

A GH127 β-l-arabinofuranosidase from G. stearothermophilus T6 (Ara127N) has been purified and crystallized in two different crystal forms. A full X-ray diffraction data set was measured for the wild-type enzyme in a P212121 crystal form (2.3 Å resolution) and complete MAD data were measured for the selenomethionine derivative in a P21 crystal form (2.5 Å resolution) to be used for the detailed three-dimensional structural analysis of the Ara127N protein.
Keywords: Geobacillus stearothermophilus; glycoside hydrolase; β-arabinofuranosidase, GH127; arabinose; arabinofuranoside; arabinan utilization; selenomethionine; MAD; SAD
Abstract
The l-arabinan utilization system of Geobacillus stearothermophilus T6 is composed of five transcriptional units that are clustered within a 38 kb DNA segment. One of the transcriptional units contains 11 genes, the last gene of which (araN) encodes a protein, Ara127N, that belongs to the newly established GH127 family. Ara127N shares 44% sequence identity with the recently characterized HypBA1 protein from Bifidobacterium longum and thus is likely to function similarly as a β-l-arabinofuranosidase. β-l-Arabinofuranosidases are enzymes that hydrolyze β-l-arabinofuranoside linkages, the less common form of such linkages, a unique enzymatic activity that has been identified only recently. The interest in the structure and mode of action of Ara127N therefore stems from its special catalytic activity as well as its membership of the new GH127 family, the structure and mechanism of which are only starting to be resolved. Ara127N has recently been cloned, overexpressed, purified and crystallized. Two suitable crystal forms have been obtained: one (CTP form) belongs to the monoclinic space group P21, with unit-cell parameters a = 104.0, b = 131.2, c = 107.6 Å, β = 112.0°, and the other (RB form) belongs to the orthorhombic space group P212121, with unit-cell parameters a = 65.5, b = 118.1, c = 175.0 Å. A complete X-ray diffraction data set has been collected to 2.3 Å resolution from flash-cooled crystals of the wild-type enzyme (RB form) at −173°C using synchrotron radiation. A selenomethionine derivative of Ara127N has also been prepared and crystallized for multi-wavelength anomalous diffraction (MAD) experiments. Crystals of selenomethionine Ara127N appeared to be isomorphous to those of the wild type (CTP form) and enabled the measurement of a three-wavelength MAD diffraction data set at the selenium absorption edge. These data are currently being used for detailed three-dimensional structure determination of the Ara127N protein.
1. Introduction
The natural degradation of plant cell-wall polysaccharides is an essential step in the carbon cycle and is carried out mainly by microorganisms that can be found either free or as part of the digestive system of higher animals (Warnecke et al., 2007 ▶). The plant cell wall is made of three major polysaccharides, cellulose, hemicellulose and pectin, which are rigidified by lignin, a heterogeneous aromatic polymer (Gilbert, 2010 ▶; Scheller & Ulvskov, 2010 ▶). Pectin is a complex polysaccharide and may account for up to 30% of the dry weight of the plant cell wall (Mohnen, 2008 ▶). Arabinan is a pectic polysaccharide primarily consisting of a backbone of α-1,5-linked l-arabinofuranosyl units, which are further decorated mainly with α-1,2- and α-1,3-linked arabinofuranosides (Mohnen, 2008 ▶). Arabinose is present in plant cell-wall polysaccharides, primarily in arabinans and arabinogalactans, usually in the form of α-l-arabinofuranoside. The natural occurrence of the β-l-arabinofuranoside configuration is significantly less common and can be found mainly in hydroxyproline-rich glycoproteins (HRGPs). HRGPs are part of the plant cell-wall proteins, which are widespread throughout the plant kingdom and are implicated in diverse aspects of plant growth and development (Ogawa-Ohnishi et al., 2013 ▶). The HRGP family includes extensins, arabinogalactan proteins, proline-rich proteins, solanaceous lectins and other related proteins (Ogawa-Ohnishi et al., 2013 ▶; Lamport et al., 2011 ▶). β-l-Arabinofuranosidases are enzymes that hydrolyze the β-l-arabinofuranoside linkages, a unique enzymatic activity that has only recently been identified. The first β-l-arabinofuranosidase to be biochemically characterized (Fujita et al., 2014 ▶), coined HypBA1, was purified from Bifidobacterium longum and its crystal structure has very recently been resolved (Ito et al., 2014 ▶). Based on its amino-acid sequence, the protein was assigned to a new glycoside hydrolase (GH) family (Henrissat & Davies, 1997 ▶), GH127. On the basis of biochemical, structural and computational analyses of HypBA1, it was suggested that the enzyme utilizes a cysteine residue as the catalytic nucleophile. Since most of the known glycoside hydrolases employ two acidic residues (Asp or Glu) as both the catalytic acid/base and the catalytic nucleophile, the proposed mechanism represents a new and unique catalytic mode of action for glycoside hydrolases (Ito et al., 2014 ▶).
Geobacillus stearothermophilus T6 is a thermophilic, Gram-positive soil bacterium which has an extensive system for the utilization of plant cell-wall polysaccharides, including xylan, arabinan and galactan (Shulami et al., 1999 ▶, 2007 ▶, 2011 ▶; Tabachnikov & Shoham, 2013 ▶). The bacterium secretes a small number of extracellular enzymes that cleave the high-molecular-weight polysaccharides into shorter decorated oligosaccharides. The resulting oligosaccharides are transported into the cell via specialized ABC transporters (Rees et al., 2009 ▶) and are further hydrolyzed into sugar monomers by a battery of specific intracellular glycoside hydrolases. The most studied and the better characterized of these complex utilization systems is that of xylan, in which the bacterium secretes an extracellular xylanase (Gat et al., 1994 ▶; Teplitsky et al., 1997 ▶, 2004 ▶; Bar et al., 2004 ▶) that partially degrades xylan into short decorated xylo-oligosaccharides, which are then imported into the cell via dedicated ABC sugar transporters (Shulami et al., 2007 ▶). Inside the cell, the decorated xylo-oligosaccharides are hydrolyzed by several side-chain-cleaving enzymes, including α-arabinofuranosidases (Shallom, Belakhov, Solomon, Gilead-Gropper et al., 2002 ▶; Shallom, Belakhov, Solomon, Shoham et al., 2002 ▶; Hövel, Shallom, Niefind, Belakhov et al., 2003 ▶), an α-glucuronidase (Teplitsky et al., 1999 ▶; Golan, Shallom et al., 2004 ▶; Zaide et al., 2001 ▶; Shallom et al., 2004 ▶), acetyl-esterases (Alalouf et al., 2011 ▶; Lansky, Alalouf, Solomon et al., 2013 ▶, 2014 ▶) and finally by an intracellular xylanase (Teplitsky et al., 2000 ▶; Solomon et al., 2007 ▶) and several xylosidases (Bravman et al., 2001 ▶, 2003 ▶; Shallom et al., 2005 ▶; Brüx et al., 2006 ▶; Ben-David et al., 2007 ▶, 2008 ▶).
