 mRNA and protein in grafts manifesting IF/TA [57–59]; (ii) increased expression of microRNA-21 capable of targeting Smad-7, a negative regulator of TGF-β1-signaling, in grafts manifesting IF/TA [60]; (iii) increased expression of several proteins, induced by TGF-β1 and involved in fibrosis (e.g., vimentin) in kidney grafts showing IF/TA [61, 62]; and (iv) the ability of calcineurin inhibitors to induce TGF-β1 [50–56].
Figure 1. A TGF-beta1- centric hypothesis for chronic rejection/chronic allograft nephropathy/chronic allograft damage.

In this formulation, both immune and nonimmune factors are responsible for the allograft damage and the attendant repair, and the relative contributions of immune and nonimmune forces vary based on genetic determinants and clinical interventions. In this TGF-β-centric hypothesis, intrarenal over-expression of TGF-β represents a critical and self-perpetuating pathogenetic event for the progressive damage to the kidney allograft and attendant decline in graft function. A therapeutic implication of this conceptualization is that TGF-beta1 blockade may be of benefit in minimizing kidney allograft damage. However, TGF-beta1 is also a potent immunosuppressant and likely contributes to allograft graft protection and/or tolerance and TGF-beta blockade could lead to allograft rejection. Selective blockade (i.e., blocking the fibrogenic arm but not the immunoregulatory arm) is desirable but remains an unmet challenge.
Our TGF-β1 centric hypothesis has received support from a number of interventional studies including the recent findings in a preclinical model. In the male Fischer rat to male Lewis rat kidney transplants, neutralizing activities of TGF-β using an anti-TGF-β antibody successfully reduced the severity of chronic rejection of kidney allografts. Compared with a control antibody-treated group, administration of anti-TGF-β monoclonal antibody was associated with a significant reduction of proteinuria, serum creatinine, and prevented decline of creatinine clearance. Moreover there was reduction of the severity of cellular infiltration, tubular injury, and graft tissue fibrosis as well as the expression of fibrosis-associated genes in renal allografts [63].
Interrogation of Immune Rejection by Urinary Cell mRNA profiling
Acute rejection is diagnosed using the invasive needle core biopsy procedure. While the procedure has become safer over the years, challenges and complications still exist including sampling error, inter-observer variability, patient being on anticoagulant therapy, bleeding, arteriovenous fistula, graft loss, and even death [64–66]. The financial costs of the procedure are substantial as well and reported to average about $3,000 [67]. With an estimated incidence of 0.4 biopsies/patient during the first year of transplantation, about 7,000 biopsies at a cost of $21 million are estimated to have been done during the first year of transplantation in the 17,671 patients who received their kidney grafts in 2011 [1, 68].
Development of urinary cell mRNA profiling as a noninvasive predictive and diagnostic tool
Because ACR is distinguished by interstitial inflammation and tubulitis and given that graft infiltrating cells gain entry into the kidney tubular space, we reasoned that the allograft may function as an “in-vivo flow cytometer” and sort cells involved in rejection into the urine (Figure 2) [69]. We therefore hypothesized that mRNA profiling of urinary cells could provide a noninvasive means of diagnosing ACR. We developed quantitative polymerase chain reaction (PCR) assays and mRNA profiling protocols and demonstrated in a series of single center studies that urinary cell levels of mRNA encoding proteins implicated in allograft rejection are predictive and/or diagnostic of ACR in human renal allografts [70–75]. Our original observations and those of others [76] served as the stimuli for the Clinical Trials in Organ Transplantation-04 (CTOT-04) multicenter study.
Figure 2. Conceptualization of the kidney allograft as an in-vivo flow cytometer.
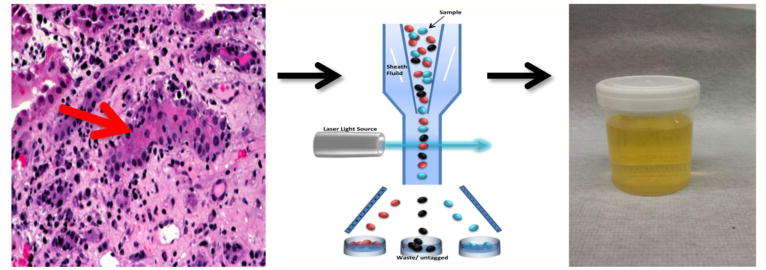
Interstitial inflammation and tubilitis are the histological hallmarks of acute cellular rejection in the kidney allograft (left panel). Since acute cellular rejection involves the exit of T-lymphocytes from the intravascular space and gain of entry into the tubular space, the kidney allograft could be considered to function as an in-vivo flow cytometer (middle panel) facilitating the entry of graft destructive/protective T-lymphocytes, other immune cells and graft parenchymal cells into the urinary space and become available for gene expression profiling.
Weill Cornell Studies of Urinary Cell mRNA Profiling
We developed highly reproducible methods for urinary cell mRNA profiling [77, 78] and quantification of mRNA abundance [79] and examined whether levels of mRNA encoding cytotoxic attack molecules – perforin and granzyme B – are informative of human kidney allograft status. With the use of competitive quantitative PCR assays and gene specific novel DNA competitors designed and developed in our laboratory we found that urinary cell levels of mRNA for perforin predicted ACR with a sensitivity of 83% and a specificity of 83% while granzyme B mRNA levels predicted ACR with a sensitivity of 79% and a specificity of 77% [70].
In our subsequent studies, we modified the real time quantitative PCR assay to quantify absolute levels of mRNA [80]. We developed a “universal” standard curve (i.e., a single standard curve for multiple mRNAs of interest) using a synthetic amplicon for absolute quantification of transcripts in the urinary cells. With the use of real time quantitative PCR assays, we found that not only levels of perforin and granzyme B are higher in urinary cells from patients with AR compared to those without AR but also the levels of serine proteinase inhibitor-9 (PI-9), an endogenous antagonist of granzyme B [72].
Intraepithelial homing of T cells is contingent upon the cell surface expression of CD103 protein [81, 82]. Since acute cellular rejection is exemplified by tubulitis with intraepithelial homing of alloreactive T cells [83], we reasoned that urinary cell levels of CD103 would be diagnostic of ACR in the kidney allograft. This hypothesis was validated by the finding that urinary cell levels of CD103 mRNA are higher in patients with ACR compared to those without ACR [71].
Chemokines and their receptors play a primary role in the trafficking of cells to inflammatory sites [84]. We hypothesized that urinary cell level of IP-10 and its receptor CXCR3 would help capture intragraft inflammation. Our research hypothesis was also based on elegant pre-clinical experimental studies implicating IP-10 and CXCR3 in inflammatory diseases including allograft rejection [85, 86]. We found that AR is indeed predicted with a sensitivity of 100% and a specificity of 78% using urinary cell levels of IP-10 mRNA, and with a sensitivity of 63% and a specificity of 83% using urinary cell levels of CXCR3 mRNA [73].
In view of the dominant role of regulatory T cells (Treg) cells in the maintenance of self-tolerance [87], and in view of the suppressive role of Treg in experimental models of transplantation tolerance [88, 89], we reasoned that measurement of mRNA for FoxP3, a specification factor of Treg, in urinary cells might provide insight into the outcome of an episode of AR. Due to low abundance of RNA present in urinary cells, we further modified the standard quantitative real time (RT)-PCR assay to a two-step PCR assay involving an initial pre-amplification step that enables measurement of multiple mRNA species from very small quantities of total RNA. With the use of pre-amplification enhanced PCR assays, we identified that FoxP3 mRNA levels are not only increased in patients with AR but also predicted reversal of AR with 90% sensitivity and 73% specificity as well as graft failure within 6-months after the incident episode of AR [74].
Co-stimulatory receptors and negative regulatory receptors on immune cells play a critical role in allograft rejection and tolerance. The OX40/OX40L proteins have also been implicated in regulating the emergence of Tregs and modulating their suppressive activity. A novel T-cell inhibitory pathway involves PD-1 and its ligands, PD-L1 and PD-L2 [90, 91]. In studies designed to characterize the expression patterns of co-stimulatory/inhibitory molecules in urinary cells during an episode of ACR, we found that urinary cell mRNA levels of OX40, OX40L, PD-1 but not the levels of PD ligand 1 or PD ligand 2 are significantly higher in the urinary cells from the AR group than the stable group. Within the AR group, levels of mRNA for OX40, OX40L and Foxp3 predicted AR reversal, while OX40 mRNA levels predicted graft failure after AR [75].
