

SUMMARY
Proteasome inhibitor (PI) resistance mechanisms in multiple myeloma (MM) remain controversial. We report the existence of a progenitor organization in primary MM that recapitulates maturation stages between B cells and plasma cells and that contributes to clinical PI resistance. Xbp1s− tumor B cells and pre-plasmablasts survive therapeutic PI, preventing cure, while maturation-arrest of MM before plasmablast stage enables progressive disease on PI treatment. Mechanistically, suppression of Xbp1s in MM is shown to induce bortezomib resistance via de-comittment to plasma cell maturation and immunoglobulin production, diminishing endoplasmic reticulum (ER) front loading and cytotoxic susceptibility to PI-induced inhibition of ER-associated degration (ERAD). These results reveal the tumor progenitor structure in MM and highlight its role in therapeutic failure.
INTRODUCTION
Multiple Myeloma (MM) is a late-stage lymphoid malignancy characterized by the accumulation of immunoglobulin (Ig) secreting plasma cells within the bone marrow. Existing therapies for MM, including the proteasome inhibitors (PIs) bortezomib (BTZ) and carfilzomib, can extend survival but are not curative (Alexanian et al., 2012; Richardson et al., 2005). To understand the failure to cure, drug resistance mechanisms must be characterized. Although the binding target of PIs has been recognized since their inception, the mechanisms of PI cytotoxicity and resistance in MM remain controversial. While some studies have linked PI cytotoxicity to stabilization of tumor suppressors (such as p53), stabilization of pro-apoptotic proteins (such as Noxa, Bim and Bik) or to stabilization of inhibitors of anti-apoptotic proteins (such as NF-κB) (Chen et al., 2010; McConkey and Zhu, 2008), others have identified induction of endoplasmic reticulum (ER) stress as the critical mediator of anti-tumor activity (Lee et al., 2003; Obeng et al., 2006).
Whereas each of the pleiotropic effects of PIs may cause cellular cytotoxicity in a specific context, induction of ER stress likely accounts for the unique sensitivity of MM to PIs in the clinic (Kim et al., 2008; Lee et al., 2003; Obeng et al., 2006). In plasma cells the ER is expanded to accommodate the synthesis of secretory Ig. Physiologic ER stress, which can be very heavy in professional secretory cells, is counteracted via adaptations known collectively as the unfolded protein response (UPR). Three ER resident transmembrane proteins (Ire1, Perk and Atf6) activate overlapping components of the UPR, which counters unfolded protein stress by suppressing global mRNA translation whilst selectively upregulating pathways that promote protein folding or degradation (Ron and Walter, 2007). PIs impede ER homeostasis by inhibiting proteasome-assisted ER-associated degradation (ERAD) (Kim et al., 2008), causing ER stress. As a consequence, PI-treated tumor cells characteristically inactivate Eif2α, increase Atf4 and upregulate the expression of UPR genes such as CHOP (DDIT3) and HSPA5 (Chen et al., 2010; Lee et al., 2003; Obeng et al., 2006; Zhu et al., 2010).
The mechanisms by which tumor cells escape the multiple cytotoxic effects of PIs are not implicit. One compelling possibility is that proteasome inhibition is prevented by mutation of the PI-binding site. Indeed, mutations of the BTZ-binding site on proteasome subunit β5 (PSMB5), or over expression of PSMB5, have been observed in various tumor cell lines; and have been asserted to be the mechanism of PI resistance in both MM and leukemia (Balsas et al., 2012; de Wilt et al., 2012; Franke et al., 2011; Oerlemans et al., 2008; Ruckrich et al., 2009). Notably, however, BTZ resistance in these studies was developed in vitro, using cell lines adapted to PI exposure, and the legitimacy of these models for the representation of drug resistance in patients remains unverified. To the contrary, multiple sequencing studies of primary MM tumors have failed to identify any proteasome mutations to account for PI-resistance (Chapman et al., 2011; Lichter et al., 2012; Politou et al., 2006; Wang et al., 2008), suggesting that such mutations, though capable of mediating PI-resistance in vitro, are infrequent in the clinic. In the present study we sought to define mechanisms of PI resistance in MM patients.
RESULTS
Ire1-Xbp1 signaling is dispensable for MM tumor cell viability but required for BTZ-induced cytotoxicity in vitro
We have previously conducted kinome- and genome-scale siRNA studies in the KMS11 MM cell line treated with BTZ, to functionally identify synthetic lethal chemo-sensitizing targets (Tiedemann et al., 2012; Tiedemann et al., 2010; Zhu et al., 2011). We re-analyzed these results to identify genes whose silencing protected MM cells from BTZ-induced cytotoxicity (Figure 1A,B). While relatively few BTZ-rescue genes were detected, “endoplasmic reticulum to nucleus signalling 1” (ERN1), better known as IRE1, appeared critical for BTZ response in both studies. On kinome screening IRE1 was the kinase whose loss was most associated with BTZ resistance, whereas in genome-scale siRNA studies IRE1 ranked at the top 1% of genes required for BTZ-induced cell death.
Figure 1. Loss of Ire1 or Xbp1 Reduces the Cytotoxic Activity of BTZ in MM.
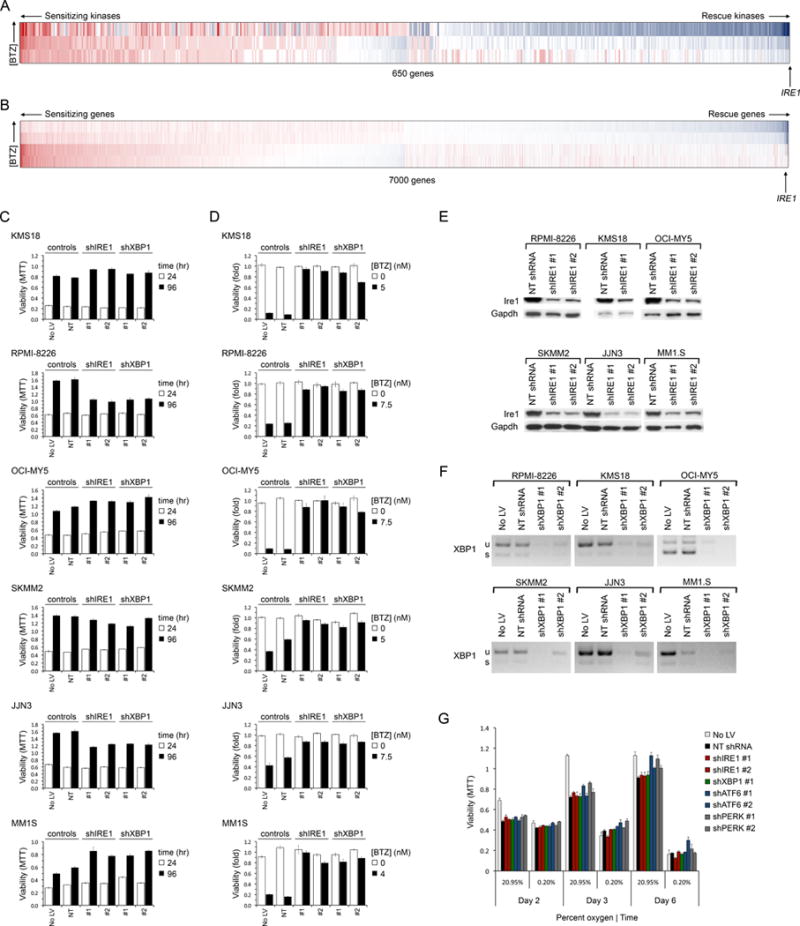
(A–B) Synthetic lethal BTZ-siRNA screens of the kinome (A) and ‘druggable’ genome (B), conducted in KMS11 MM cells, showing genes ranked by the mean Bliss independence score of siRNA at BTZ IC90. Synthetic lethal genes whose RNAi caused greater than additive cytoxicity (in red) with BTZ are to the left; rescue genes whose RNAi reduced BTZ cytotoxcity (in blue) are to the right. IRE1 ranking is shown.
(C) Viability (MTT assay) of MM cell lines expressing shIRE1 or shXBP1 for the indicated time following lentiviral (LV) infection. Controls included uninfected cells and cells expressing non-targeted (NT) shRNA.
(D) Viability (MTT) of MM cell lines infected with LV to express shIRE1 or shXBP1 for 1 day and then treated with vehicle or BTZ at approximately IC75 (4–7.5 nM) for 3 days.
(E) Western blot of Ire1 levels in shIRE1-treated and control NT shRNA-treated MM cells.
(F) RT-PCR analysis of XBP1 mRNA in shXBP1-treated and control MM cells. u=unspliced; s=spliced.
(G) Viability of OCI-MY5 MM cells treated with UPR gene shRNA, following culture in normoxia (20.95% O2) or extreme hypoxia (0.2% O2).
Error bars represent ±SEM (n=3).
See also Figure S1.
Identification of IRE1 in this context was surprising because Ire1 knockdown prevents activation of one branch of the homeostatic UPR pathway, and accordingly its loss might be predicted to diminish a cell’s ability to respond to ER stress – rendering it more sensitive to BTZ – contrary to our observation. Therefore, to verify this result and to explore whether Ire1 suppression induces BTZ resistance in other MM subtypes, Ire1 was silenced in 6 other MM cell lines (Figure 1C–E); cells were then grown in the presence or absence of BTZ. In the absence of drug, loss of Ire1 was well tolerated (Figure 1C), albeit that mild reductions in proliferation were observed for RPMI-8226 and JJN3 cells. Conspicuously, however, suppression of Ire1 (Figure 1E) induced relative BTZ resistance in all MM lines tested (Figures 1D and S1A–D), confirming our initial observation.
