

Abstract
Mutations in five unconventional myosin genes have been associated with genetic hearing loss (HL). These genes encode the motor proteins myosin IA, IIIA, VI, VIIA and XVA. To date, most mutations in myosin genes have been found in the Caucasian population. In addition, only a few functional studies have been performed on the previously reported myosin mutations. We performed screening and functional studies for mutations in the MYO1A and MYO6 genes in Korean cases of autosomal dominant non-syndromic HL. We identified four novel heterozygous mutations in MYO6. Three mutations (p.R825X, p.R991X and Q918fsX941) produce a premature truncation of the myosin VI protein. Another mutation, p.R205Q, was associated with diminished actin-activated ATPase activity and actin gliding velocity of myosin VI in an in vitro analysis. This finding is consistent with the results of protein modelling studies and corroborates the pathogenicity of this mutation in the MYO6 gene. One missense variant, p.R544W, was found in the MYO1A gene, and in silico analysis suggested that this variant has deleterious effects on protein function. This finding is consistent with the results of protein modelling studies and corroborates the pathogenic effect of this mutation in the MYO6 gene.
Keywords: myosin, mutation, ATPase, protein structure, gene
2. Introduction
Myosins comprise a large family of motor proteins in eukaryotic tissues. While originally described in muscle tissue, another form of myosin, called unconventional myosin, has been found in many other tissues [1,2]. Unconventional myosins are associated with a diverse number of functions, including endocytosis, the regulation of ion channels and the movement of vesicles in the cytoplasm [3]. In the inner ear, myosins are expressed in the stereocilia and the cell body of the inner and outer hair cells. They bind actin filaments and hydrolyse adenosine triphosphate (ATP) to generate force and movement as well as to anchor hair cell stereocilia of the inner ear [4]. This arrangement enables the stereocilia to bend to sound waves, thereby opening ion channels, which allows for the transduction of sound (the conversion of sound waves to nerve impulses) [5].
To date, mutations in five different types of unconventional myosins—myosin IA (MYO1A), myosin IIIA (MYO3A), myosin VI (MYO6), myosin VIIA (MYO7A) and myosin XVA (MYO15A)—have been identified in hereditary hearing loss (HL) [6–13]. Among these, the MYO1A (OMIM 601478) and MYO6 (OMIM 600970) genes have been identified as the cause of autosomal dominant non-syndromic hearing loss (ADNSHL) [10,13]. MYO1A, which is also known as the brush border myosin-I gene, is located at the DFNA48 locus on chromosome 12, and thus far, eight different mutations have been identified in Italy in patients with HL [13,14]. The role of MYO1A in the inner ear and the impact of MYO1A mutations on HL in general, however, remain unclear. MYO6 is located on chromosome 6 [10,12]. Mutations in the MYO6 gene cause both ADNSHL (DFNA22) and autosomal recessive non-syndromic hearing loss (DFNB37). Previous studies have reported that myosin VI transports cargo molecules at the base of stereocilia, anchors actin filaments to the membrane of stereocilia by cargo molecules in the inner ear and is a mechanism for HL in myosin VI-null mice [15,16].
To date, most missense mutations among the previously reported mutations in the myosin family have only been described at the genome level. All known missense mutations are important for the protein's structure and function because their residue positions are highly conserved among many species. Therefore, a functional evaluation in vitro or in vivo is required for an accurate characterization of the mutated proteins.
In this study, we identified MYO1A and MYO6 mutations in patients with ADNSHL in Korea and analysed the molecular reasons for the pathogenic effect of novel mutations using in silico and in vitro analyses.
3. Results
3.1. Mutation screening of MYO6 and MYO1A
We sequenced the MYO6 and MYO1A genes in 53 unrelated Korean ADNSHL patients. Table 1 shows the four novel heterozygous mutations that were identified in the coding regions of the MYO6 gene. Two nonsense mutations, p.R825X and p.R991X, were identified in exons 24 and 28 of MYO6, respectively. The p.R825X mutation was detected in patient II-5 of the SR-149 family (figure 1a). This mutation is a C to T transition at nucleotide position 2473 (c.2473 C > T), which results in a stop codon in the IQ domain of the neck region. The p.R991X mutation was identified in the YS-052 family (figure 1b). The single-nucleotide change of C to T at nucleotide position 2971 (c.2971 C > T) introduced a stop codon corresponding to amino acid position 991. This stop codon causes the protein to lose part of the globular domain of the tail region. Patient II-2 of the SR-157 family had a single-nucleotide insertion at position 2752 (c.2752insA) in exon 26 (figure 1c). This mutation causes a frameshift that first results in the introduction of 23 new amino acids after the amino acid at position 917 (p.Q918fsX941) and then results in a truncated form of myosin VI. A fourth mutation was detected in patient III-1 of the SR-107 family (figure 1d). This missense mutation was a G to A nucleotide change at position 614 (c.614 G > A), which changes an Arg residue to a Gln at amino acid position 205 (p.R205Q) (figure 1d). The arginine at position 205 is conserved among myosins from many different species [17]. The previously solved X-ray structure of Dictyostelium myosin II suggested that Arg238 (corresponding to Arg205 of human myosin VI) in the switch I region [18] forms a salt bridge with Glu459 in the switch II region [19]. Therefore, it is anticipated that the p.R205Q mutation in human myosin VI disrupts this salt bridge, which will hamper motor activity. To predict whether this non-synonymous amino acid variant is likely to have a deleterious effect on the phenotype, two prediction programs, PolyPhen2 and SIFT, were applied. The results of both programs indicated that the variation would have a deleterious effect, with a PolyPhen2 score of 1.000 and a SIFT score of 0.00.
