

Abstract
Objective:
To determine whether phosphodiesterase type 5 (PDE5) inhibition can alleviate exercise-induced skeletal muscle ischemia in boys with Duchenne muscular dystrophy (DMD).
Methods:
In 10 boys with DMD and 10 healthy age-matched male controls, we assessed exercise-induced attenuation of reflex sympathetic vasoconstriction, i.e., functional sympatholysis, a protective mechanism that matches oxygen delivery to metabolic demand. Reflex vasoconstriction was induced by simulated orthostatic stress, measured as the decrease in forearm muscle oxygenation with near-infrared spectroscopy, and performed when the forearm muscles were rested or lightly exercised with rhythmic handgrip exercise. Then, the patients underwent an open-label, dose-escalation, crossover trial with single oral doses of tadalafil or sildenafil.
Results:
The major new findings are 2-fold: first, sympatholysis is impaired in boys with DMD—producing functional muscle ischemia—despite contemporary background therapy with corticosteroids alone or in combination with cardioprotective medication. Second, PDE5 inhibition with standard clinical doses of either tadalafil or sildenafil alleviates this ischemia in a dose-dependent manner. Furthermore, PDE5 inhibition also normalizes the exercise-induced increase in skeletal muscle blood flow (measured by Doppler ultrasound), which is markedly blunted in boys with DMD.
Conclusions:
These data provide in-human proof of concept for PDE5 inhibition as a putative new therapeutic strategy for DMD.
Classification of evidence:
This study provides Class IV evidence that in patients with DMD, PDE5 inhibition restores functional sympatholysis.
Duchenne muscular dystrophy (DMD) is a devastating X-linked muscle wasting disease for which there is no specific treatment.1,2 Glucocorticoids can prolong ambulation by 2 to 3 years, reduce scoliosis, and temper pulmonary and cardiac decline in the second decade of life.1,3 However, glucocorticoids cause well-known side effects, which are intolerable in more than 25% of patients.1,2,4 Thus, a disease-specific treatment is a major unmet need.
The nitric oxide (NO)–cyclic guanosine 3′,5′-monophosphate (cGMP) pathway constitutes a putative new drug target for DMD. A compelling body of preclinical research shows that phosphodiesterase type 5 (PDE5) inhibitors, which prolong the half-life of cGMP—the downstream target of NO in vascular smooth muscle—benefit limb, respiratory, and cardiac muscles in mdx mice5–11 and prolong survival in dystrophin-deficient zebrafish.12 Remarkably, even a single dose of sildenafil or tadalafil, 2 common PDE5 inhibitors, prevents muscle ischemia, as well as injury and fatigue, when mdx mice are subjected to a brief bout of exercise.5
The challenge remains to translate this promising preclinical work to actual human patients with DMD. Thus, we first performed a case-control study to establish that blood flow regulation is impaired in boys with DMD despite contemporary background therapy with glucocorticoids and cardiac prophylactic medication. Then, we conducted an acute dose-finding study to test whether tadalafil can eliminate functional muscle ischemia in boys with DMD. Moreover, to test for a drug class effect, we compared tadalafil with sildenafil, another PDE5 inhibitor with a very different chemical structure.13
METHODS
Standard protocol approvals, registrations, and patient consents.
The study was approved by the Institutional Review Board at Cedars-Sinai Medical Center. Each subject gave pediatric assent and their parents gave written informed consent to participate. An investigational new drug exemption was granted by the US Food and Drug Administration (FDA) to administer tadalafil and sildenafil for the purposes of this pediatric research study (NCT01580501).
Subjects.
Between 2012 and 2013, we studied ambulatory boys with DMD, 8 to 13 years of age, with normal left ventricular ejection fraction (>50%), and healthy age-matched male controls. All patients had a preexisting clinical diagnosis of DMD based on rate of skeletal muscle disease progression, and all had confirmed DMD mutation.
All subjects were screened for eligibility by clinical examination, 12-lead ECG, and 2-dimensional transthoracic echocardiogram. Potential subjects, both cases and controls, were excluded from study if they had a history of hypertension or measured blood pressure >120/80 mm Hg; diabetes mellitus; heart failure by history, physical examination, or left ventricular ejection fraction <50% by 2-dimensional transthoracic echocardiography; required nocturnal ventilator support; or had any contraindication to PDE5 inhibitors (use of nitrates, α-adrenergic blockers, other PDE5A inhibitors, or potent inhibitors of cytochrome P450 3A4).
Hemodynamic measurements.
Subjects were studied in the supine position. Heart rate was measured continuously by ECG and blood pressure by automated oscillometric sphygmomanometry, using a validated monitor (Welch Allyn Vital Signs Monitor 300 Series; Welch Allyn, Inc., Skaneateles Falls, NY).
Skeletal muscle oxygenation by near-infrared spectroscopy.
Forearm muscle tissue oxygenation was measured with near-infrared (NIR) spectroscopy, as previously described.14 Briefly, optodes were placed over the flexor digitorum profundus, the main muscle recruited during handgrip. The optodes were housed in a customized flexible rubber casing and secured to the skin with adhesive. The optodes were covered with an optically dense, black vinyl sheet, to minimize interference from external light and loss of NIR transmitted light.