Ire1 activates the UPR via the bZIP transcription factor, Xbp1. When stimulated by ER stress, Ire1 splices 25 nucleotides from the unspliced XBP1 mRNA (XBP1u) to yield a frame-shifted mRNA (XBP1s) that encodes the active transcription factor (Calfon et al., 2002). As XBP1 was not tested in our original siRNA studies, its effect on BTZ-response was assessed next. Surprisingly, silencing of XBP1 (Figure 1F), like IRE1, was well tolerated in MM cells (Figure 1C) even under conditions of extreme hypoxia (Figure 1G), contrary to previous reports that XBP1 is required for myeloma pathogenesis (Carrasco et al., 2007). Strikingly, however, loss of Xbp1 induced BTZ resistance in all 6 MM cell lines tested, mirroring Ire1 suppression (Figure 1D). Similar results were obtained with 4 distinct shRNA. Therefore the Ire1-Xbp1 pathway is dispensable for MM cell viability but promotes BTZ cytotoxicity, at least in MM cell lines grown in vitro.
Ire1-Xbp1 Signaling is Suppressed in BTZ-Refractory Primary MM
To determine if attenuation of Ire1-Xbp1 signaling occurs in primary MM and plays a role in BTZ resistance, we next examined the gene expression of pretreatment tumor samples from MM patients who developed either a complete response (CR) or progressive disease (PD) with single-agent BTZ treatment (Mulligan et al., 2007; Richardson et al., 2005). Conspicuously, almost all Xbp1 target genes (Shaffer et al., 2006) were expressed at lower levels in PD tumors (Figure 2A), concordant with our observation that Ire1-Xbp1 signaling modulates BTZ sensitivity in MM cell lines. However, the difference in Xbp1-target gene expression between BTZ CR and PD primary MM tumors was modest, suggesting that, pre-treatment, Xbp1s activity may be suppressed in only a subpopulation of cells in BTZ-refractory tumors.
Figure 2. Ire1-Xbp1 Signaling is Suppressed or Mutated in BTZ-Refractory Primary Tumors from MM Patients.
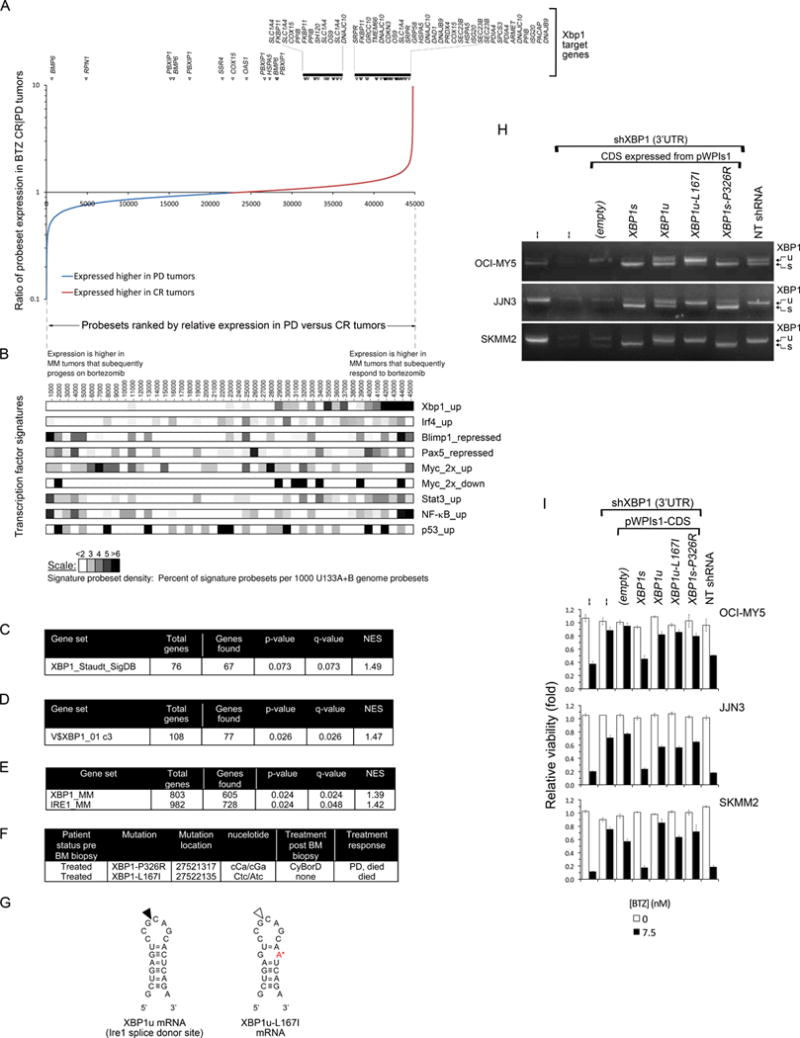
(A) Comparative gene expression of BTZ-responsive and -resistant primary MM from patients enrolled on the APEX 039 clinical study. Affymetrix probe sets are plotted by rank-order of differential mean expression in tumors that responded completely (CR) or that showed progressive disease (PD) following BTZ therapy. The vertical axis shows the log ratio of mean tumor expression intensity in the two response groups. Xbp1s target genes are identified above the plot.
(B) Heatmaps comparing the relative expression of an Xbp1 gene signature with that of other B cell transcription factor signatures in BTZ-sensitive (CR) versus BTZ-resistant (PD) primary MM tumors, using the same probe set rank-order as in (A). To normalize for signature sizes results are plotted as the % of signature probe sets per 1000 rank-ordered probe sets. The suffixes up, down, repressed and 2x reflect the selection criteria used to define genes in the transcription factor signatures.
(C–E) Gene set enrichment analysis (GSEA) for Xbp1 target genes in primary MM, amongst genes preferentially expressed pre-treatment in BTZ-responsive versus BTZ-refractory tumors. Four distinct Xbp1 signatures were used, derived either by overexpression of Xbp1 in B cells (C), by identification of genes with Xbp1-binding motifs within their promoters (D), or by shRNA knockdown of either Xbp1 or Ire1 in MM cells (E). NES=normalized enrichment score.
(F) XBP1 mutations identified on initial genome sequencing of 38 patient MM, and subsequent patient treatment response. CyBorD = cyclophosphamide, BTZ, dexamethasone.
(G) The wild-type (left) and L167I mutation (right, indicated by the red letter) XBP1u mRNA sequences at the Ire1 splice site (indicated by the black arrow on the left) hairpin.
(H) RT-PCR analysis of XBP1u and XBP1s mRNA in three MM cell lines engineered to express mutant XBP1 CDSs recapitulating mutations identified in primary tumors. Endogenous XBP1 mRNA was suppressed using a 3′UTR-targeted shXBP1.
(I) Relative viability (MTT) of the MM cell lines in (H) following treatment with BTZ or vehicle for 3 days (mean ± SEM).
See also Figure S2.
Notably, from a library of lymphoid transcription factor signatures, only Xbp1 correlated closely with clinical BTZ response. By comparison, no association was observed between BTZ response and tumor expression of p53, NF-κB or Stat3 (Figure 2B). Similarly, no positive association was observed between BTZ resistance and proteasome expression in primary MM; instead most proteasome subunits including PSMB5 are down-regulated in BTZ-resistant MM (Figure S2), contrary to in vitro resistance models.
The preferential expression of Xbp1 target genes in BTZ-sensitive MM tumors, relative to BTZ-resistant tumors, was confirmed by gene set enrichment analysis (GSEA) using three independent Xbp1 gene signatures, derived by distinct methods. Whereas an Xbp1 signature derived from over-expression of Xbp1 in B cells (Shaffer et al., 2004) was moderately enriched in BTZ-sensitive primary MM tumors (q=0.07) (Figure 2C), a second Xbp1 signature representing genes with Xbp1-binding promoters, was significantly enriched (q=0.026) (Figure 2D). A third Xbp1 signature, derived from profiling of MM cells following Xbp1 knockdown, most closely matched the biological context in which Xbp1 activity was being assessed and showed the most significant enrichment in BTZ-sensitive MM (q=0.024) (Figure 2E). In addition, a fourth related Ire1 gene signature, derived from MM cells with Ire1 knockdown, was also enriched in BTZ-sensitive primary MM (q=0.048) (Figure 2E), consistent with the requirement for Ire1 activation of Xbp1. Thus, GSEA of primary MM tumors from patients treated with single-agent BTZ confirm that down-regulation of the Ire1-Xbp1 pathway occurs in primary myeloma and correlates with therapeutic PI resistance.
XBP1 mutations in MM
Suppression of Xbp1 signaling may occur through a variety of mechanisms, including XBP1 gene mutation. Notably, while genome sequencing of 20 treated MM tumors did not identify any proteasome PSMB5 mutations, this initial genomic analysis of MM did identify two mutations in XBP1 (Chapman et al., 2011), both in treatment-refractory tumors (Figure 2F). Conspicuously, the first mutation, XBP1-L167I, maps to the Ire1 splice site on XBP1u and, we predicted, might prevent splice activation to XBP1s (Figure 2G). The second mutation, XBP1s-P326R, involves a non-conservative missense mutation within the trans-activation domain of Xbp1s, where loss of proline’s conformational rigidity may affect tertiary structure and influence transcriptional activity.
To confirm if either or both of these mutations are inactivating, identical XBP1 mutants were generated and expressed in MM cell lines (Figure 2H,I); simultaneously endogenous XBP1 was silenced using a 3′UTR-targeted shXBP1. As predicted, the mutation, XBP1-L167I, consistently prevented splicing of XBP1u mRNA to XBP1s, whereas unmutated XBP1u mRNA, expressed in parallel, could be processed. While both XBP1u and XBP1s are translated, only Xbp1s protein is transcriptionally active; Xbp1u, which has a functional N-terminal leucine zipper motif, instead acts as an inhibitor of Xbp1s (Lee et al., 2003). L167I therefore inhibits XBP1 both by decreasing Xbp1s and by increasing Xbp1u.