The l-arabinan utilization system of G. stearothermophilus T6, which has been relatively less studied compared with the corresponding xylan system, is composed of five transcriptional units that are clustered within a 38 kb DNA segment. One of the transcriptional units contains 11 genes (abnEFJ-abnA-abfBA-araJKLMN). This operon encodes an arabino-oligosaccharide transporter system, two α-l-arabinofuranosidases (AbfA and AbfB) (Gilead & Shoham, 1995 ▶; Shallom, Belakhov, Solomon, Gilead-Gropper et al., 2002 ▶; Shallom, Belakhov, Solomon, Shoham et al., 2002 ▶; Hövel, Shallom, Niefind, Baasov et al., 2003 ▶; Hövel, Shallom, Niefind, Belakhov et al., 2003 ▶) and a cluster of genes which presumably constitute an alternative arabinose-utilization pathway. Based on its sequence, the last gene in this operon (araN) encodes a protein that belongs to the GH127 family and was hence termed Ara127N. Ara127N shares 44% sequence identity with the HypBA1 protein from B. longum (based on a 642-amino-acid overlap), including the key catalytic residues around the active site, and is thus likely to function similarly as a β-l-arabinofuranosidase.
In this report, we describe the cloning, purification, crystallization and preliminary crystallographic characterization of the Ara127N protein from G. stearothermophilus T6 (EC 3.2.1.185). We also describe the complete multi-wavelength anomalous diffraction (MAD) data measurement of the fully substituted selenomethionine derivative of Ara127N, which is expected to lead to detailed structure determination of this protein. As only one other structure of a GH127 protein has currently been reported, the expected structure of Ara127N should be highly important not only for understanding the mechanism of this particular enzyme but also the general mode of catalysis of the entire GH127 family.
2. Experimental
2.1. Cloning, overexpression and purification of wild-type Ara127N (Ara127N-WT)
The His-tag-fused araN gene was amplified from G. stearothermophilus genomic DNA by PCR using the forward primer 5′-CAGAACATATGGGACATCACCATCACCATCACGTGGAAAAAGTAGCAACA-3′ and the reverse primer 5′-TGATCAGGATCCTCACTTCTCATTCACCCACACCA-3′. The digested (NdeI, BamHI) PCR product was ligated into a linearized (NdeI, BamHI) pET9a vector (Novagen, Madison, Wisconsin, USA) and the resulting plasmid was transformed into Escherichia coli strain BL21(DE3). Transformed E. coli cells were grown overnight (500 ml culture in 2 l baffled shake flasks shaken at 230 rev min−1 at 37°C) in Terrific Broth medium supplemented with kanamycin (25 µg ml−1) to an optical density of ∼24 at 600 nm. Cells were harvested by centrifugation, the cell pellet was suspended in about 70 ml of buffer (20 mM imidazole, 500 mM NaCl, 20 mM phosphate buffer pH 7.0), disrupted by two passages through an EmulsiFlex-C3 homogenizer (Avestin Inc., Ottawa, Canada) and centrifuged (14 000 rev min−1 for 30 min). The soluble fraction was heat-treated (50°C, 20 min), centrifuged (14 000 rev min−1 for 30 min) and loaded onto a 5 ml HisTrap column (GE Healthcare Life Sciences) mounted on an ÄKTA Avant fast protein liquid chromatography system (GE Healthcare Life Sciences). The His-tag-fused protein was eluted in the same buffer and a linear gradient of imidazole to 500 mM over ten column volumes. The protein was further purified by gel filtration on a HiLoad 26/600 Superdex 200 pg column (GE Healthcare Life Sciences) with 50 mM Tris–HCl buffer pH 7.0, 100 mM NaCl, 0.02% sodium azide. This procedure yielded about 115 mg protein from 1 l culture. Based on gel filtration (Superose 12 HR column, GE Healthcare Life Sciences) the enzyme appeared to be a dimer in solution (data not shown).
2.2. Production and purification of selenomethionine-substituted Ara127N (Ara127N-Se)
Selenomethionine-substituted protein was prepared in the E. coli B834(DE3) (Met−) background following the general procedure described previously (Mechaly et al., 2000 ▶). Briefly, the culture was grown on defined medium consisting of 2 g l−1 NH4Cl, 6 g l−1 KH2PO4, 12 g l−1 Na2HPO4, 0.5 g l−1 MgSO4.7H2O, 0.02 g l−1 FeSO4, supplemented with vitamins (riboflavin, pyridoxine and thiamine) at 1 µg ml−1 each, 5 g l−1 glucose, amino acids (Asn, Asp, Cys, Glu, Ala, Arg, Val, Gln, Gly, His, Ile, Leu, Phe, Trp, Tyr, Lys, Pro, Ser, Thr) at 50 µg ml−1 each, 50 µg ml−1 seleno-l-methionine and 25 µg ml−1 kanamycin. The starter culture was first grown in 50 ml medium to a turbidity of ∼4.5 OD600 nm and then transferred into 450 ml fresh medium containing 0.4 mM IPTG (isopropyl β-d-1-thiogalactopyranoside) and grown to a final OD600 nm of ∼5. The purification process was the same as for Ara127N-WT, but with a 1 ml HisTrap column, providing a total yield of about 30 mg protein from 1 l culture.