The NIR signal was sampled at a rate of 5 Hz, converted to oxyhemoglobin (HbO2) + oxymyoglobin (MbO2) concentration with validated algorithms, displayed as the running average of 50 consecutive samples, and stored digitally for analysis (PowerLab; ADInstruments, Boulder, CO). Before each experiment, absorption and scattering coefficients were calibrated against an external standard. After each experiment, a pneumatic cuff was inflated on the upper arm to suprasystolic pressure (200 mm Hg) to establish the total labile signal (TLS, the difference between the baseline and nadir in muscle tissue oxygenation). Changes in forearm muscle tissue oxygenation were expressed as a percentage of TLS.
Brachial artery blood flow by Doppler ultrasound.
Brachial artery mean blood velocity was measured using pulsed-Doppler ultrasonography (Siemens iE33). Data were acquired continuously with a 9-MHz probe with a 60° angle of insonation. The ultrasound gate was optimized to ensure complete insonation of the entire vessel cross-section with constant intensity. The Doppler audio signal was converted to a real-time flow velocity signal using a validated Doppler audio converter,15 and recorded using a PowerLab data acquisition system (ADInstruments). Brachial artery diameter was measured by B-mode imaging. Brachial artery blood flow was calculated as mean blood velocity (cm/s)·πr2·60, where r is radius of the brachial artery.
Handgrip exercise.
Handgrip exercise was performed with a dynamometer (Smedley Hand Dynamometer modified by Stoelting). To determine maximal voluntary contraction (MVC), each subject was asked to grip the dynamometer as hard as possible 3 separate times; the greatest force output was taken as MVC. Force output was displayed on a computer screen to provide visual feedback for subjects. Subjects performed intermittent isometric handgrip (20 handgrips per minute, 50% duty cycle) at 20% MVC for 7 minutes. This mild level of handgrip exercise was chosen because it does not activate sympathetic outflow to skeletal muscle but causes sympatholysis in healthy boys.16,17
Functional sympatholysis.
The subjects' lower body was enclosed in a negative pressure chamber to the level of the iliac crest as previously described.16 The pressure in the chamber was measured by a Statham transducer (Gould Inc., Oxnard, CA). Lower-body negative pressure (LBNP) at −20 to −30 mm Hg simulates mild orthostatic stress caused by transition from the supine to the seated position. This technique mainly unloads the low-pressure cardiopulmonary baroreceptors, producing highly reproducible reflex increases in sympathetic vasoconstrictor drive to the skeletal muscle vasculature without changing systemic blood pressure.16 To measure exercise-induced attenuation of reflex vasoconstriction (i.e., functional sympatholysis), the LBNP was (1) applied at rest, and then (2) superimposed on mild rhythmic handgrip at 20% MVC. Reflex vasoconstriction was measured as the LBNP-induced decrease in forearm muscle oxygenation by NIR spectroscopy. Using the pressure inside the LBNP chamber as a trigger, the PowerLab software was programed to mean the HbO2 + MbO2 signal for 20 seconds before the onset of LBNP and for 20 seconds before the offset of LBNP; the difference between these mean values was taken as the LBNP-induced change in forearm muscle tissue oxygenation.18 Blood pressure, heart rate, and handgrip force were also recorded in response to 2 minutes of LBNP applied at rest and during the third to fifth minutes of the 7-minute exercise protocol.
Exercise-induced hyperemia.
Brachial artery blood flow was measured at rest and for 60 seconds postexercise to evaluate exercise-induced changes in skeletal muscle blood flow, defined as the percentage increase in brachial artery blood flow from rest to postexercise. Vascular conductance was calculated as forearm blood flow divided by mean arterial pressure.
Specific protocols.
Case-control study.
Functional sympatholysis (and exercise-induced hyperemia) was compared in 12 boys with DMD and 10 age-matched healthy male controls. The screening visit included a practice session to familiarize the boys with the protocol. Baseline data were acquired the next day. Background therapy of DMD with glucocorticoids and/or prophylactic cardiac medication was continued throughout the study.
Acute dose-finding study.
Boys with DMD (but not healthy controls) then participated in the PDE5 inhibitor dose-finding study. Patients received either single doses of sildenafil or tadalafil in an open-label crossover design with a 2-week washout period before crossover (accounting for the 17.5-hour elimination half-life of tadalafil). The order of sildenafil or tadalafil was random. Patients received 0.5 mg/kg (not to exceed 20 mg) of either oral sildenafil or oral tadalafil on day 1, followed by 1.0 mg/kg of sildenafil or tadalafil (not to exceed 40 mg) on day 2. Experiments were performed 1 hour after administration of sildenafil or 3 hours after administration of tadalafil to match the expected peak blood levels. Patients were queried about potential side effects throughout the study visits.
Pharmacokinetics.
Serum blood samples for pharmacokinetic determinations were collected from patients with DMD at time 0, 0.25, 0.5, 1, 2, 4, 8, and 24 hours after each dose of both sildenafil and tadalafil. Blood levels were measured using high-performance liquid chromatography–tandem mass spectrometry (NMS Labs, Willow Grove, PA).
Statistics.
The prespecified primary outcome was a 50% restoration of functional sympatholysis (i.e., exercise-induced attenuation of reflex vasoconstriction). Based on recent work from our laboratory,18 the sample size was chosen to achieve a power of 80% to detect an effect size of 0.9, based on a 2-sided t test.