Table 1.
Mutations in the MYO6 gene (n.a., not available).
| exon | nucleotide change | amino acid change | time of onset | phenotype | shape of audiogram |
|---|---|---|---|---|---|
| 8 | c.614G > A | p.R205Q | first decade | progressive, mild to moderate | U-shaped or flat |
| 24 | c.2473C > T | p.R825X | n.a. | progressive, moderate to profound | down-sloping |
| 26 | c.2752insA | p.Q918fsX941 | fifth decade | progressive, moderate | flat |
| 28 | c.2971C > T | p.R991X | second decade | progressive, mild to moderate | down-sloping |
Figure 1.

Pedigree and genetic information for patients with novel mutations in the MYO6 and MYO1A genes. The lineages (a) SR-149, (b) YS-052, (c) SR-157 and (d) SR-107 exhibit autosomal dominant inheritance patterns. Two nonsense mutations, one frameshift mutation and one missense mutation were identified in these families by direct sequencing (c: reverse sequence). The arginine at amino acid position 205 of myosin VI is highly conserved among several species. (e) DNA sequence shows the p.R544W variant in the MYO1A gene. A comparison of amino acid sequences of myosin IA between several species. The asterisk indicates the mutation site. Squares, males; circles, females; slashes, deceased; shaded, affected. The arrows indicate the proband of each family.
In the MYO1A gene, we identified one variant with a G to A substitution at nucleotide position 1630 (c.1630 G > A) in patient FTM-01. This variant converts a highly conserved Arg to a Trp at amino acid position 544 (p.R544W; figure 1e). The results of both prediction programs showed that the variation would have a deleterious effect, with a PolyPhen2 score of 0.998 and a SIFT score of 0.00. None of the five aforementioned variants identified in this study were detected in any of the hundred normal-hearing Korean participants who were examined as controls.
Overall, we found one and four mutations in MYO1A and MYO6 in this study, respectively. Figure 2 shows the locations of these mutations.
Figure 2.
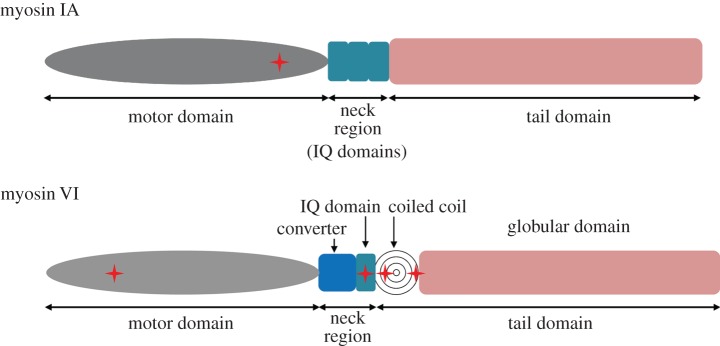
Diagram structure of myosin IA and myosin VI. Schematic of myosin IA and VI show the location of the mutations (cross shape).
3.2. Protein structure of mutant myosin VI and IA
We inspected the MYO6 homology model to determine the structural differences between the wild-type and mutant types, p.R205Q. Arg205 has hydrogen bonds and ionic interactions with several surrounding residues, but the p.R205Q mutation loses all of the ionic interactions because Gln is an uncharged residue (figure 3a). Because of the smaller size of Gln, most of the hydrogen bonds are also likely to be lost. In addition, we inspected the MYO1A homology model to determine the possible molecular effects of the p.R544W variant. The Arg544 residue in the motor domain of myosin 1A is hydrophilic and forms hydrogen bonds with several surrounding residues (figure 3b). The larger and hydrophobic side chain that is introduced by p.R544W cannot form these hydrogen bonds and thus destabilizes the motor domain.