Clinical Trials in Organ Transplantation-04 (CTOT-04) study
CTOT-04 is a National Institutes of health (NIH) sponsored prospective observational multicenter study designed to investigate whether urinary cell levels of mRNA encoding for the CD3ε, perforin, granzyme B, PI9, CD103, IP-10 and CXCR3, ascertained at the time of biopsy, are diagnostic of AR and to determine whether mRNA profiles of sequential urine specimens obtained at clinically stable time points predict the development of AR. We measured two additional transcripts (TGF-β1 mRNA and 18S rRNA) for quality control. In CTOT-04, 497 kidney transplant recipients were enrolled at 5 clinical sites and 485 of the 497 provided 4300 urine specimens for urinary cell mRNA profiling. Among the 485 patients, 220 had at least one renal allograft biopsy for clinical (for-cause) or surveillance reason. We designed CTOT-04 as an observational study with each of the 5 participating sites being able to use site-specific immunosuppression and infection protocols with the rationale that urine mRNA profiling results should be generalizable to the kidney transplant population and not be restricted by the immunosuppressive regimen used in any one clinic. We used WCMC protocol to obtain urine samples (~50 ml) sequentially with a pre-specified schedule for collection (post-transplant days 3, 7, 15 and 30 and months 2, 3, 4, 5, 6, 9 and 12, and at the time of any kidney allograft biopsy- before treatment and at 2 weeks after biopsy). Urine cell pellets were prepared at the clinical sites, stored at −80°C and shipped in batches to our Gene Expression Monitoring Core at Weill Cornell.
RNA yield from urinary sediments
We isolated total RNA from the urinary cell pellets using a commercial kit (PureLink™ Micro-to-Midi total RNA purification system, Invitrogen) and measured the RNA quantity (absorbance at 260 nm) by NanoDrop® ND-1000 UV spectrophotometer. We examined whether the amount of RNA isolated from the urinary sediment varied by allograft diagnosis and/or clinical status of the graft recipient. In the CTOT-04 study, urine collection, urine sediment preparation and RNA isolation were all performed using the protocol developed in our laboratory and all RNA quantification was carried out using NanoDrop® ND-1000 UV spectrophotometer. Our data analysis showed that the total amount of RNA isolated from the urine sediment does not vary by allograft diagnosis and/or clinical status. The amount of total RNA isolated from the urine sediment (median and 25th percentile and 75th percentile) was 582 ng (381 ng and 973 ng) in the 43 biopsy-matched urine specimens collected from 34 kidney graft recipients with biopsies showing ACR (ACR Biopsy group), 476 ng (309 ng and 760 ng) in the 163 biopsy-matched urine specimens collected from 126 kidney graft recipients with biopsies showing no rejection (No Rejection Biopsy group), and 534 ng (307 ng and 918 ng)in the 1540 urine specimens from the 202 kidney graft recipients with stable graft function (Stable Function group) (P=0.662, one-way ANOVA).
Reverse transcription to cDNA
Prior to reverse transcription of the RNA to cDNA, and irrespective of the amount of RNA isolated from an individual urine specimen, the RNA isolated from all urine specimens were adjusted to a concentration of 1μg in 100μl volume. This adjustment of RNA to a consistent concentration prior to reverse transcription nullifies any differences in urinary sediment size or RNA yield contributing to differences in mRNA copy number ascertained in the PCR assays. On the other hand, the type of cells (immune cells vs. renal parenchymal cells), the activation status of the cells and their state of differentiation could contribute to the differences in mRNA copy number in the urine of patients with ACR compared to stable graft function group. Transcripts were then quantified by quantitative RT-PCR assay and expressed as copies/μg total RNA. Urine samples were considered quality control passed if in a given sample the 18S rRNA copy number was ≥ 5×107 and TGF-β1 mRNA copy number was ≥1×102 copies/μg RNA. With these parameters, 3559 (83%) of the 4300 urine samples were classified as quality control passed.
Urinary cell mRNA levels diagnostic of ACR
We investigated the hypothesis that urinary cell levels of mRNA are diagnostic of ACR. We tested this by measuring the 8 pre-specified mRNAs in 43 urine samples matched to 43 ACR biopsies from 34 patients and comparing the levels to those found in 163 samples matched to 163 biopsies without rejection (No Rejection Biopsy group, 126 patients) and 1540 samples from the Stable (no Biopsy) group (201 patients with urine specimens). In this aspect of CTOT-04 study, our objective was to examine whether the urine sample would serve as a surrogate for the invasive biopsy procedure. Thus, we used matched urine samples in those who underwent a biopsy. In those who did not undergo a biopsy (Stable [no biopsy] group), all QC-passed samples were analyzed. In accord with the research hypothesis, 18S-normalized levels of mRNA for CD3ε, perforin, granzyme B and IP-10 in urinary cells differed significantly among the ACR, No Rejection Biopsy, and Stable (no biopsy) groups (P<0.0001 for all 4 mRNAs). Pairwise group comparisons showed that the levels of mRNA for CD3ε, perforin, granzyme B and IP-10 in the ACR group were higher than the levels in the No Rejection Biopsy group (P<0.0001 for all 4 mRNAs) and the Stable (no biopsy) group (P<0.0001 for all 4 mRNAs). The 18s rRNA normalized levels of mRNA for CXCR3 (P=0.06), CD103 (P=0.13) and PI-9 (P=0.38) were not associated with ACR, in contrast to our single center studies. The reasons may include analysis of levels of mRNA for association with ACR without 18S rRNA normalization in our earlier single center studies versus analysis of levels of mRNA for association with ACR after 18S rRNA normalization in the CTOT-04 study. This is critical as non-normalized urinary cell levels of mRNA for CD103 (P<0.0001), CXCR3 (P<0.0001) and PI-9 (P=0.002) were all significantly associated with ACR in the CTOT-04 study in accord with our previous studies.
Development of a model for the diagnosis of ACR
We next developed a prediction model comprised of linear combination of mRNAs to accurately diagnose ACR. A 3-gene model of 18S-normalized CD3ε mRNA, 18S-normalized IP-10 mRNA and 18S rRNA (all log10-transformed) emerged as the best-fitting parsimonious model, yielding a diagnostic signature (−6.1487+0.8534 log10(CD3ε/18S)+0.6376 log10(IP-10/18S)+1.6464 log10(18S)). This 3-gene signature had an area under the curve (AUC) of 0.85 (95%CI 0.78 to 0.91, P<0.0001). Our 3-gene formula incorporated 18S-normalized CD3ε mRNA and IP-10 mRNA and 18S rRNA. The rationale for incorporating both 18S-normalized mRNAs measures and 18S rRNA in the equation were (i) normalization ensures that the levels of target mRNAs (e.g., IP-10 mRNA) are altered above and beyond the alteration observed with the reference gene in a given sample; (ii) normalization is a recommended guideline for reporting PCR assay results [92]; and (iii) the formula for reporting the levels of mRNA measured using real time PCR assays incorporates delta CT that is derived by subtracting the CT of reference mRNA from the CT of the target mRNA [93]. In view of these considerations, we normalized each of the mRNAs measured using the reference gene, 18S rRNA, and examined for differences in the normalized measures among the three diagnostic groups: ACR Biopsy group, the No Rejection Biopsy group and the Stable Function group (Figure 2 and Table S6A in [17]). The absolute quantification protocol used in our study enabled the discovery that the levels of 18S rRNA, reported to vary less than most genes recommended as reference genes [94], are also significantly different between the ACR Biopsy group and the No Rejection Biopsy Group or the Stable Function group (Table S6B in [17]). We entered all normalized mRNA measures and the 18S rRNA in a in a full linear logistic regression model and found that following backward elimination ACR is best predicted by the parsimonious model of normalized measures of CD3ε mRNA and IP-10 mRNA and 18S rRNA. In an internal cross validation using bootstrap resampling, we also found that 18S rRNA enters the model 99% of the time. We were cognizant that the incorporation of 18S normalized measures and 18S rRNA in the same equation mostly cancels out normalization of target mRNA with 18S rRNA. Nevertheless, we retained 18S rRNA in the 3-gene formula since normalized mRNA measures were used in our comparisons of three diagnostic groups and because a non-normalized, 2-gene formula of CD3ε mRNA and IP-10 mRNA performed in an identical fashion to the 3-gene formula containing normalized measures of CD3ε mRNA and IP-10 mRNA and 18S rRNA (AUC 0.8447 vs. 0.8454).