We next assessed whether these XBP1 mutations have effects on drug response by testing the mutant XBP1 cell lines for BTZ sensitivity (Figure 2I). Consistent with our earlier observations, knockdown of endogenous Xbp1 in OCI-MY5, JJN3 or SKMM2 control cells, using an XBP1 3′UTR-directed shRNA, attenuated BTZ cytotoxicity. Constitutive expression of an shRNA-resistant XBP1s CDS however re-established sensitivity to BTZ, despite endogenous XBP1 silencing, verifying that BTZ sensitivity in MM cells is modulated specifically by Xbp1s. In contrast, constitutive expression of the mutant XBP1s-P326R or XBP1u-L167I CDS in MM cells failed to re-sensitize shXBP1-treated MM cells to BTZ, indicating that both mutations are functionally inactivating and promote BTZ resistance. These studies indicate that primary MM tumors tolerate inactivating mutations in XBP1 and that PIs therapy may exert a selective pressure for the emergence of cells with suppression of this pathway.
Of UPR stress transducers, Only Ire1-Xbp1, and Not Atf6 or Perk, Regulate BTZ Sensitivity
Ire1 is one of three ER transmembrane proteins that transduce stress signals to the nucleus to induce the UPR. To determine if loss of either of the other ER stress transducers, Perk or Atf6, induced BTZ resistance, each transducer was knocked down separately using two shRNA in RPMI-8226 and SKMM2 MM cells (Figure 3A). However, suppression of Atf6 or Perk, unlike suppression of Ire1-Xbp1, had no effect on BTZ sensitivity (Figures 3B, 3C and S3), indicating that amongst the ER stress pathways BTZ sensitivity maps specifically to Ire1-Xbp1.
Figure 3. Only Ire1 among ER Stress Transducers Regulates BTZ Sensitivity in MM.
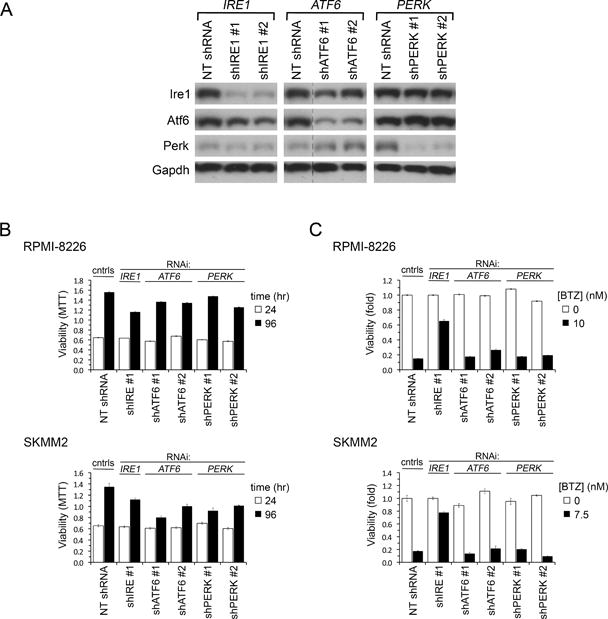
(A) Immunoblot of indicated proteins in RPMI-8226 MM cells 1.5 days after LV infection to express shIRE1, shATF6, shPERK or control NT shRNA. In the ATF6 panel irrelevant lanes were removed between NT shRNA and shATF6 lanes for all four blots; all samples were run on the same gel.
(B) Growth of MM cell lines expressing shIRE1, shATF6, shPERK or control NT shRNA measured by sequential MTT assays 24 and 96 hr after LV infection.
(C) Viability (MTT assay) of MM cell lines infected with LV for 24 hr to express shIRE, shATF6 or shPERK and then treated with BTZ at IC90 or vehicle for 3 days.
Error bars show ±SEM.
See also Figure S3.
BTZ Resistance is Not Due to Increases in Perk or Atf6 or Loss of an Xbp1s Death Signal
Whereas BTZ cytotoxicity in MM has been linked to the induction of overwhelming ER stress, we show here that loss of the Ire1-Xbp1 branch of the ER homeostasis apparatus reduces BTZ lethality, contrary to expectation. To address this dilemma, and to clarify the mechanism of PI resistance, we hypothesized that loss of Ire1-Xbp1 signaling in MM cells might cause a compensatory increase in Atf6 or Perk signaling (and in downstream UPR gene expression) that might ‘pre-adapt’ MM cells to better withstand proteasome inhibition. To investigate this possibility, we generated MM cells in which both Ire1 and Atf6, or both Ire1 and Perk, were concurrently suppressed (Figure 4A,B) Notably, double knockdown of Ire1 and Perk, or of Ire1 and Atf6 together, in MM cells, failed to diminish the induction of BTZ resistance by Ire1 silencing (Figure 4C,D), indicating that neither of these parallel pathways, individually, mediates the BTZ resistance seen on Ire1 suppression. Conceivably, however, Ire1-Xbp1 suppression might cause compensatory increases in both Atf6 and Perk, either of which might then redundantly prime cells to survive proteasome inhibition. Therefore, to test this hypothesis, we also generated triple knockdown MM cells in which Ire1, Atf6 and Perk were all concurrently suppressed (Figure 4E). Surprisingly, these cells proved viable in tissue culture, though demonstrated a growth disadvantage (Figure 4F). Notably however simultaneous silencing of both Atf6 and Perk failed to diminish BTZ resistance induced on Ire1 silencing (Figure 4G). Therefore BTZ resistance arising from Ire1-Xbp1 loss is not mediated via parallel UPR pathways.
Figure 4. BTZ Resistance Induced by Ire1-Xbp1 Suppression is Not Due to Adaptive Increases in Perk or Atf6.
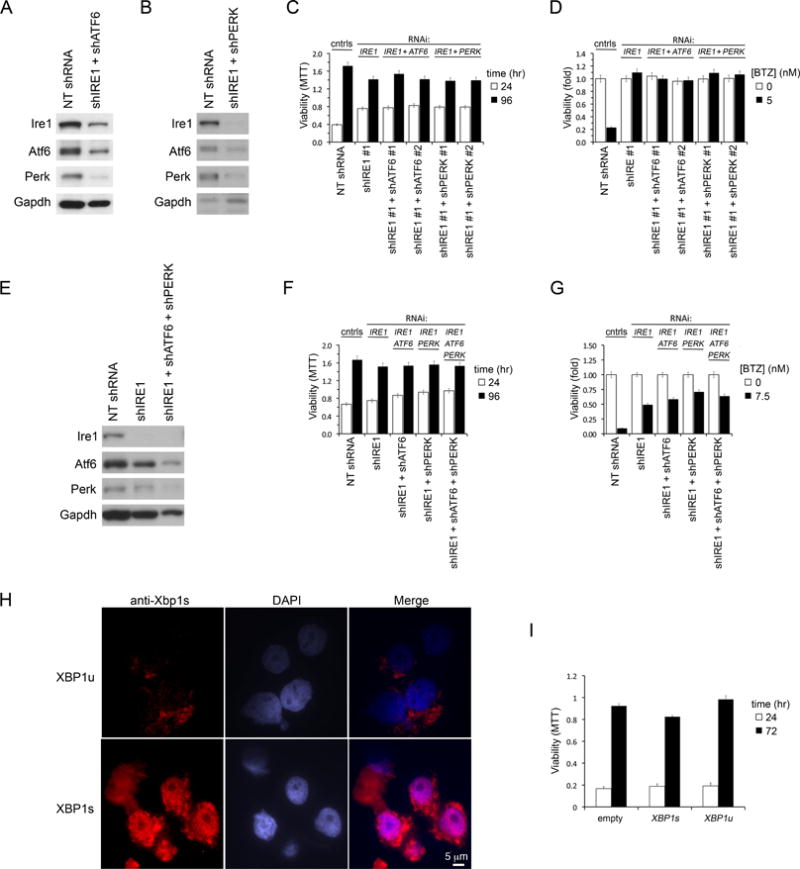
(A) Immunoblot of Ire1, Atf6 and Perk in JJN3 MM cells expressing concurrently shIRE1 and shATF6, 2 days post infection. Control cells expressed NT shRNA.
(B) Same as (A) except that MM cells concurrently expressed shIRE1 and shPERK.
(C) Growth of JJN3 MM cells following knockdown of Ire1, or dual knockdowns of Ire1+Atf6 or Ire1+Perk, measured by sequential MTT assays 1 and 4 days after infection.
(D) Viability (MTT) of JJN3 MM cells infected with indicated LV shRNA combinations for 1 day and then treated with BTZ or vehicle for 3 days.
(E) Immunoblot of indicated proteins in JJN3 MM cells infected with LV to concurrently express shRNA targeting all 3 ER UPR transducers Ire1, Atf6 and Perk. Control cells expressed NT shRNA.
(F) Growth of JJN3 MM cells following triple knockdown of Ire1+Atf6+Perk, compared with single or dual gene knockdowns, measured by MTT assays 1 and 4 days after LV infection.
(G) Same as (D) but includes JJN3 MM cells with simultaneous triple knockdown of Ire1, Atf6 and Perk.
(H) Immunofluorescence staining for Xbp1s in OCI-MY5 MM stably overexpressing XBP1u or XBP1s CDS. Nuclei are stained with DAPI. Scale is identical for all micrographs.
(I) Growth of OCI-MY5 cells over-expressing XBP1u or XBP1s, compared with control cells infected with empty expression vector, measured by repeat MTT assays at 0 and 3 days.
Error bars represent ±SEM. See also Figure S4.
We next speculated that the overwhelming ER stress generated by PI therapy might induce a lethal signal transmitted via Ire1-Xbp1 and that loss of this death signal might underlie BTZ resistance. However, MM cells forced to constitutively over-express Xbp1s protein, the final common effector of Ire1-Xbp1 signaling, remain viable in vitro (Figures 4H,I and S4), and in vivo (Carrasco et al., 2007), providing no evidence for the existence of an Xbp1s death signal in these cells.
Ire1-Xbp1 Suppression in MM Blocks Commitment to Plasma Cell Differentiation and lowers ER stress
XBP1 is essential for differentiation of B lymphocytes to plasma cells, particularly for the terminal maturation of proliferating plasmablasts to plasma cells, for induction of Ig secretion, and for bone marrow colonization (Carrasco et al., 2007; Hu et al., 2009; Reimold et al., 2001). As the role of Ire1-Xbp1 signaling in BTZ resistance remained unaccounted, we next surmised that loss of Ire1-Xbp1 signaling might cause de-commitment of MM cells to terminal plasma cell differentiation and that tumor cell immaturity might account for cellular BTZ resistance.
