

Abstract
Fcabs (Fc antigen binding) are crystallizable fragments of IgG where the C-terminal structural loops of the CH3 domain are engineered for antigen binding. For the design of libraries it is beneficial to know positions that will permit loop elongation to increase the potential interaction surface with antigen. However, the insertion of additional loop residues might impair the immunoglobulin fold. In the present work we have probed whether stabilizing mutations flanking the randomized and elongated loop region improve the quality of Fcab libraries. In detail, 13 libraries were constructed having the C-terminal part of the EF loop randomized and carrying additional residues (1, 2, 3, 5 or 10, respectively) in the absence and presence of two flanking mutations. The latter have been demonstrated to increase the thermal stability of the CH3 domain of the respective solubly expressed proteins. Assessment of the stability of the libraries expressed on the surface of yeast cells by flow cytometry demonstrated that loop elongation was considerably better tolerated in the stabilized libraries. By using in silico loop reconstruction and mimicking randomization together with MD simulations the underlying molecular dynamics were investigated. In the presence of stabilizing stem residues the backbone flexibility of the engineered EF loop as well as the fluctuation between its accessible conformations were decreased. In addition the CD loop (but not the AB loop) and most of the framework regions were rigidified. The obtained data are discussed with respect to the design of Fcabs and available data on the relation between flexibility and affinity of CDR loops in Ig-like molecules.
Abbreviations: IgG1, immunoglobulin G class 1; IgG1-Fc, crystallizable fragment of immunoglobulin G class 1; Fc-wt, recombinant wild-type human IgG1-Fc; Fab, antigen binding fragment; mAb, monoclonal antibody; scFv, single-chain variable fragment; FcγRI, Fcγ-receptor I (also termed CD64); ADCC, antibody dependent cell-mediated cytotoxicity; CDC, complement dependent cytotoxicity; aCH2, antibody recognizing the intact fold of the CH2-domain of human IgG1; FACS, fluorescence activated cell sorting; DSC, differential scanning calorimetry; MD, molecular dynamics; DSSP algorithm, Define Secondary Structure of Proteins algorithm
Keywords: Therapeutic antibody fragment, Fcab, Protein engineering, Yeast surface display, Loop reconstruction, Molecular dynamics simulation
Graphical abstract
Highlights
-
•
Characterization of EF loop libraries of IgG1-Fc displayed on yeast surface.
-
•
Artificial stable stem regions increase tolerance to amino acid insertions.
-
•
Combination of in silico loop elongation with MD simulations.
-
•
Analysis of loop dynamics and conformational variability.
-
•
Pronounced impact of loop stabilization on domain and loop dynamics.
1. Introduction
The immunoglobulin-like fold (Ig-like) is one of the most common structural motifs, exhibiting a β-sandwich structure of two interacting antiparallel β-sheets with a Greek Key topology [1]. Immunoglobulin domains evolved an outstanding capacity to tolerate variability in the length of loops (that connect the β-strands), the amino acid sequence as well as loop conformation while maintaining their overall structure and function. This is most evident for the three CDR-loops (CDR1, CDR2 and CDR3) of variable domains of antibodies, but is generally observed in protein domains belonging to the immunoglobulin fold family [2]. Besides this natural loop variation, it could be demonstrated that artificially introduced sequences in loops can also be tolerated by Ig-like folds and used for the design of specific binders. This was shown for example with a fibronectin type III domain [3], the CH2 domain of IgG1-Fc [4] and the CH3 domain in the context of the crystallizable fragment (Fc) of IgG1 [5,6]. The latter turned out to be a promising starting scaffold for the design of a novel antibody-based therapeutic format called Fcab, i.e. antigen binding Fc fragment. The Fc protein has – except for an antigen-binding site – all properties of a full-size IgG1, i.e. the ability to bind Fcγ-receptors, the complement activator C1q and the neonatal Fc receptor (FcRn). Upon engineering the C-terminal structural loops of the CH3 domains in IgG1-Fc, small 50 kDa HER2/neu-binding homodimeric Fcabs could be designed which elicited potent antibody-dependent cell-mediated cytotoxicity in vitro and which have a long half-life in vivo [6].
Successful introduction of novel binding sites in the C-terminal AB-, CD- and EF-loops of the IgG1-Fc CH3 domains needs detailed information about the correlation between primary and tertiary structure and stability. For the design of libraries that ideally contain a high percentage of well-folded clones to guarantee the efficient selection of binders, it is important to know those amino acids that can be randomized without significantly decreasing the conformational stability of resulting variants. Individual loop residues may exhibit important non-covalent interaction(s) with the β-strands or with other loops and thus should not be mutated. Additionally, those sites in loops have to be identified that allow the insertion of additional amino acids in order to increase loop length and thus the potential interaction surface with antigen. The optimization of Fcab libraries according to these criteria was reported recently [7]. However, no information on the conformation and dynamics of elongated structural loops of constant domains has been reported so far.
In the work described in this paper we constructed 13 yeast surface libraries in which the C-terminal part of the EF-loop of the CH3 domain of IgG1-Fc was randomized and additional residues were inserted. We evaluate how the insertion of stabilizing mutations in the EF loop may support the preservation of the overall stability of the respective libraries. Rapid stability assessment on a library scale together with novel in silico loop reconstruction and molecular dynamics simulations is demonstrated. This gives valuable information on the effect that the insertion of additional residues, in the absence and presence of stabilizing stem residues, has on the fluctuation between conformations of the EF-loop itself as well as on neighboring structural loops and the dynamics of the immunoglobulin framework. Results are discussed with respect to the impact that these findings will have on the selection of Fcabs.
2. Materials and methods
2.1. Differential scanning calorimetry
Differential scanning calorimetry (DSC) was used to analyze the thermal stability of wild-type IgG1-Fc and the variants Q418L and S424T. After adjusting the protein concentration to 5 μM, samples were degassed and measured on a VP–DSC Capillary Cell MicroCalorimeter (MicroCal, Northampton, MA) at a temperature range of 20 °C to 110 °C and a heating rate of 1 °C/min. The baseline was recorded by performing a rescan under the same conditions. After the subtraction of the baseline, data were normalized for protein concentration and fitted according to a non-2-state thermal unfolding model using the software MicroCal addin for OriginLab (Origin Lab Corporation, Northampton, MA).
2.2. Cloning and library construction
The gene encoding the human IgG1-Fc fragment (comprising hinge-region, CH2- and CH3-domains) was codon-optimized for the expression in yeast and cloned into the vector pYD1 (Invitrogen, Carlsbad, CA, USA) for expression as a fusion protein with Aga2p on the surface of Saccharomyces cerevisiae using BamHI and NotI [8]. A stop codon was introduced at the 3′ end of the region coding for the CH3 domain to exclude any C-terminal tags present on pYD1. To construct yeast cell surface display libraries, two novel BsmBI restriction sites were introduced upstream of the region coding for the CD-loop of the CH3 domain and downstream of the EF loop (QuikChange Lightning Site-Directed Mutagenesis Kit, Agilent Technologies, Santa Clara, CA, USA). In accordance with that, a stuffer fragment (non-coding) was amplified containing two BsmBI restriction sites. Vector as well as stuffer fragment were digested with BsmBI and the fragment was ligated into the CIAP treated linearized vector using the T4 DNA ligase. The utilization of the resulting vector pYD1-2BN would therefore not lead to surface expression of wild-type IgG1-Fc as a consequence of incomplete BsmBI-digest or religation.
A multi-step polymerase chain reaction (PCR) was performed to randomize loop sequences by saturated mutagenesis using NNK-oligonucleotides (N codes for a mixture of all four nucleotides, whereas K represents a mixture of G and T; ordered from Sigma, St. Louis, MO), resulting in fragments comprising regions of homology to the linearized pYD1 backbone flanking the regions coding for the EF-loop for homologous recombination in yeast. The stabilizing mutations Q418L and/or S424T (see Results Section) were introduced in the respective libraries by the modification of the oligonucleotide sequences (Supplementary Table 1).
A multi-step polymerase chain reaction (PCR) was performed to randomize loop sequences by saturated mutagenesis using NNK-oligonucleotides (N codes for a mixture of all four nucleotides, whereas K represents a mixture of G and T; ordered from Sigma, St. Louis, MO), resulting in fragments comprising regions of homology to the linearized pYD1 backbone flanking the regions coding for the EF-loop for homologous recombination in yeast. The stabilizing mutations Q418L and/or S424T (see Results Section) were introduced in the respective libraries by the modification of the oligonucleotide sequences (Supplementary Table 1).