Performance characteristics of the model
Using the cutpoint of −1.213, which maximized Youden’s index, our diagnostic signature was 79% sensitive and 78% specific to distinguish ACR biopsies from No Rejection biopsies. Besides the good discrimination, the model was well calibrated as well (Hosmer-Lemeshow χ2=4.84, p=0.77). The inclusion of granzyme B or perforin did not significantly improve the diagnostic signature based on IP-10, CD3ε and 18s. This should not detract from the fact that when considered alone, 18S-normalized levels of granzyme B and perforin are strongly associated with ACR in CTOT-04. Similarly the AUC for the combination of 18S-normalized mRNAs of perforin, IP-10 and 18S rRNA was 0.84 (95% CI: 0.78 to 0.90; P<0.0001), only slightly lower than that of our best diagnostic signature, and this combination discriminated ACR biopsies with a sensitivity of 77% and a specificity of 76%. This signature correlated very highly (r=0.93) with our 3-gene diagnostic signature.
Validation of the model
We used bootstrap validation to internally validate our model. In an initial set of 500 bootstrap samples, 18S-normalized CD3ε, 18S-normalized IP-10 and 18S remained in the final backwards elimination models more often than any of the other 6 mRNA measures. Bootstrap validation of this 3-gene model yielded an optimism-corrected estimate of the AUC of 0.83, an estimate of the expected value of the AUC in independent samples not used to derive the diagnostic signature. Thus, the diagnostic model was generalizable and did not show overfitting.
Urinary Cell 3-Gene Signature Predicts Future Development of ACR
The CTOT-04 study was also designed to determine whether mRNA profiles of sequential urine specimens obtained at clinically stable time points predict (anticipate) the development of AR. This analysis showed that the 3-gene signature predicted the future development of AR as well. Figure 3 shows the average within-person trajectories of the diagnostic signature, looking backwards from the time of biopsy (time=0), for the ACR biopsy group and the No Rejection Biopsy group. The trajectories between the two groups were statistically significant (P<0.0001). In the group of patients in whom the biopsy findings showed no rejection, the score of the diagnostic signature remained relatively flat and well below the diagnostic threshold prior to the biopsy. It did not increase at the time of biopsy or at anytime during the 270 days preceding the biopsy. However in the group of patients in whom the first biopsy specimen showed ACR, the score of the diagnostic signature started to increase around 130 days prior to the biopsy with further marked increase during the 20-day period prior to the biopsy diagnosis of ACR.
Figure 3. Noninvasive Diagnosis of Acute Cellular Rejection: Retrospective Trajectory of Three-Gene Diagnostic Signature as a Function of Time Prior to Biopsy Diagnosis of Acute Cellular Rejection.

The LOESS-smoothed average within-person retrospective trajectory of the diagnostic signature (i.e., the trajectory as a function of the time before biopsy) in urine samples obtained at or before biopsy that passed quality control are shown for the group of 38 kidney graft recipients with first biopsy specimens showing acute cellular rejection (201 urine samples) (Panel A) and the group of 113 patients with specimens showing no rejection (833 urine samples) (Panel B). The diagnostic signature remained relatively flat and well below the 1.213 threshold that was diagnostic of acute cellular rejection during the 270 days before biopsy in the group of patients with biopsy findings showing no rejection (Panel C). There was a significant difference in the trajectories between the acute cellular rejection group and no rejection biopsy group, with a marked increase in the diagnostic signature during the 20-day period before the first specimen showing acute cellular rejection (P<0.001) (Panel D). The y-axis values are diagnostic-signature scores without intrinsic units of measurement and they were calculated from the logistic-regression equation (6.1487 +0.8534 log10[CD3ε/18S] + 0.6376 log10[IP-10/18S] + 1.6464 log10[18S]). Absolute levels of CD3ε mRNA, IP-10 mRNA, and 18S rRNA in the cells from each urine sample were measured by polymerase-chain-reaction assay, with the units of measurement being copies per microgram of total RNA for each mRNA measure and copies (×10−6) per microgram of total RNA for 18S rRNA. The mRNA copy numbers were 18S-normalized by dividing the mRNA copy number by the 18S rRNA copy number in the same sample, and the ratio was log10-transformed. In all panels the colored bands represent the 95% confidence interval for the LOESS-smoothed average.
Additional Attributes of the Urinary Cell 3-Gene signature
The 3-gene diagnostic signature was specific for ACR. The signature had an AUC of 0.78 (95%CI: 0.68 to 0.89, P<0.0001) to distinguish the 43 biopsy confirmed ACR from the 31 biopsy confirmed other rejections (Borderline changes, antibody mediated rejection [AMR] or CAN). We next examined whether the 3-gene signature functions as a response-predictive biomarker. Among 43 ACR biopsies with paired urine samples, 32 had quality control-passed urine samples collect at 14±7 days after the incident ACR biopsy. A comparison of the diagnostic signature values showed a significant decrease from −0.37 at the time of ACR biopsy to −0.86 at 14±7 days post-biopsy (P=0.05). We further compared the decrease in the value for the diagnostic signature according to whether the patient was a “responder”, defined as the follow-up estimated glomerular filtration rate (eGFR) being no higher than it was at the time of the ACR biopsy or a non-responder to the initial anti-rejection therapy. eGFR data were available at the time of biopsy and at the follow-up time for 25 of the 32 samples and among these 25, 21 were responders and the remaining 4 were non-responders based on the above criterion. In the 21 responders, the value for the diagnostic signature decreased significantly from the baseline value of −0.43 to −1.08 at 14±7 days post biopsy (P=0.05); in the small number of non-responders, the decrease in the value from +0.12 to −0.80 was even greater than in the responders (0.92 vs. 0.65), but was not statistically significant (P=0.20). Of note, the non-responders had higher levels of the diagnostic signature at the time of biopsy and two weeks later, and among the 21 responders, 7 did not show a decrease in the value of the diagnostic signature.
The 3-gene signature was diagnostic in patients who underwent for-cause biopsies as well as in those who underwent surveillance biopsies and was similarly diagnostic across transplantation sites. The score of the diagnostic signature was not associated with the Banff grade for ACR. The urine samples from patients who received induction therapy with interleukin-2 receptor antibodies, as compared with those from patients who received T-cell–depleting antibodies, had a higher diagnostic score (P<0.001) especially during the first month after transplantation, but the signature was diagnostic of ACR with either type of induction therapy. The diagnostic signature was not associated with urinary tract infection, blood infection or CMV infection (all P>0.05) but was associated with BK virus infection (P = 0.03). The mean diagnostic score at 4 to 6 months was associated with a decline of 30% or more in renal allograft function from 6 to 12 months (odds ratio, 2.66; 95% CI, 1.45 to 4.87; P = 0.002).
Urinary Cell mRNA Profiles Predictive of Chronic Rejection/IF/TA
We have developed a molecular signature based on urinary cell mRNA profile for the noninvasive diagnosis of kidney allograft fibrosis (IF/TA) [95]. We collected 114 urine samples at the time of an allograft biopsy from 114 kidney allograft recipients; 48 biopsies from 48 patients were classified as IF/TA, and 66 biopsies from 66 patients were classified as normal biopsies. The biopsies classified as IF/TA were clinically indicated biopsies and the normal biopsies were surveillance biopsies. We specifically chose a pristine control group–urine samples from patients with normal biopsy findings in their surveillance biopsies, as the use biopsies with AR, calcineurin toxicity, or BK virus nephropathy as controls, may have compromised the development of a robust biomarker that distinguishes biopsy specimens with IF/TA from biopsy specimens without IF/TA as each of these conditions may be associated with some degree of fibrosis.
The biopsy-matched urine samples were centrifuged and RNA was extracted from the urine cell pellet and reverse-transcribed to cDNA. We designed oligonucleotide primers and fluorogenic probes for the measurement, by preamplification enhanced RT-PCR assay, of absolute levels of 22 mRNAs of genes associated with fibrosis and the endogenous control 18S rRNA. Our urinary cell mRNA panel was designed with due consideration that multiple cell types and an inflammatory milieu contribute to the development of fibrosis and that fibrosis is characterized by the accumulation of extracellular matrix proteins. Our study cohort was portioned in to a discovery cohort and a validation cohort and statistical analysis, in addition to protecting against type I error, involved the use of LOESS (locally weighted scatterplot smoothing) methods to elucidate the potentially non-linear relationship between two variables since it has the advantage of fitting segments of data without pre-specifying a specific, usually linear, global function.
