

Abstract
Background
Protein Z (PZ) has been reported to promote the inactivation of factor Xa (FXa) by PZ-dependent protease inhibitor (ZPI) three orders of magnitude. Previously, we prepared a chimeric PZ in which its C-terminal pseudo-catalytic domain was grafted on FX light-chain (Gla and EGF-like domains) (PZ/FX-LC). Characterization of PZ/FX-LC revealed the ZPI interactive-site is primarily located within PZ pseudo-catalytic domain. Nevertheless, the cofactor function and apparent Kd of PZ/FX-LC for interaction with ZPI remained impaired ~6–7-fold, suggesting PZ contains a ZPI interactive-site outside pseudo-catalytic domain. X-ray structural data indicates Tyr-240 of ZPI interacts with EGF2-domain of PZ. Structural data further suggests 3 other ZPI surface loops make salt-bridge interactions with PZ pseudo-catalytic domain. To identify ZPI interactive-sites on PZ, we grafted the N-terminal EGF2 subdomain of PZ onto PZ/FX-LC chimera (PZ-EGF2/FX-LC) and also generated two compensatory charge reversal mutants of PZ pseudo-catalytic domain (Glu-244 and Arg-212) and ZPI surface loops (Lys-239 and Asp-293).
Methods
PZ chimeras were expressed in mammalian cells and ZPI derivatives were expressed in E.coli.
Results
The PZ EGF2 subdomain fusion restored the defective cofactor function of PZ/FX-LC. The activities of PZ and ZPI mutants were all impaired if assayed individually, but partially restored if the compensatory charge reversal mutants were used in the assay.
Conclusions
PZ EGF2 subdomain constitutes an interactive-site for ZPI. Data with compensatory charge reversal mutants validates structural data that the identified residues are part of interactive-sites.
General significance
Insight is provided into mechanisms through which specificity of ZPI-PZ-FXa complex formation is determined.
1. Introduction
Protein Z (PZ)1 is a vitamin K-dependent plasma protein which promotes the inactivation rate of factor Xa (FXa) by the PZ-dependent proteinase inhibitor (ZPI) on negatively charged phospholipids (PC/PS) in the presence of Ca2+ by more than three orders of magnitude [1–3]. It has a genetic organization identical to vitamin K-dependent coagulation zymogens [4]. However, PZ has no enzymatic activity, but instead functions as a cofactor to regulate the proteolytic activity of FXa by ZPI on PC/PS vesicles in the presence of Ca2+ [1–3]. Similar to other vitamin K-dependent coagulation proteins, PZ has an N-terminal γ-carboxyglutamic acid (Gla) domain that is followed by two epidermal growth factor (EGF)-like domains (light chain homologue) and a C-terminal pseudo-catalytic domain [4]. ZPI is a 72 kDa serpin which binds to the active-site of FXa via its P1-Tyr on the reactive center loop (RCL), thereby trapping it in the form of an inactive and covalently modified serpin-protease complex, a property shared by other inhibitory serpins [1–3,5]. In addition to FXa, ZPI is also a specific inhibitor of factor XIa, in this case however, ZPI does not require PZ and thus effectively inhibits the protease, independent of a cofactor [6]. We recently investigated the mechanism of the cofactor function of PZ by constructing a chimeric PZ derivative in which the pseudo-catalytic domain of the molecule was grafted on the light chain of factor X (PZ/FX-LC). Analysis of the cofactor function and the ZPI-binding properties of PZ/FX-LC chimera indicated that the primary ZPI-interactive site on PZ is located within the C-terminal pseudo-catalytic domain of the cofactor [7]. However, the chimeric cofactor exhibited ~7-fold weaker affinity for ZPI which was also associated with ~6-fold decreased maximal cofactor function in the FXa inhibition assay on the negatively charged phospholipid vesicles in the presence of Ca2+ [7]. The molecular basis for the decreased cofactor activity of the PZ/FX-LC chimera was not investigated but the results raised the possibility that there is another interactive-site for ZPI outside the pseudo-catalytic domain of the cofactor.
Recently, the x-ray crystal structure of ZPI in complex with PZ was resolved by two groups [8–10]. Structural data supports our mutagenesis data demonstrating that ZPI makes extensive salt-bridge and hydrophobic interactions with 4 surface loops within the pseudo-catalytic domain of PZ [10]. Interestingly, the structural data further revealed that a hydrophobic residue on ZPI (Tyr-240) is oriented toward the EGF2 domain of PZ, interacting with a hydrophobic cavity in the interface between this domain and the pseudo-catalytic domain of the cofactor [10]. To validate the structural data and identify the site on PZ EGF2 domain that may constitute an interactive-site for ZPI, we grafted the first subdomain (residues forming the first 2 disulfide-stabilized loops) of PZ back onto PZ/FX-LC chimeric cofactor (Fig. 1). Moreover, we substituted the first subdomain of PZ EGF2 domain with the corresponding loops of FXa EGF2 domain. Since an interaction between the Gla-domain of PZ and FXa on PC/PS vesicles has been postulated [2,7], we also prepared a PZ chimera in which the Gla-domain of the cofactor was replaced with the corresponding Gla-domain of FXa (Fig. 1). Characterization of these PZ chimeras in kinetic assays indicates that ZPI interacts with a hydrophobic cavity formed by the first subdomain of PZ EGF2 domain and that the Gla-domain of FXa can functionally substitute for the Gla-domain of PZ on PC/PS vesicles in the presence of Ca2+. Moreover, we mapped the proposed salt-bridge mediated interactive-site of ZPI with the pseudo-catalytic domain of PZ by a compensatory mutagenesis approach, validating the structural data that the interaction of several charged residues of PZ with complementary sites of ZPI contribute to the binding affinity of the cofactor-serpin inhibitory complex formation.
