

Abstract
Objective:
To identify the genetic cause of a complex syndrome characterized by autophagic vacuolar myopathy (AVM), hypertrophic cardiomyopathy, pigmentary retinal degeneration, and epilepsy.
Methods:
Clinical, pathologic, and genetic study.
Results:
Two brothers presented with visual failure, seizures, and prominent cardiac involvement, but only mild cognitive impairment and no motor deterioration after 40 years of disease duration. Muscle biopsy revealed the presence of widespread alterations suggestive of AVM with autophagic vacuoles with sarcolemmal features. Through combined homozygosity mapping and exome sequencing, we identified a novel p.Gly165Glu mutation in CLN3.
Conclusions:
This study expands the clinical phenotype of CLN3 disease. Genetic testing for CLN3 should be considered in AVM with autophagic vacuoles with sarcolemmal features.
Autophagic vacuolar myopathies (AVMs) are a heterogeneous group of disorders that include Danon disease, X-linked myopathy with excessive autophagy, and infantile autophagic vacuolar myopathy.1 A number of genes causing AVM have been identified, including LAMP2 and VMA212,3; however, in many cases of AVM, a genetic cause is not found.
CLN3 disease, the classic juvenile form of neuronal ceroid lipofuscinosis, is an autosomal recessive condition clinically characterized by visual impairment at 6 to 10 years of age, psychomotor deterioration, and seizures, and it is caused by mutations in the CLN3 gene.4,5 In CLN3 disease, skeletal muscle is not clinically involved so muscle biopsy is rarely performed.
We report the case of 2 siblings with a complex multiorgan syndrome caused by a novel p.Gly165Glu mutation in CLN3, causing protracted CLN3 disease with widespread alteration on muscle biopsy suggestive of AVM.
METHODS
Patients.
The patients were referred to the epilepsy clinic of National Neurologic Institute C. Mondino. They underwent thorough neurologic, cardiologic, and ophthalmologic investigations.
Standard protocol approvals, registrations, and patient consents.
The institutional review board approved the study, and all examined family members gave written informed consent.
Muscle pathology.
Both subjects underwent biceps brachii muscle biopsy. Specimens were processed according to standard procedures.6 Ultrastructural examination of muscle sample and lymphocytes was performed as previously described.6
Genetic analysis.
Genome-wide, single nucleotide polymorphism genotyping was performed in the 2 affected individuals (V-1 and V-2) and the mother. Each individual was assayed on an Illumina chip (HumanOmniExpress-12v1_H; Illumina, Inc., San Diego, CA). Data were analyzed using the GenomeStudio 2010.3 Genotyping Module (Illumina) and BeadStudio suite.
Whole-exome sequencing was performed on individuals V-1 and V-2 on a HiSeq2000 next-generation sequencing platform (Illumina) and data processed through Novoalign and SAMtools 0.18.
Primers for PCR amplification of the exon containing the mutation are available upon request.
RESULTS
Clinical features.
The patients (V-1, now aged 48 years, and V-2, aged 41 years) were born into a consanguineous family (figure 1A). There is no family history of neurologic disease. Both siblings presented at the age of 7 years with visual loss rapidly progressing to total blindness. A diagnosis of retinitis pigmentosa was made. They had normal psychomotor development and, after graduating from high school, started to work as switchboard operators. No other symptoms were reported until the age of 36 (V-1) and 29 (V-2), when they presented with generalized tonic-clonic seizures responsive to wide-spectrum antiepileptic drugs. Brain MRI showed the presence of mild cerebral and cerebellar vermian atrophy (figure 1B). At the same time, they were diagnosed with sick sinus syndrome and cardiac conduction defect and treated with pacemaker implantation and, later, implantable cardiac defibrillator. Cardiac echo scan showed the presence of concentric hypertrophic cardiomyopathy. Cardiac function progressively deteriorated over 5 years to cardiac failure (New York Heart Association class III). Both patients are now waiting for cardiac transplant. At the age of 42 (V-1) and 37 (V-2), neurologic examination was strictly normal apart from blindness. Neuropsychological examination was uneventful apart from mild impairment of verbal fluency and executive functions. Ophthalmologic fundus evaluation was uninformative because of bilateral lens opacity. EEG showed normal background activity and rare isolated diffuse spikes and sharp waves. Increased creatine kinase (up to 600 U/L) was detected in seizure-free periods. EMG showed the presence of mild myogenic features.