Levels of 12 of the 22 mRNAs (vimentin, HGF, α-SMA, fibronectin 1, perforin, PAI1, TGF-β1, TIMP1, granzyme B, FSP1, CD103, and collagen 1A1) were significantly associated (P<0.05) with the diagnosis of IF/TA. Of the individual mRNAs, vimentin and HGF had the highest AUCs and a multigene prediction model of IF/TA was developed around vimentin because of vimentin’s potential to contribute to the pathogenesis of fibrosis. Interestingly, after controlling for vimentin mRNA level and the quadratic relationship of 18S rRNA level, we found that none of the previously significant predictors but two of the previously non-significant predictors, mRNA for tubular transporter NKCC2 and tubular epithelial cell adhesion molecule E-cadherin, became statistically significant. The composite score based on the 4-gene signature of vimentin, NKCC2, E-cadherin and 18S rRNA was highly associated with the diagnosis of IF/TA. The AUC for 4-gene signature was 0.95 (95%CI: 0.90 to 0.99, P<0.0001) and with the use of the optimal cutpoint of 4.5, the composite score predicted IF/TA with a sensitivity of 93.8% (95%CI: 85.4 to 99.9%) and a specificity of 84.1% (95%CI: 73.3 to 94.9%).
We then validated the final diagnostic equation predicting IF/TA in an independent validation set of 38 kidney graft recipients, 16 with biopsy-proven IF/TA and 22 with normal allograft biopsy results. This 4-gene signature was accurately diagnostic of IF/TA; the AUC for the diagnosis of fibrosis in the independent validation set was 0.89 (95%CI: 0.78 to 0.99, P<0.0001). At the composite score cutpoint of 4.5, derived in the discovery set, IF/TA was diagnosed in the validation set with a sensitivity of 87.5% (95%CI: 71.3 to 99.9%) and a specificity of 77.3% (95%CI: 59.8 to 94.8%). We examined the fit of the prediction model by dividing the discovery and validation sets into sextile of the composite score. Based on the Hosmer-Lemeshow test, the fit between the observed and the predicted number of subjects with IF/TA in each of the sextile was excellent (P=0.69) in the discovery set. For the validation set, the P-value was 0.04, suggesting a good fit, given that this set was not involved in the estimation of the model (Figure 4).
Figure 4. Noninvasive Diagnosis of IF/TA in the Kidney Allograft: Discovery and Validation of Urinary Cell Four-Gene Composite Score.

To predict IF/TA in the Discovery set, we calculated a composite score based on a logistic model, from levels of vimentin mRNA, NKCC2 mRNA and E cadherin mRNA and the 18S rRNA (4-gene signature) in urine samples obtained from the 32 kidney graft recipients with biopsy-confirmed IF/TA and 44 subjects with stable graft function and with normal allograft biopsy results. The composite score of the 4-gene signature predicted IF/TA with high accuracy. Panel A shows the predicted probability of IF/TA (Y-axis) as a logistic function of the composite score of the 4-gene signature (X-axis). The blue band represents the 95% confidence interval of the composite score. Panel B shows the receiver-operating-characteristic curve for the diagnosis of IF/TA using the composite score. The diagnostic model had an area under the curve of 0.95 (95%CI: 0.90 to 0.99, P<0.0001). At a cutpoint or 4.5, IF/TA was diagnosed with a specificity of 84.1% (95%CI: 73.3 to 94.9%) and a sensitivity of 93.8% (95%CI: 85.4 to 99.9%) in the discovery set. We used the final prediction equation derived from the discovery set to calculate the predicted probability of IF/TA in the validation set of 38 kidney transplant recipients; 16 with biopsy-confirmed IF/TA and 22 with stable graft function and normal allograft biopsy results. Panel C shows the receiver-operating characteristic curve of the composite score (derived from the equation used in the discovery set) applied to the validation set for the diagnosis of IF/TA. The area under the curve for the diagnosis of IF/TA in the validation set was 0.89 (95%CI: 0.78 to 0.99, P<0.0001). At the original composite score cutpoint of 4.5 derived from the discovery set, IF/TA was diagnosed in the validation set with a specificity of 77.3% (95%CI: 59.8 to 94.8%) and a sensitivity of 87.5% (95%CI: 71.3 to 99.9%). Panel D shows the predicted and observed number of kidney graft recipients with IF/TA for each sextile of the composite score within the discovery and validation sets. Based on the Hosmer-Lemeshow test, the fit between the observed and the predicted number of subjects with IF/TA in each of the sextiles was excellent (P=0.69) in the discovery set (left half of Fig. 4D). For the validation set (right half of Fig. 4D), the P value was 0.04, suggesting a good fit, given that this set was not involved in the estimation of the model.
Concluding Remarks
Our urinary cell mRNA profiling studies have shown considerable robustness in diagnosing AR and IF/TA in the kidney allograft and has the potential to provide mechanistic insights regarding allograft rejection and IF/TA (Figure 5) [69]. It is noteworthy that the multi-center CTOT-04 trial has not only validated the accuracy of urinary cell mRNA profiling for the detection of ACR in single center studies but also the utility of urinary cell mRNA profiles for the prediction of future occurrence of an ACR. It is also noteworthy that the reproducibility of the urinary cell mRNA protocols across laboratories has been examined and the overall, gene expression measurements had a correlation greater than 0.938 for the samples in this independent study [96].
Figure 5. Key Molecular Events During Acute Cellular Rejection of the Kidney Allograft.

The immune repertory contributing to acute cellular rejection involves multiple cell types, cytokines and chemokines and their ligands. T-lymphocytes are the principal effector cells and dendritic cells are the primary antigen presenting cells. In this minimalist model, the chemokine IP-10 and its receptor CXCR3, expressed on T-lymphocytes, facilitate the trafficking of alloreactive T-lymphocytes in to the kidney graft. Intraepithelial homing of the T-lymphocytes is then effected by the physical interaction between CD103 expressed on the T-lymphocytes and E-cadherin displayed on the kidney tubular epithelial cells. The activated/alloreactive T-lymphocytes then employ perforin and granzyme to mediate parenchymal cell damage, which is antagonized in part by the endogenous antagonist proteinase inhibitor-9 (PI-9). Counter-regulatory forces are exerted by FoxP3 expressing regulatory T-lymphocytes (Tregs) and help dampen the anti-allograft response and the ultimate outcome of an episode of acute rejection is determined in part by the balance between the graft destructive cells such as cytotoxic T cells and graft protective Tregs. Although the cells with different functional attributes are depicted here to have distinct pedigrees, transition from one functional type to another (plasticity) is feasible based on the cells versatility and environmental cues. In this T-lymphocyte -centric formulation deterministic of allograft destiny, the antigen-experienced T-lymphocytes provide help to B-lymphocytes and facilitate antibody-mediated rejection. Acute rejection is a precursor of chronic rejection manifested histologically by interstitial fibrosis/tubular atrophy (IF/TA). Acute rejection-associated tissue injury, de-differentiation and repair (epithelial/endothelial-mesenchymal transition) contribute to the pathogenesis of IF/TA and progressive loss of allograft function. The levels of mRNA encoding vimentin, αSMA, E-cadherin and NKCC2 in urinary cells reflect the cellular events associated with IF/TA quantitatively. The mRNAs, colored red in the schematic illustration, have all been detected and quantified using urinary cell mRNA profiling and found to be associated with human kidney allograft status.
Several challenges remain regarding urinary cell mRNA profiling; can it replace the use of renal biopsy in many instances where a for-cause biopsy or a surveillance biopsy is currently performed; can we utilize a standardized urinary gene mRNA profiling protocol as a monitoring tool to prevent the development of overt AR; can we develop a urinary mRNA gene signature that will predict the response to anti-rejection treatment? Should in-vivo immune status of the kidney allograft be reflected accurately by urinary cell mRNA profiles, transition from the current “one-size fits all” immunosuppressive regimen to a personalized immunosuppressive regimen could be realized [97]. Recent technical advances such as massively parallel sequencing should help identify additional candidate biomarkers informative of diagnosis and prognosis of allograft status and inform gene expression studies performed in our laboratory and elsewhere.