To investigate this possibility, we first assessed MM cell lines for evidence of reversion of plasma cell commitment following RNAi-induced silencing of IRE1 or XBP1. Following knockdown of Ire1 or Xbp1, OCI-MY5 cells appeared smaller and rounder with less cytoplasm and peri-nuclear hof (Figure 5A), resembling pre-plasmablasts (Jourdan et al., 2011). Profiling of gene expression in IRE1- or XBP1-silenced MM cell lines demonstrated partial repression of markers of plasma cell maturation such as SDC1 (CD138), CD38, IL6R and IL6ST in these cells and simultaneous weak mRNA expression of B cell markers such as CD20 and PAX5 (Figure 5B). Diminished expression of CD138 ± CD38 surface antigens on XBP1-silenced RPMI-8226 and JJN3 MM cells was confirmed by flow cytometry (Figure 5C), while up regulation of Pax5 protein in Ire1-Xbp1 inactivated MM cells was confirmed by immunoblot (Figure 5D). As Pax5 is expressed in B cells but is suppressed in plasma cells (Nutt et al., 2011), these findings are consistent with the hypothesis that Ire1-Xbp1 loss in MM cells causes a modest reversal of plasma cell maturation.
Figure 5. Ire1-Xbp1 Suppression and BTZ Resistance in MM are Associated with De-Commitment to Plasma Cell Differentiation.
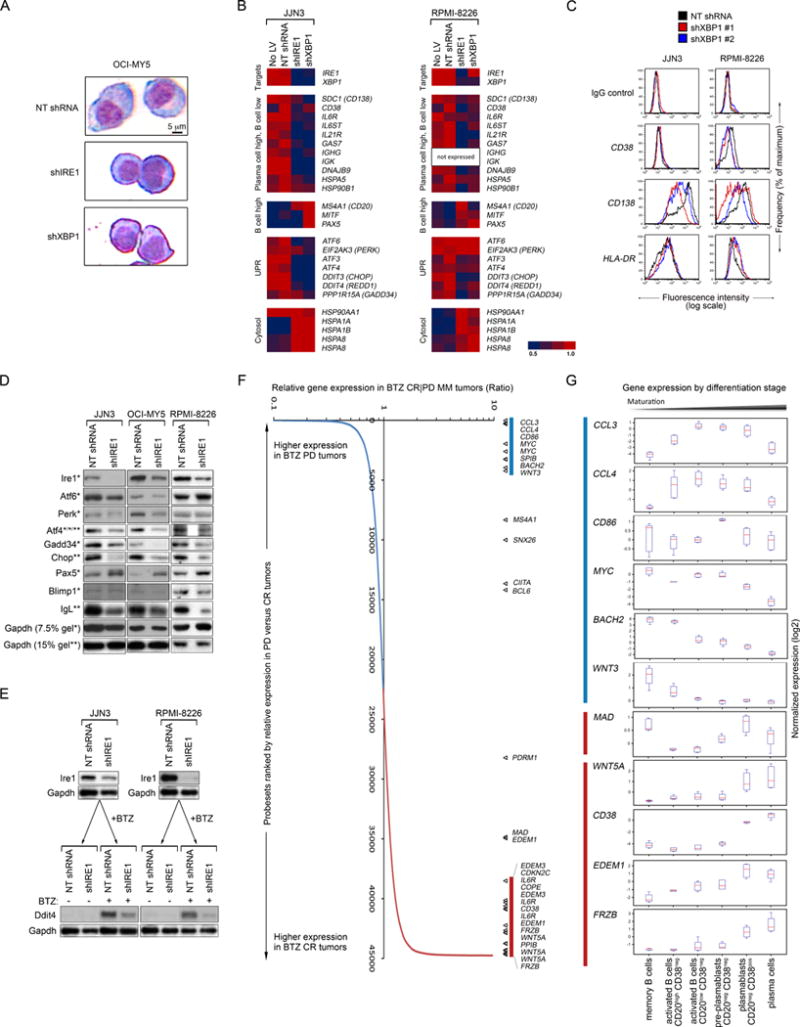
(A) Giemsa stain of OCI-MY5 cells following expression of NT shRNA, shIRE1 or shXBP1. Scale is identical for all micrographs.
(B) Relative mRNA abundance of indicated genes in JJN3 and RPMI-8226 MM cell lines expressing shIRE1, shXBP1 or control NT shRNA.
(C) Flow cytometric analysis of surface CD38 and CD138 on JJN3 and RPMI-8226 MM cell lines expressing shXBP1 or control NT shRNA.
(D) Immunoblot analysis of indicated proteins in JJN3, OCI-MY5 and RPMI-8226 MM cell lines 3 days after LV infection to express shIRE1. Proteins were separated on either 7.5%(*) or 15%(**) polyacrylamide gels. IgL reflects κ for JJN3 and λ for RPMI-8226 and OCI-MY5.
(E) Immunoblot of Ddit4 induction by BTZ (100 nM, 6 hr) in JJN3 and RPMI-8226 MM cells expressing either shIRE1 or control shRNA.
(F) Relative expression of indicated B cell and plasma cell maturation genes amongst primary MM that respond (with CR) or fail to respond (with PD) to clinical BTZ treatment. Data are from pre-BTZ samples from patients treated on the APEX 039 study.
(G) Box-whisker plots of relative expression of indicated genes from (E) during physiological B cell to plasma cell maturation, with median shown in red.
We next assessed Ig production in Xbp1s-silenced MM cells. By gene expression profiling, loss of Xbp1 or Ire1 was associated with decreased expression of both IgG heavy chain (IGHG) and Igκ-light chain (IGK), in IgGκ-secreting JJN3 cells, as well as decreased expression of IL21R, a regulator of IgG production (Ozaki et al., 2002) (Figure 5B). A marked decline in Ig light chain protein production in Ire1-Xbp1s silenced MM cells was also observed by immunoblot (Figure 5D). XBP1 is a known pre-requisite for establishing Ig production by B cells (Reimold et al., 2001) and our data indicate that MM plasma cells remain dependent on Xbp1s for ongoing Ig synthesis.
We next hypothesized that by arresting or reversing their secretory plasma cell maturation, MM tumor cells may lessen their ER front loading by secretory Ig and thereby diminish both their basal ER stress and their risk of lethal ER stress arising from PI-induced ERAD inhibition, providing a mechanism by which Xbp1s loss (and other forms of maturation arrest) might cause PI resistance. Consistent with this, MM cell lines in which IRE1 or XBP1 were silenced showed reduced expression of UPR genes such as ATF4, DDIT3 (CHOP), DDIT4 (REDD1), PPP1R15A (GADD34) and HSPA5 (BiP); and of downstream chaperones HSP90B1 and DNAJB9 (Figure 5B). Notably, this diminished expression of UPR genes in Ire1-Xbp1s inactivated MM cells reflected reduced ER stress rather than simple loss of Xbp1s-driven gene transcription as Ire1-Xbp1s silencing was accompanied by reduced or stable expression of genes in parallel ER stress pathways such as EIF2AK3 (PERK), ATF4 and PPP1R15A (GADD34) (Figure 5B,D). If loss of Xbp1s-driven gene expression alone accounted for the decline in CHOP and chaperones, without a concurrent decrease in ER stress, these parallel UPR homeostasis pathways would instead be activated as they assumed the burden of ER homeostasis. A significant decline in basal expression of Perk-responsive UPR elements Atf4, Gadd34 and Chop in shIRE1-treated MM cells was confirmed by immunoblot (Figures 5D and S5A,B). Furthermore, suppression of Ire1 caused diminished induction of the Atf4 response gene DDIT4 (REDD1) following either BTZ- or thapsigargin-induced ER stress (Figures 5E and S5C). As Ire1-Xbp1s suppression causes de-commitment to Ig production and a decline in ER stress; and as PI cytotoxicity in MM has been linked to exacerbation of ER stress, which in plasma cells is driven by Ig production (Obeng et al., 2006), these data provide a mechanistic link between Ire1-Xbp1 silencing in MM cells and PI-insensitivity.
To determine if BTZ-refractory MM tumors in the clinic show evidence of plasma cell de-commitment or maturation arrest, we next examined the gene expression of tumor samples from MM patients who developed either a CR or PD following treatment with BTZ (Figure 5F). We compared the differential gene expression of the two tumor groups with programmed changes in gene expression seen during the differentiation of B cells to plasma cells (Figure 5G and Table S1) (Jourdan et al., 2011). Gene sets defining expression differences between CD20+ memory B cells, activated CD20low CD38− B-cells, CD20− CD38− pre-plasmablasts, CD20− CD38+ CD138− plasmablasts and plasma cells were derived, using the expression profile of mature plasma cells as a common denominator. Notably, by GSEA, expression of plasma cell maturation genes was significantly enriched in BTZ-responsive tumors (q=0.03, NES +1.5) and reduced in BTZ-resistant tumors (q<0.01, NES −1.5), suggesting that while BTZ responsive tumors contain a high proportion of mature plasma cells, BTZ resistant MM tumors may contain greater proportions of less differentiated cells.
Included in the leading edge of differentiation genes whose expression was diminished in BTZ-resistant primary MM were FRZB, WNT5A, IL6R, CD38, EDEM1 and EDEM3 (Figure 5F; red bar), which are all expressed at higher levels in plasma cells than in activated B cells or pre-plasmablasts (Figure 5G); their relative under-expression in BTZ-refractory MM therefore supports the presence of maturation arrest in PI-resistant primary tumors. Conversely, genes that appeared to be up regulated in BTZ-resistant MM tumors included the chemokine ligands CCL3 and CCL4, CD86, MYC, BACH2 and WNT3; which are expressed at higher levels in activated B cells and pre-plasmablasts than in plasma cells (Figure 5F), suggesting that BTZ-refractory primary MM may be associated with increases in these progenitor subpopulations. Additional gene expression analyses of B cell maturation support the presence of a “sweet spot” maturation stage intermediate between B cells and plasma cells that may be intrinsically less susceptible to PI-induced cytotoxic ER stress (Figure S5D,E).