Figure 1.
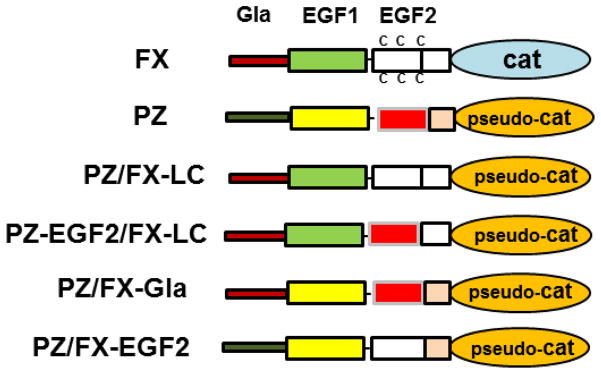
Cartoons of factor X and PZ chimeras used in the study. Cat represents factor X catalytic domain; pseudo-Cat represents PZ pseudo-catalytic domain; LC represents factor X light chain.
2. Materials and Methods
2.1. Construction, Mutagenesis and Expression of Recombinant Proteins
Wild-type PZ and a PZ chimera in which the pseudo-catalytic domain of the cofactor was grafted on the light chain of factor X (PZ/FX-LC) were expressed in a mammalian expression/purification vector system as described [7]. A PZ chimera was constructed in which the first EGF2 subdomain of PZ/FX-LC was replaced with the corresponding subdomain of PZ EGF2 domain (Fig. 1). Two other PZ chimeras were prepared. In the first construct, the first EGF2 subdomain of wild-type PZ was replaced with the corresponding FXa EGF2 subdomain. In the second construct, the Gla-domain of PZ was replaced with the corresponding Gla-domain of FXa. The charge reversal mutants of PZ including Glu-244 to Lys (PZ-E244K), Arg-212 to Asp (PZ-R212D) and Arg-298 to Asp (PZ-R298D) were constructed using the same vector system. All mutations were introduced by the PCR mutagenesis approach and the accuracy of all constructs was confirmed by DNA sequencing. The expression vectors, containing a neomycin gene for selection in mammalian cells with G418, were transfected to human embryonic kidney (HEK-293) cells. Several G418 resistant clones were selected and examined for PZ expression by an ELISA using the HPC4 monoclonal antibody and a polyclonal anti-PZ antibody (Haematologic Technologies Inc. Essex Junction, VT) as described [7]. A high expressing clone for each PZ derivative was identified and 20 liters of cell culture supernatant were collected, concentrated and purified by a combination of HPC4 immunoaffinity and Mono Q ion exchange chromatography as described [7]. Concentrations of PZ derivatives were calculated from their absorbance at 280 nm using a molar absorption coefficient of 74,400 M−1 cm−1 as described [7,11]. The purity of all recombinant cofactors was ensured by SDS-PAGE under non-reducing conditions and the protein preparations were frozen at -80 °C in small aliquots until use.
Wild-type ZPI was prepared in E. coli using the SUMO fusion expression/purification system and characterized as described [12]. A ZPI mutant in which an Ala substituted the native P1-Tyr-387 of the serpin (ZPI-Y387A) was constructed by standard PCR mutagenesis methods and expressed using the same vector system. The compensatory charge reversal mutants of ZPI including Lys-239 to Glu (ZPI-K239E) and Asp-293 to Arg (D293R) were constructed and expressed in the same vector system. Concentrations of ZPI derivatives were calculated from their absorbance at 280 nm using a molar absorption coefficient of 31,525 M−1 cm−1 as described [3].
Human plasma FXa was purchased from Haematologic Technologies Inc. (Essex Junction, VT). Phospholipid vesicles containing 80% phosphatidylcholine and 20% phosphatidylserine (PC/PS) were prepared as described [13]. The chromogenic substrate Spectrozyme FXa (SpFXa) was purchased from American Diagnostica (Greenwich, CT).
2.2. Inhibition assay
A discontinuous assay method was used to measure the second-order association rate constants (k2) for the ZPI inhibition of FXa under pseudo-first-order conditions as a function of increasing concentrations of the PZ chimeras as described [7,12]. Briefly, FXa (0.75 nM) was incubated with ZPI (5 nM) and different concentrations of each PZ derivative (0.3–20 nM) on PC/PS vesicles (25 μM) in 0.1 M NaCl, 0.02 M Tris-HCl (pH 7.5), 0.1% polyethylene glycol 8000 (PEG 8000), 0.1 mg/mL bovine serum albumin (BSA) and 5 mM Ca2+ (TBS/Ca2+) for 1.5–8 min in 50 μL volumes in 96-well polystyrene plates at room temperature. The inhibition reactions were stopped by the addition of 50 μL SpFXa (0.2 mM final) in TBS containing 50 mM EDTA and the remaining enzyme activity was measured with a Vmax Kinetics Microplate Reader (Molecular Devices, Menlo Park, CA) at 405 nm. The cofactor concentration dependence of the ZPI inhibition of FXa indicated that the PZ concentrations used are saturating under these conditions. The observed pseudo-first-order (kobs) rate constants were calculated from a first-order rate equation and the second-order rate constants (k2) were calculated from the slope of plots of kobs values versus PZ-ZPI complex concentrations as described [7,12]. The apparent dissociation constants (Kd(app)) of the PZ derivatives for interaction with ZPI were estimated from the hyperbolic dependence of kobs values on PZ concentrations in the presence of a fixed concentration of ZPI (5 nM) on PC/PS vesicles in TBS/Ca2+.