Acknowledgments
Adapted from the published Weill Cornell Medical Center single center studies and the published (N Eng J Med 2013) multicenter study
Supported in part by awards from:
the National Institutes of Health to T. Muthukumar (K08-DK087824) and M. Suthanthiran (UO1A163589 PI-A.Shaked, R37-AI051652 PI-M.Suthanthiran),
the National Center for Advancing Translational Sciences at the National Institutes of Health through the Weill Cornell Clinical and Translational Science Center to J.R. Lee (KL2-TR000458),
Clinical and Translational Science Center award from the National Institutes of Health to Weill Cornell Medical College (ULI RR024996) and
the Qatar National Research Foundation to M. Suthanthiran (NPRP 08-503-3-11).
Abbreviations
- AR
Acute rejection
- ACR
Acute cellular rejection
- AMR
Acute antibody mediated rejection
- AUC
Area under the curve
- CAN
Chronic allograft nephropathy
- cDNA
Complementary deoxy ribonucleic acid
- CTOT-04
Clinical Trials in Organ Transplantation-04
- ESRD
End stage kidney disease
- IF/TA
Interstitial fibrosis/tubular atrophy
- LOESS
Locally weighted scatterplot smoothing
- mRNA
messenger ribonucleic acid
- NIH
National Institutes of health
- RT-PCR
Real-time polymerase chain reaction
- TGF-β1
Transforming growth factor beta 1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.U.S. Renal Data System. USRDS 2013 Annual Data Report: Atlas of End-Stage Renal Disease in the United States. National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases; Bethesda, MD: 2013. [Google Scholar]
- 2.Anand S, Bitton A, Gaziano T. The gap between estimated incidence of end-stage renal disease and use of therapy. PLoS One. 2013;8:e72860. doi: 10.1371/journal.pone.0072860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kidney disease statistics for the United States. National Kidney and Urologic Diseases Information Clearinghouse, National Institutes of Health; [Google Scholar]
- 4.Tonelli M, Wiebe N, Knoll G, Bello A, Browne S, Jadhav D, Klarenbach S, Gill J. Systematic review: kidney transplantation compared with dialysis in clinically relevant outcomes. Am J Transplant. 2011;11:2093–2109. doi: 10.1111/j.1600-6143.2011.03686.x. [DOI] [PubMed] [Google Scholar]
- 5.Wolfe RA, Ashby VB, Milford EL, Ojo AO, Ettenger RE, Agodoa LY, Held PJ, Port FK. Comparison of mortality in all patients on dialysis, patients on dialysis awaiting transplantation, and recipients of a first cadaveric transplant. N Engl J Med. 1999;341:1725–1730. doi: 10.1056/NEJM199912023412303. [DOI] [PubMed] [Google Scholar]
- 6.Matas AJ, Smith JM, Skeans MA, Lamb KE, Gustafson SK, Samana CJ, Stewart DE, Snyder JJ, Israni AK, Kasiske BL. OPTN/SRTR 2011 Annual Data Report: kidney. Am J Transplant. 2013;13(Suppl 1):11–46. doi: 10.1111/ajt.12019. [DOI] [PubMed] [Google Scholar]
- 7.Pomfret EA, Sung RS, Allan J, Kinkhabwala M, Melancon JK, Roberts JP. Solving the organ shortage crisis: the 7th annual American Society of Transplant Surgeons’ State-of-the-Art Winter Symposium. Am J Transplant. 2008;8:745–752. doi: 10.1111/j.1600-6143.2007.02146.x. [DOI] [PubMed] [Google Scholar]
- 8.Pallardo Mateu LM, Sancho Calabuig A, Capdevila Plaza L, Franco Esteve A. Acute rejection and late renal transplant failure: risk factors and prognosis. Nephrol Dial Transplant. 2004;19(Suppl 3):iii38–42. doi: 10.1093/ndt/gfh1013. [DOI] [PubMed] [Google Scholar]
- 9.Meier-Kriesche HU, Ojo AO, Hanson JA, Cibrik DM, Punch JD, Leichtman AB, Kaplan B. Increased impact of acute rejection on chronic allograft failure in recent era. Transplantation. 2000;70:1098–1100. doi: 10.1097/00007890-200010150-00018. [DOI] [PubMed] [Google Scholar]
- 10.Cole EH, Johnston O, Rose CL, Gill JS. Impact of acute rejection and new-onset diabetes on long-term transplant graft and patient survival. Clin J Am Soc Nephrol. 2008;3:814–821. doi: 10.2215/CJN.04681107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Faravardeh A, Eickhoff M, Jackson S, Spong R, Kukla A, Issa N, Matas AJ, Ibrahim HN. Predictors of graft failure and death in elderly kidney transplant recipients. Transplantation. 2013;96:1089–1096. doi: 10.1097/TP.0b013e3182a688e5. [DOI] [PubMed] [Google Scholar]
- 12.Shabir S, Halimi JM, Cherukuri A, Ball S, Ferro C, Lipkin G, Benavente D, Gatault P, Baker R, Kiberd B, Borrows R. Predicting 5-Year Risk of Kidney Transplant Failure: A Prediction Instrument Using Data Available at 1 Year Posttransplantation. Am J Kidney Dis. 2013 doi: 10.1053/j.ajkd.2013.10.059. [DOI] [PubMed] [Google Scholar]
- 13.Suthanthiran M. Acute rejection of renal allografts: mechanistic insights and therapeutic options. Kidney Int. 1997;51:1289–1304. doi: 10.1038/ki.1997.176. [DOI] [PubMed] [Google Scholar]
- 14.Suthanthiran M, Strom TB. Renal transplantation. N Engl J Med. 1994;331:365–376. doi: 10.1056/NEJM199408113310606. [DOI] [PubMed] [Google Scholar]
- 15.Nankivell BJ, Alexander SI. Rejection of the kidney allograft. N Engl J Med. 2010;363:1451–1462. doi: 10.1056/NEJMra0902927. [DOI] [PubMed] [Google Scholar]
- 16.Wood KJ, Zaitsu M, Goto R. Cell mediated rejection. Methods Mol Biol. 2013;1034:71–83. doi: 10.1007/978-1-62703-493-7_3. [DOI] [PubMed] [Google Scholar]
- 17.Suthanthiran M, Schwartz JE, Ding R, Abecassis M, Dadhania D, Samstein B, Knechtle SJ, Friedewald J, Becker YT, Sharma VK, Williams NM, Chang CS, Hoang C, Muthukumar T, August P, Keslar KS, Fairchild RL, Hricik DE, Heeger PS, Han L, Liu J, Riggs M, Ikle DN, Bridges ND, Shaked A. Urinary-cell mRNA profile and acute cellular rejection in kidney allografts. N Engl J Med. 2013;369:20–31. doi: 10.1056/NEJMoa1215555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hricik DE, Nickerson P, Formica RN, Poggio ED, Rush D, Newell KA, Goebel J, Gibson IW, Fairchild RL, Riggs M, Spain K, Ikle D, Bridges ND, Heeger PS. Multicenter validation of urinary CXCL9 as a risk-stratifying biomarker for kidney transplant injury. Am J Transplant. 2013;13:2634–2644. doi: 10.1111/ajt.12426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.El Ters M, Grande JP, Keddis MT, Rodrigo E, Chopra B, Dean PG, Stegall MD, Cosio FG. Kidney allograft survival after acute rejection, the value of follow-up biopsies. Am J Transplant. 2013;13:2334–2341. doi: 10.1111/ajt.12370. [DOI] [PubMed] [Google Scholar]
- 20.Strom TB, Tilney NL, Carpenter CB, Busch GJ. Identity and cytotoxic capacity of cells infiltrating renal allografts. N Engl J Med. 1975;292:1257–1263. doi: 10.1056/NEJM197506122922402. [DOI] [PubMed] [Google Scholar]
- 21.Williams LE, Merino GE, Goren G, Najarian JS, Loken MK. Detection of canine kidney allograft rejection with 51Cr-labeled lymphocytes. Radiology. 1975;115:205–206. doi: 10.1148/115.1.205. [DOI] [PubMed] [Google Scholar]
- 22.Shresta S, Pham CT, Thomas DA, Graubert TA, Ley TJ. How do cytotoxic lymphocytes kill their targets? Curr Opin Immunol. 1998;10:581–587. doi: 10.1016/s0952-7915(98)80227-6. [DOI] [PubMed] [Google Scholar]