Figure 1. Family pedigree and genetic investigations.

(A) Family pedigree and Sanger sequencing validation study of the c.494G>A mutation in the carrier mother (IV-1) and the 2 affected patients (V-1 and V-2). (B) T1-weighted brain MRI of V-1 performed at the age of 38 years showing mild cerebral and cerebellar atrophy. (C) Clustal Omega multiple alignment showing that glycine at position 165 (green highlighted) is conserved across very distant species and is part of a highly conserved small stretch of amino acids. (D) Schematic representation of CLN3 protein showing the relative position of the mutations in the second luminal loop of CLN3. The second luminal loop of CLN3 contains 12 pathogenic mutations, including 6 other missense mutations, suggesting that it is functionally or structurally important to CLN3 function. The transmembrane domains of CLN3 are shown as blue boxes and the cytosolic and lysosomal facing domains as lines. The mutations previously reported in the literature are shown in black. The novel p.Gly165Glu mutation reported here is shown in red.
Muscle pathology.
Muscle biopsy alterations were similar in both brothers. Light microscopy examination was suggestive of an AVM. The most common finding was the presence of vacuoles involving approximately 50% of fibers. Autophagic vacuoles showed a strong reaction for lysosomal acid phosphatase activity. Immunohistochemical analysis revealed a positive binding at vacuolar membrane level, using dystrophin and human leukocyte antigen class 1 (HLA1) antibodies (figure 2). There was also clear autofluorescence (data not shown).
Figure 2. Muscle biopsy of CLN3 disease presents alterations suggestive of autophagic vacuolar myopathy with autophagic vacuoles with sarcolemmal features.
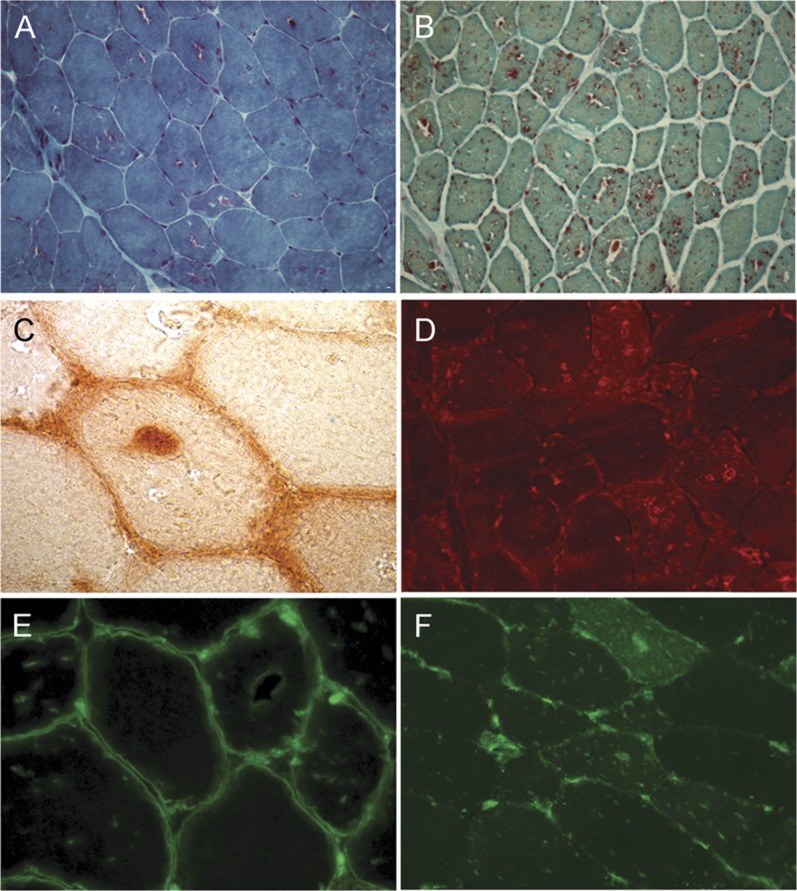
Representative light microscopy images of muscle biopsy from patients V-1 and V-2. (A) Gomori trichrome staining shows vacuoles with fuchsinophil rims. (B) All vacuoles show intense positive reaction for acid phosphatase. (C) Some vacuoles show acetylcholinesterase activity and a clear autofluorescence (data not shown). (D) LAMP-2 immunoreactivity is present at vacuole membrane level. (E) Vacuoles contain dystrophin-positive membrane debris; moreover, some vacuoles show dystrophin-positive membrane. (F) Positive staining for membrane attack complex (data not shown) and human leukocyte antigen class 1 (HLA1) is present in the cytoplasm of most muscle fibers; in addition, HLA1 is localized at membrane level. Original magnification: (A) 250×, (B) 100×, (C, E) 1,000×, (D, F) 400×.