Similar methods were employed to evaluate the PC/PS dependence of the cofactor effect of each PZ derivative (20 nM) in catalyzing the ZPI (5 nM) inhibition of FXa (0.75 nM) in the presence of 5 mM Ca2+. All values are presented as the average of at least 3 independent measurements ±S.D.
2.3. Competitive inhibition assay
The same inhibition assay was used to evaluate the affinity of PZ derivatives for interaction with an RCL mutant of ZPI (ZPI-Y387A), which cannot inhibit FXa, by the ability of the serpin mutant to compete with wild-type ZPI for interaction with the cofactor. In this assay, the PZ-mediated ZPI inhibition of FXa (0.75 nM) was monitored by incubating FXa with fixed concentrations of PZ (5 nM) and ZPI (5 nM) on PC/PS vesicles (2 μM) in the presence of increasing concentrations of the ZPI mutant (0–500 nM) in TBS/Ca2+. Following a 3–12 min incubation at room temperature, 50 μL SpFXa (200 μM final) was added to each well and the remaining FXa activity was measured as described above. Under these experimental conditions ~80–90% of the FXa activity in the presence of the competitor (ZPI-Y387A) was recovered. Kd apparent values for interaction of the PZ derivatives with the ZPI mutant were determined by non-linear regression analysis of the saturable ZPI-Y387A concentration dependence of recovery of FXa activity according to a hyperbolic equation as described [12].
2.4. Stoichiometry of inhibition (SI)
The SI values for the ZPI inhibition of FXa in the presence and absence of PZ derivatives were determined by titrating 10–100 nM active-site titrated FXa with increasing concentrations of ZPI (5-fold molar excess) in complex with PZ (equimolar with ZPI) as described [12]. Following incubation at room temperature allowing for sufficient time to reach maximal inhibition based on measured k2 values, the residual activity of FXa was monitored from the hydrolysis of the chromogenic substrate SpFXa at 405 nm as described above. SI values were determined from the x-intercept of the linear regression fit of the residual activities plotted versus the serpin/protease ratios as described [14].
3. Results
3.1. Expression and characterization of PZ derivatives
Wild-type and PZ chimeras (Fig. 1) were expressed in HEK-293 cells using an expression/purification vector system as described [7]. We have already demonstrated that recombinant wild-type PZ has essentially identical ZPI-affinity and cofactor function as the plasma-derived PZ [7]. Wild-type PZ exhibited Kd(app) of ~1 nM for ZPI in the FXa inhibition assay on PC/PS vesicles in the presence of Ca2+ (Fig. 2A and Table 1). In agreement with our previous results [7], PZ/FX-LC chimera exhibited ~7-fold lower affinity for ZPI, thus yielding Kd(app) of 7.2 nM for the serpin (Fig. 2A, Table 1). These results indicate that PZ may have an interactive-site for ZPI outside the C-terminal pseudo-catalytic domain. Structural data suggests that this site may be located in the EGF2 domain of the cofactor [10]. This domain, like other EGF domains of vitamin K-dependent coagulation proteins, contains 6 Cys residues forming three disulfide bonds divided into a primary subdomain stabilized by two disulfides and a second subdomain stabilized by the last disulfide bond [4,16]. To determine whether EGF2 domain of PZ contains a binding site for ZPI, we grafted the N-terminal primary subdomain of PZ EGF2 back onto the PZ/FX-LC chimera (Fig. 1, fourth construct). Interestingly, this construction strategy restored both the defective ZPI-affinity and the cofactor function of the chimeric cofactor. Thus, the resulting chimera (PZ-EGF2/FX-LC) exhibited a wild-type like apparent Kd for ZPI and normal cofactor function in accelerating the ZPI inhibition of FXa on PC/PS vesicles in the presence of Ca2+ (Fig. 2A, Table 1).
Figure 2.

Concentration-dependence of PZ-mediated ZPI inhibition of FXa on PC/PS vesicles. (A) Inhibition of FXa (0.75 nM) by ZPI (5 nM) was monitored in the presence of increasing concentrations of PZ derivatives (x-axis) on PC/PS vesicles (25 μM) in TBS/Ca2+ as described under “Materials and Methods”. kobs values were calculated and plotted as a function of different concentrations of PZ derivatives. The symbols are: PZ-WT (○), PZ-FX-Gla (●), PZ/FX-EGF2 (□), PZ/FX-LC (■), and PZ-EGF2/FX-LC (△). Non-linear regression analysis of kinetic data using a hyperbolic equation yielded Kd(app) and k2 values that are presented in Table 1. (B) The same as panel A except that the inhibition of the FXa by fixed concentrations of PZ (20 nM) and ZPI (5 nM) was monitored as a function of increasing concentrations of PC/PS vesicles (x-axis). The symbols are the same as those described in panel A. Non-linear regression analysis of kobs values using a hyperbolic equation yielded Kd(app) for the interaction of PZ derivatives with PC/PS: PZ-WT (0.95 ±0.1), PZ-FX-Gla (2.1 ±0.3), PZ/FX-EGF2 (1.5 ±0.2), PZ/FX-LC (2.8 ±0.4), and PZ-EGF2/FX-LC (2.4 ±0.2). Data in both panels are derived from at least three independent measurements ±S.D.
Table 1.