2.3. Crystallization experiments
Crystallization experiments were set up immediately after the last purification step of the Ara127N-WT protein. The purified protein was concentrated using Centricon centrifugal concentrators (Millipore, Massachusetts, USA), so that the resulting solution contained approximately 3.8 mg ml−1 protein, 50 mM Tris–HCl pH 7.0, 100 mM NaCl, 0.02% NaN3. This protein solution was used directly for the initial crystallization experiments, which were performed by the sitting-drop vapour-diffusion method using an extensive series of different factorial screening solutions (‘screens’; Jancarik & Kim, 1991 ▶) at a constant temperature of 20°C. The first crystallization experiments were set up manually in 4 × 6 Cryschem crystallization plates (Hampton Research, California, USA) using a number of commercially available screens. These experiments were carried out with 6 µl crystallization drops consisting of 3 µl protein solution and 3 µl reservoir solution, suspended over a 400 µl reservoir of the screen solution. Additional screens were set up in 96-3 iQ TTP Labtech plates using a Mosquito LCP crystallization robot (TTP Labtech), with 0.6 µl crystallization drops consisting of 0.3 µl protein solution and 0.3 µl reservoir solution, suspended over a 55 µl reservoir of the screen solution. The robot-driven crystallization screening experiments were performed in the Technion Center for Structural Biology (TCSB, Technion, Haifa, Israel) using our in-house macromolecular crystallographic setup. In general, once positive results were obtained (i.e. crystals or micro-crystals), further refinement experiments of these crystallization conditions were performed (Almog et al., 1993 ▶, 1994 ▶; Teplitsky et al., 1997 ▶, 1999 ▶, 2000 ▶; Gilboa et al., 1998 ▶). This refinement was performed with specially prepared solutions, optimizing various parameters of the initial conditions, such as pH, ionic strength, protein concentration, temperature, precipitating additives, protein drop volume and drop-to-reservoir ratio (Bar et al., 2004 ▶; Golan, Zharkov et al., 2004 ▶; Reiland et al., 2004 ▶; Lansky, Alalouf, Solomon et al., 2013 ▶; Lansky, Salama, Solomon et al., 2013 ▶; Lansky, Alalouf, Salama et al., 2014 ▶; Lansky, Zehavi et al., 2014 ▶; Solomon et al., 2013 ▶). Two different crystal forms were obtained from these screens and both were found to be suitable for further crystallographic analysis (see below).
All diffraction data measurements were performed at the BM14 beamline of the European Synchrotron Research Facility (ESRF; Grenoble, France). Processing, reduction, indexing, integration and scaling of the diffraction data were conducted using iMosflm (Battye et al., 2011 ▶) and the HKL-2000 suite (Otwinowski & Minor, 1997 ▶).
3. Results and discussion
3.1. Crystal forms of Ara127N-WT
The screening experiments produced two different crystal forms for AraN-WT: the first was obtained from the initial manually set-up screens and the second was obtained at a later stage from the robotically set-up screens. The first crystal form was obtained from screening solution No. 48 of the commercial PEG/Ion 2 screen (Hampton Research, California, USA), consisting of 20% polyethylene glycol (PEG) 3350, 1%(w/v) tryptone, 0.1 M HEPES sodium buffer pH 7.0. These crystals could be initially observed after only 5–10 d of equilibration over the well solution and usually appeared as clusters of thin plates (CTP), which grew to their final size in an additional 1–2 weeks. Typically, one to three such clusters would appear in the crystallization drop, with plates of final dimensions in the range 0.1–0.3 × 0.1–0.3 × 0.01–0.02 mm (Fig. 1 ▶ a). The second crystal form was obtained from screening solution No. 2 of the PEGS Suite (Qiagen, Hilden, Germany) consisting of 30% PEG 300, 0.1 M sodium acetate buffer pH 4.60. These crystals could be initially observed after about 3 d of equilibration and appeared as relatively small rectangular boxes (RB). The crystals grew to their final size in an additional 4–8 d, with typical final dimensions of about 0.02 × 0.04 × 0.02 mm (Fig. 1 ▶ b).
Figure 1.

Typical crystals of Ara127N. (a) A cluster of crystals of the CTP form of Ara127N-WT. The larger plates of the cluster are about 0.3 × 0.3 × 0.01 mm in size. (b) Crystals of the RB form of Ara127N-WT, as obtained from the initial screen. Typical dimensions of these crystals are about 0.02 × 0.04 × 0.02 mm. (c) The Ara127N-WT crystal (CTP form) used for full data collection to 3.50 Å resolution. (d) The Ara127N-Se crystal (CTP form) used for full MAD data collection to 2.25–2.70 Å resolution. Scale bars are shown in green at the bottom left in (c) and (d). The X-ray beam cross-section is shown as a blue circle. As the crystal in (d) is about 250 µm in length and the beam size is about 75 µm, this allowed three different ‘fresh’ positions for the four data sets measured from this crystal.
After these initial crystals had been obtained, a series of optimization experiments were performed in order to reproduce and improve both crystal forms. Nevertheless, multiple optimization experiments with different purification batches of the active protein were generally unsuccessful and were unable to improve the initial crystals obtained from the commercial screens in both cases. In fact, the only crystals which could be reasonably reproduced were those of the CTP habit, and surprisingly (and somewhat scientifically embarrassing) even these crystals could only be obtained with the original screening solution (No. 48) from PEG/Ion 2. The reasons for such unexpected behaviour is as yet unclear, but probably reflect the high sensitivity of the Ara127N protein to very small differences in its host solutions, at least with respect to the conformational stability needed for crystallization. The best crystals of the CTP habit were obtained and reproduced using the procedure described above, with hanging drops formed from 40% of the original protein solution and 60% of the commercial (PEG/Ion 2 No. 48) well solution (Figs. 1 ▶ a and 1 ▶ c).