Xbp1s− B cell progenitors in MM
Our observations that Ire1-Xbp1 suppression correlates with BTZ resistance in MM, and that B cell and pre-plasmablast gene set expression is increased in BTZ-refractory MM, lead us to examine primary MM tumors for the existence of Xbp1s− progenitor populations. Using FACS, viable plasma cells and B-lineage cells were isolated from patient bone marrows and then segregated according to CD38/CD138 status (Figures 6A and 6B). Tumor cells within these subpopulations were identified by the presence of tumor clone FISH abnormalities, and were further characterized for Xbp1s and CD20 expression by IF-FISH (Figures 6C and S6). Using this technique we isolated five subpopulations of MM tumor cells from primary MM samples, including CD20+ CD38− CD138− Xbp1s− B cells, CD20low CD38− CD138− Xbp1s− activated B cells, CD20− CD38− CD138− Xbp1s− pre-plasmablasts, CD38+ CD138low Xbp1s+ plasmablasts and CD38+ CD138+ Xbp1s+ plasma cells. The existence of these tumor progenitors cells was observed directly by IF-FISH and was confirmed by the greater-than background rate of cells positive for the tumor FISH abnormality within sorted subpopulations. Virtually all CD38− CD138− tumor cells were Xbp1s− while the majority of tumor B cells were CD27+ (Figure 6C), consistent with a post germinal centre memory phenotype. In some samples tumor B cells accounted for the majority of bone marrow memory B cells (not shown). Xbp1s− tumor B cells and pre-plasmablasts express lower levels of Chop than Xbp1s+ tumor plasma cells and plasmablasts (Figure 6D), suggesting that Xbp1s− tumor progenitors, like Xbp1s-silenced MM cell lines, have lower basal ER stress than tumor cells that have undergone plasma cell maturation.
Figure 6. Tumor Progenitor Subpopulations in a Newly Diagnosed MM Patient.
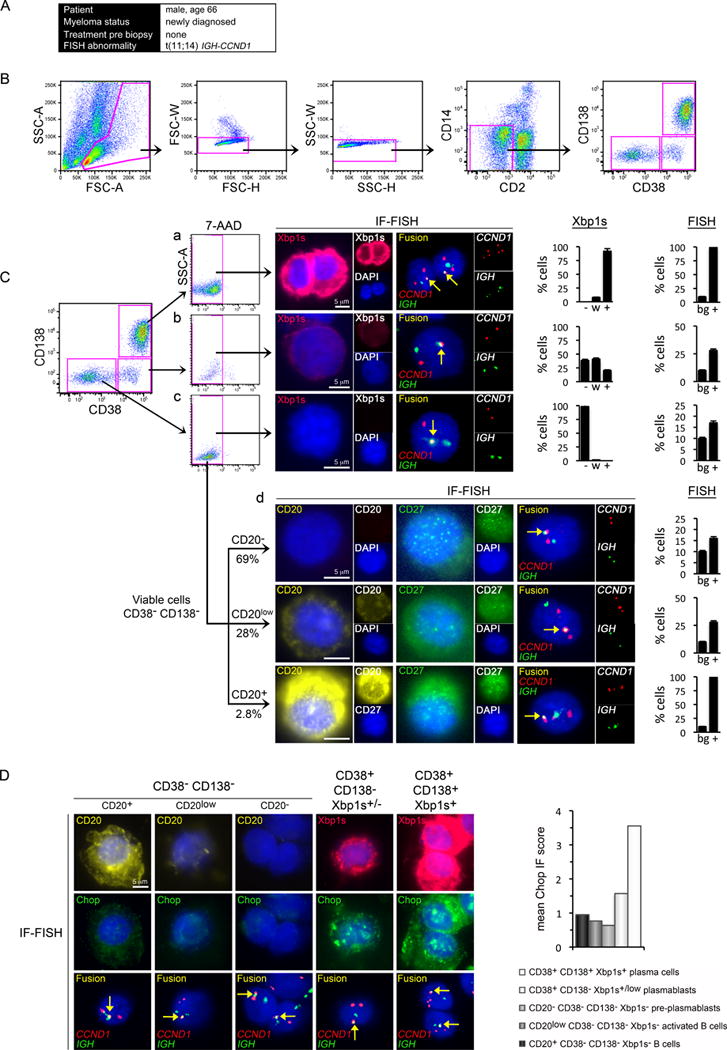
(A) The clinical information of the patient whose bone marrow was examined.
(B) Bone marrow MM cells were enriched by sequential FACS for lymphoid FSC/SSC (left panel), by removal of aggregates (second and third panels) and by negative selection for CD2 (T and NK cells) and CD14 (monocytes). Gates are shown in pink.
(C) Tumor cells gated into subpopulations by CD38 and CD138 status. Single viable CD38+ CD138+ plasma cells (a), CD38+ CD138− plasmablasts and B cells (b), and CD38− CD138− B cells (c) were examined for concurrent Xbp1s protein expression and for IGH-CCND1 gene fusion (arrows) by IF-FISH on single cells. Viable CD38− CD138− B cells from (Cc) were further examined for concurrent CD20 and CD27 expression and for IGH-CCND1 by IF-FISH (d). Each row illustrates a single cell. Scale bars (5μm) are identical for IF and FISH; monochrome insets are shown at 0.5x scale. The overall proportion of cells in each subpopulation with strong (+), weak (w) or no (−) expression of Xbp1s and the proportion of cells positive (+) for IGH-CCND1, compared with the background rate (bg), are quantified and shown in the bar graphs.
(D) Ddit3 (Chop) protein expression in primary MM tumor progenitors. Progenitor stages (left to right) were characterized by CD20, Xbp1s and Chop IF-FISH. Each column illustrates single cells. Scale is consistent across micrographs. The mean Chop IF score by MM progenitor stage is quantified and shown in the bar graph.
See also Figure S6.
Xbp1s− MM B cell and pre-plasmablast progenitors survive therapeutic BTZ and are enriched in BTZ refractory MM
In newly diagnosed MM and in patients relapsing following treatment discontinuation the predominating tumor cells are Xbp1s+ plasma cells or plasmablasts (Figures 6, 7A, and 7D), although progenitor subpopulations including CD20− CD38− CD138− Xbp1s− pre-plasmablasts and CD20low CD38− CD138− Xbp1s− activated B cells were consistently identified (Figures 6Cd and 7Af). Progenitor CD20+ CD38− CD138− Xbp1s− B cells, with strong CD20 staining were identified in nearly all samples but were comparatively rare.
Figure 7. Tumor Progenitor Subpopulations in Relapsed or Refractory MM.
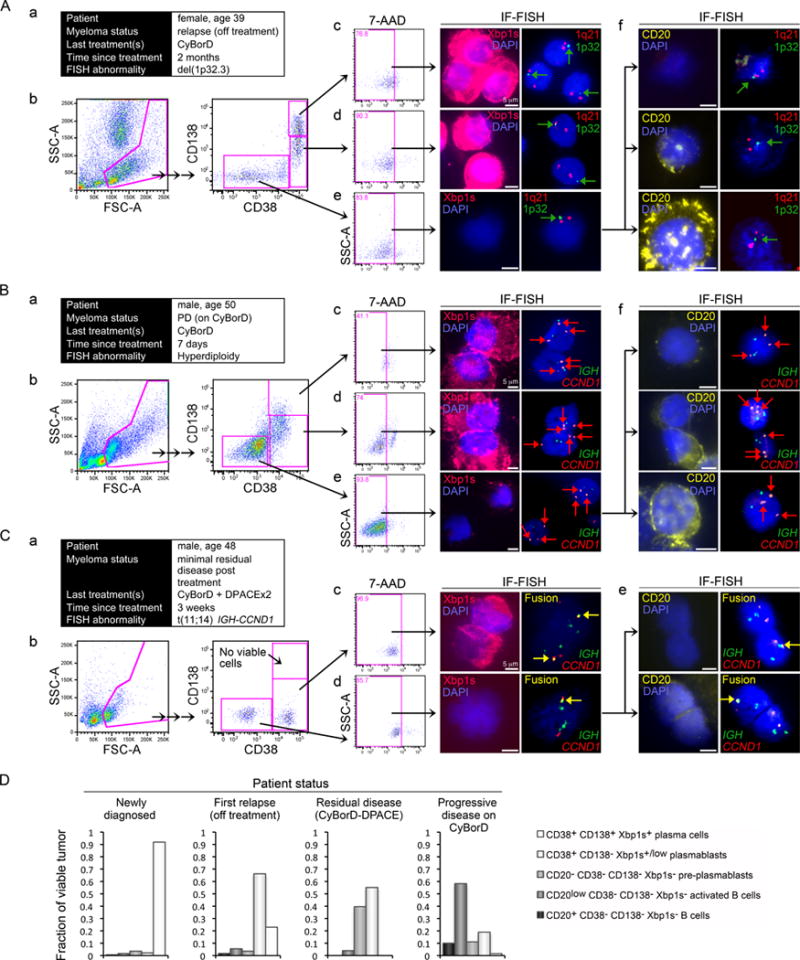
(A) Findings from a patient with relapsed MM following BTZ discontinuation. (a) The clinical information of the patient whose bone marrow was examined. (b) Patient bone marrow was FACS sorted as in Figure 6. Viable CD38+ CD138+ plasma cells (c), CD38+ CD138−/low plasmablasts (d), and CD38− CD138− cells (e) were examined for the presence of Xbp1s by IF and for the loss of chromosome 1p32 (arrows), which contains the CDKN2C locus, by FISH. (f) Viable CD38− CD138− B cells were also examined for CD20 expression and for chromosome 1p32 loss by IF-FISH.
(B) Findings from a MM patient with progression on BTZ therapy. (a) The clinical information of the patient. (b) The patient’s bone marrow was FACS sorted as in Figure 6. Viable CD38+ CD138+ plasma cells (c), CD38+ CD138−/low plasmablasts (d), and CD38− CD138− cells (e) were examined for the presence of Xbp1s by IF and for trisomy of chromosome 11, which contains the CCND1 locus (arrows), by FISH. (f) Viable CD38− CD138− B cells were also examined for CD20 expression and for gain of CCND1 by IF-FISH.