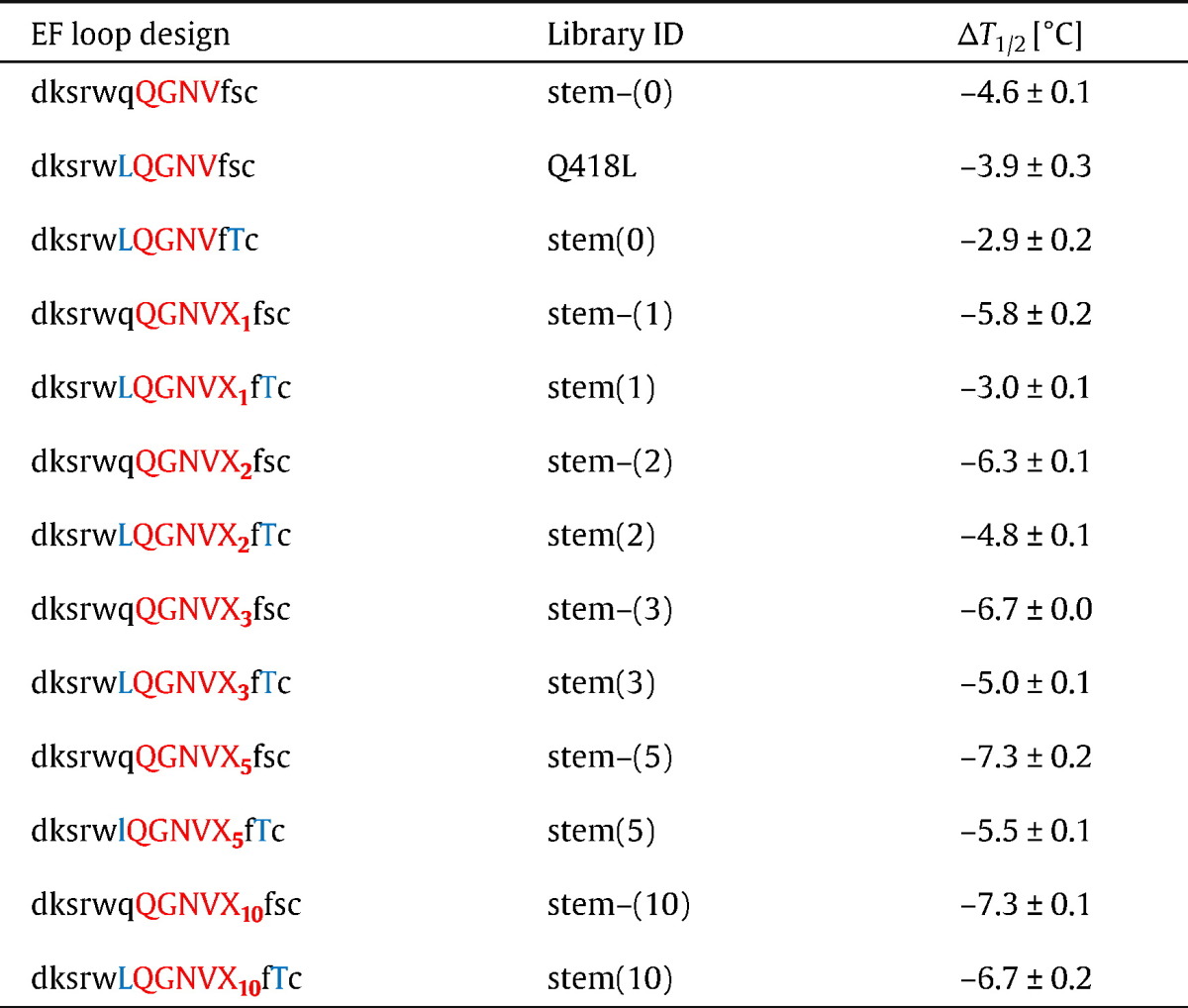
S. cerevisiae EBY100 (Invitrogen, Carlsbad, CA, USA) were transformed with purified library inserts and BsmBI-digested pYD1-2BN using the lithium-acetate method [9]. Gap repair driven homologous recombination in S. cerevisiae due to the presence of homologous regions on inserts and BsmBI-digested pYD1-2BN resulted in reconstitution of the plasmids. The S. cerevisiae-libraries were cultured in SD-CAA medium [20 g/L glucose, 0.1 M KH2PO4/K2HPO4, pH 6, 10 g/L (NH4)2SO4, 0.1 g/L l-leucine (all Sigma, St. Louis, MO), 3.4 g/L yeast nitrogen base, 10 g/L bacto casamino acids (all Difco, BD, Franklin Lakes, NJ)] at 28 °C for 48 h while shaking at 180 rpm. The isolation of pYD1 vector DNA for Escherichia coli transformation and ensuing sequencing was done using the Zymoprep Yeast Plasmid Miniprep Kit II (Zymo Research, Orange, CA). In total, 13 libraries were constructed as described in Table 1: Library stem-(0) consists of IgG1-Fc variants without stabilizing mutations or additional inserts, but with parts of the EF loop randomized (419–422). Library stem(0) is constructed similarly, but with two additional stabilizing mutations flanking the randomized region. In libraries stem-(1), stem-(2), stem-(5) and stem-(10), 1,2,3,5 or 10 additional residues are inserted into the randomized EF loop, without stabilizing mutations, while the EF loops in the corresponding libraries stem(1), stem(2), stem(3), stem(5) and stem(10) are again flanked by two stabilizing mutations.
Table 1.
Library design, library identity (ID) and experimentally determined temperatures of half-maximal irreversible denaturation. Black lowercase letters in column ‘EF loop design’ represent amino acids that were kept constant in the design,  represent sites of stabilizing mutations (Q418
represent sites of stabilizing mutations (Q418 , S424
, S424 ),
),  represent residues that have been randomized in the respective design and
represent residues that have been randomized in the respective design and  represent the number of inserted random amino acids at the respective position.
represent the number of inserted random amino acids at the respective position.
2.3. Library expression
Overnight SD-CAA-cultures of S. cerevisiae-libraries were passaged to an OD600 of 1, incubated at 28 °C for 4 h while shaking at 180 rpm, centrifuged and resuspended in an SGR-CAA-medium [20 g/L galactose, 10 g/L raffinose, 0.1 M KH2PO4/K2HPO4, pH 6, 10 g/L (NH4)2SO4, 0.1 g/L l-leucine (all Sigma, St. Louis, MO), 3.4 g/L yeast nitrogen base, 10 g/L bacto casamino acids (all Difco, BD, Franklin Lakes, NJ)] to an OD600 of 1. The induction of surface expression was done at 20 °C for 18–20 h. Cultures were centrifuged and resuspended in PBS/BSA [0.2 g/L KCl, 0.2 g/L KH2PO4, 8 g/L NaCl, 1.15 g/L Na2HPO4, 20 g/L bovine serum albumin (Sigma, St. Louis, MO)] to an OD600 of 3. Cell suspensions were aliquoted, transferred to microcentrifuge tubes and incubated for 30 min on ice or at 50 °C, 55 °C, 60 °C, 65 °C, 70 °C or 75 °C, respectively, in thermomixers (Eppendorf, Hamburg, Germany) while shaking at 300 rpm. Temperature-treated aliquots were cooled on ice. 50 μL were transferred to a round-bottom 96-well-plate (TPP, Trasadingen, Switzerland) followed by an incubation step at 4 °C for 30 min. Following centrifugation, cells were stained with 2.5 μg/mL anti-Xpress antibody (Invitrogen, Carlsbad, CA, USA) conjugated to allophycocyanin (APC) using the LYNX Rapid APC Antibody Conjugation Kit (AbD Serotec, Kidlington, UK) and with 1 μg/mL His-tagged FcγRI (R&D Systems, Abingdon, UK). Subsequently, cells were washed with PBS/BSA and stained with 1 μg/mL anti-His antibody conjugated to Alexa Fluor 488 (QIAGEN, Venlo, Netherlands). All stainings were performed in PBS/BSA at 4 °C for 30 min while shaking at 225 rpm. After a final washing step, cells were resuspended in PBS/BSA and analyzed on a FACSCanto II (BD, Franklin Lakes, NJ).
2.4. Flow cytometric determination of temperatures of half-maximal irreversible denaturation
In flow cytometric experiments, a total of 10,000 events were measured and gated according to the desired morphology, presence as singlets and binding of the fluorescently labeled anti-Xpress antibody to the expression tag (Xpress epitope at the N-terminus of the Fc). The resulting population was then probed for the binding of the respective fluorescence-labeled ligand to the displayed Fc protein. Data were analyzed using the FACSDiva software (BD). Overall library stability was determined from flow cytometric data by calculating the temperatures of half-maximal irreversible denaturation (T1/2) for each distinct library as described previously [7]. Briefly, the mean fluorescence intensity (MFI) of the FcγRI-signal measured for the population of Xpress-positive yeast cells displaying wild-type IgG1-Fc on the cell surface after incubation at 0 °C was set to 100% relative MFI, while the lowest MFI of a measurement series was set to 0%. Data were fitted according to , where x corresponds to the incubation temperature, y corresponds to the residual MFI after heat incubation, a and b correspond to the maximum and minimum y values as defined by the model and x0 corresponds to T1/2. All measurements were performed in triplicates.