Figure 3.

Protein structures of missense mutations in myosin VI and myosin Ia. These images show protein modelling of (a) WT and p.R205Q of myosin VI as well as (b) WT and p.R544W in myosin IA. These images show a close-up of the mutation site. (a) Green, arginine; red, glutamine; magenta, ATP; orange, glutamic acid and serine. (b) Green, arginine; red, tryptophan.
3.3. Actin-activated ATPase activity and in vitro motility activity of missense mutation in MYO6
We examined the effect of the p.R205Q mutation on the ATP hydrolysis-associated motor activity of full-length human myosin VI (M6Full). The p.R205Q myosin VI hydrolysed ATP in the presence of Mg2+ at a constant rate similar to that of the wild-type in the absence of actin. The results suggest that the p.R205Q mutation abolishes neither ATP binding nor hydrolysis. The actin-activated ATPase activity of the wild type (M6Full WT) in 0.1 mM CaCl2 showed a KATPase value that was similar to the tail-truncated mouse myosin VI (M6S1) previously reported [20], and the Vmax was approximately half of the Vmax of M6S1. However, the KATPase of M6Full WT in ethylene glycol tetraacetic acid (EGTA) was more than 10-fold higher than that in 0.1 mM CaCl2 and that of M6S1 in EGTA [20]. These results suggest that the tail portion of the molecule interferes with the interaction with actin in the presence of EGTA. The p.R205Q mutation, however, significantly diminished the actin-activated ATP hydrolysis cycle, which is coupled to the mechanical function of myosin VI. The major effect of the mutation was the significant decrease in Vmax rather than a change in KATPase (figure 4). In addition, we tested in vitro motility activity of M6Full WT and M6Full R205Q myosin VI full length to study interaction of F-actin and myosin. As a result, the velocity of rhodamine-phalloidin labelled F-actin movement was significantly decreased by p.R205Q mutation (figure 5). These results indicate that p.R205Q mutation causes a decrease in the actin-activated ATPase activity, and hampers F-actin movement.
Figure 4.
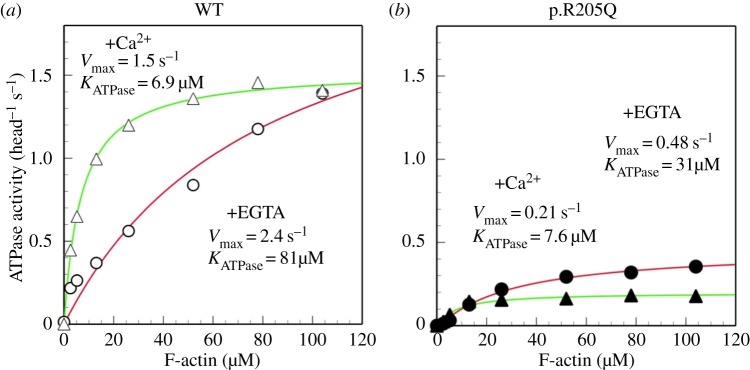
ATPase activity of missense variant (p.R205Q) of myosin VI. This image is F-actin-activated ATPase activity of WT and p.R205Q. Triangles, reaction buffer with 0.1 mM CaCl2; circles, reaction buffer with 1 mM EGTA; open symbols, ATPase activity of WT; solid symbols, ATPase activity of (a) WT and (b) p.R205Q.
Figure 5.
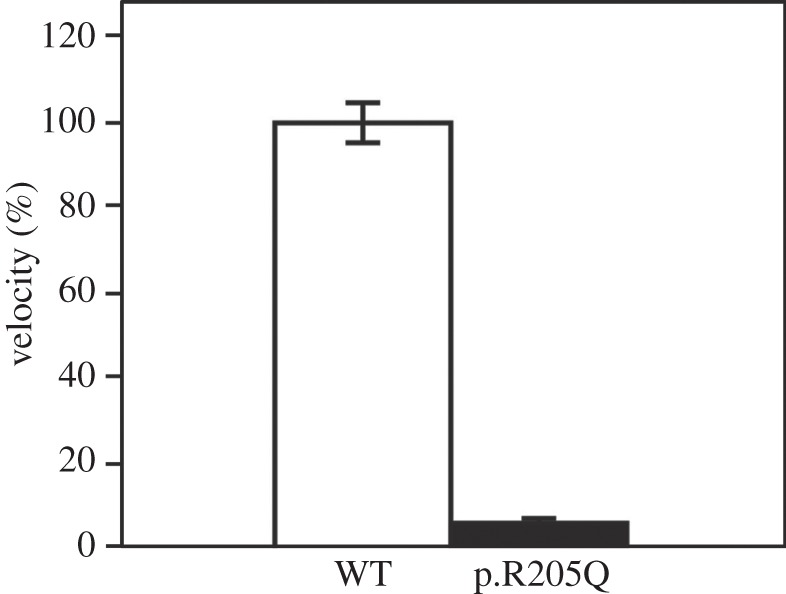
Multi-molecule in vitro motility activity of wild-type and p.R205Q mutant of myosin VI. This image shows in vitro motility activity of WT (open bar) and p.R205Q (closed bar). Error bars show mean ± s.e. (n = 6).