Baseline characteristics of patients and controls were compared using Student t test. Exercise-induced attenuation of reflex vasoconstriction (functional sympatholysis) was assessed by comparing the LBNP-induced ΔHbO2 + MbO2 (%TLS) at rest vs the LBNP-induced decrease in ΔHbO2 + MbO2 during handgrip. Student t test was used to test the following: group differences in ΔHbO2 + MbO2; drug treatment effect on LBNP-induced ΔHbO2 + MbO2 (%TLS) at rest vs the LBNP-induced decrease in ΔHbO2 + MbO2 during handgrip; group difference in the %Δ in brachial artery blood flow; and drug treatment effect on the %Δ in brachial artery blood flow. The apparent elimination half-life (t1/2) was determined using linear regression of ≥3 natural logarithm-transformed data points in the terminal phase. Data are expressed as mean ± SEM, unless otherwise specified.
Primary research question and classification of evidence.
Does PDE5 inhibition alleviate functional muscle ischemia in boys with DMD? This study provides Class IV evidence that in patients with DMD, PDE5 inhibition restores functional (i.e., exercise-induced) sympatholysis.
RESULTS
Data are presented for 10 boys with DMD and 10 healthy controls; movement artifacts precluded data analysis in 2 boys with DMD. Baseline characteristics and indices of disease severity of these 10 patients are shown in table 1. All patients were ambulatory, although some often used a wheelchair or scooter. All patients with DMD were receiving background therapy with glucocorticoids (either deflazacort or prednisone); 5 of the 10 patients were also receiving prophylactic cardiac medication (either lisinopril or losartan).
Table 1.
Baseline characteristics of patients with Duchenne muscular dystrophy
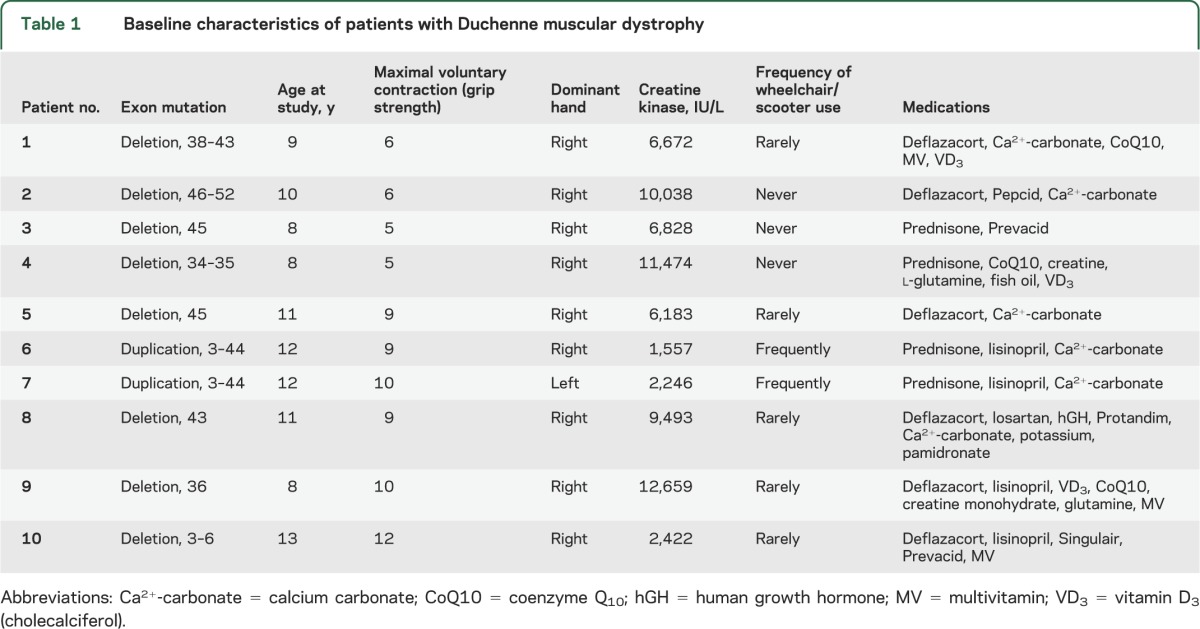
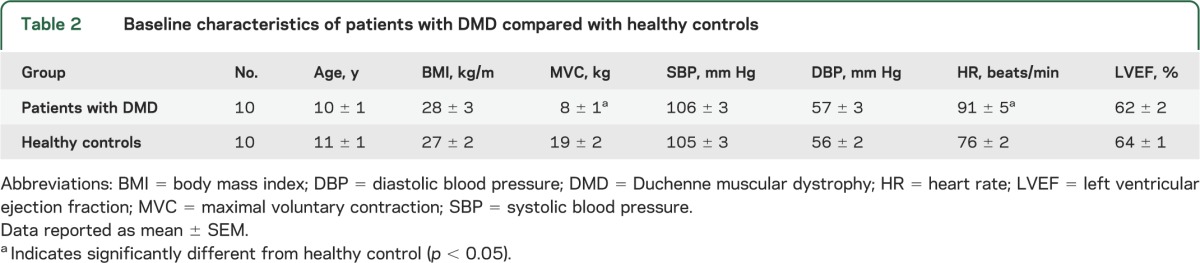
Patients and controls were well matched for age, body mass index, blood pressure, and left ventricular ejection fraction (table 2). As expected, resting heart rate was higher in the boys with DMD (p < 0.05 vs healthy controls), and MVC (i.e., grip strength) was much lower (p < 0.05).