Kinetic constants and SI values for the inhibition of FXa by ZPI-WT in the absence and presence of PZ derivatives on PC/PS vesicles
| k2(app) (M−1s−1) | SI (mol I/mol E) | k2(app)×SI (M−1s−1) | Kd(app) (nM) | |
|---|---|---|---|---|
| ZPI-WT | 3.0 ± 0.3× 103 | 4.2 ± 0.30 | 1.2 ± 0.2× 104 | - |
| PZ-WT | 2.8 ± 0.2× 106 | 3.1 ± 0.26 | 8.7 ± 1.2× 106 | 1.0 ± 0.2 |
| PZ/FX-LC | 0.4 ± 0.1× 106 | 3.8 ± 0.25 | 1.6 ± 0.3× 106 | 7.2 ± 1.5 |
| PZ-EGF2/FX-LC | 3.0 ± 0.2× 106 | 2.8 ± 0.27 | 8.4 ± 1.3× 106 | 0.75± 0.1 |
| PZ/FX-EGF2 | 0.8 ± 0.1× 106 | 3.7 ± 0.21 | 3.0 ± 0.5× 106 | 8.5 ± 1.8 |
| PZ/FX-Gla | 2.9 ± 0.3× 106 | 2.7 ± 0.18 | 7.8 ± 1.1× 106 | 1.3 ± 0.2 |
The apparent second-order rate constants (k2(app)) and SI values for the inhibition of FXa by ZPI in the absence and presence of PZ derivatives on PC/PS vesicles were determined in TBS/Ca2+ by a discontinuous assay as described in Materials and Methods. The product k2(app) x SI represents the second-order rate constant corrected for the SI values. Kd(app) values for the interaction of ZPI with PZ derivatives were measured from the hyperbolic dependence of kobs values on PZ concentrations on PC/PS vesicles in FXa inhibition assays as described in Materials and Methods.
To provide further support for the hypothesis that the primary subdomain of PZ contains a binding site for ZPI, we substituted this subdomain with the corresponding subdomain of FXa. In agreement with our conclusion, this PZ chimera (PZ/FX-EGF2) also exhibited the same cofactor properties of PZ/FX-LC. Thus, the apparent affinity of this chimera for ZPI was decreased more than 8-fold and its extent of cofactor function was impaired ~2-3-fold (Fig. 2A, Table 1). Previous results have indicated that the interaction of the Gla-domain of PZ with FXa contributes to its cofactor function on PC/PS vesicles [2,7]. To further investigate this question, we substituted the Gla-domain of PZ with the corresponding domain of FXa (PZ/FX-Gla). Analysis of the cofactor properties of this PZ chimera indicated that the Gla-domain of FXa can functionally substitute for the Gla-domain of the cofactor since the chimera exhibited wild-type like properties in the FXa inhibition assay (Fig. 2A, Table 1). To ensure that alterations in the interaction of the chimeric cofactors with PC/PS vesicles do not affect the results presented above, the PC/PS concentration dependence of all chimeric cofactors was evaluated in the same FXa inhibition assay. The results presented in Fig. 2B suggest that the PC/PS affinities of the PZ chimeras are minimally affected and that the concentration of PC/PS vesicles used in the inhibition assays is not limiting.
3.2. Contribution of the ZPI reactive center loop for its affinity in the inhibitory complex
Next, we investigated the extent to which the Kd(app) values for the interaction of PZ derivatives with ZPI in the inhibition assays are affected by the RCL-dependent interaction of ZPI with the active-site of FXa. Thus, we constructed a ZPI mutant in which the P1-Tyr-387 of the RCL was replaced with an Ala (ZPI-Y387A). The ZPI-Y387A mutant did not exhibit a detectable reactivity with FXa, thus it was used as a competitive inhibitor of PZ interaction with ZPI on PC/PS vesicles in the inhibition assay. This strategy facilitated the estimation of the extent of the contribution of the exosite-dependent interaction of ZPI from its RCL-dependent interaction with PZ in the inhibitory complex. The ZPI mutant was a relatively effective competitive inhibitor of wild-type PZ, PZ/FX-Gla and PZ-EGF2/FX- LC, exhibiting Kd(app) values of ~40 nM for the three PZ derivatives (Fig. 3). The ZPI mutant exhibited ~3-fold lower affinity for the PZ/FX-EGF2 chimera, thus inhibiting its interaction with ZPI with Kd(app) of 124 nM (Fig. 3 and its legend). By contrast, the ZPI mutant was the poorest competitive inhibitor of PZ/FX-LC chimera, exhibiting a Kd(app) of 227 nM for the mutant cofactor. These results suggest that the exosite-dependent interaction of ZPI with PZ in the inhibitory complex makes significant contributions to the mechanism of FXa inhibition by the serpin. The results further support the conclusion that ZPI has an interactive-site outside the pseudo-catalytic domain of PZ.
Figure 3.
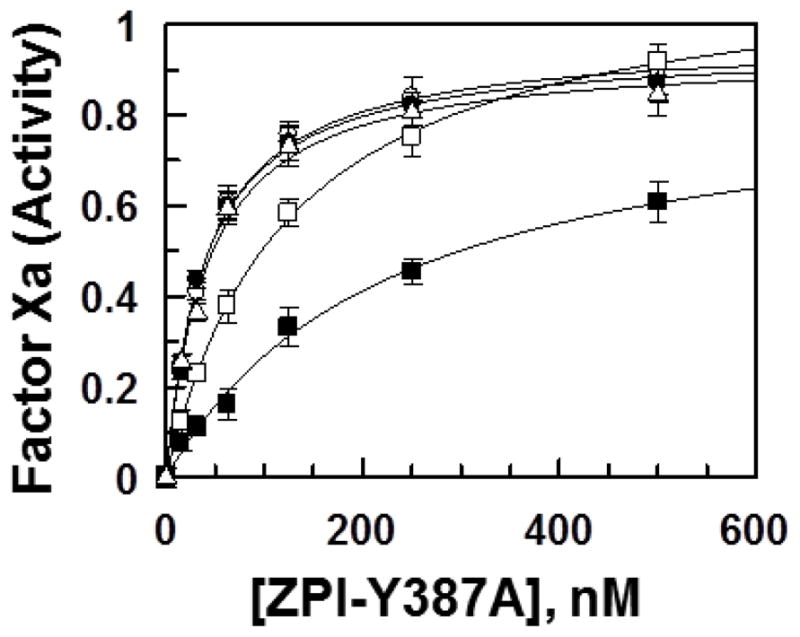
Competitive effect of ZPI-Y387A on the ZPI inhibition of FXa in the presence of PZ derivatives. The competitive effect of the ZPI derivative (0–500 nM) on the inhibition of FXa (0.75 nM) by ZPI-WT (5 nM) was monitored in the presence of different PZ derivatives (5 nM) for 3–12 min in TBS/Ca2+ on PC/PS vesicles (2 μM) as described under “Materials and Methods”. The symbols are:PZ-WT (○), PZ-FX-Gla (●), PZ/FX-EGF2 (□), PZ/FX-LC (■), and PZ-EGF2/FX-LC (△). Non-linear regression analysis of kinetic data using a hyperbolic equation yielded Kd(app) values of ~40 nM for PZ-WT, PZ/FX-Gla and PZ-EGF2/FX-LC. The same values for PZ/FX-EGF2 and PZ/FX-LC were 124 nM and 227 nM, respectively. Data are derived from at least three independent measurements ±S.D.