(C) Findings from a MM patient following intensive BTZ and DPACE chemotherapy with clinical response. (a) The clinical information of the patient. (b) The patient’s bone marrow was FACS sorted as in Figure 6. Viable CD38+ CD138+ plasma cells were not identified. Viable CD38+ CD138−/low plasmablasts (c), and CD38− CD138− B cells (d) were examined for concurrent Xbp1s protein expression by IF and for IGH-CCND1 gene fusion (arrows), by FISH. (e) Viable CD38− CD138− B cells were also examined for CD20 expression and for IGH-CCND1 by IF-FISH.
(D) MM tumor progenitor frequency summarized by patient status for the bone marrow examples presented.
Scale bar: 5 μM.
See also Figure S7.
Supporting our conclusion that MM tumor cells with arrested secretory maturation are BTZ resistant, in some primary MM tumors with PD on BTZ-therapy (Figure 7B) substantial expansion of tumor CD20low CD38− CD138− Xbp1s− activated B cell- and CD20− Xbp1s− pre-plasmablast subpopulations was observed, causing activated CD20low B cells or CD20− pre-plasmablasts to become the predominant subpopulations in these tumors (Figures 7Be, 7Bf and 7D). The marked expansion of these subpopulations in patients progressing on PI therapy is consistent both with a partial tumor maturation arrest and with relative invulnerability of these progenitor stages to BTZ. CD20+ B cell progenitors were also detectable in these tumors and appeared viable and enriched despite BTZ-therapy (Figures 7Bf, lower panels, and 7D). In contrast, plasmablasts and plasma cells were reduced; their numbers, 7-AAD staining and appearance (Figures 7Bb–d and 7D) suggested continued susceptibility of these more mature subpopulations to BTZ therapy.
As MM is incurable in the vast majority of patients, even with intensive multi-agent therapy, we evaluated the minimal residual disease in patients treated with BTZ-based therapy. Figure 7C shows a representative marrow sample obtained 3 weeks after sequential BTZ-based treatment (CyBorD) and DPACE chemotherapy. This sample shows an almost empty marrow with delayed recovery of normal marrow elements post chemotherapy. At this sampling, no viable MM plasma cells could be detected by flow. However, small residual populations of tumor cells were identified, with phenotypes compatible with activated CD20low B cells, pre-plasmablasts and early CD138− Xbp1slow plasmablasts. As Xbp1s− cells preferentially survive BTZ (Figure 1) the presence of CD138− Xbp1slow tumor plasmablasts in this marrow may reflect differentiation from earlier progenitors in the post treatment interval.
In each of the MM patient bone marrow samples examined in Figure 7 the proportion of CD38− CD138− CD20+ B cells, CD38− CD138− CD20low activated B cells and CD38− CD138− CD20− pre-plasmablasts that showed tumor-specific clonal FISH abnormalities, such as loss of chromosome 1p32, gain of CCND1 or fusion of IGH-CCND1 genes, consistently exceeded the background FISH positive rate, confirming the presence of tumor progenitor cells within these isolated subpopulations, as in Figure 6. Similar tumor progenitor subpopulations were also identified in hyperdiploid MM and in MM tumors bearing t(4;14) IGH-FGFR3 gene rearrangement (Figure 8A), demonstrating that MM B cell and pre-plasmablast progenitors exist across a broad spectrum of MM genetic subtypes. Overall, our examination of primary MM tumors at various treatment stages suggests that persistence of tumor progenitor subpopulations accounts for the failure to cure MM with PI-based treatment strategies, even when complete response may be attained within the plasma cell compartment.
Figure 8. Tumor Progenitor Cells in Multiple Myeloma and Their Role in Therapeutic Resistance.
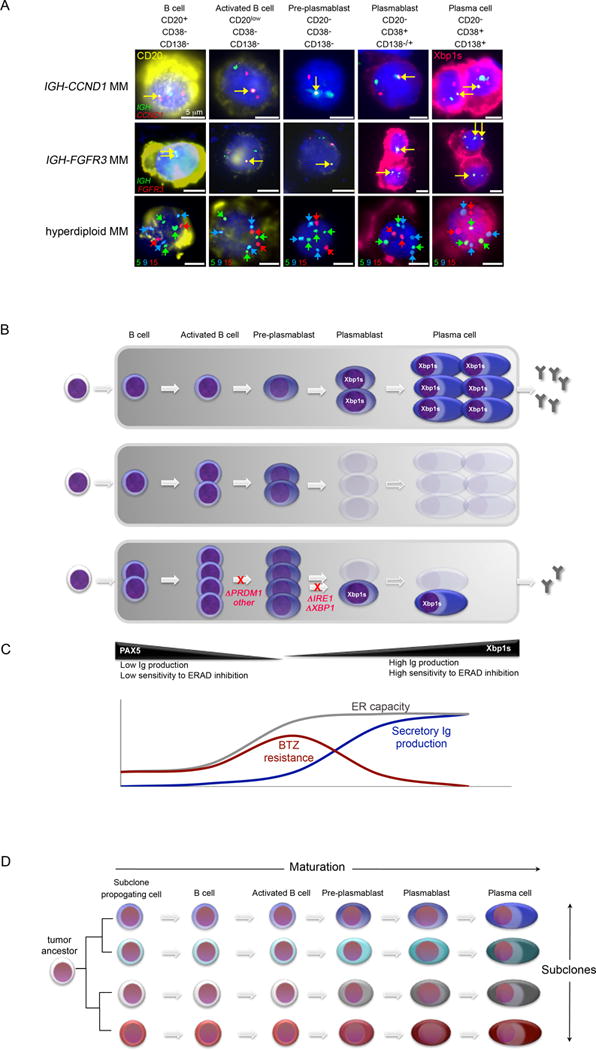
(A) Summary of primary MM progenitor stages, which correspond to physiological stages of B cell to plasma cell maturation (from left to right), as observed in MM genetic subtypes with t(11;14) IGH-CCND1 (Top) or t(4;14) IGH-FGFR3/MMSET (middle) or with hyperdiploid karyotype (bottom). Progenitors were characterized by IF-FISH as in Figure 6. CD20 protein expression is shown in yellow; Xbp1 protein is shown in pink. Gene fusions between IGH (Top and middle, green) and CCND1 (top, red) or FGFR3 loci (middle, red) are indicated by a yellow arrow. In the bottom row, trisomies of chromosomes 5 (green) and 9 (blue) and diploidy of chromosome 15 (red) are indicated by arrows. Scale bars: 5 μm.
(B) Schema showing tumor progenitor organization in newly diagnosed MM (Top). Tumor B cell and pre-plasmablast progenitors are intrinsically resistant to PI, preventing cure, even in patients that achieve plasma cell complete response (CR) (middle). MM progression on PI treatment is achieved in some MM tumors by maturation arrest at pre-plasmablast or earlier B cell stages, preventing secretory commitment. Mutations identified in MM that may mediate maturation arrest are shown in red (bottom). Cells eliminated by PI-therapy are shown as ghosts.
(C) Schema comparing the relative ER capacity and ER burden of plasma cell precursors. A ‘sweet spot’ appears to exist at the activated B cell and pre-plasmablast stage, where ER maturation precedes secretory Ig production, contributing to the reduced susceptibility of these cells to lethal ER stress following PI-induced ERAD inhibition. Whereas some MM tumors develop PI-resistance by maturation arrest at pre-plasmablast stage, mantle cell lymphoma is instead reported to achieve PI-resistance by promoting maturation towards this state.
(D) A model of intra-tumor cellular diversity, which likely contributes to the failure to cure MM. A plethora of tumor cell phenotypes arise from the presence of multiple genetic subclones and from multiple maturation stages within each subclone.
DISCUSSION
Although PIs can substantially debulk MM, these drugs fail to cure patients. We show here that primary MM tumors consist not only of Xbp1s+ plasma cells and plasmablasts but also of subpopulations of Xbp1s− pre-plasmablasts and of earlier CD20+ B cell progenitors (Figure 8A) that are intrinsically PI-insensitive. Whereas BTZ is potently cytotoxic to the predominating MM plasma cell and plasmablast compartments, tumor progenitor subpopulations including Xbp1s− pre-plasmablasts and B cells persist in BTZ treated patients (Figure 8B). We propose that the inability of PIs such as BTZ to address these non-secretory tumor cell progenitors substantially accounts for the failure of PI therapy to cure MM.
The malignant plasma cells encountered in MM are professional secretory cells typified by massive protein synthesis, an expanded ER network and elevated basal ER stress. This likely explains the unique sensitivity of MM to PIs, which exacerbate ER stress by inhibiting ERAD (Elkabetz et al., 2004; Lee et al., 2003; Obeng et al., 2006). Xbp1s− progenitors however lack full secretory status and produce less Ig (Jourdan et al., 2011; Reimold et al., 2001). XBP1-silenced MM plasma cells similarly produce less Ig. As the foremost activity of the ER in plasma cells is Ig synthesis, pre-plasmablasts and earlier progenitors, which produce less Ig, have a lower ER pre-load than plasma cells, rendering them predictably less vulnerable to lethal ER stress when ERAD is inhibited.
The finding that suppression of a branch of the ER homeostatic UPR mediates PI resistance, when PIs induce cytotoxicity in MM via the induction of ER stress, is notably contrary to any expectation from consideration of the UPR alone. However, whereas PIs cause activation of Atf6 and Perk in response to ER stress, Xbp1s activity is only minimally enhanced, perhaps because PIs stabilize ubiquitinated Xbp1u, which can act as a dominant negative (Davenport et al., 2007; Lee et al., 2003; Obeng et al., 2006). As Xbp1 fails to contribute to ER homeostasis following PI treatment, its loss likely has no negative effect on the survival of PI-treated cells. Instead, the overriding effect for MM cells of Xbp1 suppression is a de-commitment to secretory Ig production, which reduces ER loading and ultimately enables the cell to better withstand ERAD inhibition.