2.5. In silico introduction of stabilizing mutations and loop reconstruction
Based on the X-ray crystal structure of human IgG1-Fc, solved in complex with the ZZ domain of protein A (Protein Data Bank entry 1OQO), computational design of CH3 domains of the stabilized variants Q418L, S424T and the respective double mutant was performed. Using the RepairPDB option of the FoldX protein design algorithm, side chain conformations of amino acids exhibiting unfavorable torsion angles, Van der Waals clashes or high energy in the crystal structure were replaced by lower energy rotamers [10]. In the next step, using the FoldX option BuildModel, the side chains of the three following constructs were modeled: The single mutants (i) Q418L and (ii) S424T and (iii) the double mutant Q418L / S424T.
To mimic randomization in libraries and create the systems representative for the libraries stem(0), stem-(0), stem(5) and stem-(5), we used the software package LoopX [11]. LoopX is a MySQL database dependent algorithm written in C++ and is specifically designed to graft loop backbone coordinates into protein crystal structures using two residues as anchor points. The LoopX database contains the coordinates of backbone atoms (N, Cα, O, C) of loops derived from in silico digestion of proteins from the ASTRAL95 dataset (14,525 protein crystal structures with less than 95% sequence identity). Depending on the secondary structural element that the anchor residues contribute to, combinations are sorted in four major elements, thereby almost entirely covering the three-dimensional loop space: H-H, H-E, E-H and E-E, where H corresponds to an α-helix and E to a β-strand [12].
The LoopX algorithm retrieves and evaluates fragments from the loop database in three steps. First, residues 419–422 were deleted from the structure to create a gap ready for bridging by four residue fragments from the LoopX database. Residues 418 and 423 were used as anchor points, where the DSSP information was H and E, respectively. Renumbering of the residues 423–444 according to the number of inserted alanines in the starting structures increases the gap size forcing the algorithm to look for fragments of the corresponding length. Second, positioning of the loop is done by the superimposition of the first and last loop residue on the anchor residues of the structure to bridge the created gap. In order to reduce the number of possible candidates and increase their quality this database query step applied the following filters: Backbone entropy filter, backbone ω-filter, steric clash filter, soft energy potential filter, stability filter, and entropy sort filter. Finally, Cβ information was restored for the retrieved loops using the FoldX-command BuildModel that is integrated in the LoopX algorithm, leading to alanine-only loops. Scoring and ranking of loop candidates were done using the FoldX stability command that estimates the free energy of folding (ΔΔG).
2.6. Molecular dynamics simulation setup
We applied the GROMOS11 package for molecular dynamics (MD) simulation [13]. Using the GROMOS++ software [14] eight different systems were set up for simulation based on the starting structures prepared by using FoldX and LoopX as described above (Table 2). The force-field parameters for the protein were taken from the GROMOS 54A7 united-atom force field [15]. Each system was simulated twice using different random seeds.
Table 2.
Overview of simulated systems.
| Water molecules | Box edge length [nm] | Total number of atoms | |
|---|---|---|---|
| IgG1-Fc-CH3 wild-type | 10,245 | 6.91 | 31,812 |
| Q418L | 9387 | 6.83 | 30,585 |
| S424T | 9813 | 6.81 | 30,517 |
| Q418L/S424T | 9833 | 6.83 | 30,574 |
| stem(0) | 9879 | 6.84 | 30,700 |
| stem-(0) | 9808 | 6.82 | 30,489 |
| stem(5) | 11,010 | 7.01 | 34,123 |
| stem-(5) | 11,151 | 7.11 | 34,548 |
The CH3 domains were solvated in cubic simulation boxes with a minimum solute to wall distance of 1.0 nm and simple point charge (SPC) water molecules [16]. All systems contained 2 sodium ions. Dimensions and the numbers of SPC water molecules of each system are detailed in Table 2.
Before the thermalization step, velocities that correspond to an initial temperature of 60 K were randomly assigned to all atoms of each system. During thermalization, the systems were gradually heated to 300 K in 5 discrete simulation steps of 20 ps with ΔT = 60 K. At the same time the force constant of imposed harmonic position restraints on the solute atoms was reduced from 2.5 × 104 to 0 kJ·mol− 1·nm− 2.
Simulations were run with a constant number of particles, at a constant pressure of 1 atm and at a constant temperature of 300 K. After thermalization, each simulation was extended for an additional 20 ns. Degrees of freedom of solvent and solute were separately coupled to two temperature baths using a relaxation time of 0.1 ps according to the weak-coupling method [17]. The pressure was kept constant accordingly, using a relaxation time of 0.5 ps and an estimated isothermal compressibility of 4.575 kJ mol− 1 nm− 1 [17]. The leap-frog algorithm [18] with a timestep of 2 fs was used. The SHAKE algorithm was used to constrain all bonds to their minimum energy values [19]. The calculation of non-bonded interactions was performed according to a triple range cut-off scheme. Short-range interactions up to a distance of 0.8 nm were calculated at every timestep from a pairlist being updated every fifth step. At pairlist construction [20], intermediate range interactions of up to 1.4 nm were calculated as well and kept constant between the updates. A reaction field contribution [21] was added to forces and energies in order to account for a dielectric continuum with a relative permittivity of 61 beyond the cut-off sphere of 1.4 nm [22]. Solvent–solvent interactions were calculated using the graphics processing unit (GPU) solvent loop that was implemented in GROMOS11 and that significantly accelerates calculations [23].
2.7. Molecular dynamics simulation data analysis
For analysis, only coordinate trajectories generated after the thermalization were used. Coordinates that are separated by a time interval of 2 ps were included. The GROMOS++ software for the analysis of biomolecular simulation trajectories [14] was used for analyzing the trajectories.
The atom-positional root-mean-square deviations (RMSD), after a roto-translational least-squares fit, of (i) backbone atoms (C, Cα and N) of the CH3 framework regions (i.e. excluding N- and C-terminal loops) and (ii) backbone atoms (C, Cα and N) of the EF loop were calculated as a function of time with respect to the initial structure. Atom-positional root-mean-square fluctuations (RMSF) of all atoms were calculated using a similar procedure.
Hydrogen-bond analysis was performed using a maximum hydrogen-acceptor distance of 0.25 nm and a minimum donor-hydrogen-acceptor angle of 135° as the geometric criteria. Each hydrogen bond between a pair of distinct atoms was considered unique. The prevalence of hydrogen bonds was calculated as the percentage of time in which the hydrogen bond was active during the entire simulation.
The radius of gyration (rgyr) was calculated for all simulated systems. Solvent accessible surface areas (SASA) [24] of all systems were determined by moving a water-sized probe, with a radius of 0.14 nm, over the protein surface and recording individual atomic contributions. Changes in secondary structure of the CH3 domains were quantified using the Define Secondary Structure of Proteins algorithm (DSSP) [25].
Configurations were clustered based on the pairwise RMSD of backbone atoms (C, Cα and N) of the EF loop after a least-squares superposition based on the backbone atoms of the entire protein. Every fifth frame of the pooled trajectories of the 2 individual simulations, i.e. separated by 10 ps, was considered for clustering. The cut-off for clustering was set such that approx. 80–90% of all clustered structures would be distributed among the first 10 clusters, i.e. 0.15 nm for clustering of the EF loop configurations of each system alone and 0.1 nm for combined clustering. All structures differing by an RMSD that is less than the specified cut-off of 0.15 nm were considered as structural neighbors and the structure with the largest number of neighbors was considered the central-member structure of the first cluster. This structure, along with all its structural neighbors was removed from the pool of structures and the same procedure was repeated to determine the second, third, etc. clusters [26]. Lifetime-analysis was performed with a lifetime limit equal to 2, meaning that at least two subsequent structures of a cluster that differ from the cluster of interest have to occur in order to record a transition to a different cluster.
3. Results
3.1. Design and construction of libraries
Recently, we have described the directed evolution of stabilized IgG1-Fc scaffolds by an application of strong heat shock to IgG1-Fc libraries displayed on yeast [27]. After four rounds of selection 17 variants with intact FcRn, CD16a and Protein A binding and increased thermal stability could be selected by FACS. The stabilizing mutations were located exclusively in the CH3 domain including two positions within (Q418L) and at the base (S424T) of the EF-loop (D413-V422), the most prominent structural loop at the C-terminus of the CH3 domain (Fig. 1). The EF-loop has a peculiar symmetry since R416 and W417 act as a central stabilizing motif. R416 forms an ionic interaction with E388 in the CD-loop, whereas the indole ring of W417 points to the hydrophobic core of the β-barrel of the CH3 domain thereby contributing to its hydrophobic packing (Fig. 1). Mutational analyses demonstrated that the exchange of both R416 and W417 significantly destabilized the resulting variants [28] suggesting that they should not be included in library design.