4. Discussion
Several studies have reported that mutations in unconventional myosin proteins cause HL in the Caucasian population [6–13]. HL caused by a mutation in the MYO6 gene was first reported in Snell's waltzer mouse. This strain carries a spontaneous recessive mutation that results in protein deficiency by frameshift mutation (130 bp deletion) [15]. Since then, seven different mutations in the MYO6 gene have been reported to be associated with DFNA22 deafness in Caucasian families: three missense mutations (p.H246R, p.C442Y and p.R1204W), a nonsense mutation (p.R849X), two splicing mutations (c.897G > T and c.2416 + 2321T > G) and an overexpression mutation [10,21–25]. In addition, three unrelated Pakistani families exhibited homozygosity for three different mutations associated with DFNB37 deafness (c.36insT, p.E216V and p.R1166X) [12]. A number of studies have revealed MYO6 mutations in the Caucasian and Pakistani populations; however, mutations have not previously been reported in East Asian populations. Our study of Korean patients with ADNSHL identified four novel mutations in the MYO6 gene that appear to be causally related to deafness. These mutations were neither observed in normal-hearing Korean controls nor in the 1000 Genomes Database and are thus far unique to the Korean deaf population.
Myosin VI is expressed in the early stage of hair cell differentiation [26]. It is located in the hair cell body, the cuticular plate, the pericuticular necklace of hair cells, and between actin and the membrane of stereocilia of hair cells of the organ of Corti [5,27]. The myosin VI dimer can move with a cargo molecule towards the minus end of an actin filament [28]. Moreover, it has been suggested that myosin VI plays an important role in the generation and maintenance of hair cell stereocilia [29], which has been shown in several in vivo studies using myosin VI null mice with stereocilia fusion and growth of giant hair cell stereocilia in the cochlea [15,16].
Myosin VI has three functional domains: an N-terminal motor domain or head domain, a C-terminal tail domain and a so-called neck region. The N-terminal motor domain has ATPase activity and binds to actin molecules, whereas the tail domain contributes to the dimerization of myosin molecules and cargo binding. The neck region is located between the head and tail domains, and contains light chain- or calmodulin-binding IQ motifs [3,30,31]. Myosin VI is a motor protein that uses energy derived from ATP hydrolysis by actin-activated ATPase [31], and half of all myosin VI mutations have been found in the motor domain [10,12,21,23]. Because Snell's waltzer mouse with an alternatively spliced, truncated myosin VI protein has shown disturbing dimerization [15,23], we expect that the two nonsense mutations and the frameshift mutation disrupt dimerization in a similar manner, resulting in a myosin VI null phenotype. In addition, the p.R238E mutation of Dictyostelium myosin II [32–34] and the p.R247A/p.R247E mutation of smooth muscle myosin II [35] were previously reported to abolish actin-activated ATPase activity. The present functional studies revealed that mutation of the conserved Arg205 to Gln does not abolish the actin activity of myosin VI but significantly decreases Vmax. Because the Gln side chain contains the –NH2 group, it is possible that it can still form a hydrogen bond with the Glu side chain in switch II, but we cannot exclude the possibility that the active site pockets are slightly different between myosin II and myosin VI. Taken together, these findings indicate that the p.R205Q mutation causes a myosin VI null function via reduced ATPase activity in the motor domain.
Myosin IA is one of the most abundant components of the enterocyte brush border, and plays critical roles in the interface between the membrane and actin cytoskeleton [36]. The p.E385D missense mutation is considered causative largely because it was demonstrated to disrupt actin movement by lowering ATPase activity [37]. We observed one variant, p.R544W, in the MYO1A gene. This Arg544 residue is located in a conserved region of the myosin IA motor domain in vertebrates. Our in silico analyses suggest that p.R544W may be a disease-causing variant, because this mutation causes destabilization of the motor domain because hydrogen bonds cannot be formed. However, further in vivo or in vitro analyses are required to determine whether this variant is truly causative or whether it is a rare polymorphism.