Ultrastructural examination (figure 3) showed vacuoles containing free glycogen, lysosomal glycogen sacs, and frequent autophagic debris including myelin-like figures in several muscle fibers. There were also intermyofibrillar and subsarcolemmal accumulations of electron-dense material that in some cases were arranged to form structures similar to curvilinear bodies.
Figure 3. Ultrastructural examination of muscle tissue and blood lymphocytes.
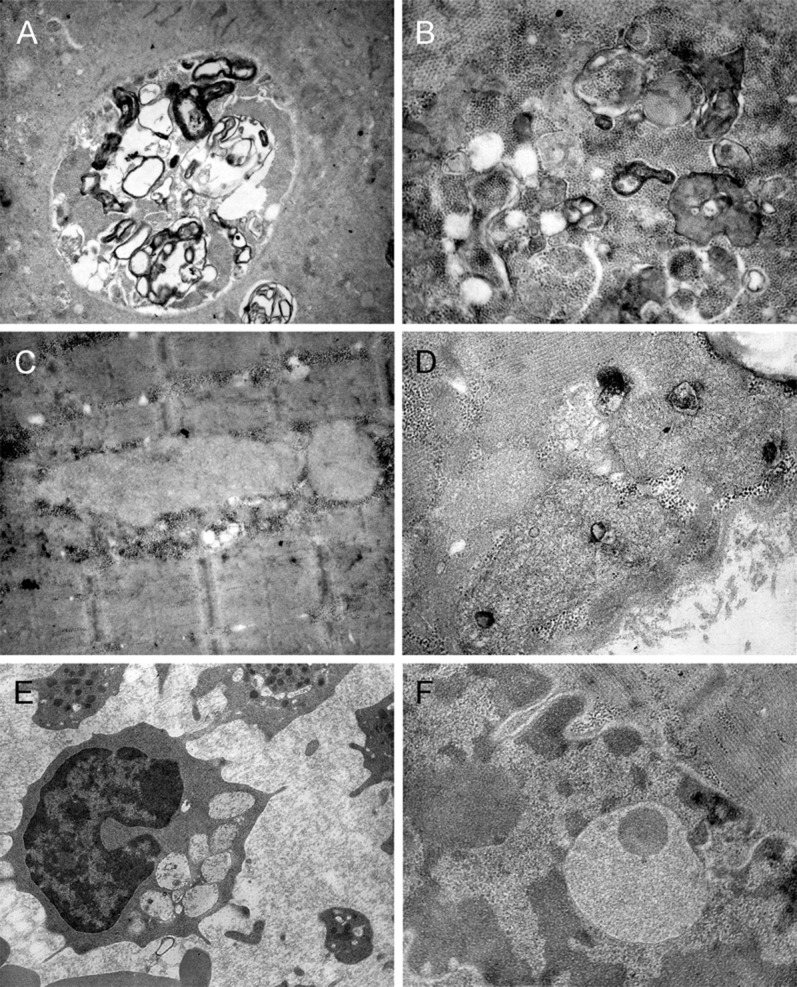
Representative electron microscopy images of muscle biopsy from patients V-1 and V-2. (A, B) Large autophagic vacuoles containing myelin figures, cytoplasmic debris, and lysosomal glycogen sacs. (C, D) Intermyofibrillar and subsarcolemmal accumulations of electron-dense material that in some cases (D) are arranged to form structures similar to curvilinear bodies. (E) Blood lymphocyte containing multiple vacuoles of large size, some of which are empty, while other vacuoles contain osmiophilic material. (F) Vacuoles similar to those observed in blood lymphocytes have been recognized in some nuclei. Original magnification: (A) 4,000×, (C, E) 12,000×, (B, D, F) 20,000×.