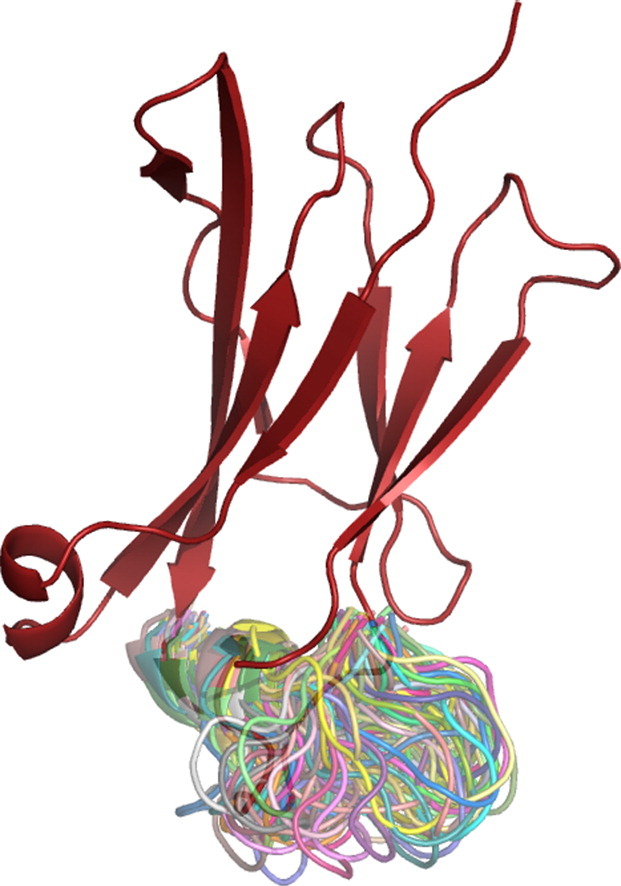
Fig. 1.

Graphical representation of IgG1-CH3-Fc. Residues of interest are shown in blue, sites of randomization and insertion in red (A and B). Most prevalent structures of two systems according to cluster analyses are shown. (A) The variant stem(0), bearing the two stabilizing mutations Q418L and S424T. Residues 419–422 were replaced by alanines. (B) The variant stem(5), differing from stem(0) between residues 419 and 422, where 5 additional alanines were inserted. (C) The variant Q418L/S424T. Amino acids contributing to a hydrophobic cluster on the C-terminal part of the CH3 domain are highlighted in blue. The mutation Q418L is shown in yellow.
Fig. 2 compares the thermograms of wild-type IgG1-Fc and the two variants Q418L and S424T. All proteins were recombinantly produced in Pichia pastoris in soluble form. As described recently [27], wild-type IgG1-Fc produced in P. pastoris shows at least three transitions that represent unfolding of the CH2 domain at 65.7 °C and of the CH3 domain at 78.1 °C (Tm2) and 82.6 °C (Tm3). In both variants the CH3 domain was stabilized [Q418L: 81.0 ± 0.1 °C (Tm2) and 86.3 ± 0.1 (Tm3); S424T: 81.0 ± 0.1 °C (Tm2) and 85.9 ± 0.1 (Tm3)] (Fig. 2). Q418 is located at the central part of the EF loop adjacent to R416 and W417. S424 is located between F423 which is also involved in hydrophobic packing and C425 which forms the intra-CH3-disulfide bond. Since the point mutations Q418L and S424T, together with these residues, create stabilizing patches within and at the base of the EF-loop we hypothesized that they would lead to better tolerance to randomization of residues in between them as well as allow for the insertion of additional random amino acids.
Fig. 2.
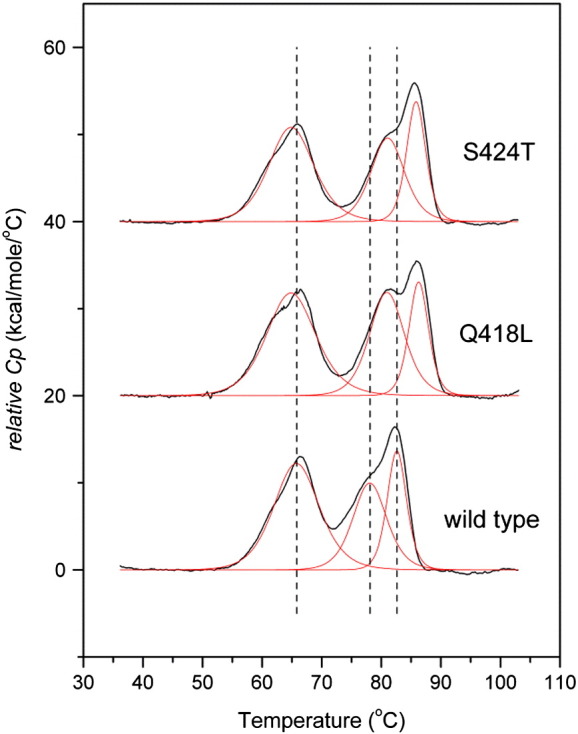
Differential scanning calorimetric analysis of wild-type IgG-Fc and the variants Q418L and S424T. Protein solutions were adjusted to a concentration of 5 μM in PBS buffer. Temperature range was from 20 to 110 °C. Fits of the obtained endotherms representing distinct unfolding events are depicted in red. The three thermal transitions of the wild-type protein are represented by dashed lines for comparison.
Based on these findings, a total of 13 yeast surface display libraries were constructed having residues 419–422 randomized including N additional residues inserted after amino acid 422 (N = 0, 1, 2, 3, 5, 10). In libraries ‘stem(N)’, both Q418L and S424T were included, whereas in libraries ‘stem-(N)’ none of the two stabilizing mutations was introduced. Additionally, in one library only ‘Q418L’ was included.
3.2. Characterization of libraries
Next, we compared the overall stability of the 13 libraries in order to analyze whether Q418L and S424T increase the tolerance to randomization of residues 419–422 as well as to insertion of additional amino acids after position 422. We applied a recently described method [7] that allows for the determination of the overall stability of whole libraries displayed on the surface of yeast by flow cytometry. The effect of point mutations in the context of the insertion of additional residues can be analyzed accordingly. ΔT1/2-values were determined which represent the change of temperatures of half-maximal irreversible denaturation of the distinct libraries with respect to the wild-type IgG1-Fc protein.
Fig. 3 depicts the effect of the destabilization of randomization of residues 419–422 and the insertion of additional amino acids compared to surface expressed wild-type IgG1-Fc. As hypothesized, including leucine and threonine at positions 418 and 424 increases tolerance to randomization and the inclusion of additional residues (Fig. 3 and Table 1). Compared to library stem-(0) (wild-type Q418 and S424, destabilization by 4.6 °C), library stem(0) with Q418L and S424T was more tolerant to randomization (ΔT1/2 = − 2.9 °C). The stabilizing effect of including only Q418L (ΔT1/2 = − 3.9 °C) was smaller. Results obtained for libraries carrying additional loop residues followed a similar scheme (Fig. 3 and Table 1) clearly suggesting that L418 and T424 should be included in the design of EF-loop libraries. With an increasing number of inserted residues the differences in ΔT1/2 between stem(N) and stem-(N) became smaller. But still the insertion of the relatively large number of 10 additional residues was better tolerated in the presence of mutations Q418L and S424T (ΔT1/2 = − 6.7 °C compared to − 7.3 °C).
Fig. 3.

ΔT1/2 values for stabilized and non-stabilized libraries. Library designs and nomenclature are shown in Table 1. Constructed libraries were expressed on the surface of yeast and cell suspensions were incubated at increasing temperatures. After cooling cells were probed for binding to FcγRI by using flow cytometry. Obtained ΔT1/2 values describe the overall destabilization of a distinct library with respect to the yeast-displayed wild-type protein. Error bars are the standard errors of the mean of three replicate experiments.
3.3. In silico loop reconstruction
In a previous publication we could demonstrate that the isolated CH3 domain can be used for molecular dynamics simulations as long as the regions of interest in the protein do not contribute to the CH3–CH3 and CH2–CH3 interfaces [29]. The data suggested that the C-terminal loops of one CH3 domain within homodimeric IgG1-Fc do not significantly interact with the loops of the other CH3 domain and the individual contributions to the antigen binding site seem to be independent. Therefore, we have constructed CH3-based model systems as described in the Materials and methods Section.
FoldX-aided introduction of the stabilizing mutation Q418L resulted in a decrease in the free energy of folding (ΔΔG = − 1.55 kcal·mol− 1 or 6.49 kJ·mol− 1) while for the variant S424T no change was calculated. When both stabilizing mutations were introduced consecutively, the resulting ΔΔG value was − 1.48 kcal·mol− 1 (6.19 kJ·mol− 1).
For the construction of in silico stabilized library mimics (stem(0) and stem(5)), residues 419–422 were deleted from the structure. By using LoopX, the resulting gap was bridged by compatible loop backbone structures from the LoopX protein fragment database and the respective residues were mutated to alanine. Reconstruction of stem(0) and stem-(0) with a total loop length of 6 (residues 418–423) yielded considerably higher numbers of compatible fragments (n = 123) than the reconstruction of stem(5) and stem-(5) with a total loop length of 11 (n = 3). Structures exhibiting the lowest values for free energy of folding were used for setting up MD simulations.
3.4. Molecular dynamics simulations
Molecular dynamics simulations were run in duplicates for 20 ns. The resulting trajectories were analyzed to gain insight into the stabilizing effect of mutations Q418L and S424T in different setups. The introduction of leucine at position 418 and serine at position 424 did not alter the total number of hydrogen bonds (i.e. 73 bonds) or the number of hydrogen bonds formed by residues 418 and 424 (on average 3.6 bonds). Also, the radius of gyration (1.46 nm) and the solvent accessible surface area (53 nm2) were constant throughout both independent simulations of each system. No significant influence on the secondary structure content of the domain as a consequence of stabilization or randomization could be observed in the DSSP analysis.