Table 2.
Baseline characteristics of patients with DMD compared with healthy controls
Functional sympatholysis is impaired in DMD and restored by PDE5 inhibition.
In resting forearm muscle, LBNP evoked comparable decreases in forearm muscle oxygenation (HbO2 + MBO2) in patients and controls (figure 1). When LBNP was superimposed on handgrip in healthy controls, the reflex decrease in muscle oxygenation was attenuated by 54% ± 8% (ΔHbO2 + MbO2: −18% ± 3% at rest vs −9% ± 2% during handgrip, p < 0.01), indicating functional sympatholysis. In contrast, no such attenuation was observed in the patients with DMD (ΔHbO2 + MbO2: −14% ± 2% at rest vs −13% ± 2% during handgrip), indicating functional muscle ischemia (figure 1). In boys with DMD, sympatholysis was similarly impaired in those treated (n = 5) or not treated (n = 5) with prophylactic cardiac medication (figure e-1 on the Neurology® Web site at Neurology.org).
Figure 1. Functional sympatholysis is impaired in patients with DMD and rescued by acute PDE5 inhibition.
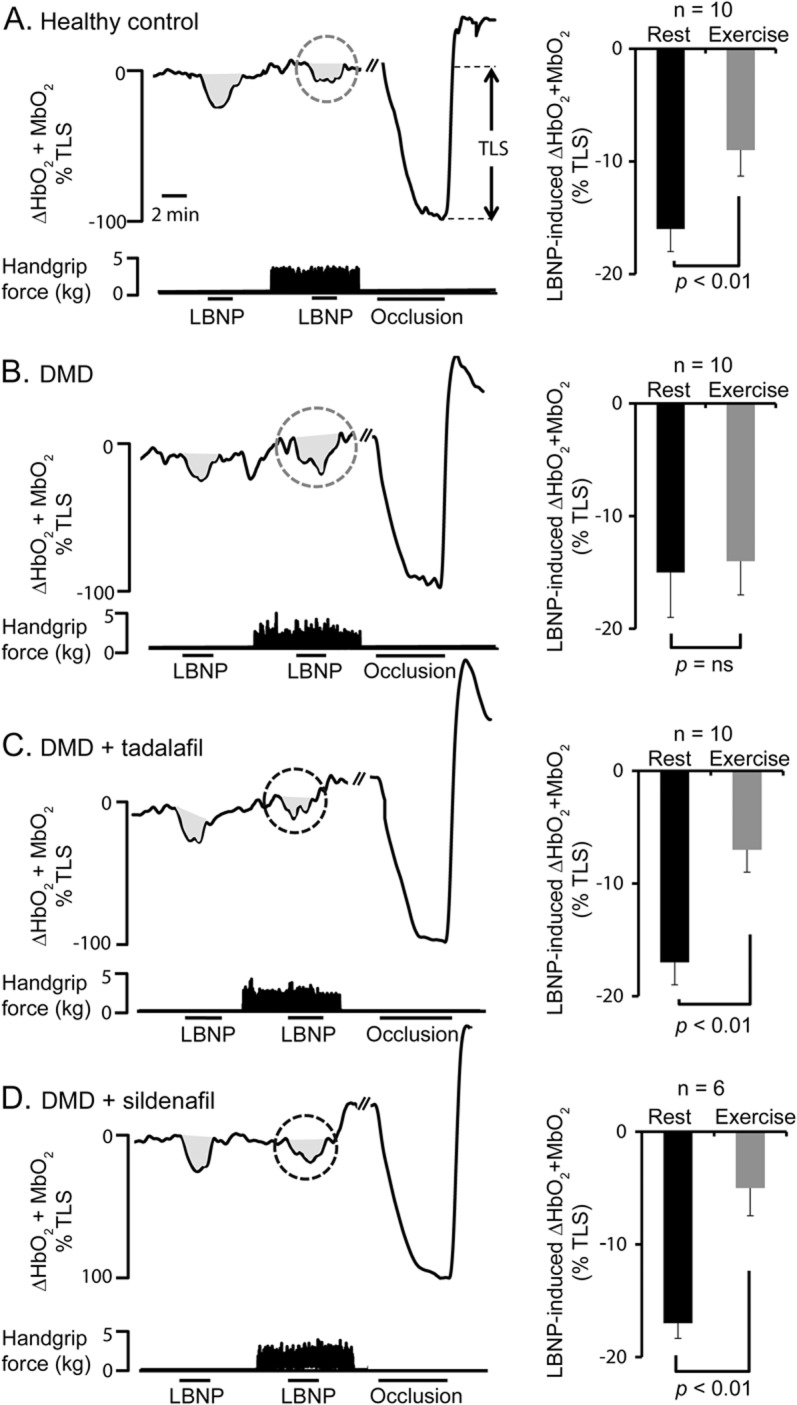
(A) Left panel shows a representative tracing from a healthy control subject, showing that the lower-body negative pressure (LBNP)-induced decrease in forearm muscle oxygenation (HbO2 + MbO2) is greatly attenuated during mild handgrip exercise (gray dashed circle), demonstrating functional sympatholysis. At the end of each experiment, an arm cuff was inflated to suprasystolic pressure to occlude the forearm circulation, producing a maximal decrease in muscle oxygenation to calculate the total labile signal (TLS). Right panel shows summary forearm muscle oxygenation data from the 10 healthy control subjects studied at rest and during exercise expressed as a percentage of TLS. (B) Representative tracings from a patient with Duchenne muscular dystrophy (DMD) at baseline (pretreatment), showing that sympatholysis is impaired because handgrip fails to attenuate the LBNP response (gray dashed circle). Summary data are shown in the right panel for 10 boys with DMD. (C) Representative tracing from a subject with DMD treated with tadalafil, showing that the LBNP-induced decrease in forearm muscle oxygenation is greatly attenuated during mild handgrip exercise (black dashed circle). Summary data are shown in the right panel for 10 patients with DMD. (D) Representative tracing from a subject with DMD treated with sildenafil, showing that the LBNP-induced decrease in forearm muscle oxygenation is greatly attenuated during mild handgrip exercise (black dashed circle). Summary data are shown in the right panel for 6 patients with DMD. Data are reported as mean ± SEM. ns = not significant.