Notably the existence of PI-insensitive Xbp1s− tumor progenitors within primary MM tumors likely produces class-effect PI resistance that is independent of drug identity, in contrast to resistance produced by PI-binding site PSMB5 mutations. Therefore primary MM tumors that are resistant to BTZ via Xbp1s− tumor progenitors may also prove to be cross-resistant to proteasome inhibition induced by carfilzomib, the second-in-class PI recently approved by the FDA, consistent with early clinical findings (Siegel et al., 2012).
Xbp1 has been reported to be essential for MM pathogenesis (Carrasco et al., 2007). Because Ire1 directly regulates Xbp1, Ire1 inhibitors are currently under development for the treatment of MM (Koong et al., 2006; Kotz, 2011; Papandreou et al., 2011). However, we find that Xbp1 is not required for MM tumor cell survival, only for secretory maturation, and that its absence promotes PI resistance. This suggests that Ire1 inhibitors used outside of carefully construed combination strategies could prove ineffectual for the treatment of MM and may be deleterious if combined with PIs.
Maturation arrest likely represents a final common pathway that unifies a spectrum of genetic or epigenetic changes within MM tumors that escape therapeutic pressures or the limitations of the microenvironment. Notably, PRDM1, like XBP1, is essential for plasma cell maturation and for Ig production (Shapiro-Shelef et al., 2003); and is also recurrently mutated in MM tumors (Chapman et al., 2011), suggesting that mutation of genes other than IRE1 or XBP1 may drive maturation arrest in MM. Furthermore, mutations of IRE1 and XBP1 can be identified in MM cell lines that have not been exposed to BTZ (data not shown), suggesting that such mutations can serve purposes other than BTZ resistance. Expression profiling of BTZ-resistant MM tumors suggests that these tumors have reduced IL-6 sensitivity (with reduced IL6R and IL6ST), which can be directly attributed to suppression of Xbp1s. This suggests that MM maturation arrest not only enables PI resistance but may also facilitate cytokine independence and extra-medullary growth. In addition, maturation arrest in MM likely contributes to the emergence of oligosecretory disease. Therefore PIs appear to exert a selective pressure on MM tumors that may alter the clinical picture of relapsed MM encountered by modern physicians, contributing to a higher incidence of extra-medullary oligosecretory disease.
While we demonstrate that MM cells achieve BTZ resistance via de-commitment to terminal plasma cell maturation, others have instead found that MCL tumor cells attain BTZ resistance by plasmacytic differentiation (Perez-Galan et al., 2011). However, BTZ-resistant MCLs fail to acquire Xbp1s or Ig secretion, indicating that such cells are not full-fledged plasma cells and suggesting that their differentiation state resembles that of BTZ-resistant MM cells, namely activated B cells or pre-plasmablasts. We propose that a maturation “sweet spot” exists between pre-germinal center B cells and plasma cells, exemplified by cells with low ER burden that are intrinsically less susceptible to the lethal effects of PI, because such cells express the machinery of the adaptive UPR, but have not yet committed to Ig production (Figure 8C).
The role of tumor progenitors and maturation arrest in therapeutic resistance described here may be illustrative for other drugs and cancers, particularly for therapies targeted towards distinctive characteristics of well-differentiated tissues such as secretion, hormone responsiveness or antigen expression. Such treatments, like PIs, may fail to address less distinctive progenitor populations; and may thereby fail to provide cure.
The biology of MM is revealed to be closely related to that of other B cell Non Hodgkin lymphomas in demonstrating a discernible trail of tumor cell maturation from B cells. Whereas most MM contain multiple genetic subclones that respond to treatment in ‘clonal tides’ (Keats et al., 2012), we propose that within each subclone there exists a spectrum of progenitors, that markedly contribute to intra-tumor diversity and to the ability of MM tumors to endure treatment (Figure 8D). As tumor progenitors contribute to treatment failure in MM, these subpopulations must be addressed by future therapeutic strategies to produce a pervasive cure for patients.
EXPERIMENTAL PROCEDURES
RNAi screens
High throughput siRNA studies in MM cells were conducted as described (Tiedemann et al., 2012; Tiedemann et al., 2010; Zhu et al., 2011). To identify BTZ resistance mechanisms genes were ranked by mean siRNA Bliss independence scores at BTZ IC90.
Gene Expression Data
MM tumor profiles from patients enrolled on the APEX Study 039 of BTZ were obtained from NCBI GEO (GSE9782) (Mulligan et al., 2007). Three Xbp1 signature gene sets were used: XBP1_Staudt_SigDB reflects Xbp1 over expression in B cells (Shaffer et al., 2006) and together with other lymphoid transcription factor signatures was obtained from http://lymphochip.nih.gov/signaturedb/; V$XBP1_01 represents genes with promoters containing a conserved Xbp1 motif and is from the Broad MSigDB at http://www.broadinstitute.org/gsea/msigdb/index.jsp; XBP1_MM and the related Ire1 signature, IRE1_MM, were generated by shRNA knockdown of Xbp1 or Ire1 in MM cells. Plasma cell progenitor profiles were obtained from ArrayExpress (http://www.ebi.ac.uk/microarray-as/ae/) (E-MEXP-3034 and E-MEXP-2360)(Jourdan et al., 2011).
Lentiviral Transductions
Viruses were prepared in 293T cells. MM cells were transduced using 293T supernatant diluted to 30–50% in media and 8 μg/ml polybrene.
XBP1 constructs
Human XBP1u and XBP1s cDNA (OriGene) were cloned into pWPIs1. Mutations were generated by PCR.
Gene Expression Profiling
Samples were hybridized on Affymetrix HG-U133 Plus 2.0 GeneChip microarrays (Affymetrix) and analyzed using GeneSpring GX 12.1 (Agilent Technologies).
MM Samples
All patient samples were obtained following informed consent in compliance with the Declaration of Helsinki and a protocol approved by the University Health Network Research Ethics Board.
IF-FISH
Slides were fixed in 4% paraformaldehyde, permeabilized with 0.5% Triton X-100, incubated with primary and secondary antibodies and then mounted in Vectashield (Vector Laboratories). IF images were acquired. Cover slips were removed and cells were treated with 0.005% pepsin in 0.01N HCl, 1% formaldehyde, and with ethanol and were then hybridized with Vysis LSI or Cytocell Aquarius DNA Probes. FISH images were acquired at IF image coordinates.
Supplementary Material
SIGNIFICANCE.
PI, including bortezomib, are a mainstay of treatment for MM but fail to cure. Previously-reported in vitro resistance mechanisms have not been validated in the clinic and reflect an artefact of cell culture. An alternative PI resistance mechanism is described here that occurs in MM patients; as this differs from in vitro resistance reports, the need for clinical confirmation of in vitro drug resistance models is highlighted. Our results reveal that MM cells tolerate XBP1 inactivation, which contributes to therapeutic resistance, suggesting that IRE1 inhibitors may prove ineffectual in MM. Furthermore, an extensive progenitor organization is revealed in primary MM. Our results suggest that to achieve cure, treatment strategies must better address early MM progenitors.
Acknowledgments
This research was supported by the Canadian Cancer Society (grant #701464), the Molly and David Bloom Chair in Multiple Myeloma Research, the Arthur Macaulay Cushing Estate and the Princess Margaret Cancer Foundation. We thank the patients, Dr Chris Chen, Dr Ade Olujohungbe and the Manitoba Tumor Bank for providing samples and Dr Brad Wouters and Dr Leif Bergsagel for expert comments.
Footnotes
ACCESSION NUMBERS
Microarray results reported in this paper are available at NCBI GEO (accession GSE44968).
SUPPLEMENTAL INFORMATION
Supplemental information includes seven figures, one table and supplemental experimental procedures.