The ultrastructural examination of blood lymphocytes revealed cytoplasmic vacuolization characteristic of CLN3 disease. Electron microscopy did not show the presence of fingerprint profiles in either skeletal muscle or lymphocytes.
Genetic investigation.
Mitochondrial DNA and LAMP2 genetic testing were normal. Whole-genome genotyping identified 3 shared homozygosity segments (2 regions on chromosome 16 of 5 and 9 Mb, respectively, and a region of 1 Mb on chromosome 13). Exome sequencing identified 62 homozygous variants in the critical 9-Mb region. After filtering against the Single Nucleotide Polymorphism Database and synonymous polymorphism, 3 novel homozygous variants were identified: one missense in the SH2B1 gene (c.175G>A:p.Gly59Arg), one 2–base pairs deletion in the ASPHD1 gene (c.515_516insTGG:p.Gly172delinsGlyGly), and one missense in the CLN3 gene.
Because mutations in the latter are known to cause an autosomal recessive neurodegenerative disease that presents with visual failure, we further investigated the novel mutation in this gene. The mutation occurred in exon 7 of CLN3 (c.494G>A), causing a glycine to glutamic acid substitution at amino acid residue 165 (p.Gly165Glu) on the second luminal loop domain of CLN3 protein.7 By Sanger sequencing, we confirmed the presence of the homozygous c.494G>A mutation in the siblings and in one allele of the mother (figure 1A). No other living relatives were available for genetic testing. The effect of p.Gly165Glu mutation on gene function was predicted to be “probably damaging” by PolyPhen (PSIC [position-specific independent counts] score difference +2.89) and “damaging” by SIFT.
DISCUSSION
Classic juvenile CLN3 disease is an autosomal recessive inherited progressive encephalopathy of childhood due to disruption of the function of CLN3.8 The most common mutation is a 1-kb deletion that removes exons 7 and 8.9 Our cases showed a very protracted disease course without motor deterioration and only slight cognitive impairment after 40 years of disease duration. Atypical phenotypes, including some less severe, have previously been reported in individuals who are compound heterozygotes for the 1-kb deletion and some homozygous mutations.5,9,10 Multiple genetic evidences point to a pathogenic role of the novel CLN3 p.Gly165Glu mutation reported here, including in silico approaches, conservation of glycine residue at position 165 among very distant species (figure 1C), and previous identification of 6 other missense mutations in flanking regions (figure 1D).5,7
Of note, pathologic hallmarks of CLN3 disease, such as autofluorescent material (data not shown), electron-dense storage material resembling curvilinear bodies, and vacuolated lymphocytes were also present.
Although fingerprint profiles are reportedly difficult to demonstrate in muscle tissue of patients with CLN3 disease,11 their absence at lymphocytes electron microscopy examination could represent a variant in our reported cases. Also, a modifier effect of SH2B1 and ASPHD1 mutations on the pathologic and clinical phenotype cannot be excluded in our patients.
Cardiac function impairment in our patients is consistent with recently recognized cardiac involvement at later stages of the disease in protracted cases of CLN3 disease.12
Of note, muscle biopsy of both patients was characterized by the presence of autophagic vacuoles. The striking discrepancy between their normal skeletal muscle strength and the widespread vacuolar changes parallels recent experimental evidence of a possible protective role of autophagy in muscle in CLN3 disease.13,14
Although autophagic vacuoles did not show acetylcholinesterase activity on their membranes, as described in typical autophagic vacuoles with unique sarcolemmal features, the histologic features of the biopsies present striking similarities with other AVMs with autophagic vacuoles with unique sarcolemmal features, namely, dystrophin and HLA1-positive autophagic vacuoles. This finding highlights the importance of considering a protracted CLN3 disease and, if appropriate, performing CLN3 genetic testing in the diagnostic workup of individuals with AVM, particularly if there is visual impairment and cardiac muscle is involved.