Tadalafil restored functional sympatholysis in boys with DMD (figure 1) in a dose-dependent manner. With the 0.5 mg/kg dose of tadalafil, the reflex decrease in muscle oxygenation was attenuated by 45% ± 8% (ΔHbO2 + MbO2: −20% ± 2% at rest vs −11% ± 1% during handgrip, p < 0.01), a response that is indistinguishable from normal (p = not significant vs healthy controls). The 1.0 mg/kg dose caused a super-normal 63% ± 5% attenuation (ΔHbO2 + MbO2: −17% ± 1% at rest vs −7% ± 1% during handgrip, p < 0.01). In a subset of patients (n = 6), we confirmed these results with sildenafil (figures 1 and 2). Individual data are illustrated in figure e-2.
Figure 2. Sildenafil equally restores functional sympatholysis compared with tadalafil.
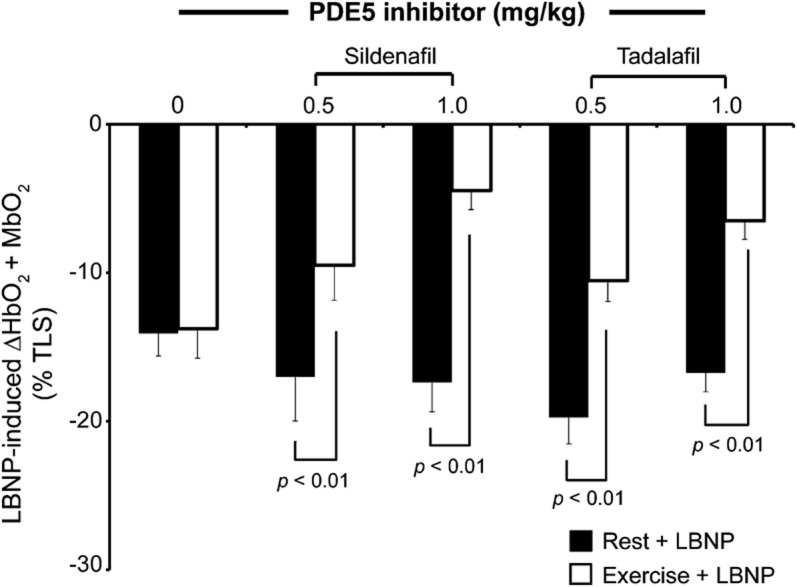
Lower-body negative pressure (LBNP)-induced decreases on forearm muscle oxygenation by near-infrared spectroscopy at rest and during mild rhythmic handgrip exercise (n = 6). Data expressed as a percentage of the total labile signal (TLS). Data reported as mean ± SEM. PDE5 = phosphodiesterase type 5.
Exercise-induced hyperemia is blunted in boys with DMD and restored by tadalafil.
Handgrip exercise increased brachial artery blood flow by 78% ± 12% over baseline in healthy controls but only by 32% ± 5% in boys with DMD (p < 0.05, figure 3). Remarkably, in DMD, tadalafil restored exercise-induced hyperemia in a dose-dependent manner (figure 3, table e-1). A similar tendency was seen with sildenafil, but the treatment effect was not statistically significant (figure e-3).
Figure 3. Postexercise hyperemia is impaired in DMD and rescued by tadalafil.
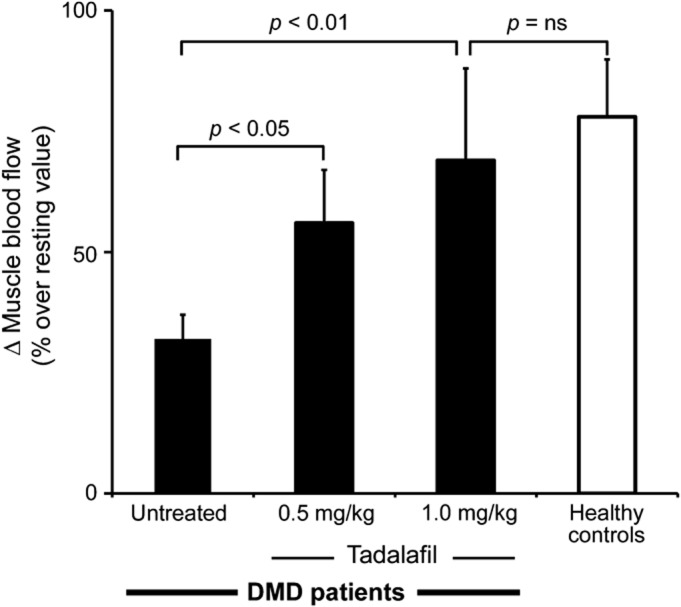
Exercise-induced change in skeletal muscle blood flow, from rest to postexercise, in 10 patients with Duchenne muscular dystrophy (DMD) and 10 healthy controls. Data expressed as mean ± SEM. ns = not significant.