3.3. Charge-reversal mutants of PZ and ZPI
Analysis of structural data indicates that three surface loops on ZPI make salt-bridge/hydrogen bond interactions with the C-terminal pseudo-catalytic domain of PZ [10]. Thus, residues His-210, Arg-212 and Arg-298 of PZ are three dimensionally located at a distance to contact Asp-293 of ZPI (Fig. 4). Other similar contacts of PZ with ZPI include Glu-244 with Lys-239; His-250 with Asp-238; Thr-297 with Met-71; Arg-350 with Asp-74; and Gln-357 with Asn-261 [10]. To validate the structural data we took a compensatory mutagenesis approach and reversed the charges of Arg-212, Glu-244 and Arg-298 (R212E, E244K and R298D) in PZ individually in three different constructs and expressed the mutants in HEK-293 cells. PZ-R298D was not expressed to a sufficient quantity for characterization. However, adequate quantities of the other two cofactors were expressed and purified. Similarly, we reversed the charges of Asp-293 and Lys-239 (D293R and K239E) individually in ZPI and expressed the ZPI derivatives in E. coli as described [12]. Analysis of the inhibitory properties of the ZPI mutants toward FXa suggested that the serpin derivatives inactivate FXa with normal rate constants in the absence of PZ. However, the affinity of the charge reversal ZPI-K239E mutant for interaction with PZ-WT was decreased ~5-fold (Table 2). Thus, in contrast to a Kd(app) of ~1 nM for the ZPI-WT interaction with PZ-WT, the same value was increased to ~5 nM for the interaction of ZPI-K239E with the wild-type cofactor (Table 2). Interestingly, this trend in the cofactor affinity for the serpin mutant was reversed if the Kd(app) for ZPI-K239E was measured with the compensatory PZ-E244K mutant (Table 2). In this case, the mutant serpin exhibited a Kd(app) of 2.2 nM for the mutant cofactor, however, wild-type ZPI had an elevated Kd(app) of 4.4 nM for the mutant cofactor (Table 2). The PZ-R212D mutant exhibited a Kd(app) of ~5 nM for wild-type ZPI (elevated 5-fold relative to wild-type PZ) and its capacity to function as a cofactor was significantly impaired (Fig. 5A, Table 2). While the reactivity of the compensatory ZPI mutant (ZPI-D293R) with FXa was normal in the absence of a cofactor, its reactivity with FXa was not promoted by wild-type PZ, suggesting the cofactor function of PZ with this ZPI mutant is completely abolished (Fig. 5B, Table 2). By contrast, the compensatory PZ mutant (PZ-R212D) promoted the reactivity of ZPI-D293R with FXa ~2.5-fold (Fig. 5B, Table 2), supporting structural data that Arg-212 of PZ interacts with Asp-293 of ZPI. These results are in line with structural data that Asp-293 is one of the most critical residues of ZPI, interacting with at least two other sites (Fig. 4) and making the greatest contribution to the free energy of PZ binding [10].
Figure 4.

Crystal structure of the PZ-ZPI complex. The structures of ZPI and PZ in ribbon representations are shown in cyan and green, respectively. The relative three dimensional locations of the side-chains of selected charged residues of the interface under study are shown in blue for basic residues and in red for acidic residues. The coordinates obtained from Protein Data Bank accession code (PDB ID: 3H5C) were used to prepare the figure [10].
Table 2.