- 23.Russell JH, Ley TJ. Lymphocyte-mediated cytotoxicity. Annu Rev Immunol. 2002;20:323–370. doi: 10.1146/annurev.immunol.20.100201.131730. [DOI] [PubMed] [Google Scholar]
- 24.Sharma VK, Bologa RM, Li B, Xu GP, Lagman M, Hiscock W, Mouradian J, Wang J, Serur D, Rao VK, Suthanthiran M. Molecular executors of cell death--differential intrarenal expression of Fas ligand, Fas, granzyme B, and perforin during acute and/or chronic rejection of human renal allografts. Transplantation. 1996;62:1860–1866. doi: 10.1097/00007890-199612270-00031. [DOI] [PubMed] [Google Scholar]
- 25.Lipman ML, Stevens AC, Strom TB. Heightened intragraft CTL gene expression in acutely rejecting renal allografts. J Immunol. 1994;152:5120–5127. [PubMed] [Google Scholar]
- 26.Suthanthiran M. Molecular analyses of human renal allografts: differential intragraft gene expression during rejection. Kidney Int Suppl. 1997;58:S15–21. [PubMed] [Google Scholar]
- 27.Strehlau J, Pavlakis M, Lipman M, Maslinski W, Shapiro M, Strom TB. The intragraft gene activation of markers reflecting T-cell-activation and -cytotoxicity analyzed by quantitative RT-PCR in renal transplantation. Clin Nephrol. 1996;46:30–33. [PubMed] [Google Scholar]
- 28.Williams GM, Hume DM, Hudson RP, Jr, Morris PJ, Kano K, Milgrom F. “Hyperacute” renal-homograft rejection in man. N Engl J Med. 1968;279:611–618. doi: 10.1056/NEJM196809192791201. [DOI] [PubMed] [Google Scholar]
- 29.Ahern AT, Artruc SB, DellaPelle P, Cosimi AB, Russell PS, Colvin RB, Fuller TC. Hyperacute rejection of HLA-AB-identical renal allografts associated with B lymphocyte and endothelial reactive antibodies. Transplantation. 1982;33:103–106. doi: 10.1097/00007890-198201000-00023. [DOI] [PubMed] [Google Scholar]
- 30.Takemoto SK, Zeevi A, Feng S, Colvin RB, Jordan S, Kobashigawa J, Kupiec-Weglinski J, Matas A, Montgomery RA, Nickerson P, Platt JL, Rabb H, Thistlethwaite R, Tyan D, Delmonico FL. National conference to assess antibody-mediated rejection in solid organ transplantation. Am J Transplant. 2004;4:1033–1041. doi: 10.1111/j.1600-6143.2004.00500.x. [DOI] [PubMed] [Google Scholar]
- 31.Worthington JE, Martin S, Al-Husseini DM, Dyer PA, Johnson RW. Posttransplantation production of donor HLA-specific antibodies as a predictor of renal transplant outcome. Transplantation. 2003;75:1034–1040. doi: 10.1097/01.TP.0000055833.65192.3B. [DOI] [PubMed] [Google Scholar]
- 32.Hourmant M, Cesbron-Gautier A, Terasaki PI, Mizutani K, Moreau A, Meurette A, Dantal J, Giral M, Blancho G, Cantarovich D, Karam G, Follea G, Soulillou JP, Bignon JD. Frequency and clinical implications of development of donor-specific and non-donor-specific HLA antibodies after kidney transplantation. J Am Soc Nephrol. 2005;16:2804–2812. doi: 10.1681/ASN.2004121130. [DOI] [PubMed] [Google Scholar]
- 33.Terasaki PI, Ozawa M. Predictive value of HLA antibodies and serum creatinine in chronic rejection: results of a 2-year prospective trial. Transplantation. 2005;80:1194–1197. doi: 10.1097/01.tp.0000174338.97313.5a. [DOI] [PubMed] [Google Scholar]
- 34.Gailiunas P, Jr, Suthanthiran M, Busch GJ, Carpenter CB, Garovoy MR. Role of humoral presenitization in human renal transplant rejection. Kidney Int. 1980;17:638–646. doi: 10.1038/ki.1980.75. [DOI] [PubMed] [Google Scholar]
- 35.Matignon M, Muthukumar T, Seshan SV, Suthanthiran M, Hartono C. Concurrent acute cellular rejection is an independent risk factor for renal allograft failure in patients with C4d-positive antibody-mediated rejection. Transplantation. 2012;94:603–611. doi: 10.1097/TP.0b013e31825def05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gaston RS. Our evolving understanding of late kidney allograft failure. Curr Opin Organ Transplant. 2011;16:594–599. doi: 10.1097/MOT.0b013e32834c23a7. [DOI] [PubMed] [Google Scholar]
- 37.Josephson MA. Late Kidney Dysfunction in a Kidney Transplant Recipient. Clin J Am Soc Nephrol. 2013 doi: 10.2215/CJN.07390713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Farris AB, Colvin RB. Renal interstitial fibrosis: mechanisms and evaluation. Curr Opin Nephrol Hypertens. 2012;21:289–300. doi: 10.1097/MNH.0b013e3283521cfa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bosmans JL, Ysebaert DK, Verpooten GA. Chronic allograft nephropathy: what have we learned from protocol biopsies? Transplantation. 2008;85:S38–41. doi: 10.1097/TP.0b013e318169c5d0. [DOI] [PubMed] [Google Scholar]
- 40.Amer H, Griffin MD. Modulating kidney transplant interstitial fibrosis and tubular atrophy: is the RAAS an important target? Kidney Int. 2014;85:240–243. doi: 10.1038/ki.2013.400. [DOI] [PubMed] [Google Scholar]
- 41.Jevnikar AM, Mannon RB. Late kidney allograft loss: what we know about it, and what we can do about it. Clin J Am Soc Nephrol. 2008;3(Suppl 2):S56–67. doi: 10.2215/CJN.03040707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li C, Yang CW. The pathogenesis and treatment of chronic allograft nephropathy. Nat Rev Nephrol. 2009;5:513–519. doi: 10.1038/nrneph.2009.113. [DOI] [PubMed] [Google Scholar]
- 43.Gago M, Cornell LD, Kremers WK, Stegall MD, Cosio FG. Kidney allograft inflammation and fibrosis, causes and consequences. Am J Transplant. 2012;12:1199–1207. doi: 10.1111/j.1600-6143.2011.03911.x. [DOI] [PubMed] [Google Scholar]
- 44.Vitalone MJ, Naesens M, Sigdel T, Li L, Hseih S, Sarwal MM. The dual role of epithelial-to-mesenchymal transition in chronic allograft injury in pediatric renal transplantation. Transplantation. 2011;92:787–795. doi: 10.1097/TP.0b013e31822d092c. [DOI] [PubMed] [Google Scholar]
- 45.Park WD, Griffin MD, Cornell LD, Cosio FG, Stegall MD. Fibrosis with inflammation at one year predicts transplant functional decline. J Am Soc Nephrol. 2010;21:1987–1997. doi: 10.1681/ASN.2010010049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mannon RB, Matas AJ, Grande J, Leduc R, Connett J, Kasiske B, Cecka JM, Gaston RS, Cosio F, Gourishankar S, Halloran PF, Hunsicker L, Rush D. Inflammation in areas of tubular atrophy in kidney allograft biopsies: a potent predictor of allograft failure. Am J Transplant. 2010;10:2066–2073. doi: 10.1111/j.1600-6143.2010.03240.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Heilman RL, Devarapalli Y, Chakkera HA, Mekeel KL, Moss AA, Mulligan DC, Mazur MJ, Hamawi K, Williams JW, Reddy KS. Impact of subclinical inflammation on the development of interstitial fibrosis and tubular atrophy in kidney transplant recipients. Am J Transplant. 2010;10:563–570. doi: 10.1111/j.1600-6143.2009.02966.x. [DOI] [PubMed] [Google Scholar]
- 48.Heilman RL, Chakkera HA, Reddy KS, Colby TV, Moss AA, Williams JW, Mazur MJ, Petrides S, Mulligan DC. Clinical factors associated with graft fibrosis in kidney-transplant recipients on steroid-avoidance immunosuppression. Clin Transplant. 2008;22:309–315. doi: 10.1111/j.1399-0012.2007.00786.x. [DOI] [PubMed] [Google Scholar]