In conclusion, we identified five novel mutations in MYO1A and MYO6, and analysed their characteristics by in vitro and in silico analyses. We explained in detail that the p.R205Q mutation in myosin VI decreases ATPase activity significantly.
5. Material and methods
5.1. Subjects and clinical evaluations
We selected 53 unrelated Korean patients with non-syndromic HL whose families exhibited an autosomal dominant inheritance pattern. All subjects were recruited from the Yonsei University Hospital, Seoul, Korea, the Soree Ear Clinic, Seoul, Korea or the Patima Hospital, Daegu, Korea. Following a physical and otoscopic examination, pure tone audiometry (PTA) was performed in a sound-attenuated room. We determined the average thresholds for a frequency range of 250–8000 Hz. One hundred unrelated Koreans diagnosed as normal by PTA test were recruited as control subjects.
5.2. Genetic analysis
Genomic DNA was extracted from peripheral blood using a FlexiGene DNA extraction kit (Qiagen, Hilden, Germany) or from buccal cells using a Puregene Buccal Cell Core kit (Qiagen). To analyse the MYO1A and MYO6 gene sequences, all of the exons and exon–intron boundary regions in each gene were amplified by polymerase chain reaction (PCR) using specifically designed primer pairs. We performed cycling PCR using an ABI Big Dye Terminator v. 3.1 Cycle Sequencing kit (Applied Biosystems, Foster City, CA). PCR products were sequenced using an ABI 3130XL genetic analyser (Applied Biosystems). The sequence data were analysed using Chromas Pro (v. 1.5) software (Technelysium, Pty Ltd., Tewantin, Queensland, Australia) and Seqscape (v. 2.5) software (Applied Biosystems), and the sequences were compared with the corresponding sequences in GenBank (accession no. NM_001256041.1 for MYO1A and accession no. NM_004999.3 for MYO6). The amino acid sequences of various vertebrate species were aligned using the Clustal W2 program (http://www.ebi.ac.uk/Tools/msa/clustalw2/) to evaluate whether amino acid substitutions occur in conserved regions of the proteins. The pathogenic effect of these substitutions was predicted using SIFT (http://sift.jcvi.org/) and PolyPhen2 (http://genetics.bwh.harvard.edu/pph2).
5.3. Protein modelling
Because no experimentally solved structures exist for either MYO1A or MOY6, we built homology models for these proteins. The YASARA and WHAT IF Twinset was used (with default parameters) for model building and subsequent analyses [38]. The model for MYO1A was constructed using the PDB file 2dfs [39] as a template. This file contains the structure for myosin V from chicken, which is 37% identical to MYO1A over 770 residues. The modelled domain of MYO1A contains the location of the newly identified mutation p.R544W. The model for myosin VI was constructed from myosin VI of wild boar, which is 98% identical to the human sequence over 784 residues [40]. The point mutations in MYO6 were analysed using this model.
5.4. Generation of myosin VI constructs and isolation of the proteins
Human myosin VI cDNA clones containing nucleotides 1–1285 were obtained from a human kidney cDNA library and inserted into the pFastbac1 (Invitrogen, Carlsbad, CA) baculovirus transfer vector at the polylinker region. For wild-type recombinant myosin VI (M6FullWT), a c-myc and FLAG tag sequence (ACGCGTGAGCAAAAGCTCATTTCTGAAGAGGACTTGTCGCGTGATTATAAAGATGATGATGATAAA) was introduced between the 3′ end of the myosin VI cDNA and the stop codon to aid purification of the recombinant protein. The p.R205Q mutation of myosin VI was generated by PCR-based site-directed mutagenesis [41] using Pfu DNA polymerase. The presence of the mutation was confirmed by sequencing. Expression and purification of the recombinant myosin VI was performed as described previously [20].
5.5. ATPase assay
ATPase activity was measured at 25°C in a buffer that contained 30 mM KCl, 20 mM MOPS-KOH (pH 7.5), 3 mM MgCl2, 2 mM ATP, 5 µM CaM, 40 unit ml−1 pyruvate kinase, 2.5 mM phosphoenolpyruvate, 1 mM EGTA or 0.2 mM CaCl2, 11–14 µg ml−1 M6Full WT or the mutant, and various concentrations of F-actin ranging from 0 to 104 µM. The liberated pyruvate was determined as described previously [42]. To calculate the ATPase activity, protein concentrations of M6Full WT and the mutant were determined using Coomassie Plus Assay Reagent (Thermo Scientific, Waltham, MA). The molecular weight of M6Full WT was assumed to be 185 kDa (151.4 kDa M6 heavy chain and 2 × 16.8 kDa calmodulin light chains).