3.2. X-ray diffraction data for the CTP habit of Ara127N-WT
Several crystals of both forms obtained either directly from the screen (in the case of the RB crystal form; Fig. 1 ▶ b) or from the screen solution (in the case of the CTP crystal form; Figs. 1 ▶ a and 1 ▶ c) were used for detailed crystallographic characterization and measurement of X-ray diffraction data under cryogenic conditions. These experiments were all performed using X-ray synchrotron radiation (λ = 0.95–0.98 Å) and a CCD area detector (MAR-225, Rayonix, USA) on the BM14 beamline at the ESRF synchrotron facility. The crystal-cooling procedure used for these experiments included a short soak of the target crystal (for about 20–60 s) in a cryoprotecting solution based on the original crystallization mother liquor (see below). Each of these pre-soaked crystals was submitted to a flash-cooling procedure involving immediate immersion in liquid nitrogen followed by quick transfer into the centre of a cold nitrogen-gas stream (−173°C) flowing around the crystal throughout the X-ray data collection.
The CTP crystals of Ara127N-WT were soaked briefly (for about 30 s) in a cryoprotecting solution consisting of 26% PEG 4K, 0.8% tryptone, 80 mM HEPES sodium buffer pH 7 and were then flash-cooled for X-ray data collection. The observed diffraction pattern of some of these crystals extended beyond 4.0 Å resolution and indicated that the crystals belonged to a primitive monoclinic crystal system (space group P21), with average crystallographic unit-cell parameters a = 104.0, b = 131.2, c = 107.6 Å, β = 112.0°.
One of these crystals (Fig. 1 ▶ c) was used for a complete X-ray diffraction data measurement at 3.50 Å resolution (Fig. 2 ▶). An oscillation data set (Δϕ = 1.0°, 20 s exposure per frame, 270 frames) was measured on the BM14 beamline at ESRF (λ = 0.978 Å, −173°C). The raw CCD diffraction images were processed with iMosflm (Battye et al., 2011 ▶). A total of 172 201 accepted reflections [I > −4.0σ(I)] were measured in the 59.20–3.50 Å resolution range, resulting in 33 226 independent reflections with 97.9% completeness to 3.50 Å resolution (96.1% for the highest resolution shell of 3.67–3.50 Å). The overall multiplicity of the data set was 5.2, the overall mosaicity was 1.65°, 〈I/σ(I)〉 was 8.3 and the final R merge for the whole data was 17.8% (57.6% for the highest resolution shell) (Table 1 ▶). The relatively high R merge originated probably from the rather weak diffraction pattern and its relatively high degree of anisotropy. While reasonable diffraction could be measured to 3.5 Å resolution for some of the crystal orientations (Fig. 2 ▶), relatively weak diffraction was observed in other crystal orientations (especially in the outer diffraction shells).
Figure 2.

X-ray diffraction pattern of Ara127N-WT (CTP form) obtained using a synchrotron source (BM14, ESRF). The outer circle corresponds to 3.5 Å resolution. The insets represent magnified views of the sections indicated by the corresponding squares (bottom, low resolution; top, medium resolution).
Table 1. Representative parameters from the crystallographic data measurement of Ara127N-WT (CTP and RB crystal forms).
Values in parentheses are for the outer diffraction shell.
| Ara127N-WT (CTP form) | Ara127N-WT (RB form) | |
|---|---|---|
| Source | BM14, ESRF | BM14, ESRF |
| Wavelength (Å) | 0.978 | 0.978 |
| Rotation range (°) | 270 | 240 |
| Crystal-to-detector distance (mm) | 386 | 268 |
| Space group | P21 | P212121 |
| Unit-cell parameters | ||
| a (Å) | 104.0 | 65.5 |
| b (Å) | 131.2 | 118.1 |
| c (Å) | 107.6 | 175.0 |
| β (°) | 112.0 | |
| Resolution (Å) | 59.20–3.50 (3.67–3.50) | 30.00–2.30 (2.34–2.30) |
| No. of reflections | ||
| Total | 172201 | 558797 |
| Unique | 33226 | 61543 |
| Multiplicity | 5.2 (4.9) | 9.1 (4.9) |
| 〈I/σ(I)〉 | 8.3 (3.5) | 8.5 (2.4) |
| Mosaicity (°) | 1.65 | 0.78 |
| Completeness (%) | 97.9 (96.1) | 99.9 (98.6) |
| R merge † (%) | 17.8 (57.6) | 10.7 (52.9) |
R
merge = 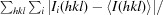
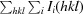 , where I
i(hkl) is the intensity of observation i of reflection hkl.
, where I
i(hkl) is the intensity of observation i of reflection hkl.
At this point, it was possible to estimate the protein content of the crystallographic unit cell by a rough calculation of the solvent content in the crystal and the specific ratio of volume to protein (Matthews coefficient, V M). The range of V M values originally observed for soluble protein crystals was 1.68–3.5 Å3 Da−1 with a mean of 2.17 Å3 Da−1 (based on 116 proteins; Matthews, 1968 ▶). A wider range and a mean of 2.69 Å3 Da−1 was subsequently calculated based on 10 471 proteins (Kantardjieff & Rupp, 2003 ▶). These mean V M values correspond to mean solvent contents of 43 and 47%, respectively. The volume of the Ara127N-WT CTP crystallographic unit cell, as determined from the mean value of the unit-cell dimensions at −173°C, is 1.40 × 106 Å3. Assuming that the V M value here is within the normal range, there should be between three and five Ara127N monomers (642 amino acids; molecular mass 73 642 Da) in the crystallographic asymmetric unit. With three molecules in the CTP asymmetric unit (six in the unit cell), the calculated V M is 3.17 Å3 Da−1 and the corresponding solvent content in the crystals is 61.2%. With four molecules in the asymmetric unit (eight in the unit cell) the calculated V M is 2.38 Å3 Da−1 and the corresponding solvent content in the crystals is 48.3%. With five molecules in the asymmetric unit (ten in the unit cell), the calculated V M is 1.90 Å3 Da−1 and the corresponding solvent content in the crystals is 35.4%. These three different possibilities all seem reasonable considering their calculated V M values and solvent contents; however, the second possibility (four monomers per asymmetric unit) gives values that are significantly closer to the mean values observed, making four monomers per asymmetric unit the most likely solution. Such a content of the crystallographic asymmetric unit is also consistent with gel-filtration experiments, which suggests that Ara127N is active as a dimer (see above), making two dimers in the asymmetric unit the more reasonable option.