References
- Alexanian R, Delasalle K, Wang M, Thomas S, Weber D. Curability of multiple myeloma. Bone Marrow Res. 2012;2012:916479. doi: 10.1155/2012/916479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balsas P, Galan-Malo P, Marzo I, Naval J. Bortezomib resistance in a myeloma cell line is associated to PSMbeta5 overexpression and polyploidy. Leuk Res. 2012;36:212–218. doi: 10.1016/j.leukres.2011.09.011. [DOI] [PubMed] [Google Scholar]
- Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- Carrasco DR, Sukhdeo K, Protopopova M, Sinha R, Enos M, Carrasco DE, Zheng M, Mani M, Henderson J, Pinkus GS, et al. The differentiation and stress response factor XBP-1 drives multiple myeloma pathogenesis. Cancer Cell. 2007;11:349–360. doi: 10.1016/j.ccr.2007.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman MA, Lawrence MS, Keats JJ, Cibulskis K, Sougnez C, Schinzel AC, Harview CL, Brunet JP, Ahmann GJ, Adli M, et al. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011;471:467–472. doi: 10.1038/nature09837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Blank JL, Peters T, Liu XJ, Rappoli DM, Pickard MD, Menon S, Yu J, Driscoll DL, Lingaraj T, et al. Genome-wide siRNA screen for modulators of cell death induced by proteasome inhibitor bortezomib. Cancer Res. 2010;70:4318–4326. doi: 10.1158/0008-5472.CAN-09-4428. [DOI] [PubMed] [Google Scholar]
- Davenport EL, Moore HE, Dunlop AS, Sharp SY, Workman P, Morgan GJ, Davies FE. Heat shock protein inhibition is associated with activation of the unfolded protein response pathway in myeloma plasma cells. Blood. 2007;110:2641–2649. doi: 10.1182/blood-2006-11-053728. [DOI] [PubMed] [Google Scholar]
- de Wilt LH, Jansen G, Assaraf YG, van Meerloo J, Cloos J, Schimmer AD, Chan ET, Kirk CJ, Peters GJ, Kruyt FA. Proteasome-based mechanisms of intrinsic and acquired bortezomib resistance in non-small cell lung cancer. Biochem Pharmacol. 2012;83:207–217. doi: 10.1016/j.bcp.2011.10.009. [DOI] [PubMed] [Google Scholar]
- Elkabetz Y, Shapira I, Rabinovich E, Bar-Nun S. Distinct steps in dislocation of luminal endoplasmic reticulum-associated degradation substrates: roles of endoplamic reticulum-bound p97/Cdc48p and proteasome. J Biol Chem. 2004;279:3980–3989. doi: 10.1074/jbc.M309938200. [DOI] [PubMed] [Google Scholar]
- Franke NE, Niewerth D, Assaraf YG, van Meerloo J, Vojtekova K, van Zantwijk CH, Zweegman S, Chan ET, Kirk CJ, Geerke DP, et al. Impaired bortezomib binding to mutant beta5 subunit of the proteasome is the underlying basis for bortezomib resistance in leukemia cells. Leukemia. 2011 doi: 10.1038/leu.2011.256. [DOI] [PubMed] [Google Scholar]
- Hu CC, Dougan SK, McGehee AM, Love JC, Ploegh HL. XBP-1 regulates signal transduction, transcription factors and bone marrow colonization in B cells. EMBO J. 2009;28:1624–1636. doi: 10.1038/emboj.2009.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jourdan M, Caraux A, Caron G, Robert N, Fiol G, Reme T, Bollore K, Vendrell JP, Le Gallou S, Mourcin F, et al. Characterization of a transitional preplasmablast population in the process of human B cell to plasma cell differentiation. J Immunol. 2011;187:3931–3941. doi: 10.4049/jimmunol.1101230. [DOI] [PubMed] [Google Scholar]
- Keats JJ, Chesi M, Egan JB, Garbitt VM, Palmer SE, Braggio E, Van Wier S, Blackburn PR, Baker AS, Dispenzieri A, et al. Clonal competition with alternating dominance in multiple myeloma. Blood. 2012;120:1067–1076. doi: 10.1182/blood-2012-01-405985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2008;7:1013–1030. doi: 10.1038/nrd2755. [DOI] [PubMed] [Google Scholar]
- Koong AC, Chauhan V, Romero-Ramirez L. Targeting XBP-1 as a novel anti-cancer strategy. Cancer Biol Ther. 2006;5:756–759. doi: 10.4161/cbt.5.7.2973. [DOI] [PubMed] [Google Scholar]
- Kotz J. Cancer stress tests. SciBX. 2011;4:1–2. [Google Scholar]
- Lee AH, Iwakoshi NN, Anderson KC, Glimcher LH. Proteasome inhibitors disrupt the unfolded protein response in myeloma cells. Proc Natl Acad Sci U S A. 2003;100:9946–9951. doi: 10.1073/pnas.1334037100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichter DI, Danaee H, Pickard MD, Tayber O, Sintchak M, Shi H, Richardson PG, Cavenagh J, Blade J, Facon T, et al. Sequence analysis of beta-subunit genes of the 20S proteasome in patients with relapsed multiple myeloma treated with bortezomib or dexamethasone. Blood. 2012;120:4513–4516. doi: 10.1182/blood-2012-05-426924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConkey DJ, Zhu K. Mechanisms of proteasome inhibitor action and resistance in cancer. Drug Resist Updat. 2008;11:164–179. doi: 10.1016/j.drup.2008.08.002. [DOI] [PubMed] [Google Scholar]
- Mulligan G, Mitsiades C, Bryant B, Zhan F, Chng WJ, Roels S, Koenig E, Fergus A, Huang Y, Richardson P, et al. Gene expression profiling and correlation with outcome in clinical trials of the proteasome inhibitor bortezomib. Blood. 2007;109:3177–3188. doi: 10.1182/blood-2006-09-044974. [DOI] [PubMed] [Google Scholar]
- Nutt SL, Taubenheim N, Hasbold J, Corcoran LM, Hodgkin PD. The genetic network controlling plasma cell differentiation. Semin Immunol. 2011;23:341–349. doi: 10.1016/j.smim.2011.08.010. [DOI] [PubMed] [Google Scholar]
- Obeng EA, Carlson LM, Gutman DM, Harrington WJ, Jr, Lee KP, Boise LH. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood. 2006;107:4907–4916. doi: 10.1182/blood-2005-08-3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oerlemans R, Franke NE, Assaraf YG, Cloos J, van Zantwijk I, Berkers CR, Scheffer GL, Debipersad K, Vojtekova K, Lemos C, et al. Molecular basis of bortezomib resistance: proteasome subunit beta5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood. 2008;112:2489–2499. doi: 10.1182/blood-2007-08-104950. [DOI] [PubMed] [Google Scholar]
- Ozaki K, Spolski R, Feng CG, Qi CF, Cheng J, Sher A, Morse HC, 3rd, Liu C, Schwartzberg PL, Leonard WJ. A critical role for IL-21 in regulating immunoglobulin production. Science. 2002;298:1630–1634. doi: 10.1126/science.1077002. [DOI] [PubMed] [Google Scholar]
- Papandreou I, Denko NC, Olson M, Van Melckebeke H, Lust S, Tam A, Solow-Cordero DE, Bouley DM, Offner F, Niwa M, Koong AC. Identification of an Ire1alpha endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood. 2011;117:1311–1314. doi: 10.1182/blood-2010-08-303099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Galan P, Mora-Jensen H, Weniger MA, Shaffer AL, 3rd, Rizzatti EG, Chapman CM, Mo CC, Stennett LS, Rader C, Liu P, et al. Bortezomib resistance in mantle cell lymphoma is associated with plasmacytic differentiation. Blood. 2011;117:542–552. doi: 10.1182/blood-2010-02-269514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Politou M, Karadimitris A, Terpos E, Kotsianidis I, Apperley JF, Rahemtulla A. No evidence of mutations of the PSMB5 (beta-5 subunit of proteasome) in a case of myeloma with clinical resistance to Bortezomib. Leuk Res. 2006;30:240–241. doi: 10.1016/j.leukres.2005.06.014. [DOI] [PubMed] [Google Scholar]
- Reimold AM, Iwakoshi NN, Manis J, Vallabhajosyula P, Szomolanyi-Tsuda E, Gravallese EM, Friend D, Grusby MJ, Alt F, Glimcher LH. Plasma cell differentiation requires the transcription factor XBP-1. Nature. 2001;412:300–307. doi: 10.1038/35085509. [DOI] [PubMed] [Google Scholar]
- Richardson PG, Sonneveld P, Schuster MW, Irwin D, Stadtmauer EA, Facon T, Harousseau JL, Ben-Yehuda D, Lonial S, Goldschmidt H, et al. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med. 2005;352:2487–2498. doi: 10.1056/NEJMoa043445. [DOI] [PubMed] [Google Scholar]
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Ruckrich T, Kraus M, Gogel J, Beck A, Ovaa H, Verdoes M, Overkleeft HS, Kalbacher H, Driessen C. Characterization of the ubiquitin-proteasome system in bortezomib-adapted cells. Leukemia. 2009;23:1098–1105. doi: 10.1038/leu.2009.8. [DOI] [PubMed] [Google Scholar]
- Shaffer AL, Shapiro-Shelef M, Iwakoshi NN, Lee AH, Qian SB, Zhao H, Yu X, Yang L, Tan BK, Rosenwald A, et al. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity. 2004;21:81–93. doi: 10.1016/j.immuni.2004.06.010. [DOI] [PubMed] [Google Scholar]
- Shaffer AL, Wright G, Yang L, Powell J, Ngo V, Lamy L, Lam LT, Davis RE, Staudt LM. A library of gene expression signatures to illuminate normal and pathological lymphoid biology. Immunol Rev. 2006;210:67–85. doi: 10.1111/j.0105-2896.2006.00373.x. [DOI] [PubMed] [Google Scholar]
- Shapiro-Shelef M, Lin KI, McHeyzer-Williams LJ, Liao J, McHeyzer-Williams MG, Calame K. Blimp-1 is required for the formation of immunoglobulin secreting plasma cells and pre-plasma memory B cells. Immunity. 2003;19:607–620. doi: 10.1016/s1074-7613(03)00267-x. [DOI] [PubMed] [Google Scholar]
- Siegel DS, Martin T, Wang M, Vij R, Jakubowiak AJ, Lonial S, Trudel S, Kukreti V, Bahlis N, Alsina M, et al. A phase 2 study of single-agent carfilzomib (PX-171-003-A1) in patients with relapsed and refractory multiple myeloma. Blood. 2012;120:2817–2825. doi: 10.1182/blood-2012-05-425934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiedemann RE, Zhu YX, Schmidt J, Shi CX, Sereduk C, Yin H, Mousses S, Stewart AK. Identification of molecular vulnerabilities in human multiple myeloma cells by RNA interference lethality screening of the druggable genome. Cancer Res. 2012;72:757–768. doi: 10.1158/0008-5472.CAN-11-2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiedemann RE, Zhu YX, Schmidt J, Yin H, Shi CX, Que Q, Basu G, Azorsa D, Perkins LM, Braggio E, et al. Kinome-wide RNAi studies in human multiple myeloma identify vulnerable kinase targets, including a lymphoid-restricted kinase, GRK6. Blood. 2010;115:1594–1604. doi: 10.1182/blood-2009-09-243980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Kumar S, Fridley BL, Kalari KR, Moon I, Pelleymounter LL, Hildebrandt MA, Batzler A, Eckloff BW, Wieben ED, Greipp PR. Proteasome beta subunit pharmacogenomics: gene resequencing and functional genomics. Clin Cancer Res. 2008;14:3503–3513. doi: 10.1158/1078-0432.CCR-07-5150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu K, Dunner K, Jr, McConkey DJ. Proteasome inhibitors activate autophagy as a cytoprotective response in human prostate cancer cells. Oncogene. 2010;29:451–462. doi: 10.1038/onc.2009.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu YX, Tiedemann R, Shi CX, Yin H, Schmidt JE, Bruins LA, Keats JJ, Braggio E, Sereduk C, Mousses S, Stewart AK. RNAi screen of the druggable genome identifies modulators of proteasome inhibitor sensitivity in myeloma including CDK5. Blood. 2011;117:3847–3857. doi: 10.1182/blood-2010-08-304022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.