Time series of the atom-positional RMSD of framework regions were analyzed (Supplementary Fig. S1). There is no constant increase in RMSD during most of the simulations but we observed fluctuations around average RMSD values with the possible exception of run 2 of S424T. We calculated average RMSD values of 0.12/0.15 nm (MD simulations 1/MD simulation 2) for the wild-type CH3 domain, 0.12/0.18 nm for the variant Q418L, 0.15/0.15 nm for the variant S424T and 0.15/0.15 nm for the double mutant Q418L/S424T. For the systems stem(0) and stem-(0), very similar average RMSD values of 0.13/0.13 nm and 0.14/0.15 nm were determined (Supplementary Fig. S2). These systems are the ‘randomized’ equivalents of the wild-type CH3 domain [stem-(0)] and the double mutant Q418L/S424T [stem(0)].
Time series of the atom-positional RMSD of framework regions were analyzed (Supplementary Fig. S1). There is no constant increase in RMSD during most of the simulations but we observed fluctuations around average RMSD values with the possible exception of run 2 of S424T. We calculated average RMSD values of 0.12/0.15 nm (MD simulations 1/MD simulation 2) for the wild-type CH3 domain, 0.12/0.18 nm for the variant Q418L, 0.15/0.15 nm for the variant S424T and 0.15/0.15 nm for the double mutant Q418L/S424T. For the systems stem(0) and stem-(0), very similar average RMSD values of 0.13/0.13 nm and 0.14/0.15 nm were determined (Supplementary Fig. S2). These systems are the ‘randomized’ equivalents of the wild-type CH3 domain [stem-(0)] and the double mutant Q418L/S424T [stem(0)].
The insertion of five additional alanines [stem(5), stem-(5)] resulted in average RMSD values of 0.15/0.13 nm and 0.17/0.20 nm, respectively. Here, the presence of the stabilizing mutations caused the RMSD to remain relatively constant, while for the non-stabilized structure RMSD values were still increasing at the end of the simulation. This leads to the assumption that mutations Q418L and S424T preserve the overall structure of the domain even after considerable elongation of the EF loop (Supplementary Fig. S2).
The insertion of five additional alanines [stem(5), stem-(5)] resulted in average RMSD values of 0.15/0.13 nm and 0.17/0.20 nm, respectively. Here, the presence of the stabilizing mutations caused the RMSD to remain relatively constant, while for the non-stabilized structure RMSD values were still increasing at the end of the simulation. This leads to the assumption that mutations Q418L and S424T preserve the overall structure of the domain even after considerable elongation of the EF loop (Supplementary Fig. S2).
RMSD analysis of the EF loop backbone of the simulated systems resulted in averages of 0.07/0.07 nm for the wild-type CH3 domain, 0.05/0.07 nm and 0.05/0.06 nm for the single mutants Q418L and S424T and 0.05/0.04 nm for the double mutant Q418L/S424T (Supplementary Fig. 3). For all three systems involving the stabilizing mutations, RMSD values temporarily increased but leveled off toward the end of the simulation. RMSD values for the EF loop backbone of stem(0) and stem-(0) were 0.05/0.10 nm and 0.13/0.9 nm, respectively (Supplementary Fig. 4). The elongated loops of stem(5) and stem-(5) undergo significant conformational changes with respect to the reference structures, as the average RMSD values are in the range of 0.18/0.28 nm for stem(5) and 0.28/0.23 nm for stem-(5).
RMSD analysis of the EF loop backbone of the simulated systems resulted in averages of 0.07/0.07 nm for the wild-type CH3 domain, 0.05/0.07 nm and 0.05/0.06 nm for the single mutants Q418L and S424T and 0.05/0.04 nm for the double mutant Q418L/S424T (Supplementary Fig. 3). For all three systems involving the stabilizing mutations, RMSD values temporarily increased but leveled off toward the end of the simulation. RMSD values for the EF loop backbone of stem(0) and stem-(0) were 0.05/0.10 nm and 0.13/0.9 nm, respectively (Supplementary Fig. 4). The elongated loops of stem(5) and stem-(5) undergo significant conformational changes with respect to the reference structures, as the average RMSD values are in the range of 0.18/0.28 nm for stem(5) and 0.28/0.23 nm for stem-(5).
Additionally, we calculated the change in root-mean-square fluctuation of all Cα atoms as a measure of altered backbone flexibility following mutation according to ΔRMSF = RMSF (mutant) — RMSF (wild-type). The average absolute deviations between the two simulations calculated for all systems amount to an average of 0.03 nm, giving an estimate of the convergence of these data. Fig. 4A depicts the calculated differences between averages over the RMSF values obtained from analysis of the two separate simulations of the variants and the average of the two separate simulations of the wild-type CH3 domain. Supplementary Fig. S5 shows the RMSF for the two independent simulations of the wild-type CH3 domain. Interestingly, the single mutations Q418L and S424T increased the flexibility of parts of the CD loop (Q386-E388) and part of the G-strand (T437-Q438). At the same time the mutations rigidify part of the D-strand (K392-T394) as well as the C-terminal part of the EF loop (R416-V422). Upon the inclusion of both stabilizing mutations (Q418L/S424T) no additional flexibility in the CD loop and the G-strand was observed and the rigidification of the D-strand was less pronounced. In contrast to the single mutations the flexibility in the EF loop was slightly increased.
Fig. 4.

Atom-positional root-mean-square fluctuation for Cα atoms. (A) ΔRMSF for each residue with respect to the wild-type CH3 domain is shown for Q418L (red), S424T (green) and Q418L/S424T. (B) Change in ΔRMSF for stem(0) with respect to stem-(0). (C) Change in RMSF for stem(5) with respect to stem-(5). For nomenclature see Table 1.
Additionally, we calculated the change in root-mean-square fluctuation of all Cα atoms as a measure of altered backbone flexibility following mutation according to ΔRMSF = RMSF (mutant) — RMSF (wild-type). The average absolute deviations between the two simulations calculated for all systems amount to an average of 0.03 nm, giving an estimate of the convergence of these data. Fig. 4A depicts the calculated differences between averages over the RMSF values obtained from analysis of the two separate simulations of the variants and the average of the two separate simulations of the wild-type CH3 domain. Supplementary Fig. S5 shows the RMSF for the two independent simulations of the wild-type CH3 domain. Interestingly, the single mutations Q418L and S424T increased the flexibility of parts of the CD loop (Q386-E388) and part of the G-strand (T437-Q438). At the same time the mutations rigidify part of the D-strand (K392-T394) as well as the C-terminal part of the EF loop (R416-V422). Upon the inclusion of both stabilizing mutations (Q418L/S424T) no additional flexibility in the CD loop and the G-strand was observed and the rigidification of the D-strand was less pronounced. In contrast to the single mutations the flexibility in the EF loop was slightly increased.
Fig. 4B depicts the change in root-mean-square fluctuation between stem(0) and stem-(0), i.e. ΔRMSF = RMSF [stem(0)] — RMSF [stem-(0)]. The rigidification of the CD-loop, the D-strand and the EF-loop is observed, which could be assigned to the fact that, even though stem-(0) is not stabilized, the substitution of residues 419–422 and especially G421 by alanine reduces flexibility in the first place. Stabilization by Q418L and S424T added even more rigidity in these regions.
As a reference point for the calculation of the ΔRMSF values at positions 422a–422e in insertion-variants stem(5) and stem-(5), an average RMSF value was calculated from all C-terminal loop residues of the wild-type CH3 domain. In these variants the stabilizing mutations Q418L/S424T do not hold the EF loop in place but allow for considerable flexibility of the elongated loop and possibly a larger number of available conformations (Fig. 4C). At the same time the flexibilities of parts of the CD-loop, F-strand (M428-H429), FG-loop (E430-H435) and G-strand (Y436-T437) are largely reduced.