5.6. In vitro motility assay
In vitro motility of Rhodamine-labelled F-actin was measured using a cooled-CCD camera (Sensi CamQE, Cooke Co.) equipped with an Olympus IX-51-based fluorescence microscope [20]. The assay was carried out in a buffer containing 25 mM KCl, 20 mM MOPS-KOH (pH7.5), 5 mM MgCl2, 1 mM EGTA, 2 mM ATP, 216 µg ml−1 glucose oxidase, 36 µg ml−1 catalase, 4.5 mg ml−1 glucose, 10 mM dithiothreitol, 5 µM calmodulin and 15 nM F-actin at 21°C. Each of wild-type and the mutant M6Full with the appropriate concentration was attached to the same coverslip separated with double-adhesive tape, and F-actin movements were captured at every 2 (wild-type) or 8 (p.R205Q) seconds. The velocity was calculated from the distance and the elapsed time in the video images.
Acknowledgement
The authors are grateful to the families for their collaboration in this study. None of the authors declare any conflict of interest.
This study was approved by the Institutional Review Board of the Yonsei University College of Medicine, and written informed consent was obtained from all participants.
Funding statement
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (MEST) (no. 2011–0028066) and the Korea Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (HI12C1004). H.V. receives funding from the EU thematic area KBBE-2011–5, contract no. 289350.
References
- 1.Pollard TD, Korn ED. 1973. Acanthamoeba myosin. I. Isolation from Acanthamoeba castellanii of an enzyme similar to muscle myosin. J. Biol. Chem. 248, 4682–4690. [PubMed] [Google Scholar]
- 2.Cheney RE, Mooseker MS. 1992. Unconventional myosins. Curr. Opin. Cell Biol. 4, 27–35. (doi:10.1016/0955-0674(92)90055-H) [DOI] [PubMed] [Google Scholar]
- 3.Friedman TB, Sellers JR, Avraham KB. 1999. Unconventional myosins and the genetics of hearing loss. Am. J. Med. Genet. 89, 147–157. (doi:10.1002/(SICI)1096-8628(19990924)89:3<147::AID-AJMG5>3.0.CO;2-6) [DOI] [PubMed] [Google Scholar]
- 4.Mermall V, Post PL, Mooseker MS. 1998. Unconventional myosins in cell movement, membrane traffic, and signal transduction. Science 279, 527–533. (doi:10.1126/science.279.5350.527) [DOI] [PubMed] [Google Scholar]
- 5.Hasson T, Gillespie PG, Garcia JA, MacDonald RB, Zhao Y, Yee AG, Mooseker MS, Corey DP. 1997. Unconventional myosins in inner-ear sensory epithelia. J. Cell Biol. 137, 1287–1307. (doi:10.1083/jcb.137.6.1287) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu XZ, Walsh J, Mburu P, Kendrick-Jones J, Cope MJ, Steel KP, Brown SD. 1997. Mutations in the myosin VIIA gene cause non-syndromic recessive deafness. Nat. Genet. 16, 188–190. (doi:10.1038/ng0697-188) [DOI] [PubMed] [Google Scholar]
- 7.Liu XZ, Walsh J, Tamagawa Y, Kitamura K, Nishizawa M, Steel KP, Brown SD. 1997. Autosomal dominant non-syndromic deafness caused by a mutation in the myosin VIIA gene. Nat. Genet. 17, 268–269. (doi:10.1038/ng1197-268) [DOI] [PubMed] [Google Scholar]
- 8.Weil D, Kussel P, Blanchard S, Levy G, Levi-Acobas F, Drira M, Ayadi H, Petit C. 1997. The autosomal recessive isolated deafness, DFNB2, and the Usher 1B syndrome are allelic defects of the myosin-VIIA gene. Nat. Genet. 16, 191–193. (doi:10.1038/ng0697-191) [DOI] [PubMed] [Google Scholar]
- 9.Wang A, et al. 1998. Association of unconventional myosin MYO15 mutations with human nonsyndromic deafness DFNB3. Science 280, 1447–1451. (doi:10.1126/science.280.5368.1447) [DOI] [PubMed] [Google Scholar]