- 49.Gelens MA, Steegh FM, van Hooff JP, van Suylen RJ, Nieman FH, van Heurn LW, Peutz-Kootstra CJ, Christiaans MH. Immunosuppressive regimen and interstitial fibrosis and tubules atrophy at 12 months postrenal transplant. Clin J Am Soc Nephrol. 2012;7:1010–1017. doi: 10.2215/CJN.09030911. [DOI] [PubMed] [Google Scholar]
- 50.Prashar Y, Khanna A, Sehajpal P, Sharma VK, Suthanthiran M. Stimulation of transforming growth factor-beta 1 transcription by cyclosporine. FEBS Lett. 1995;358:109–112. doi: 10.1016/0014-5793(94)01382-b. [DOI] [PubMed] [Google Scholar]
- 51.Ahuja SS, Shrivastav S, Danielpour D, Balow JE, Boumpas DT. Regulation of transforming growth factor-beta 1 and its receptor by cyclosporine in human T lymphocytes. Transplantation. 1995;60:718–723. doi: 10.1097/00007890-199510150-00018. [DOI] [PubMed] [Google Scholar]
- 52.Khanna A, Kapur S, Sharma V, Li B, Suthanthiran M. In vivo hyperexpression of transforming growth factor-beta1 in mice: stimulation by cyclosporine. Transplantation. 1997;63:1037–1039. doi: 10.1097/00007890-199704150-00026. [DOI] [PubMed] [Google Scholar]
- 53.Shin GT, Khanna A, Ding R, Sharma VK, Lagman M, Li B, Suthanthiran M. In vivo expression of transforming growth factor-beta1 in humans: stimulation by cyclosporine. Transplantation. 1998;65:313–318. doi: 10.1097/00007890-199802150-00003. [DOI] [PubMed] [Google Scholar]
- 54.Khanna A, Cairns V, Hosenpud JD. Tacrolimus induces increased expression of transforming growth factor-beta1 in mammalian lymphoid as well as nonlymphoid cells. Transplantation. 1999;67:614–619. doi: 10.1097/00007890-199902270-00021. [DOI] [PubMed] [Google Scholar]
- 55.McMorrow T, Gaffney MM, Slattery C, Campbell E, Ryan MP. Cyclosporine A induced epithelial-mesenchymal transition in human renal proximal tubular epithelial cells. Nephrol Dial Transplant. 2005;20:2215–2225. doi: 10.1093/ndt/gfh967. [DOI] [PubMed] [Google Scholar]
- 56.Slattery C, Ryan MP, McMorrow T. Protein kinase C beta overexpression induces fibrotic effects in human proximal tubular epithelial cells. Int J Biochem Cell Biol. 2008;40:2218–2229. doi: 10.1016/j.biocel.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 57.Sharma VK, Bologa RM, Xu GP, Li B, Mouradian J, Wang J, Serur D, Rao V, Suthanthiran M. Intragraft TGF-beta 1 mRNA: a correlate of interstitial fibrosis and chronic allograft nephropathy. Kidney Int. 1996;49:1297–1303. doi: 10.1038/ki.1996.185. [DOI] [PubMed] [Google Scholar]
- 58.Pribylova-Hribova P, Kotsch K, Lodererova A, Viklicky O, Vitko S, Volk HD, Lacha J. TGF-beta1 mRNA upregulation influences chronic renal allograft dysfunction. Kidney Int. 2006;69:1872–1879. doi: 10.1038/sj.ki.5000328. [DOI] [PubMed] [Google Scholar]
- 59.Nogare AL, Dalpiaz T, Pedroso JA, Montenegro RM, Pegas KL, Veronese FV, Goncalves LF, Manfro RC. Expression of fibrosis-related genes in human renal allografts with interstitial fibrosis and tubular atrophy. J Nephrol. 2013;26:1179–1187. doi: 10.5301/jn.5000274. [DOI] [PubMed] [Google Scholar]
- 60.Ben-Dov IZ, Muthukumar T, Morozov P, Mueller FB, Tuschl T, Suthanthiran M. MicroRNA sequence profiles of human kidney allografts with or without tubulointerstitial fibrosis. Transplantation. 2012;94:1086–1094. doi: 10.1097/TP.0b013e3182751efd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kers J, Xu-Dubois YC, Rondeau E, Claessen N, du MMI, Roelofs JJ, Bemelman FJ, ten Berge IJ, Florquin S. Intragraft tubular vimentin and CD44 expression correlate with long-term renal allograft function and interstitial fibrosis and tubular atrophy. Transplantation. 2010;90:502–509. doi: 10.1097/TP.0b013e3181e86b42. [DOI] [PubMed] [Google Scholar]
- 62.Hertig A, Anglicheau D, Verine J, Pallet N, Touzot M, Ancel PY, Mesnard L, Brousse N, Baugey E, Glotz D, Legendre C, Rondeau E, Xu-Dubois YC. Early epithelial phenotypic changes predict graft fibrosis. J Am Soc Nephrol. 2008;19:1584–1591. doi: 10.1681/ASN.2007101160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Guan Q, Li S, Gao S, Chen H, Nguan CY, Du C. Reduction of chronic rejection of renal allografts by anti-transforming growth factor-beta antibody therapy in a rat model. Am J Physiol Renal Physiol. 2013;305:F199–207. doi: 10.1152/ajprenal.00665.2012. [DOI] [PubMed] [Google Scholar]
- 64.Wilczek HE. Percutaneous needle biopsy of the renal allograft. A clinical safety evaluation of 1129 biopsies. Transplantation. 1990;50:790–797. doi: 10.1097/00007890-199011000-00010. [DOI] [PubMed] [Google Scholar]
- 65.Schwarz A, Gwinner W, Hiss M, Radermacher J, Mengel M, Haller H. Safety and adequacy of renal transplant protocol biopsies. Am J Transplant. 2005;5:1992–1996. doi: 10.1111/j.1600-6143.2005.00988.x. [DOI] [PubMed] [Google Scholar]
- 66.Furness PN, Philpott CM, Chorbadjian MT, Nicholson ML, Bosmans JL, Corthouts BL, Bogers JJ, Schwarz A, Gwinner W, Haller H, Mengel M, Seron D, Moreso F, Canas C. Protocol biopsy of the stable renal transplant: a multicenter study of methods and complication rates. Transplantation. 2003;76:969–973. doi: 10.1097/01.TP.0000082542.99416.11. [DOI] [PubMed] [Google Scholar]
- 67.Gloor JM, Cohen AJ, Lager DJ, Grande JP, Fidler ME, Velosa JA, Larson TS, Schwab TR, Griffin MD, Prieto M, Nyberg SL, Sterioff S, Kremers WK, Stegall MD. Subclinical rejection in tacrolimus-treated renal transplant recipients. Transplantation. 2002;73:1965–1968. doi: 10.1097/00007890-200206270-00023. [DOI] [PubMed] [Google Scholar]
- 68.U.S. Renal Data System. USRDS 2007 Annual Data Report: Atlas of End-Stage Renal Disease in the United States. National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases; Bethesda, MD: 2007. [Google Scholar]
- 69.Lee JR, Muthukumar T, Dadhania D, Ding R, Sharma VK, Schwartz JE, Suthanthiran M. Urinary cell mRNA profiles predictive of human kidney allograft status. Immunol Rev. 2014;258:218–240. doi: 10.1111/imr.12159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li B, Hartono C, Ding R, Sharma VK, Ramaswamy R, Qian B, Serur D, Mouradian J, Schwartz JE, Suthanthiran M. Noninvasive diagnosis of renal-allograft rejection by measurement of messenger RNA for perforin and granzyme B in urine. N Engl J Med. 2001;344:947–954. doi: 10.1056/NEJM200103293441301. [DOI] [PubMed] [Google Scholar]
- 71.Ding R, Li B, Muthukumar T, Dadhania D, Medeiros M, Hartono C, Serur D, Seshan SV, Sharma VK, Kapur S, Suthanthiran M. CD103 mRNA levels in urinary cells predict acute rejection of renal allografts. Transplantation. 2003;75:1307–1312. doi: 10.1097/01.TP.0000064210.92444.B5. [DOI] [PubMed] [Google Scholar]