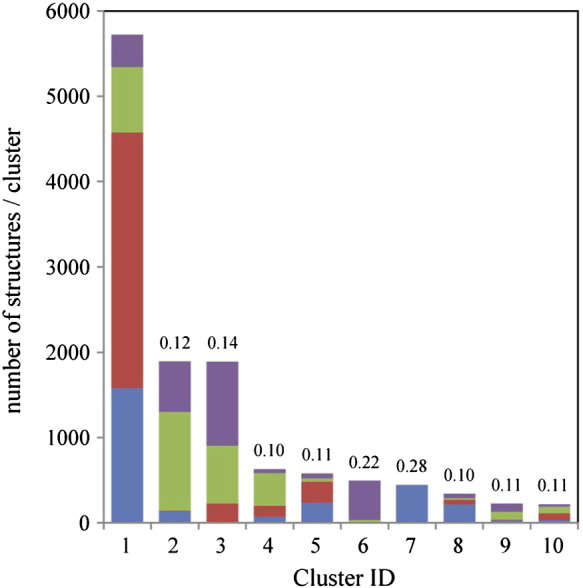
Next, in order to gain a better understanding of the different conformations the EF loop can adopt in the analyzed systems, we performed a detailed cluster analysis of the EF loop backbone configurations. The trajectories from both independent simulations were considered for this analysis. Every fifth frame was used for the analysis, resulting in a total of 4000 structures. Table 3 shows how the total number of clusters determined for systems with non-elongated EF loops is decreased by the introduction of stabilizing mutations. Lifetime-analysis of the wild-type CH3 domain and the mutants Q418L, S424T and Q418L/S424T revealed that not only the total number of clusters (i.e. distinctly different conformations) but also the fluctuation between these conformations is reduced by the stabilization of the domain. In stabilized variants, cluster switches occur less frequently as the number of visits of the clusters is reduced (Fig. 5). At the same time the lifetime is increased i.e. the time of residency after a cluster switch. The same trend is observed for the variants stem(0) and stem-(0) (Fig. 5). For variants stem(5) and stem-(5) the loop is elongated and the number of clusters using the same cutoff value is increased to 102 and 96 clusters, respectively. However, the number of visits to the first ten clusters is lower for the stabilized system [stem(5)] and the average lifetimes are extended.
Table 3.
Number and size of clusters. Configurations were clustered based on the RMSD of backbone atoms C, Cα and N of the EF loop. A configuration belongs to a different cluster if its RMSD with respect to the first structure in the cluster is larger than the specified cut-off of 0.15 nm.
| System |
ID Fig. 5 |
# of clusters |
Size cluster 1 |
Size cluster 2 |
||
|---|---|---|---|---|---|---|
| # of structures | % | # of structures | % | |||
| Wild-type | A | 18 | 2654 | 66.4 | 898 | 22.5 |
| Q418L | B | 12 | 3413 | 85.3 | 292 | 7.3 |
| S424T | C | 9 | 3696 | 92.4 | 202 | 5.1 |
| Q418L / S424T | D | 6 | 3958 | 99.0 | 18 | 0.5 |
| stem-(0) | E | 18 | 2362 | 59.1 | 880 | 22.0 |
| stem(0) | F | 13 | 2880 | 72.0 | 879 | 22.0 |
| stem-(5) | G | 96 | 1330 | 33.3 | 411 | 10.3 |
| stem(5) | H | 102 | 936 | 23.4 | 572 | 14.3 |
Fig. 5.

Cluster lifetime analysis. The 10 most populated clusters are shown for each simulated system in the following order: (A) wild-type CH3 domain, (B) Q418L, (C) S424T, (D) Q418L / S424T, (E) stem-(0), (F) stem(0), (G) stem-(5), (H) stem(5) (see Table 3). White bars represent how often a cluster was visited in the course of 2 separate 20 ns simulations. Gray bars represent the average lifetime of a cluster, i.e. the average number of snapshots in the simulation observed in the cluster before a cluster switch occurs.
Visual inspection of the central member structure of the most populated cluster found for the variant Q418L/S424T revealed that the mutation Q418L possibly contributes to a hydrophobic cluster at the C-terminal part of the inner core of the CH3 domains together with residues L358, V363, L365, the aliphatic part of the side chain of K414, W417, F423 and L441 (Fig. 1C).
Finally, combined clustering of the wild-type CH3 domain and Q418L/S424T together with their ‘randomized’ counterparts stem-(0) and stem(0) was performed to find the configurations that the EF loops of these systems have in common. Fig. 6 illustrates that the most prevalent configuration is adopted by all four systems, which clearly indicates an overlap in the conformational sampling. One configuration that the wild-type CH3 domain adopts for ~ 10% of the time is not available to the other variants (Cluster 7). In terms of RMSD, this conformation exhibits the strongest deviation from the most prevalent one (Cluster 1). Also, clusters being most different from cluster 1 are mainly populated by stem-(0) (Clusters 3 and 6). The fact that cluster 1 represents the most prevalent configuration of the variant Q418L/S424T, supports the observation that a stabilized domain without insertions limits the fluctuation of the EF loop between different configurations. Furthermore, cluster 2, which is mainly populated by stem(0), exhibits a very small deviation from cluster 1, illustrating that the configurations adopted by the stabilized variants are highly similar. Fig. 7 shows a graphical summary of structural deviation between clusters. Similarities (i.e. population of the same clusters) of stem(0) and stem-(0) can probably be assigned to the preferred conformations of a loop carrying only alanines in the sequence.
Fig. 6.
Combined EF loop clustering of corresponding systems. Stacked bars represent combined clusters from two separate simulations of the wild-type CH3 domain (blue), the double mutant Q418L/S424T (red), the stabilized, ‘randomized’ variant stem(0) (green) and the non-stabilized, randomized variant stem-(0) (purple). RMSD (in nm) of cluster central member structures with respect to the central member structure of cluster 1 are shown on top of bars.
Fig. 7.
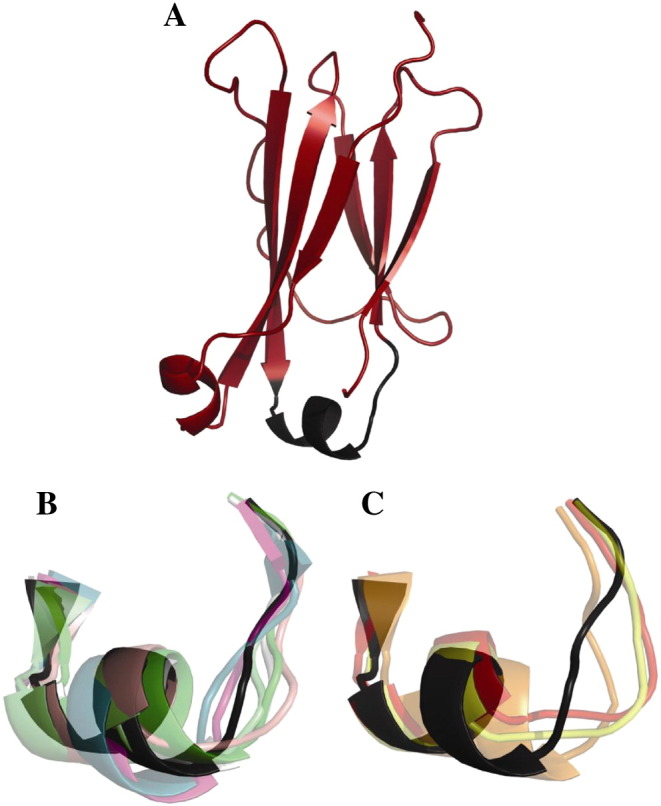
Graphical representation of the combined EF loop clustering of wild-type CH3 domain, Q418L/S424T, stem(0) and stem-(0). The EF loop of the central member structure of cluster 1 is shown in black. (A) Central member structure of cluster 1. (B) Superposition of central member structure EF loops for the structurally similar clusters 1, 2, 4, 5, 8, 9 and 10. (C) Superposition of central member structure EF loops for the structurally deviating clusters 3, 6 and 7.
4. Discussion
Antigen-binding IgG1-Fc fragments (Fcabs) are a promising new class of potential antibody therapeutics. However, even though the Fcab is an intrinsically stable protein, the manipulation of the EF loop and insertion of multiple residues to enlarge the binding surface can negatively affect the Fcab structure to different degrees, which is a well known issue in protein engineering and not specific to the Fcab system. Studies have been performed to optimize the engineering process and thereby further improve the already highly robust platform of modular antibody engineering: Wozniak et al. described the stabilization of Fcabs through introducing several artificial disulfide bonds [30,31]; and in vitro directed evolution approaches were used to (i) improve the biophysical properties of an Fcab binding to Her2/neu by loop-specific stability maturation [32] and (ii) identify stabilizing mutations in the CH3 domain in order to enforce the structural integrity of the engineered Fc fragment [27]. Two of these stabilizing mutations, Q418L and S424T, are located within or in close proximity to the EF loop of the CH3 domain. Recently, we developed a flow cytometry-based method to determine the overall stability of entire libraries of IgG1-Fc variants displayed on the surface of yeast in order to identify optimum sites for randomization and/or the insertion of additional residues in the C-terminal loops of the CH3 domain [7]. In the present work, we applied this method to systematically evaluate whether the introduction of the aforementioned stabilizing mutations Q418L and S424T could increase the tolerance to loop insertions and improve the overall stability of the respective libraries. This in turn would allow us to mimic CDR loop length variation and enlarge the binding surface available for antigen recognition [33].
Yeast surface display libraries were constructed and analyzed by flow cytometry to prove the hypothesis that the mutations Q418L and S424T are able to preserve the structural integrity of the domain after diversification and elongation of the loop region that is flanked by the mutations. The determination of temperatures of half-maximal irreversible denaturation revealed that a combination of the two mutations was superior to the single mutations and the insertion of additional residues was considerably better tolerated in the stabilized libraries. Thus, both mutations seem to establish stabilizing patches at the base of the randomized and elongated EF loop.