Comparisons of kinetic constants and SI values for the inhibition of FXa by wild-type PZ/ZPI complex and compensatory charge reversal mutants of PZ-E244K/ZPI-K239E and PZ-R212D/ZPI-D293R complexes on PC/PS vesicles in TBS/Ca2+
| k2(app) (M−1s−1) | SI (mol I/mol E) | k2(app)×SI (M−1s−1) | Kd(app) (nM) | |
|---|---|---|---|---|
| ZPI-WT | 3.0 ± 0.3× 103 | 4.2 ± 0.30 | 1.2 ± 0.2× 104 | - |
| ZPI-WT+PZ-WT | 2.8 ± 0.2× 106 | 3.1 ± 0.26 | 8.7 ± 1.2× 106 | 1.0 ± 0.2 |
| ZPI-K239E | 2.7 ± 0.1× 103 | 4.5 ± 0.48 | 1.2 ± 0.4× 104 | - |
| ZPI-K239E+PZ-WT | 1.6 ± 0.1× 106 | 4.3 ± 0.36 | 6.9 ± 1.4× 106 | 4.5 ± 0.5 |
| ZPI-K239E+PZ-E244K | 1.7 ± 0.2× 106 | 4.1 ± 0.31 | 7.0 ± 1.5× 106 | 2.2 ± 0.3 |
| ZPI-WT+PZ-E244K | 1.8 ± 0.1× 106 | 4.0 ± 0.27 | 7.2 ± 1.7× 106 | 4.4 ± 0.5 |
| ZPI-D293R | 3.0 ± 0.3× 103 | 4.7 ± 0.24 | 1.4 ± 0.5× 104 | - |
| ZPI-D293R+PZ-WT | 3.2 ± 0.3× 103 | 4.8 ± 0.24 | 1.5 ± 0.3× 104 | ND |
| ZPI-D293R+PZ-R212D | 7.6 ± 1.6× 103 | 4.6 ± 0.14 | 3.5 ± 0.3× 104 | ND |
| ZPI-WT+PZ-R212D | 0.6 ± 0.1× 106 | 2.7 ± 0.24 | 1.6 ± 0.1× 106 | 4.7 ± 0.6 |
The apparent second-order rate constants (k2(app)) and SI values for the inhibition of FXa by ZPI in the absence and presence of PZ derivatives on PC/PS vesicles were determined in TBS/Ca2+ by a discontinuous assay as described in Materials and Methods. The product k2(app) x SI represents the second-order rate constant corrected for the SI values. Kd(app) values for the interaction of ZPI with PZ derivatives were measured from the hyperbolic dependence of kobs values on PZ concentrations on PC/PS vesicles in FXa inhibition assays as described in Materials and Methods. ND, not determined.
Figure 5.

Inhibition of FXa by ZPI-WT in the presence of PZ-WT and PZ-R212D on PC/PS vesicles. (A) Inhibition of FXa (0.75 nM) by ZPI-WT (5 nM) was monitored in the presence of increasing concentrations of either PZ-WT (○) or PZ-R212D (●) on PC/PS vesicles (25 μM) for 1.5–8.0 min in TBS/Ca2+ as described under “Materials and Methods”. (B) The same as panel A except that the inhibition of the FXa by increasing concentrations of the compensatory ZPI-D293R mutant was monitored in the absence of PZ (○) and presence of equimolar concentrations of either PZ-WT (●) or PZ-R212 (□). Non-linear regression analysis of kobs values in panel A using a hyperbolic equation yielded Kd(app) and k2 values that are presented in Table 2. kobs values in panel B did reach saturation for measuring kinetic constants. Data in both panels are derived from at least three independent measurements ±S.D.
4. Discussion
In this study, we used “gain of function” and compensatory mutagenesis approaches to map the interactive-sites of the cofactor, PZ, with the serpin inhibitor, ZPI, on PC/PS vesicles in the presence of Ca2+. Our results suggest that 1) in addition to interaction with the C-terminal pseudo-catalytic domain of PZ, ZPI also interacts with a hydrophobic cavity in the EGF2 domain of the cofactor (PZ), 2) the interaction of Asp-293 of ZPI with a basic pocket in PZ makes the greatest contribution to the binding energy of the cofactor interaction, and 3) the N-terminal Gla-domain of the protease FXa can functionally substitute for the Gla-domain of PZ on PC/PS vesicles. Previously, we demonstrated that grafting the C-terminal pseudo-catalytic domain of PZ on the light chain of FXa (PZ/FX-LC) restores most of the cofactor function of PZ in catalyzing the ZPI inhibition of FXa on PC/PS vesicles in the presence of Ca2+ [7]. However, the apparent dissociation constant (Kd(app)) for the interaction of the PZ/FX-LC chimera was elevated ~7-fold and its cofactor function was also similarly decreased ~6-fold [7], suggesting a possible interactive site for ZPI outside the pseudo-catalytic domain of PZ. A recent x-ray crystal structure of PZ in complex with ZPI indicates that in addition to extensive interaction between ZPI and the pseudo-catalytic domain, the side chain residue of ZPI Tyr-240 orients toward a hydrophobic cavity between the pseudo-catalytic domain and EGF2 domain of PZ [10]. Structures of EGF domains in vitamin K-dependent plasma proteins consist of three disulfide bonded β sheets forming three loops divided into a primary subdomain stabilized by the first two disulfide bonds and a secondary subdomain stabilized by a single disulfide bond [4,15]. Two Pro residues (Pro-91 and Pro-103) of PZ, located in the primary subdomain of EGF2 contribute to the hydrophobicity of this cavity (Fig. 6). Several other hydrophobic residues including Phe-247 and Phe-250 which are located on the pseudo-catalytic domain of PZ also protrude into this cavity, rendering it highly hydrophobic (Fig. 6). Analysis of structural data suggests that the phenolic ring of Tyr-240 in ZPI can be stabilized in this cavity without any evidence for its hydroxyl group making a hydrophilic interaction with any one of the surrounding residues (Fig. 6). The corresponding hydrophobic residues in the FXa EGF2 domain have not been conserved [16], but rather several charged residues (in particular Glu-102 and Glu-103) would render this pocket hydrophilic and thus unsuitable for accommodating ZPI Tyr-240.
Figure 6.

Crystal structure of the PZ-ZPI complex. The structures of ZPI and PZ in ribbon representations are shown in cyan and green, respectively. The relative three dimensional locations of the side-chains of PZ and ZPI residues which form a hydrophobic cavity between EGF2 and pseudo-catalytic domain of PZ and stabilize Tyr-240 of ZPI are shown by arrows. The first EGF2 subdomain of PZ is shown in light brown color. The coordinates obtained from Protein Data Bank accession code (PDB ID: 3H5C) were used to prepare the figure [10].