Further support for the content of the asymmetric unit of the CTP crystal form comes from self-rotation function (SRF) calculations, performed with the POLARRFN program within the CCP4 suite (Winn et al., 2011 ▶). The SRF calculations of the CTP crystal data (as calculated for both the Ara127N-WT and the Ara127N-Se crystals) show four peaks in the κ = 180° section (Fig. 3 ▶). The two peaks at κ = 180° [(ω = 137.0°, ϕ = 180.0°, κ = 180.0°) and (ω = 47.0°, ϕ = 180.0°, κ = 180.0°)] with a peak height of 34.9 correspond to two twofold non-crystallographic symmetry (NCS) axes and are related by 90°. The peaks at ϕ = 90° are derived from the crystallographic twofold axis. The asymmetry of these peaks indicates the presence of a third NCS axis nearly parallel to the crystallographic axis. These results confirm that there are four protein monomers in the crystallographic asymmetric unit and that these momomers are likely to be arranged as two functional homodimers. These dimers are related by an NCS axis situated roughly parallel to the b axis of the unit cell. Obviously, the actual content of the crystallographic asymmetric unit (and the unit cell) will only be unequivocally resolved when the crystallographic protein structure of this crystal form is fully determined.
Figure 3.

Self-rotation function calculated for the CTP crystallographic data of Ara127N, showing the κ = 180° section. Peaks are scaled from 0 to 100%, with 100% representing the origin peak. Contour lines are plotted for peaks greater than 16% of the origin peak at intervals of 2%. The calculation was performed with the diffraction data in the 8–5 Å resolution range, with a search radius of 30 Å. This rotation function confirms that there are four monomers in the asymmetric unit of the CTP crystal form.
3.3. X-ray diffraction data for the RB habit of Ara127N-WT
Several Ara127N-WT crystals of the RB crystal form were submitted to crystallographic characterization directly from the initial screening experiment. For X-ray diffraction measurement, these crystals were immersed for about 40–60 s in a cryoprotecting solution consisting of 10% PEG 200, 18% PEG 300, 90 mM acetate buffer pH 4.60, followed by immediate flash-cooling in liquid nitrogen. The observed diffraction pattern of some of these crystals extended beyond 2.5 Å resolution and indicated that the crystals belonged to a primitive orthorhombic crystal system (space group P212121), with average crystallographic unit-cell parameters a = 65.5, b = 118.1, c = 175.0 Å. One of these crystals was used for the collection of a complete X-ray diffraction data set at 2.30 Å resolution (Δϕ = 0.5°, 8 s exposure per frame, 480 frames) on the BM14 beamline at the ESRF (λ = 0.978 Å, −173°C) (Fig. 4 ▶). Data processing was performed with the HKL-2000 software (Otwinowski & Minor, 1997 ▶) using a total of 558 797 accepted reflections [I > −3.0σ(I)] measured in the 30.00–2.30 Å resolution range. This processing resulted in 61 543 independent reflections with 99.9% overall completeness (98.6% for the highest resolution shell of 2.34–2.30 Å). The overall multiplicity in the final data set was 9.1, the overall mosaicity was 0.78°, 〈I/σ(I)〉 was 8.5 and the final R merge for the whole data was 10.7% (52.9% for the highest resolution shell) (Table 1 ▶).
Figure 4.

X-ray diffraction pattern of Ara127N-WT (RB form) obtained using a synchrotron source (BM14, ESRF). The outer circle corresponds to 2.3 Å resolution. The insets represent magnified views of the sections indicated by the corresponding squares (bottom, low resolution; top, high resolution).
Similarly to the CTP crystal habit, an estimate of the protein content of the crystallographic unit cell may be made at this point. The volume of the Ara127N-WT RB crystallographic unit cell, as determined from the mean value of the unit-cell dimensions at −173°C, is 1.35 × 106 Å3. In order for the V M value to be within the normal range, there should be two Ara127N monomers in the crystallographic asymmetric unit (eight in the unit cell), corresponding to a V M value of 2.30 Å3 Da−1 and a solvent content of 46.6%. Such a content is also consistent with the gel-filtration results suggesting that Ara127N is a dimer in solution, as discussed above. The V M value and solvent content for three monomers per asymmetric unit are 1.53 Å3 Da−1 and 19.92%, respectively, while the V M value and solvent content for one monomer per asymmetric unit are 4.60 Å3 Da−1 and 73.31%, respectively, making these last two possibilities unlikely as the values are well outside the normal range for protein crystals. This conclusion is supported by SRF calculations performed as described above for the CTP crystal form (Winn et al., 2011 ▶). The SRF of the RB crystal data (Fig. 5 ▶) obtained using the 2.3 Å data presented here clearly demonstrated the three expected twofold axes and revealed an additional peak (ω = 15.1°, ϕ = 180.0°, κ = 180.0°) in the κ = 180° section, suggesting the presence of a non-crystallographic twofold axis. This NCS axis probably relates the two protein molecules to each other, confirming that the asymmetric unit of this crystal form consists of two Ara127N monomers, possibly the functional dimer discussed above. Here again, the actual content of the RB crystallographic unit cell will only be unequivocally resolved when the complete structure of this crystal form is fully determined.
Figure 5.

Self-rotation function calculated for the RB crystallographic data of Ara127N, showing the κ = 180° section. Peaks are scaled from 0 to 100%, with 100% representing the origin peak. Contour lines are plotted for peaks greater than 16% of the origin peak at intervals of 2%. The calculation was performed with the diffraction data in the 8–5 Å resolution range, with a search radius of 30 Å. This rotation function confirms that there are two monomers in the asymmetric unit of the RB crystal form.
3.4. X-ray diffraction data for Ara127N-Se
At the time of data collection, no protein structure with sufficient homology to Ara127N was available, making the selenomethionine multi-wavelength or single-wavelength anomalous diffraction (MAD/SAD) procedures the best alternatives for crystallographic phase solution. A fully replaced selenomethionine derivative of Ara127N (Ara127N-Se) was therefore overexpressed and purified as described above, resulting in a protein with 15 Se atoms per protein monomer. Attempts were made to obtain Ara127N-Se crystals of both forms, starting from the conditions that produced the crystals of the wild-type protein. Nevertheless, suitable Ara127N-Se crystals were obtained only in the CTP form, under the same conditions as used for the CTP crystal habit of the wild-type enzyme. The diffraction pattern of these crystals, some of which extended beyond 2.4 Å resolution, confirmed that they are highly isomorphous with the corresponding CTP crystals of Ara127N-WT (see above), as reflected by the same space group (P21), similar self-rotation function (Fig. 3 ▶) and very similar unit-cell parameters (Table 2 ▶).