- 72.Muthukumar T, Ding R, Dadhania D, Medeiros M, Li B, Sharma VK, Hartono C, Serur D, Seshan SV, Volk HD, Reinke P, Kapur S, Suthanthiran M. Serine proteinase inhibitor-9, an endogenous blocker of granzyme B/perforin lytic pathway, is hyperexpressed during acute rejection of renal allografts. Transplantation. 2003;75:1565–1570. doi: 10.1097/01.TP.0000058230.91518.2F. [DOI] [PubMed] [Google Scholar]
- 73.Tatapudi RR, Muthukumar T, Dadhania D, Ding R, Li B, Sharma VK, Lozada-Pastorio E, Seetharamu N, Hartono C, Serur D, Seshan SV, Kapur S, Hancock WW, Suthanthiran M. Noninvasive detection of renal allograft inflammation by measurements of mRNA for IP-10 and CXCR3 in urine. Kidney Int. 2004;65:2390–2397. doi: 10.1111/j.1523-1755.2004.00663.x. [DOI] [PubMed] [Google Scholar]
- 74.Muthukumar T, Dadhania D, Ding R, Snopkowski C, Naqvi R, Lee JB, Hartono C, Li B, Sharma VK, Seshan SV, Kapur S, Hancock WW, Schwartz JE, Suthanthiran M. Messenger RNA for FOXP3 in the urine of renal-allograft recipients. N Engl J Med. 2005;353:2342–2351. doi: 10.1056/NEJMoa051907. [DOI] [PubMed] [Google Scholar]
- 75.Afaneh C, Muthukumar T, Lubetzky M, Ding R, Snopkowski C, Sharma VK, Seshan S, Dadhania D, Schwartz JE, Suthanthiran M. Urinary cell levels of mRNA for OX40, OX40L, PD-1, PD-L1, or PD-L2 and acute rejection of human renal allografts. Transplantation. 2010;90:1381–1387. doi: 10.1097/TP.0b013e3181ffbadd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kotsch K, Mashreghi MF, Bold G, Tretow P, Beyer J, Matz M, Hoerstrup J, Pratschke J, Ding R, Suthanthiran M, Volk HD, Reinke P. Enhanced granulysin mRNA expression in urinary sediment in early and delayed acute renal allograft rejection. Transplantation. 2004;77:1866–1875. doi: 10.1097/01.tp.0000131157.19937.3f. [DOI] [PubMed] [Google Scholar]
- 77.Strom TB, Suthanthiran M. Prospects and applicability of molecular diagnosis of allograft rejection. Semin Nephrol. 2000;20:103–107. [PubMed] [Google Scholar]
- 78.Medeiros M, Sharma VK, Ding R, Yamaji K, Li B, Muthukumar T, Valderde-Rosas S, Hernandez AM, Munoz R, Suthanthiran M. Optimization of RNA yield, purity and mRNA copy number by treatment of urine cell pellets with RNAlater. J Immunol Methods. 2003;279:135–142. doi: 10.1016/s0022-1759(03)00237-0. [DOI] [PubMed] [Google Scholar]
- 79.Li B, Sehajpal PK, Khanna A, Vlassara H, Cerami A, Stenzel KH, Suthanthiran M. Differential regulation of transforming growth factor beta and interleukin 2 genes in human T cells: demonstration by usage of novel competitor DNA constructs in the quantitative polymerase chain reaction. J Exp Med. 1991;174:1259–1262. doi: 10.1084/jem.174.5.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ding R, Medeiros M, Dadhania D, Muthukumar T, Kracker D, Kong JM, Epstein SR, Sharma VK, Seshan SV, Li B, Suthanthiran M. Noninvasive diagnosis of BK virus nephritis by measurement of messenger RNA for BK virus VP1 in urine. Transplantation. 2002;74:987–994. doi: 10.1097/00007890-200210150-00016. [DOI] [PubMed] [Google Scholar]
- 81.Cepek KL, Parker CM, Madara JL, Brenner MB. Integrin alpha E beta 7 mediates adhesion of T lymphocytes to epithelial cells. J Immunol. 1993;150:3459–3470. [PubMed] [Google Scholar]
- 82.Hadley GA, Bartlett ST, Via CS, Rostapshova EA, Moainie S. The epithelial cell-specific integrin, CD103 (alpha E integrin), defines a novel subset of alloreactive CD8+ CTL. J Immunol. 1997;159:3748–3756. [PubMed] [Google Scholar]
- 83.Robertson H, Wong WK, Talbot D, Burt AD, Kirby JA. Tubulitis after renal transplantation: demonstration of an association between CD103+ T cells, transforming growth factor beta1 expression and rejection grade. Transplantation. 2001;71:306–313. doi: 10.1097/00007890-200101270-00024. [DOI] [PubMed] [Google Scholar]
- 84.Hancock WW, Gao W, Faia KL, Csizmadia V. Chemokines and their receptors in allograft rejection. Curr Opin Immunol. 2000;12:511–516. doi: 10.1016/s0952-7915(00)00130-8. [DOI] [PubMed] [Google Scholar]
- 85.Hancock WW, Lu B, Gao W, Csizmadia V, Faia K, King JA, Smiley ST, Ling M, Gerard NP, Gerard C. Requirement of the chemokine receptor CXCR3 for acute allograft rejection. J Exp Med. 2000;192:1515–1520. doi: 10.1084/jem.192.10.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hancock WW, Gao W, Csizmadia V, Faia KL, Shemmeri N, Luster AD. Donor-derived IP-10 initiates development of acute allograft rejection. J Exp Med. 2001;193:975–980. doi: 10.1084/jem.193.8.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sakaguchi S. Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat Immunol. 2005;6:345–352. doi: 10.1038/ni1178. [DOI] [PubMed] [Google Scholar]
- 88.Graca L, Cobbold SP, Waldmann H. Identification of regulatory T cells in tolerated allografts. J Exp Med. 2002;195:1641–1646. doi: 10.1084/jem.20012097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lee I, Wang L, Wells AD, Dorf ME, Ozkaynak E, Hancock WW. Recruitment of Foxp3+ T regulatory cells mediating allograft tolerance depends on the CCR4 chemokine receptor. J Exp Med. 2005;201:1037–1044. doi: 10.1084/jem.20041709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Burrell BE, Lu G, Li XC, Bishop DK. OX40 costimulation prevents allograft acceptance induced by CD40-CD40L blockade. J Immunol. 2009;182:379–390. doi: 10.4049/jimmunol.182.1.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fife BT, Pauken KE, Eagar TN, Obu T, Wu J, Tang Q, Azuma M, Krummel MF, Bluestone JA. Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR-induced stop signal. Nat Immunol. 2009;10:1185–1192. doi: 10.1038/ni.1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 2009;55:611–622. doi: 10.1373/clinchem.2008.112797. [DOI] [PubMed] [Google Scholar]
- 93.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 94.Roge R, Thorsen J, Torring C, Ozbay A, Moller BK, Carstens J. Commonly used reference genes are actively regulated in in vitro stimulated lymphocytes. Scand J Immunol. 2007;65:202–209. doi: 10.1111/j.1365-3083.2006.01879.x. [DOI] [PubMed] [Google Scholar]
- 95.Anglicheau D, Muthukumar T, Hummel A, Ding R, Sharma VK, Dadhania D, Seshan SV, Schwartz JE, Suthanthiran M. Discovery and validation of a molecular signature for the noninvasive diagnosis of human renal allograft fibrosis. Transplantation. 2012;93:1136–1146. doi: 10.1097/TP.0b013e31824ef181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Keslar KS, Lin M, Zmijewska AA, Sigdel TK, Tran TQ, Ma L, Bhasin M, Rao P, Ding R, Ikle DN, Mannon RB, Sarwal MM, Strom TB, Reed EF, Heeger PS, Suthanthiran M, Fairchild RL. Multicenter evaluation of a standardized protocol for noninvasive gene expression profiling. Am J Transplant. 2013;13:1891–1897. doi: 10.1111/ajt.12284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Anglicheau D, Suthanthiran M. Noninvasive prediction of organ graft rejection and outcome using gene expression patterns. Transplantation. 2008;86:192–199. doi: 10.1097/TP.0b013e31817eef7b. [DOI] [PMC free article] [PubMed] [Google Scholar]