To test the hypothesis that the stabilization of Tyr-240 of ZPI within this hydrophobic cavity of PZ contributes to the binding affinity of complex formation and thus accounts for the 6-fold impaired cofactor function of the PZ/FX-LC chimera, we introduced the Pro residues of PZ EGF2 domain back onto the PZ/FX-LC chimeric cofactor. However, the resulting cofactor mutant did not express for characterization (data not presented). Thus, we grafted the first subdomain of PZ back on the PZ/FX-LC chimera and expressed the mutant in HEK-293 cells. Consistent with the hypothesis and structural data, the defective cofactor functions of this PZ chimera, with respect to both its ZPI-affinity and its extent of cofactor function, were restored. This was evidenced by the observation that the chimera exhibited kinetic and cofactor properties which were essentially identical to those of wild-type PZ. To estimate the contribution of this interaction to binding affinity, the Kd(app) values for PZ/FX-LC (7.2 nM), PZ-EGF2/FX-LC (0.75 nM) and PZ-WT (1.0 nM) were converted to decreases in the free energy of binding for these derivatives based on ΔGbinding = RT Ln (Kd(app)). Thus, relative to a binding free energy of −12.3 kcal/mol for PZ-WT (or PZ-EGF2/FX-LC), the corresponding value of −11.1 kcal/mol for PZ/FX-LC and -11.0 kcal/mol for PZ/FX-EGF2 (Kd(app) = 8.5 nM) would suggest that the intermolecular interaction between residue Tyr-240 of ZPI and the hydrophobic cavity of EGF2 in the cofactor PZ makes ~-1.2 to −1.3 kcal/mol for the binding energy of the interaction. These results are consistent with free energy measurements using the reactive center loop (RCL) mutant of ZPI (ZPI-YA) which is nearly inactive and does not inhibit FXa, but is expected to exhibit normal affinity for PZ. Relative to a binding free energy of −10 kcal/mol for the interaction of the ZPI-YA mutant with PZ-WT (Kd(app) = 40 nM), the corresponding value of -9 kcal/mol for PZ/FX-LC (Kd(app) = 227 nM) supports the conclusion that the interaction of ZPI with the EGF2 domain of PZ makes ~−1 kcal/mol contribution to the free energy of binding. Thus, in contrast to a previous report which observed a dramatic binding energy effect for the ZPI Tyr-240 interaction with EGF2 domain [10], our results predicts a modest contribution for this residue in stabilizing the binary ZPI-PZ complex formation. The basis for these differences in the extent of contribution of ZPI Tyr-240 to the binding energy of complex formation between the two studies is not known. The previous study replaced the EGF2 domain of PZ with the corresponding domain of protein C which has an N-linked glycosylation site (Asn-97) on the EGF2 domain [4]. This feature has not been conserved in the EGF2 domains of either FXa or PZ. The PZ/protein C chimera was a loss of function mutant, exhibiting no affinity for ZPI [10]. Whether this posttranslational modification, which most likely also existed in the PZ/protein C chimera, interfered with the interaction of ZPI with this cofactor mutant needs to be considered. Unlike the loss of function mutant of previous report, the results presented in the current manuscript are analyzed based on “gain of function” mutagenesis approaches.
By contrast to this modest contribution by Tyr-240 of ZPI to the binding energy of interaction with PZ, Asp-293 of ZPI makes a dramatic contribution to interaction with the cofactor since the charge reversal mutant of the D293R mutant of ZPI exhibited dramatically reduced affinity for wild-type PZ as evidenced by the linear dependence of the rate constants on concentrations of the ZPI mutant for up to 800 nM serpin. The compensatory PZ-R212D cofactor mutant could only restore the defective cofactor function of PZ-R212D by ~2-fold, confirming structural data that in addition to Arg-212, Asp-293 of ZPI is also involved in hydrophilic interactions with other residues of PZ including His-210 and Arg-298 in the binding interface between the two interacting molecules (Fig. 4). Since the Kd(app) for the interaction of ZPI-D293R could not be determined, no estimate for its binding free energy could be calculated, though the results designate a primary role for this residue in the ZPI-PZ interaction mechanism. This hypothesis is consistent with the literature [10].
Finally, there is evidence that the interaction of the N-terminal Gla-domain of PZ with the corresponding domain of FXa contributes to the cofactor function of PZ on PC/PS vesicles in the presence of Ca2+ [2,7]. We have demonstrated that replacing the Gla-domain of activated protein C (APC), another homologous vitamin K-dependent plasma protease, with the corresponding domain of FXa renders APC susceptible to inactivation by the PZ-ZPI complex on PC/PS vesicles [7]. In the current study we found that replacing the Gla-domain of PZ with the corresponding domain of FXa neither alters the affinity of the chimeric cofactor for ZPI nor the extent of its cofactor function in promoting the serpin inhibition of FXa on PC/PS vesicles. These results may suggest that specific protein-protein interactions between the Gla-domains of PZ and FXa may not be strictly required for the reaction, but rather the ability of the two vitamin K-dependent molecules to assemble on the same surface with similar high affinity is sufficient to facilitate the formation of an effective inhibitory complex. It should be noted that the Gla-dependent assembly of PZ and FXa makes an essential contribution to a ternary inhibitory complex formation on the PC/PS vesicles since neither Gla-domainless PZ (a PZ deletion mutant lacking the Gla-domain) has a significant cofactor function nor Gla-domainless FXa (a FXa deletion mutant lacking the Gla-domain) exhibits significant susceptibility to inhibition promoting effect of PZ on PC/PS vesicles in the presence of Ca2+ [7].
Highlights.
ZPI interacts with both pseudo-catalytic and EGF2 domains of PZ.
Several salt bridges stabilize ZPI interaction with pseudo-catalytic domain of PZ.
Tyr-240 of ZPI interacts with the N-terminal EGF2 subdomain of PZ.