In order to gain further insight in the influence the mutations have on domain dynamics, we performed multiple MD simulations. We followed a novel approach that combines in silico loop reconstruction by using the loop reconstruction algorithm LoopX and mimicked randomization by mutating residues that were diversified in the experimental setup to alanines. This approach was chosen because the truncation of all other side chains to a Cβ carbon would remove any effect the side chains could have on the backbone conformation. At the same time we accepted the fact that the mutation of glycine at position 420 to alanine would decrease overall flexibility, which of course adds some rigidification bias to the system. Apparently, any way of reducing the flexibility in the EF loop affects CD loop, D-strand, EF loop and G-strand. However, the mutation of residues 419–422 to alanines in the stabilized variant stem(0) and the non-stabilized variant stem-(0) altered the rigidity or flexibility of these parts of the domain to different degrees: The flexibility of the CD loop was increased in stem-(0) with respect to stem(0) and the rigidifying effect on the D-strand was less pronounced for the non-stabilized variant. Also, the EF loop was less flexible in stem(0) and the flexibility of the G-strand was decreased in stem(0) as compared to stem-(0). This suggests that this approach is valid, provided that suitable control systems are available.
RMSF analysis showed that upon the introduction of the stabilizing mutations Q418L and S424T the backbone of distinct regions in the CH3 domain was rigidified. The insertion of additional residues resulted in increased flexibility of the mutated loop with respect to its non-stabilized counterpart, while the framework was again rigidified. One possible explanation for this stabilizing effect on the framework could be the contribution of L418 to the hydrophobic cluster that is formed by several amino acids in the proximity of this residue. Also, the general β-sheet propensity of threonine is higher than that of serine as a consequence of its β-branched sidechain, which could explain the stabilizing effect of the β-sheet-located mutation S424T [34].
During two independent 20 ns simulations, the stabilized, ‘randomized’ variant stem(5) and its non-stabilized counterpart stem-(5) visited approximately the same number of unique clusters, and stem-(5) seemed to visit its cluster 1, the most populated cluster (i.e. the most prevalent EF loop conformation over time), more often than stem(5) (Table 3). However, lifetime-analysis revealed that, once stem(5) adopts a certain EF loop conformation, it remains in this conformation for a longer time than its non-stabilized counterpart stem-(5), meaning a higher fluctuation of EF loop conformations over time for the latter (Fig. 5). In addition, the RMSF of the EF loop backbone atoms was higher for stem(5) than for stem-(5), while at the same time the fluctuation of all the other loops as well as the framework regions of stem(5) was partly decreased, especially in the CD loop (Fig. 4C). As revealed by RMSF-analysis, the structural deviation of the EF loop conformation of stem(5) from its reference structure may be more pronounced than it is the case for stem-(5). These deviating structures are themselves stabilized as a consequence of the presence of L418 and T424, which is supported by experimental data as discussed above. The benefit this might have on the potential binding of an antigen is obvious: There is an apparent increase in the number of possible conformations the EF loop can adopt as a consequence of its support by the stabilized flanking regions without affecting the overall fold of the domain. In other words, it can be expected that the stabilized framework is more accommodating to alternative conformations of more varied sequences in the elongated EF loop. If the loop takes on a conformation suitable for antigen binding it could have more time for the recognition of the antigen before a switch to a non-binding conformation, than would be the case in the absence of L418 and T424. The AB loop, which can be engineered for the contribution to antigen binding in an Fcab as well, is hardly affected. Thus, it is suggested that the elongation of the EF loop together with the insertion of stabilizing stem residues can further improve the process of selection of Fcabs against diverse antigens.
Recently, Haidar et al. proposed that the affinity of immunoglobulin interactions is inversely proportional to the backbone flexibility of their corresponding CDR3, while it was independent of the flexibility of CDR1 and CDR2 [35]. They were able to increase the affinity of the A6 TCR for Tax-HLAA2 by a factor of 2400 through the reduction of the flexibility of CDR3 by using proline substitutions together with other mutations. Another method to restrict CDR flexibility is the systematic insertion of cysteine doublets to form intraloop disulfide bonds [36]. As in Fcabs the EF loop of the CH3 domain is known to play an important role in antigen binding the results we present here could imply a similar principle.
In summary, our work shows that the inclusion of the stabilizing mutations seems to allow for a larger versatility of the conformations of the EF loop without negatively affecting the structure of the framework regions, suggesting that this approach could be favorable for the design of novel antigen binding sites. As a next step, our stabilizing mutations will be applied in existing binders in order to evaluate their effect on affinity and stability. Also, large working libraries will be constructed according to the proposed designs to enhance the available Fcab library pool and thereby facilitate discovery of potent and stable binders.
The following are the supplementary data related to this article.
Oligonucleotide sequences.
Atom-positional root-mean-square deviations for backbone atoms of the CH3 framework with respect to the initial structure. Wild-type CH3 domain run 1 (A) and run 2 (B). Q418L run 1 (C) and run 2 (D). S424T run 1 (E) and run 2 (F). Q418L/S424T run 1 (G) and run 2 (H).
Atom-positional root-mean-square deviations for backbone atoms of the CH3 framework with respect to the initial structure. Stem(0) run 1 (A) and run 2 (B). stem-(0) run 1 (C) and run 2 (D). stem(5) run 1 (E) and run 2 (F). stem-(5) run 1 (G) and run 2 (H).
Atom-positional root-mean-square deviations for backbone atoms of the EF loop with respect to the initial structure. Wild-type CH3 domain run 1 (A) and run 2 (B). Q418L run 1 (C) and run 2 (D). S424T run 1 (E) and run 2 (F). Q418L/S424T run 1 (G) and run 2 (H).
Atom-positional root-mean-square deviations for backbone atoms of the EF loop with respect to the initial structure. Stem(0) run 1 (A) and run 2 (B). stem-(0) run 1 (C) and run 2 (D). stem(5) run 1 (E) and run 2 (F). stem-(5) run 1 (G) and run 2 (H).
Atom-positional root-mean-square fluctuation for Cα atoms from two independent simulations of the wild-type CH3 domain are shown in black (MD1) and gray (MD2).
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.bbapap.2014.04.020.
Acknowledgements
This work was supported by the Christian Doppler Research Association (Christian Doppler Laboratory for Antibody Engineering), the company F-star, Grant No. LS08-QM03 of the Vienna Science and Technology Fund (WWTF), Grant No. 260408 of the European Research Council (ERC), as well as the Austrian Science Foundation (FWF W1224 – Doctoral Program on Biomolecular Technology of Proteins – BioToP).