Pharmacokinetic data.
Nine boys with DMD completed the pharmacokinetic study. The average group response curves are presented in figure e-4. Blood levels peaked approximately 1 hour after oral sildenafil administration, with an elimination half-life of 2.4 ± 0.09 hours for the 0.5 mg/kg dose and 3.2 ± 0.5 hours for the 1.0 mg/kg dose. In contrast, blood levels peaked 2 to 4 hours after oral tadalafil administration, with an elimination half-life of 24.9 ± 3.1 hours for the 0.5 mg/kg dose and 39.5 ± 6.6 hours for the 1.0 mg/kg dose.
Safety data.
Facial flushing occurred in all boys with both doses of either PDE5 inhibitor. Blood pressure was unaffected by either drug. One patient developed a penile erection lasting 6 hours after low-dose tadalafil and was not escalated to the higher dose. A second boy experienced erection lasting 3 hours after low-dose tadalafil and 4 hours after high-dose tadalafil. In both cases, erections were nonpainful and resolved spontaneously.
DISCUSSION
In the dystrophin-deficient mdx mouse model of DMD, PDE5 inhibitors alleviate many features of the dystrophic phenotype, including spasm of skeletal muscle microvessels that can lead to muscle hypoperfusion, injury, and fatigue.5 However, the mdx mouse is an important but imperfect milder model of the virulent human disease. Thus, the challenge is to determine whether the compelling body of preclinical mouse research can be translated to boys with DMD. The present study directly addresses this challenge with 3 major new findings: (1) functional sympatholysis is impaired in pediatric patients with DMD, producing functional muscle ischemia, despite contemporary background therapy with corticosteroids alone or in combination with cardioprotective medication; (2) tadalafil alleviates this ischemia in a dose-dependent manner; and (3) sildenafil replicates the effect of tadalafil, strongly supporting PDE5 inhibition as the mechanism of action.
That sympatholysis is impaired in contemporary patients with DMD confirms and extends our earlier observations made well over a decade ago before patients with DMD were treated with any medication.19 While glucocorticoids represent the first significant advance in medical management of DMD, our new data in glucocorticoid-treated patients show that chronic treatment with neither prednisone nor deflazacort protects dystrophic muscle from functional ischemia. While angiotensin-converting enzyme inhibitors (lisinopril) and angiotensin receptor blockers (losartan) are being used increasingly in boys with DMD as preemptive therapy to delay onset of heart failure,1,18,20,21 our data show that chronic treatment with neither lisinopril nor losartan restores sympatholysis in DMD, even though these drugs can reduce reactive oxygen species that destroy NO and thereby rescue sympatholysis in common acquired adult cardiovascular diseases such as hypertension and heart failure.22–26
In contrast, the seminal finding of this study is that tadalafil rescues sympatholysis in boys with DMD, providing added putative benefit beyond that afforded by the current standard of care. The effect is marked, immediate, and dose-dependent. The lower dose used in our study is approximately equivalent to an adult dose of 20 mg, which was shown previously to alleviate functional ischemia in adults with Becker muscular dystrophy14 and is the highest dose approved by the FDA to treat erectile dysfunction. Our higher dose is approximately equivalent to the 40-mg dose approved by the FDA to treat adult pulmonary hypertension and the same dose most frequently used to treat pediatric pulmonary hypertension.27
That sildenafil, which was the main PDE5 inhibitor used in the preclinical studies, and tadalafil were virtually identical in their ability to restore sympatholysis in boys with DMD—despite having different chemical structures—strongly implicates PDE5 inhibition as the mechanism of action. Because PDE5 is a cGMP-specific phosphodiesterase, our data support the hypothesis that PDE5 inhibition boosts a residual NO-cGMP signal arising from cytosolic neuronal NO synthase, which, in the absence of dystrophin, is misplaced from the sarcolemma.5 That neither PDE5 inhibitor affected reflex vasoconstriction or blood flow in resting DMD skeletal muscle nor systemic blood pressure suggests that the exercise-specific action involves neither endothelial NOS-derived NO nor central inhibition of sympathetic outflow.
Moreover, that tadalafil normalized not only sympatholysis but also exercise-induced hyperemia, which was markedly blunted in the boys with DMD, provides further evidence for remarkably close clinical translation of seminal mdx mouse studies.5 This novel finding also suggests that NO released from exercising human skeletal muscle normally exerts a vasodilatory effect even in the absence of α-adrenergic stimulation. The blood flow data further suggest that a higher degree of PDE5 inhibition may be required to fully restore exercise-induced hyperemia than sympatholysis. Whereas sympatholysis reflects attenuation of a neurogenic vasoconstrictor response specifically in the most distal skeletal muscle microvessels, exercise-induced hyperemia involves multiple vascular segments and multiple active vasodilator mechanisms that remain incompletely understood.