This interaction makes modest contribution to binding energy of complex formation.
Most of the binding energy is contributed by the pseudo-catalytic domain of PZ.
Acknowledgments
We thank Audrey Rezaie for proofreading the manuscript and Dr. Aiwu Zhou for providing the bacterial expression plasmid for ZPI. The research discussed herein was supported by grants awarded by the National Heart, Lung, and Blood Institute of the National Institute of Health (HL 62565 HL 101917 to ARR).
Footnotes
Abbreviations– PZ, protein Z; ZPI, protein Z-dependent protease inhibitor; RCL, reactive center loop; ZPI-Y387A, a ZPI mutant in which Tyr-387 has been replaced with an Ala; FXa, activated factor X; Gla, γ-carboxyglutamic acid; EGF, epidermal growth factor; PZ/FX-LC, a PZ chimera in which its N-terminal Gla-domain has been replaced with the Gla-domain of factor X; PZ-EGF2/FX-LC, a PZ chimera in which the first subdomain of EGF2 domain in PZ/FX-LC has been replaced with the corresponding sequence of PZ; PZ/FX-Gla, a PZ chimera in which its N-terminal Gla-domain has been replaced with the Gla-domain of factor X; PZ/FX-EGF2, a PZ chimera in which the first subdomain of EGF2 domain of PZ has been replaced with the corresponding sequence of FX; PEG, polyethylene glycol; BSA, bovine serum albumin.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Han X, Fiehler R, Broze GJ., Jr Isolation a protein Z-dependent plasma protease inhibitor. Proc Natl Acad Sci USA. 1998;95:9250–9255. doi: 10.1073/pnas.95.16.9250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Han X, Fiehler R, Broze GJ., Jr Characterization of the protein Z-dependent protease inhibitor. Blood. 2000;96:3049–3055. [PubMed] [Google Scholar]
- 3.Huang X, Swanson R, Broze GJ, Jr, Olson ST. Kinetic characterization of the protein Z-dependent protease inhibitor reaction with blood coagulation factor Xa. J Biol Chem. 2008;283:29770–29983. doi: 10.1074/jbc.M805214200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stenflo J. Structure-function relationships of epidermal growth factor modules in vitamin K-dependent clotting factors. Blood. 1991;78:1637–1651. [PubMed] [Google Scholar]
- 5.Huntington JA, Read RJ, Carrell RW. Structure of a serpin-protease complex shows inhibition by deformation. Nature. 2000;407:923–926. doi: 10.1038/35038119. [DOI] [PubMed] [Google Scholar]
- 6.Tabatabai A, Fiehler R, Broze GJ., Jr Protein Z circulates in plasma in a complex with protein Z-dependent protease inhibitor. Thromb Haemostas. 2001;85:655–660. [PubMed] [Google Scholar]
- 7.Rezaie AR, Bae JSJS, Manithody C, Qureshi SH, Yang L. Protein Z-dependent protease inhibitor binds to the C-terminal domain of protein Z. J Biol Chem. 2008;283:19922–19926. doi: 10.1074/jbc.M802639200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wei Z, Yan Y, Carrell RW, Zhou A. Crystal structure of protein Z-dependent inhibitor complex shows how protein Z functions as a cofactor in the membrane inhibition of factor X. Blood. 2009;114:3662–3667. doi: 10.1182/blood-2009-04-210021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang X, Dementiev A, Olson ST, Gettins PG. Basis for the specificity and activation of the serpin protein Z-dependent proteinase inhibitor (ZPI) as an inhibitor of membrane-associated factor Xa. J Biol Chem. 2010;285:20399–20409. doi: 10.1074/jbc.M110.112748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang X, Yan Y, Tu Y, Gatti J, Broze GJ, Jr, Zhou A, Olson ST. Structural basis for catalytic activation of protein Z-dependent protease inhibitor (ZPI) by protein Z. Blood. 2012;120:1726–1733. doi: 10.1182/blood-2012-03-419598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sejima H, Hayashi T, Deyashiki Y, Nishioka J, Suzuki K. Primary structure of vitamin K-dependent human protein Z. Biochem Biophys Res Comm. 1990;171:661–668. doi: 10.1016/0006-291x(90)91197-z. [DOI] [PubMed] [Google Scholar]
- 12.Yang L, Ding Q, Huang X, Olson ST, Rezaie AR. Characterization of the heparin-binding site of the protein Z-dependent protease inhibitor. Biochemistry. 2012;51:4078–4085. doi: 10.1021/bi300353c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smirnov MD, Esmon CT. Phosphatidylethanolamine incorporation into vesicles selectively enhances factor Va inactivation by activated protein C. J Biol Chem. 1994;269:816–819. [PubMed] [Google Scholar]
- 14.Yang L, Rezaie AR. Residues of the 39-loop restrict the plasma inhibitor specificity of factor IXa. J Biol Chem. 2013;288:12692–12698. doi: 10.1074/jbc.M113.459347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ullner M, Selander M, Persson E, Stenflo J, Drakenberg T, Teleman O. Three-dimensional structure of the apo form of the N-terminal EGF-like module of blood coagulation factor X as determined by NMR spectroscopy and simulated folding. Biochemistry. 1992;31:5974–5983. doi: 10.1021/bi00141a004. [DOI] [PubMed] [Google Scholar]
- 16.Padmanabhan K, Padmanabhan KP, Tulinsky A, Park CH, Bode W, Huber R, Blankenship DT, Cardin AD, Kisiel W. Structure of human des(1–45) factor Xa at 2.2 A resolution. J Mol Biol. 1993;232:947–966. doi: 10.1006/jmbi.1993.1441. [DOI] [PubMed] [Google Scholar]