- 10.Melchionda S, et al. 2001. MYO6, the human homologue of the gene responsible for deafness in Snell's waltzer mice, is mutated in autosomal dominant nonsyndromic hearing loss. Am. J. Hum. Genet. 69, 635–640. (doi:10.1086/323156) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Walsh T, et al. 2002. From flies’ eyes to our ears: mutations in a human class III myosin cause progressive nonsyndromic hearing loss DFNB30. Proc. Natl Acad. Sci. USA 99, 7518–7523. (doi:10.1073/pnas.102091699) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ahmed ZM, et al. 2003. Mutations of MYO6 are associated with recessive deafness, DFNB37. Am. J. Hum. Genet. 72, 1315–1322. (doi:10.1086/375122) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Donaudy F, et al. 2003. Multiple mutations of MYO1A, a cochlear-expressed gene, in sensorineural hearing loss. Am. J. Hum. Genet. 72, 1571–1577. (doi:10.1086/375654) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.D'Adamo P, Pinna M, Capobianco S, Cesarani A, D'Eustacchio A, Fogu P, Carella M, Seri M, Gasparini P. 2003. A novel autosomal dominant non-syndromic deafness locus (DFNA48) maps to 12q13-q14 in a large Italian family. Hum. Genet. 112, 319–320. (doi:10.1007/s00439-002-0880-6) [DOI] [PubMed] [Google Scholar]
- 15.Avraham KB, Hasson T, Steel KP, Kingsley DM, Russell LB, Mooseker MS, Copeland NG, Jenkins NA. 1995. The mouse Snell's waltzer deafness gene encodes an unconventional myosin required for structural integrity of inner ear hair cells. Nat. Genet. 11, 369–375. (doi:10.1038/ng1295-369) [DOI] [PubMed] [Google Scholar]
- 16.Hertzano R, et al. 2008. A Myo6 mutation destroys coordination between the myosin heads, revealing new functions of myosin VI in the stereocilia of mammalian inner ear hair cells. PLoS Genet. 4, e1000207 (doi:10.1371/journal.pgen.1000207) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berg JS, Powell BC, Cheney RE. 2001. A millennial myosin census. Mol. Biol. Cell 12, 780–794. (doi:10.1091/mbc.12.4.780) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sprang SR. 1997. G protein mechanisms: insights from structural analysis. Annu. Rev. Biochem. 66, 639–678. (doi:10.1146/annurev.biochem.66.1.639) [DOI] [PubMed] [Google Scholar]
- 19.Smith CA, Rayment I. 1996. X-ray structure of the magnesium(II).ADP.vanadate complex of the Dictyostelium discoideum myosin motor domain to 1.9 A resolution. Biochemistry 35, 5404–5417. (doi:10.1021/bi952633) [DOI] [PubMed] [Google Scholar]
- 20.Sato O, White HD, Inoue A, Belknap B, Ikebe R, Ikebe M. 2004. Human deafness mutation of myosin VI (C442Y) accelerates the ADP dissociation rate. J. Biol. Chem. 279, 28 844–28 854. (doi:10.1074/jbc.M314332200) [DOI] [PubMed] [Google Scholar]
- 21.Mohiddin SA, Ahmed ZM, Griffith AJ, Tripodi D, Friedman TB, Fananapazir L, Morell RJ. 2004. Novel association of hypertrophic cardiomyopathy, sensorineural deafness, and a mutation in unconventional myosin VI (MYO6). J. Med. Genet. 41, 309–314. (doi:10.1136/jmg.2003.011973) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hilgert N, Topsakal V, van Dinther J, Offeciers E, Van de Heyning P, Van Camp G. 2008. A splice-site mutation and overexpression of MYO6 cause a similar phenotype in two families with autosomal dominant hearing loss. Eur. J. Hum. Genet. 16, 593–602. (doi:10.1038/sj.ejhg.5202000) [DOI] [PubMed] [Google Scholar]
- 23.Sanggaard KM, et al. 2008. A novel nonsense mutation in MYO6 is associated with progressive nonsyndromic hearing loss in a Danish DFNA22 family. Am. J. Med. Genet. A 146A, 1017–1025. (doi:10.1002/ajmg.a.32174) [DOI] [PubMed] [Google Scholar]
- 24.Brownstein Z, et al. 2013. Novel myosin mutations for hereditary hearing loss revealed by targeted genomic capture and massively parallel sequencing. Eur. J. Hum. Genet. 22, 768–775. (doi:10.1038/ejhg.2013.232) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oonk AM, et al. 2013. Progressive hereditary hearing impairment caused by a MYO6 mutation resembles presbyacusis. Hear Res. 299, 88–98. (doi:10.1016/j.heares.2012.12.015) [DOI] [PubMed] [Google Scholar]
- 26.Montcouquiol M, Rachel RA, Lanford PJ, Copeland NG, Jenkins NA, Kelley MW. 2003. Identification of Vangl2 and Scrb1 as planar polarity genes in mammals. Nature 423, 173–177. (doi:10.1038/nature01618) [DOI] [PubMed] [Google Scholar]