Our study has several limitations. While this was an open-label study, impaired sympatholysis was a robust finding in DMD and its correction with both PDE5 inhibitors was steeply dose-dependent. Because this was a single-dose study, we do not know whether the improved blood flow regulation will be sustained with chronic administration. However, “nitrate tolerance,” which is a common limitation of organic nitrates, simply is not a feature of PDE5 inhibition. This proof-of-concept study does not address the crucial question of whether restoring normal blood flow regulation will preserve dystrophic skeletal muscle and slow disease progression. Despite these limitations, the data herein, together with a compelling body of preclinical research, have informed the design of a pivotal multicenter clinical trial to determine whether chronic daily tadalafil can preserve muscle function in boys with DMD (NCT01865084).
Supplementary Material
GLOSSARY
- cGMP
cyclic guanosine 3′,5′-monophosphate
- DMD
Duchenne muscular dystrophy
- FDA
US Food and Drug Administration
- HbO2
oxyhemoglobin
- LBNP
lower-body negative pressure
- MbO2
oxymyoglobin
- MVC
maximal voluntary contraction
- NIR
near-infrared
- NO
nitric oxide
- PDE5
phosphodiesterase type 5
- TLS
total labile signal
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
All authors contributed significantly to the present manuscript and have approved the final version. Dr. M. Nelson was involved in the conception and design, data collection, data analysis, interpretation of the results, writing the first draft of the manuscript, and editing/revising of the manuscript. Dr. Rader was involved in data collection and editing/revising the manuscript. Ms. Tang had an integral role in data collection, data analysis, and data interpretation. Dr. Tavyev was involved in the conception and design, data collection, and editing/revising the manuscript. Dr. S. Nelson and Dr. Miceli were involved in the conception and design, interpretation of the results, and editing/revising the manuscript. Dr. Elashoff was involved in the conception and design, data/statistical analysis, and editing/revising the manuscript. Dr. Sweeney was involved in the conception and design, interpretation of the results, and editing/revising the manuscript. Dr. Victor obtained the research funding, and was involved in the conception and design, interpretation of the results, drafting the manuscript, and editing/revising the manuscript.
STUDY FUNDING
This study was funded by a research grant to Dr. Victor from Parent Project Muscular Dystrophy (PPMD). Supplemental support was provided by the NIH/National Center for Advancing Translational Sciences UCLA CTSI (UL1TR000124). Dr. Michael Nelson is the recipient of research fellowship grants from the Heart and Stroke Foundation of Canada and the Canadian Institutes of Health Research.
DISCLOSURE
M. Nelson, F. Rader, X. Tang, and J. Tavyev report no disclosures relevant to the manuscript. S. Nelson is funded by NIH grants P30 AR057230, P30 CA016042, and R01 NS073871, the California Institute of Regenerative Medicine, and Parent Project Muscular Dystrophy. He also serves as a scientific advisor for 23andMe. M. Miceli became a consultant for Eli Lilly and Company, the manufacturer of tadalafil, after these data were collected and analyzed, and is funded by NIH grant P30 AR057230 and the California Institute of Regenerative Medicine. R. Elashoff is funded by NIH grants R01 HL089901, P01 CA029605, and P30 CA016042. H. Sweeney became a consultant for Eli Lilly and Company, the manufacturer of tadalafil, after these data were collected and analyzed, he serves on the scientific advisory board for Parent Project Muscular Dystrophy, is funded by NIH grants U54 AR052646, R01 DC009100, R21 DK096463, P01 HL110869, and U54 NS058572, and received research support from Parent Project Muscular Dystrophy and the Muscular Dystrophy Association. R. Victor became a consultant for Eli Lilly and Company, the manufacturer of tadalafil, after these data were collected and analyzed, is funded by NIH grants U34 AR062893 and UL1TR000124, and received research support from the Muscular Dystrophy Association and Coalition Duchenne. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Bushby K, Finkel R, Birnkrant DJ, et al. Diagnosis and management of Duchenne muscular dystrophy: part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol 2010;9:77–93 [DOI] [PubMed] [Google Scholar]
- 2.Bushby K, Finkel R, Birnkrant DJ, et al. Diagnosis and management of Duchenne muscular dystrophy: part 2: implementation of multidisciplinary care. Lancet Neurol 2010;9:177–189 [DOI] [PubMed] [Google Scholar]
- 3.Moxley R, Ashwal S, Pandya S, et al. Practice parameter: corticosteroid treatment of Duchenne dystrophy. Report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology 2005;64:13–20 [DOI] [PubMed] [Google Scholar]
- 4.Mendell JR, Moxley RT, Griggs RC, et al. Randomized, double-blind six-month trial of prednisone in Duchenne's muscular dystrophy. N Engl J Med 1989;320:1592–1597 [DOI] [PubMed] [Google Scholar]