Table 2. Representative parameters from the crystallographic data measurements of Ara127N-Se (CTP crystal form).
Values in parentheses are for the outer diffraction shell.
| Peak | Inflection | Remote | High resolution | |
|---|---|---|---|---|
| Source | BM14, ESRF | BM14, ESRF | BM14, ESRF | BM14, ESRF |
| Wavelength (Å) | 0.978 | 0.979 | 0.954 | 0.971 |
| Space group | P21 | P21 | P21 | P21 |
| Rotation range (°) | 360 | 360 | 360 | 335 |
| Crystal-to-detector distance (mm) | 232 | 232 | 232 | 232 |
| Unit-cell parameters | ||||
| a (Å) | 105.4 | 105.4 | 105.4 | 105.4 |
| b (Å) | 132.4 | 132.4 | 132.5 | 132.3 |
| c (Å) | 109.2 | 109.3 | 109.5 | 109.0 |
| β (°) | 113.0 | 113.2 | 113.1 | 113.1 |
| Resolution (Å) | 50.00–2.50 (2.54–2.50) | 40.00–2.40 (2.44–2.40) | 40.00–2.70 (2.75–2.70) | 40.00–2.25 (2.29–2.25) |
| No. of reflections | ||||
| Total | 733903 | 820760 | 506009 | 912493 |
| Unique | 95509 | 10,062 | 77651 | 128276 |
| Multiplicity | 7.7 (6.8) | 7.7 (7.7) | 6.5 (6.6) | 7.1 (6.8) |
| 〈I/σ(I)〉 | 6.9 (2.4) | 6.5 (3.4) | 8.5 (4.9) | 6.7 (3.2) |
| Mosaicity (°) | 0.60 | 0.47 | 0.51 | 0.64 |
| Completeness (%) | 99.3 (87.3) | 100.0 (100.0) | 99.9 (99.8) | 100.0 (100.0) |
| R merge † (%) | 13.5 (80.9) | 12.3 (62.9) | 13.9 (68.7) | 12.3 (62.6) |
R
merge =
, where I
i(hkl) is the intensity of observation i of reflection hkl.
One of these Ara127N-Se crystals (Fig. 1 ▶ d) was used for the measurement of complete MAD data sets: one at the selenium absorption peak (λ = 0.978 Å, 2.50 Å resolution; Fig. 6 ▶), one at the selenium inflection point (λ = 0.979 Å; 2.40 Å) and one at a remote wavelength (λ = 0.954 Å; 2.70 Å). All three data sets were collected from the same crystal (on the BM14 beamline at the ESRF, −173°C), which was possible without significant radiation damage since the length of the crystal in one of its dimensions (about 0.25 mm) enabled data collection from different parts of the crystal along this axis (Fig. 1 ▶ d). The data set at the selenium peak included 360 oscillation frames (Δϕ = 1.0°, 4 s exposure per frame), resulting in a total of 733 903 accepted reflections [I > −3.0σ(I)] and 95 509 independent reflections in the 50.0–2.50 Å resolution range [99.3% completeness, overall multiplicity of 7.7, overall mosaicity of 0.60°, 〈I/σ(I)〉 of 6.9 and a final R merge of 13.5%]. The data set at the selenium inflection point included 360 oscillation frames (Δϕ = 1.0°, 4 s exposure per frame), resulting in a total of 820 760 accepted reflections [I > −3.0σ(I)] and 106 062 independent reflections in the 40.0–2.40 Å resolution range [100.0% completeness, overall multiplicity of 7.7, overall mosaicity of 0.47°, 〈I/σ(I)〉 of 6.5 and final R merge of 12.3%]. The data set at the remote energy included 360 oscillation frames (Δϕ = 1.0°, 10 s exposure per frame), resulting in a total of 506 009 accepted reflections [I > −3.0σ(I)] and 77 651 independent reflections in the 40.0–2.70 Å resolution range [99.9% completeness, overall multiplicity of 6.5, overall mosaicity of 0.51°, 〈I/σ(I)〉 of 8.5 and a final R merge of 13.9%] (Table 2 ▶).
Figure 6.

X-ray diffraction pattern of Ara127N-Se (CTP form, Se peak wavelength) obtained using a synchrotron source (BM14, ESRF). The outer circle corresponds to 2.5 Å resolution. The insets represent magnified views of the sections indicated by the corresponding squares (bottom, low resolution; top, high resolution).
The parameters for the three data sets indicate a full diffraction data set of relatively good quality at the corresponding resolutions, data that should be sufficient, in principle, for either MAD or SAD phase determination. Nevertheless, in order to provide an additional data set at another remote wavelength, a fourth data set was collected at λ = 0.971 Å from the same crystal, using a different ‘fresh’ position along the crystal (Fig. 1 ▶ d), as performed for the other data sets obtained from this crystal. This additional data set also allowed an extension of the overall resolution of the diffraction data to 2.25 Å resolution. This data set (335 frames, Δϕ = 1.0°, 4 s exposure per frame) included a total of 912 493 accepted reflections [I > −3.0σ(I)] and 128 276 independent reflections in the 40.0–2.25 Å resolution range, with 100.0% overall completeness, an overall multiplicity of 7.1, an overall mosaicity of 0.64°, an 〈I/σ(I)〉 of 6.7 and a final R merge of 12.3% (Table 2 ▶).