GLOSSARY
- AVM
autophagic vacuolar myopathy
- CLN3
ceroid-lipofuscinosis, neuronal 3
- HLA1
human leukocyte antigen class 1
AUTHOR CONTRIBUTIONS
Andrea Cortese and Arianna Tucci: drafted and revised the manuscript, acquired, analyzed, and interpreted the data. Giovanni Piccolo, Carlo Galimberti, Pietro Fratta, Enrico Marchioni, Giampiero Grampa, Cristina Cereda, Gaetano Grieco, Ivana Ricca, Alan Pittman, and Maurizio Moggio: acquired, analyzed, and interpreted the data and revised the manuscript. Valeria Lucchini, Laura Napoli, Patrizia Ciscato, Michela Ripolone, Raffaella Violano, and Gigliola Fagiolari: performed the morphologic, immunologic, and ultrastructural study of muscle biopsies. Sara Mole, John Hardy, and Arrigo Moglia interpreted the data and revised the manuscript.
STUDY FUNDING
Supported by Parkinson's UK, NIHR/UCLH Biomedical Research Centre, Associazione Amici del Centro Dino Ferrari, University of Milan, Telethon project GTB12001E, and EuroBioBank project QLTR-2001-02769.
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Nishino I. Autophagic vacuolar myopathy. Semin Pediatr Neurol 2006;13:90–95 [DOI] [PubMed] [Google Scholar]
- 2.Nishino I, Fu J, Tanji K, et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature 2000;406:906–910 [DOI] [PubMed] [Google Scholar]
- 3.Ramachandran N, Munteanu I, Wang P, et al. VMA21 deficiency causes an autophagic myopathy by compromising V-ATPase activity and lysosomal acidification. Cell 2009;137:235–246 [DOI] [PubMed] [Google Scholar]
- 4.Williams RE, Mole SE. New nomenclature and classification scheme for the neuronal ceroid lipofuscinoses. Neurology 2012;79:183–191 [DOI] [PubMed] [Google Scholar]
- 5.Kousi M, Lehesjoki AE, Mole SE. Update of the mutation spectrum and clinical correlations of over 360 mutations in eight genes that underlie the neuronal ceroid lipofuscinoses. Hum Mutat 2012;33:42–63 [DOI] [PubMed] [Google Scholar]
- 6.Sciacco M, Fagiolari G, Lamperti C, et al. Lack of apoptosis in mitochondrial encephalomyopathies. Neurology 2001;56:1070–1074 [DOI] [PubMed] [Google Scholar]
- 7.Nugent T, Mole SE, Jones DT. The transmembrane topology of Batten disease protein CLN3 determined by consensus computational prediction constrained by experimental data. FEBS Lett 2008;582:1019–1024 [DOI] [PubMed] [Google Scholar]
- 8.Kitzmüller C, Haines RL, Codlin S, Cutler DF, Mole SE. A function retained by the common mutant CLN3 protein is responsible for the late onset of juvenile neuronal ceroid lipofuscinosis. Hum Mol Genet 2008;17:303–312 [DOI] [PubMed] [Google Scholar]
- 9.Munroe PB, Mitchison HM, O'Rawe AM, et al. Spectrum of mutations in the Batten disease gene, CLN3. Am J Hum Genet 1997;61:310–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aberg L, Lauronen L, Hämäläinen J, Mole SE, Autti T. A 30-year follow-up of a neuronal ceroid lipofuscinosis patient with mutations in CLN3 and protracted disease course. Pediatr Neurol 2009;40:134–137 [DOI] [PubMed] [Google Scholar]
- 11.Anderson GW, Goebel HH, Simonati A. Human pathology in NCL. Biochim Biophys Acta 2013;1832:1807–1826 [DOI] [PubMed] [Google Scholar]
- 12.Ostergaard JR, Rasmussen TB, Mølgaard H. Cardiac involvement in juvenile neuronal ceroid lipofuscinosis (Batten disease). Neurology 2011;76:1245–1251 [DOI] [PubMed] [Google Scholar]
- 13.Cao Y, Espinola JA, Fossale E, et al. Autophagy is disrupted in a knock-in mouse model of juvenile neuronal ceroid lipofuscinosis. J Biol Chem 2006;281:20483–20493 [DOI] [PubMed] [Google Scholar]
- 14.Uusi-Rauva K, Kyttälä A, van der Kant R, et al. Neuronal ceroid lipofuscinosis protein CLN3 interacts with motor proteins and modifies location of late endosomal compartments. Cell Mol Life Sci 2012;69:2075–2089 [DOI] [PMC free article] [PubMed] [Google Scholar]