- 5.Kobayashi YM, Rader EP, Crawford RW, et al. Sarcolemma-localized nNOS is required to maintain activity after mild exercise. Nature 2008;456:511–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Asai A, Sahani N, Kaneki M, Ouchi Y, Martyn JAJ, Yasuhara SE. Primary role of functional ischemia, quantitative evidence for the two-Hit mechanism, and phosphodiesterase-5 inhibitor therapy in mouse muscular dystrophy. PLoS One 2007;2:e806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lai Y, Thomas GD, Yue Y, et al. Dystrophins carrying spectrin-like repeats 16 and 17 anchor nNOS to the sarcolemma and enhance exercise performance in a mouse model of muscular dystrophy. J Clin Invest 2009;119:624–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thomas GD, Ye J, De Nardi C, Monopoli A, Ongini E, Victor RG. Treatment with a nitric oxide-donating NSAID alleviates functional muscle ischemia in the mouse model of Duchenne muscular dystrophy. PLoS One 2012;7:e49350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adamo CM, Dai DF, Percival JM, et al. Sildenafil reverses cardiac dysfunction in the mdx mouse model of Duchenne muscular dystrophy. Proc Natl Acad Sci U S A 2010;107:19079–19083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Khairallah M, Khairallah RJ, Young ME, et al. Sildenafil and cardiomyocyte-specific cGMP signaling prevent cardiomyopathic changes associated with dystrophin deficiency. Proc Natl Acad Sci U S A 2008;105:7028–7033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Percival JM, Whitehead NP, Adams ME, Adamo CM, Beavo JA, Froehner SC. Sildenafil reduces respiratory muscle weakness and fibrosis in the mdx mouse model of Duchenne muscular dystrophy. J Pathol 2012;228:77–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kawahara G, Karpf JA, Myers JA, Alexander MS, Guyon JR, Kunkel LM. Drug screening in a zebrafish model of Duchenne muscular dystrophy. Proc Natl Acad Sci U S A 2011;108:5331–5336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bischoff E. Potency, selectivity, consequences of nonselectivity of PDE inhibition. Int J Impot Res 2004;16:S11–S14 [DOI] [PubMed] [Google Scholar]
- 14.Martin EA, Barresi R, Byrne BJ, et al. Tadalafil alleviates muscle ischemia in patients with Becker muscular dystrophy. Sci Transl Med 2012;4:162ra155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Herr MD, Hogeman CS, Koch DW, Krishnan A, Momen A, Leuenberger UA. A real-time device for converting Doppler ultrasound audio signals into fluid flow velocity. Am J Physiol Heart Circ Physiol 2010;298:H1626–H1632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hansen J, Thomas GD, Harris SA, Parsons WJ, Victor RG. Differential sympathetic neural control of oxygenation in resting and exercising human skeletal muscle. J Clin Invest 1996;98:584–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Victor RG, Seals DR, Mark AL. Differential control of heart rate and sympathetic nerve activity during dynamic exercise: insight from intraneural recordings in humans. J Clin Invest 1987;79:508–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duboc D, Meune C, Pierre B, et al. Perindopril preventive treatment on mortality in Duchenne muscular dystrophy: 10 years' follow-up. Am Heart J 2007;154:596–602 [DOI] [PubMed] [Google Scholar]
- 19.Sander M, Chavoshan B, Harris SA, et al. Functional muscle ischemia in neuronal nitric oxide synthase-deficient skeletal muscle of children with Duchenne muscular dystrophy. Proc Natl Acad Sci U S A 2000;97:13818–13823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duboc D, Meune C, Lerebours G, Devaux JY, Vaksmann G, Becane HM. Effect of perindopril on the onset and progression of left ventricular dysfunction in Duchenne muscular dystrophy. J Am Coll Cardiol 2005;45:855–857 [DOI] [PubMed] [Google Scholar]
- 21.Ramaciotti C, Heistein LC, Coursey M, et al. Left ventricular function and response to enalapril in patients with Duchenne muscular dystrophy during the second decade of life. Am J Cardiol 2006;98:825–827 [DOI] [PubMed] [Google Scholar]
- 22.Thomas GD, Zhang W, Victor RG. Impaired modulation of sympathetic vasoconstriction in contracting skeletal muscle of rats with chronic myocardial infarctions: role of oxidative stress. Circ Res 2001;88:816–823 [DOI] [PubMed] [Google Scholar]
- 23.Gao L, Wang W, Li YL, et al. Superoxide mediates sympathoexcitation in heart failure: roles of angiotensin II and NAD(P)H oxidase. Circ Res 2004;95:937–944 [DOI] [PubMed] [Google Scholar]
- 24.Dandona P, Dhindsa S, Ghanim H, Chaudhuri A. Angiotensin II and inflammation: the effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockade. J Hum Hypertens 2006;21:20–27 [DOI] [PubMed] [Google Scholar]
- 25.Vongpatanasin W, Wang Z, Arbique D, et al. Functional sympatholysis is impaired in hypertensive humans. J Physiol 2011;589:1209–1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao W, Swanson SA, Ye J, et al. Reactive oxygen species impair sympathetic vasoregulation in skeletal muscle in angiotensin II–dependent hypertension. Hypertension 2006;48:637–643 [DOI] [PubMed] [Google Scholar]
- 27.Takatsuki S, Calderbank M, Ivy D. Initial experience with tadalafil in pediatric pulmonary arterial hypertension. Pediatr Cardiol 2012;33:683–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.