As mentioned above, the Ara127N monomer contains 15 methionines, providing 15 Se atoms per Ara127N-Se monomer of 642 amino-acid residues. This Se:protein ratio is expected to give an anomalous signal which should be sufficient to enable crystallographic phasing via the powerful MAD methodology, using all of the Ara127N-Se data sets collected. It may also be sufficient to allow reasonable phasing via the related SAD methodology, using only the Se peak data. Whether provided by SAD or MAD procedures, such phasing should lead to structure determination of the Ara127N-Se protein at 2.50 Å resolution and its extension and refinement at 2.25 Å resolution. Once obtained, the structure of Ara127N-Se can then be used for structural analysis of Ara127N-WT (in both crystal forms) using the two Ara127N-WT crystallographic data sets described above. Such analyses are currently in progress in our laboratory.
Acknowledgments
The research leading to the results presented here received funding from the European Community’s Seventh Framework Programme (FP7/2007-2013) under BioStruct-X (grant agreement No. 283570). This work was also supported by Israel Science Foundation Grants 500/10 and 152/11, the I-CORE Program of the Planning and Budgeting Committee, the Ministry of Environmental Protection and the Grand Technion Energy Program (GTEP), and comprises part of The Leona M. and Harry B. Helmsley Charitable Trust Reports on Alternative Energy Series of the Technion, Israel Institute of Technology and the Weizmann Institute of Science. YS acknowledges partial support by the Russell Berrie Nanotechnology Institute and The Lorry I. Lokey Interdisciplinary Center for Life Science and Engineering, Technion. We thank the staff of the Cold Spring Harbor Laboratory course on Macromolecular Crystallography (CSHL, October 2013), in the framework of which the initial Ara127N-WT crystals were obtained. We thank the staff of the Technion Center for Structural Biology (TCSB, Technion, Haifa, Israel) for helpful assistance in the crystallization screening experiments. We thank the staff at the European Synchrotron Research Facility (ESRF, BM14 beamline) and EMBL for their helpful support in the X-ray synchrotron data measurement and analysis. The synchrotron experiments at the ESRF were supported also by the ESRF internal funding program. YS holds the Erwin and Rosl Pollak Chair in Biotechnology at the Technion.
References
- Alalouf, O., Balazs, Y., Volkinshtein, M., Grimpel, Y., Shoham, G. & Shoham, Y. (2011). J. Biol. Chem. 286, 41993–42001. [DOI] [PMC free article] [PubMed]
- Almog, O., Greenblatt, H. M., Spungin, A., Ben-Meir, D., Blumberg, S. & Shoham, G. (1993). J. Mol. Biol. 230, 342–344. [DOI] [PubMed]
- Almog, O., Klein, D., Braun, S. & Shoham, G. (1994). J. Mol. Biol. 235, 760–762. [DOI] [PubMed]
- Bar, M., Golan, G., Nechama, M., Zolotnitsky, G., Shoham, Y. & Shoham, G. (2004). Acta Cryst. D60, 545–549. [DOI] [PubMed]
- Battye, T. G. G., Kontogiannis, L., Johnson, O., Powell, H. R. & Leslie, A. G. W. (2011). Acta Cryst. D67, 271–281. [DOI] [PMC free article] [PubMed]
- Ben-David, A., Bravman, T., Balazs, Y. S., Czjzek, M., Schomburg, D., Shoham, G. & Shoham, Y. (2007). Chembiochem, 8, 2145–2151. [DOI] [PubMed]
- Ben-David, A., Shoham, G. & Shoham, Y. (2008). Chem. Biol. 15, 546–551. [DOI] [PubMed]
- Bravman, T., Mechaly, A., Shulami, S., Belakhov, V., Baasov, T., Shoham, G. & Shoham, Y. (2001). FEBS Lett. 495, 115–119. [DOI] [PubMed]
- Bravman, T., Zolotnitsky, G., Belakhov, V., Shoham, G., Henrissat, B., Baasov, T. & Shoham, Y. (2003). Biochemistry, 42, 10528–10536. [DOI] [PubMed]
- Brüx, C., Ben-David, A., Shallom-Shezifi, D., Leon, M., Niefind, K., Shoham, G., Shoham, Y. & Schomburg, D. (2006). J. Mol. Biol. 359, 97–109. [DOI] [PubMed]
- Fujita, K., Takashi, Y., Obuchi, E., Kitahara, K. & Suganuma, T. (2014). J. Biol. Chem. 289, 5240–5249. [DOI] [PMC free article] [PubMed]
- Gat, O., Lapidot, A., Alchanati, I., Regueros, C. & Shoham, Y. (1994). Appl. Environ. Microbiol. 60, 1889–1896. [DOI] [PMC free article] [PubMed]
- Gilbert, H. J. (2010). Plant Physiol. 153, 444–455. [DOI] [PMC free article] [PubMed]
- Gilboa, R., Bauer, A. J. & Shoham, G. (1998). Acta Cryst. D54, 1467–1470. [DOI] [PubMed]
- Gilead, S. & Shoham, Y. (1995). Appl. Environ. Microbiol. 61, 170–174. [DOI] [PMC free article] [PubMed]
- Golan, G., Shallom, D., Teplitsky, A., Zaide, G., Shulami, S., Baasov, T., Stojanoff, V., Thompson, A., Shoham, Y. & Shoham, G. (2004). J. Biol. Chem. 279, 3014–3024. [DOI] [PubMed]
- Golan, G., Zharkov, D. O., Fernandes, A. S., Zaika, E., Kycia, J. H., Wawrzak, Z., Grollman, A. P. & Shoham, G. (2004). Acta Cryst. D60, 1476–1480. [DOI] [PubMed]
- Henrissat, B. & Davies, G. (1997). Curr. Opin. Struct. Biol. 7, 637–644. [DOI] [PubMed]
- Hövel, K., Shallom, D., Niefind, K., Baasov, T., Shoham, G., Shoham, Y. & Schomburg, D. (2003). Acta Cryst. D59, 913–915. [DOI] [PubMed]
- Hövel, K., Shallom, D., Niefind, K., Belakhov, V., Shoham, G., Baasov, T., Shoham, Y. & Schomburg, D. (2003). EMBO J. 22, 4922–4932. [DOI] [PMC free article] [PubMed]
- Ito, T., Saikawa, K., Kim, S., Fujita, K., Ishiwata, A., Kaeothip, S., Arakawa, T., Wakagi, T., Beckham, G. T., Ito, Y. & Fushinobu, S. (2014). Biochem. Biophys. Res. Commun. 447, 32–37. [DOI] [PubMed]
- Jancarik, J. & Kim, S.-H. (1991). J. Appl. Cryst. 24, 409–411.