References
- 1.Perutz M.F. Introduction to protein-structure — Branden,C, Tooze. J. Nat. 1991;353:311. [Google Scholar]
- 2.Halaby D.M., Poupon A., Mornon J.P. The immunoglobulin fold family: sequence analysis and 3D structure comparisons. Protein Eng. 1999;12:563–571. doi: 10.1093/protein/12.7.563. [DOI] [PubMed] [Google Scholar]
- 3.Olson C.A., Roberts R.W. Design, expression, and stability of a diverse protein library based on the human fibronectin type III domain. Protein Sci. 2007;16:476–484. doi: 10.1110/ps.062498407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xiao X.D., Feng Y., Vu B.K., Ishima R., Dimitrov D.S. A large library based on a novel (CH2) scaffold: identification of HIV-1 inhibitors. Biochem. Biophys. Res. Commun. 2009;387:387–392. doi: 10.1016/j.bbrc.2009.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Traxlmayr M.W., Wozniak-Knopp G., Antes B., Stadlmayr G., Ruker F., Obinger C. Integrin binding human antibody constant domains-probing the C-terminal structural loops for grafting the RGD motif. J. Biotechnol. 2011;155:193–202. doi: 10.1016/j.jbiotec.2011.06.042. [DOI] [PubMed] [Google Scholar]
- 6.Wozniak-Knopp G., Bartl S., Bauer A., Mostageer M., Woisetschlager M., Antes B., Ettl K., Kainer M., Weberhofer G., Wiederkum S., Himmler G., Mudde G.C., Ruker F. Introducing antigen-binding sites in structural loops of immunoglobulin constant domains: Fc fragments with engineered HER2/neu-binding sites and antibody properties. Protein Eng. Des. Sel. 2010;23:289–297. doi: 10.1093/protein/gzq005. [DOI] [PubMed] [Google Scholar]
- 7.Hasenhindl C., Traxlmayr M.W., Wozniak-Knopp G., Jones P.C., Stadlmayr G., Ruker F., Obinger C. Stability assessment on a library scale: a rapid method for the evaluation of the commutability and insertion of residues in C-terminal loops of the CH3 domains of IgG1-Fc. Protein Eng. Des. Sel. 2013;26:675–682. doi: 10.1093/protein/gzt041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boder E.T., Wittrup K.D. Yeast surface display for screening combinatorial polypeptide libraries. Nat. Biotechnol. 1997;15:553–557. doi: 10.1038/nbt0697-553. [DOI] [PubMed] [Google Scholar]
- 9.Gietz R.D., Schiestl R.H. High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2007;2:31–34. doi: 10.1038/nprot.2007.13. [DOI] [PubMed] [Google Scholar]
- 10.Guerois R., Nielsen J.E., Serrano L. Predicting changes in the stability of proteins and protein complexes: a study of more than 1000 mutations. J. Mol. Biol. 2002;320:369–387. doi: 10.1016/S0022-2836(02)00442-4. [DOI] [PubMed] [Google Scholar]
- 11.Vanhee P., Verschueren E., Baeten L., Stricher F., Serrano L., Rousseau F., Schymkowitz J. BriX: a database of protein building blocks for structural analysis, modeling and design. Nucleic Acids Res. 2011;39:D435–D442. doi: 10.1093/nar/gkq972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oliva B., Bates P.A., Querol E., Aviles F.X., Sternberg M.J.E. An automated classification of the structure of protein loops. J. Mol. Biol. 1997;266:814–830. doi: 10.1006/jmbi.1996.0819. [DOI] [PubMed] [Google Scholar]
- 13.Schmid N., Christ C.D., Christen M., Eichenberger A.P., van Gunsteren W.F. Architecture, implementation and parallelisation of the GROMOS software for biomolecular simulation. Comput. Phys. Commun. 2012;183:890–903. [Google Scholar]
- 14.Eichenberger A.P., Allison J.R., Dolenc J., Geerke D.P., Horta B.A.C., Meier K., Oostenbrink C., Schmid N., Steiner D., Wang D.Q., van Gunsteren W.F. GROMOS plus software for the analysis of biomolecular simulation trajectories. J. Chem. Theory Comput. 2011;7:3379–3390. doi: 10.1021/ct2003622. [DOI] [PubMed] [Google Scholar]
- 15.Schmid N., Eichenberger A.P., Choutko A., Riniker S., Winger M., Mark A.E., van Gunsteren W.F. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur. Biophys. J. Biophys. Lett. 2011;40:843–856. doi: 10.1007/s00249-011-0700-9. [DOI] [PubMed] [Google Scholar]
- 16.Berendsen H.J.C., Postma J.P.M., van Gunsteren W.F., Hermans J. Interaction models for water in relation to protein hydration. Intermolecular Forces. 1981:331–342. [Google Scholar]
- 17.Berendsen H.J.C., Postma J.P.M., Vangunsteren W.F., Dinola A., Haak J.R. Molecular-dynamics with coupling to an external bath. J. Chem. Phys. 1984;81:3684–3690. [Google Scholar]
- 18.Hockney R.W. The potential calculation and some applications. Methods Comput. Phys. 1974;9:134–211. [Google Scholar]
- 19.Ryckaert J.-P., Ciccotti G., Berendsen H. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 1977;23:327–341. [Google Scholar]
- 20.Heinz T.N., Hunenberger P.H. A fast pairlist-construction algorithm for molecular simulations under periodic boundary conditions. J. Comput. Chem. 2004;25:1474–1486. doi: 10.1002/jcc.20071. [DOI] [PubMed] [Google Scholar]
- 21.Tironi I.G., Sperb R., Smith P.E., Vangunsteren W.F. A generalized reaction field method for molecular-dynamics simulations. J. Chem. Phys. 1995;102:5451–5459. [Google Scholar]
- 22.Heinz T.N., van Gunsteren W.F., Hunenberger P.H. Comparison of four methods to compute the dielectric permittivity of liquids from molecular dynamics simulations. J. Chem. Phys. 2001;115:1125–1136. [Google Scholar]
- 23.Schmid N., Botschi M., Van Gunsteren W.F. A GPU solvent–solvent interaction calculation accelerator for biomolecular simulations using the GROMOS software. J. Comput. Chem. 2010;31:1636–1643. doi: 10.1002/jcc.21447. [DOI] [PubMed] [Google Scholar]
- 24.Lee B., Richards F.M. Interpretation of protein structures — estimation of static accessibility. J. Mol. Biol. 1971;55:379. doi: 10.1016/0022-2836(71)90324-x. [DOI] [PubMed] [Google Scholar]
- 25.Kabsch W., Sander C. Dictionary of protein secondary structure - pattern-recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 26.Daura X., van Gunsteren W.F., Mark A.E. Folding–unfolding thermodynamics of a beta-heptapeptide from equilibrium simulations. Proteins Struct. Funct. Genet. 1999;34:269–280. doi: 10.1002/(sici)1097-0134(19990215)34:3<269::aid-prot1>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 27.Traxlmayr M.W., Faissner M., Stadlmayr G., Hasenhindl C., Antes B., Ruker F., Obinger C. Directed evolution of stabilized IgG1-Fc scaffolds by application of strong heat shock to libraries displayed on yeast. Biochim. Biophys. Acta. 2012;1824:542–549. doi: 10.1016/j.bbapap.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Traxlmayr M.W., Hasenhindl C., Hackl M., Stadlmayr G., Rybka J.D., Borth N., Grillari J., Ruker F., Obinger C. Construction of a stability landscape of the CH3 domain of human IgG1 by combining directed evolution with high throughput sequencing. J. Mol. Biol. 2012;423:397–412. doi: 10.1016/j.jmb.2012.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lai B., Hasenhindl C., Obinger C., Oostenbrink C. Molecular dynamics simulation of the crystallizable fragment of IgG1-insights for the design of Fcabs. Int. J. Mol. Sci. 2014;15:438–455. doi: 10.3390/ijms15010438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wozniak-Knopp G., Ruker F. A C-terminal interdomain disulfide bond significantly stabilizes the Fc fragment of IgG. Arch. Biochem. Biophys. 2012;526:181–187. doi: 10.1016/j.abb.2012.03.024. [DOI] [PubMed] [Google Scholar]
- 31.Wozniak-Knopp G., Stadlmann J., Ruker F. Stabilisation of the Fc fragment of human IgG1 by engineered intradomain disulfide bonds. PLoS One. 2012;7:e30083. doi: 10.1371/journal.pone.0030083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Traxlmayr M.W., Lobner E., Antes B., Kainer M., Wiederkum S., Hasenhindl C., Stadlmayr G., Ruker F., Woisetschlager M., Moulder K., Obinger C. Directed evolution of Her2/neu-binding IgG1-Fc for improved stability and resistance to aggregation by using yeast surface display. Protein Eng. Des. Sel. 2013;26:255–265. doi: 10.1093/protein/gzs102. [DOI] [PubMed] [Google Scholar]
- 33.Collis A.V.J., Brouwer A.P., Martin A.C.R. Analysis of the antigen combining site: correlations between length and sequence composition of the hypervariable loops and the nature of the antigen. J. Mol. Biol. 2003;325:337–354. doi: 10.1016/s0022-2836(02)01222-6. [DOI] [PubMed] [Google Scholar]
- 34.Minor D.L., Kim P.S. Measurement of the beta-sheet-forming propensities of amino-acids. Nature. 1994;367:660–663. doi: 10.1038/367660a0. [DOI] [PubMed] [Google Scholar]
- 35.J.N. Haidar, W. Zhu, J. Lypowy, B.G. Pierce, A. Bari, K. Persaud, X. Luna, M. Snavely, D. Ludwig, Z. Weng, Backbone Flexibility of CDR3 and Immune Recognition of Antigens, J. Mol. Biol. [DOI] [PubMed]
- 36.Lipovsek D., Lippow S.M., Hackel B.J., Gregson M.W., Cheng P., Kapila A., Wittrup K.D. Evolution of an interloop disulfide bond in high-affinity antibody mimics based on fibronectin type III domain and selected by yeast surface display: molecular convergence with single-domain camelid and shark antibodies. J. Mol. Biol. 2007;368:1024–1041. doi: 10.1016/j.jmb.2007.02.029. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Oligonucleotide sequences.
Atom-positional root-mean-square deviations for backbone atoms of the CH3 framework with respect to the initial structure. Wild-type CH3 domain run 1 (A) and run 2 (B). Q418L run 1 (C) and run 2 (D). S424T run 1 (E) and run 2 (F). Q418L/S424T run 1 (G) and run 2 (H).
Atom-positional root-mean-square deviations for backbone atoms of the CH3 framework with respect to the initial structure. Stem(0) run 1 (A) and run 2 (B). stem-(0) run 1 (C) and run 2 (D). stem(5) run 1 (E) and run 2 (F). stem-(5) run 1 (G) and run 2 (H).
Atom-positional root-mean-square deviations for backbone atoms of the EF loop with respect to the initial structure. Wild-type CH3 domain run 1 (A) and run 2 (B). Q418L run 1 (C) and run 2 (D). S424T run 1 (E) and run 2 (F). Q418L/S424T run 1 (G) and run 2 (H).
Atom-positional root-mean-square deviations for backbone atoms of the EF loop with respect to the initial structure. Stem(0) run 1 (A) and run 2 (B). stem-(0) run 1 (C) and run 2 (D). stem(5) run 1 (E) and run 2 (F). stem-(5) run 1 (G) and run 2 (H).
Atom-positional root-mean-square fluctuation for Cα atoms from two independent simulations of the wild-type CH3 domain are shown in black (MD1) and gray (MD2).