- 27.Rzadzinska AK, Schneider ME, Davies C, Riordan GP, Kachar B. 2004. An actin molecular treadmill and myosins maintain stereocilia functional architecture and self-renewal. J. Cell Biol. 164, 887–897. (doi:10.1083/jcb.200310055) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wells AL, Lin AW, Chen LQ, Safer D, Cain SM, Hasson T, Carragher BO, Milligan RA, Sweeney HL. 1999. Myosin VI is an actin-based motor that moves backwards. Nature 401, 505–508. (doi:10.1038/46835) [DOI] [PubMed] [Google Scholar]
- 29.Self T, Sobe T, Copeland NG, Jenkins NA, Avraham KB, Steel KP. 1999. Role of myosin VI in the differentiation of cochlear hair cells. Dev. Biol. 214, 331–341. (doi:10.1006/dbio.1999.9424) [DOI] [PubMed] [Google Scholar]
- 30.Friedman TB, Griffith AJ. 2003. Human nonsyndromic sensorineural deafness. Annu. Rev. Genomics Hum. Genet. 4, 341–402. (doi:10.1146/annurev.genom.4.070802.110347) [DOI] [PubMed] [Google Scholar]
- 31.Buss F, Spudich G, Kendrick-Jones J. 2004. Myosin VI: cellular functions and motor properties. Annu. Rev. Cell Dev. Biol. 20, 649–676. (doi:10.1146/annurev.cellbio.20.012103.094243) [DOI] [PubMed] [Google Scholar]
- 32.Furch M, Fujita-Becker S, Geeves MA, Holmes KC, Manstein DJ. 1999. Role of the salt-bridge between switch-1 and switch-2 of Dictyostelium myosin. J. Mol. Biol. 290, 797–809. (doi:10.1006/jmbi.1999.2921) [DOI] [PubMed] [Google Scholar]
- 33.Sasaki N, Shimada T, Sutoh K. 1998. Mutational analysis of the switch II loop of Dictyostelium myosin II. J. Biol. Chem. 273, 20 334–20 340. (doi:10.1074/jbc.273.32.20334) [DOI] [PubMed] [Google Scholar]
- 34.Shimada T, Sasaki N, Ohkura R, Sutoh K. 1997. Alanine scanning mutagenesis of the switch I region in the ATPase site of Dictyostelium discoideum myosin II. Biochemistry 36, 14 037–14 043. (doi:10.1021/bi971837i) [DOI] [PubMed] [Google Scholar]
- 35.Onishi H, Ohki T, Mochizuki N, Morales MF. 2002. Early stages of energy transduction by myosin: roles of Arg in switch I, of Glu in switch II, and of the salt-bridge between them. Proc. Natl Acad. Sci. USA 99, 15 339–15 344. (doi:10.1073/pnas.242604099) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mooseker MS, Cheney RE. 1995. Unconventional myosins. Annu. Rev. Cell Dev. Biol. 11, 633–675. (doi:10.1146/annurev.cb.11.110195.003221) [DOI] [PubMed] [Google Scholar]
- 37.Yengo CM, Ananthanarayanan SK, Brosey CA, Mao S, Tyska MJ. 2008. Human deafness mutation E385D disrupts the mechanochemical coupling and subcellular targeting of myosin-1a. Biophys. J. 94, L5–L7. (doi:10.1529/biophysj.107.122689) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Krieger E, Joo K, Lee J, Raman S, Thompson J, Tyka M, Baker D, Karplus K. 2009. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: four approaches that performed well in CASP8. Proteins 77(Suppl. 9), 114–122. (doi:10.1002/prot.22570) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu J, Taylor DW, Krementsova EB, Trybus KM, Taylor KA. 2006. Three-dimensional structure of the myosin V inhibited state by cryoelectron tomography. Nature 442, 208–211. (doi:10.1038/nature04719) [DOI] [PubMed] [Google Scholar]
- 40.Menetrey J, Llinas P, Mukherjea M, Sweeney HL, Houdusse A. 2007. The structural basis for the large powerstroke of myosin VI. Cell 131, 300–308. (doi:10.1016/j.cell.2007.08.027) [DOI] [PubMed] [Google Scholar]
- 41.Weiner MP, Costa GL, Schoettlin W, Cline J, Mathur E, Bauer JC. 1994. Site-directed mutagenesis of double-stranded DNA by the polymerase chain reaction. Gene 151, 119–123. (doi:10.1016/0378-1119(94)90641-6) [DOI] [PubMed] [Google Scholar]
- 42.Reynard AM, Hass LF, Jacobsen DD, Boyer PD. 1961. The correlation of reaction kinetics and substrate binding with the mechanism of pyruvate kinase. J. Biol. Chem. 236, 2277–2283. [PubMed] [Google Scholar]