- Kantardjieff, K. A. & Rupp, B. (2003). Protein Sci. 12, 1865–1871. [DOI] [PMC free article] [PubMed]
- Lamport, D. T., Kieliszewski, M. J., Chen, Y. & Cannon, M. C. (2011). Plant Physiol. 156, 11–19. [DOI] [PMC free article] [PubMed]
- Lansky, S., Alalouf, O., Salama, R., Dvir, H., Shoham, Y. & Shoham, G. (2014). Acta Cryst. F70, 476–481. [DOI] [PMC free article] [PubMed]
- Lansky, S., Alalouf, O., Solomon, V., Alhassid, A., Govada, L., Chayan, N. E., Belrhali, H., Shoham, Y. & Shoham, G. (2013). Acta Cryst. F69, 430–434. [DOI] [PMC free article] [PubMed]
- Lansky, S., Alalouf, O., Solomon, H. V., Alhassid, A., Govada, L., Chayen, N. E., Belrhali, H., Shoham, Y. & Shoham, G. (2014). Acta Cryst. D70, 261–278. [DOI] [PubMed]
- Lansky, S., Salama, R., Solomon, V. H., Belrhali, H., Shoham, Y. & Shoham, G. (2013). Acta Cryst. F69, 695–699. [DOI] [PMC free article] [PubMed]
- Lansky, S., Zehavi, A., Dann, R., Dvir, H., Belrhali, H., Shoham, Y. & Shoham, G. (2014). Acta Cryst. F70, 225–231. [DOI] [PMC free article] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- Mechaly, A., Teplitsky, A., Belakhov, V., Baasov, T., Shoham, G. & Shoham, Y. (2000). J. Biotechnol. 78, 83–86. [DOI] [PubMed]
- Mohnen, D. (2008). Curr. Opin. Plant Biol. 11, 266–277. [DOI] [PubMed]
- Ogawa-Ohnishi, M., Matsushita, W. & Matsubayashi, Y. (2013). Nature Chem. Biol. 9, 726–730. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Rees, D. C., Johnson, E. & Lewinson, O. (2009). Nature Rev. Mol. Cell Biol. 10, 218–227. [DOI] [PMC free article] [PubMed]
- Reiland, V., Funano-Hershcovitz, Y., Golan, G., Gilboa, R., Shoham, Y. & Shoham, G. (2004). Acta Cryst. D60, 2371–2376. [DOI] [PubMed]
- Scheller, H. V. & Ulvskov, P. (2010). Annu. Rev. Plant Biol. 61, 263–289. [DOI] [PubMed]
- Shallom, D., Belakhov, V., Solomon, D., Gilead-Gropper, S., Baasov, T., Shoham, G. & Shoham, Y. (2002). FEBS Lett. 514, 163–167. [DOI] [PubMed]
- Shallom, D., Belakhov, V., Solomon, D., Shoham, G., Baasov, T. & Shoham, Y. (2002). J. Biol. Chem. 277, 43667–43673. [DOI] [PubMed]
- Shallom, D., Golan, G., Shoham, G. & Shoham, Y. (2004). J. Bacteriol. 186, 6928–6937. [DOI] [PMC free article] [PubMed]
- Shallom, D., Leon, M., Bravman, T., Ben-David, A., Zaide, G., Belakhov, V., Shoham, G., Schomburg, D., Baasov, T. & Shoham, Y. (2005). Biochemistry, 44, 387–397. [DOI] [PubMed]
- Shulami, S., Gat, O., Sonenshein, A. L. & Shoham, Y. (1999). J. Bacteriol. 181, 3695–3704. [DOI] [PMC free article] [PubMed]
- Shulami, S., Raz-Pasteur, A., Tabachnikov, O., Gilead-Gropper, S., Shner, I. & Shoham, Y. (2011). J. Bacteriol. 193, 2838–2850. [DOI] [PMC free article] [PubMed]
- Shulami, S., Zaide, G., Zolotnitsky, G., Langut, Y., Feld, G., Sonenshein, A. L. & Shoham, Y. (2007). Appl. Environ. Microbiol. 73, 874–884. [DOI] [PMC free article] [PubMed]
- Solomon, H. V., Tabachnikov, O., Feinberg, H., Govada, L., Chayen, N. E., Shoham, Y. & Shoham, G. (2013). Acta Cryst. F69, 1114–1119. [DOI] [PMC free article] [PubMed]
- Solomon, V., Teplitsky, A., Shulami, S., Zolotnitsky, G., Shoham, Y. & Shoham, G. (2007). Acta Cryst. D63, 845–859. [DOI] [PubMed]
- Tabachnikov, O. & Shoham, Y. (2013). FEBS J. 280, 950–964. [DOI] [PubMed]
- Teplitsky, A., Feinberg, H., Gilboa, R., Lapidot, A., Mechaly, A., Stojanoff, V., Capel, M., Shoham, Y. & Shoham, G. (1997). Acta Cryst. D53, 608–611. [DOI] [PubMed]
- Teplitsky, A., Mechaly, A., Stojanoff, V., Sainz, G., Golan, G., Feinberg, H., Gilboa, R., Reiland, V., Zolotnitsky, G., Shallom, D., Thompson, A., Shoham, Y. & Shoham, G. (2004). Acta Cryst. D60, 836–848. [DOI] [PubMed]
- Teplitsky, A., Shulami, S., Moryles, S., Shoham, Y. & Shoham, G. (2000). Acta Cryst. D56, 181–184. [DOI] [PubMed]
- Teplitsky, A., Shulami, S., Moryles, S., Zaide, G., Shoham, Y. & Shoham, G. (1999). Acta Cryst. D55, 869–872. [DOI] [PubMed]
- Warnecke, F. et al. (2007). Nature (London), 450, 560–565.
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Zaide, G., Shallom, D., Shulami, S., Zolotnitsky, G., Golan, G., Baasov, T., Shoham, G. & Shoham, Y. (2001). Eur. J. Biochem. 268, 3006–3016. [DOI] [PubMed]