

Abstract
Abiotic stress represents a serious threat affecting both plant fitness and productivity. One of the promptest responses that plants trigger following abiotic stress is the differential expression of key genes, which enable to face the adverse conditions. It is accepted and shown that the cell wall senses and broadcasts the stress signal to the interior of the cell, by triggering a cascade of reactions leading to resistance. Therefore the study of wall-related genes is particularly relevant to understand the metabolic remodeling triggered by plants in response to exogenous stresses. Despite the agricultural and economical relevance of alfalfa (Medicago sativa L.), no study, to our knowledge, has addressed specifically the wall-related gene expression changes in response to exogenous stresses in this important crop, by monitoring the dynamics of wall biosynthetic gene expression. We here identify and analyze the expression profiles of nine cellulose synthases, together with other wall-related genes, in stems of alfalfa plants subjected to different abiotic stresses (cold, heat, salt stress) at various time points (e.g. 0, 24, 72 and 96 h). We identify 2 main responses for specific groups of genes, i.e. a salt/heat-induced and a cold/heat-repressed group of genes. Prior to this analysis we identified appropriate reference genes for expression analyses in alfalfa, by evaluating the stability of 10 candidates across different tissues (namely leaves, stems, roots), under the different abiotic stresses and time points chosen. The results obtained confirm an active role played by the cell wall in response to exogenous stimuli and constitute a step forward in delineating the complex pathways regulating the response of plants to abiotic stresses.
Introduction
The study of biological phenomena requires several sensitive analytical techniques, which can convey detailed information at different depths of organismal complexity, namely tissular, metabolic, genomic. One such type of information is represented by gene expression changes, which provide clues about transcripts dynamics, e.g. in response to exogenous stimuli.
Currently one of the most reliable and reproducible methods to perform differential gene expression profiling is quantitative reverse transcription PCR (hereafter referred to as RT-qPCR), a method which is robust enough to quantify challenging targets, as microRNAs (miRNAs) e.g. [1]. However, accurate gene expression analyses rely on several critical aspects and experimental steps (namely RNA purity and integrity, genomic DNA contamination, reverse transcription) and, in the case of relative quantification, on the identification of suitable reference genes for data normalization [2]–[3]. Those are genes whose expression is stable and not subject to fluctuations across the different conditions tested. This feature is particularly critical, as the choice of inappropriate reference genes can significantly bias the results obtained and therefore lead to misinterpretations of biological events.
The use of RT-qPCR is particularly suitable to study the response of a set of genes in plants after the application of specific stresses e.g. [4]: being sessile organisms, plants are not capable of escaping from adverse environmental conditions and are therefore characterized by a very responsive transcriptional regulation, which results in phenotypic plasticity [5]–[7]. Abiotic stresses constitute serious threats for plants, as they can affect not only their development, growth, reproduction and productivity, but can be so detrimental to cause their death. Exogenous stresses unleash a cascade of reactions, which lead to plant response and resistance, usually by means of wall fortification.
Many studies in the literature have provided a comprehensive view of gene expression changes in different plant species in response to abiotic stresses and identified a list of suitable reference genes for data normalization e.g. [8]–[13]. These studies have also shown how the expression of reference genes can vary in different plant species and conditions and how important it is to validate their stability in the specific experimental set-ups used.
Despite the agricultural and economical importance of the legume crop Medicago sativa L. (a.k.a alfalfa, or lucerne), no study has so far tested suitable reference genes for expression analysis using RT-qPCR in this plant. Suitable reference genes have been identified in Medicago truncatula [14] and potential reference genes in alfalfa have been proposed by Yang et al. [15], however their suitability for RT-qPCR studies has, to our knowledge, never been validated so far.
Alfalfa is an experimentally valuable model: it is not only suitable for the study of symbiotic interactions e.g. [16], but has also been proposed as an excellent model system to study dicot cell wall development [17]. Its stem shows indeed 2 clearly-defined regions characterized by active elongation and lignification/thickening, which provide “snapshots” of the cell wall maturation process. Although the genome of alfalfa has not yet been sequenced, several studies have shown the suitability of using the genome of the closely related barrel medic (M. truncatula) [18] for molecular analyses. These studies have delivered valuable information concerning the regulation of wall polysaccharide biosynthesis in cultivars with contrasting cell wall composition [19].
The aim of the present study is to provide a time-course analysis of cell wall-related gene expression in response to different abiotic stresses in alfalfa stems. In particular we analyzed nine cellulose synthases (hereafter named MsCesAs), identified on the basis of the sequence homology with the orthologs from M. truncatula (MtCesAs), together with other genes linked to wall biogenesis (namely sucrose synthase, SuSy; phenylalanine ammonia lyase, PAL; cinnamyl alcohol dehydrogenase, CAD; cellulose synthase-like gene, CslD4). To perform a reliable gene expression analysis, accurate data normalization is mandatory, which prompted us to identify the most suitable reference genes for expression analysis. We chose as candidate reference genes a set of known and widely used genes which have been tested on M. truncatula [14], together with candidates proposed by Yang et al. [15] and Huis et al. [20].
We decided to extend our survey not only to stems, but also to other tissues (namely roots and leaves), in order to provide a list of genes to be used in tissue- and/or growth condition-specific studies in alfalfa and which can be tested in other legume crops too. Ten candidate reference genes were chosen and their reliability for RT-qPCR studies tested at different time-points in different tissues of M. sativa plants exposed to abiotic stresses. To further validate their suitability, we studied the expression of a stress-associated kinase (SK1) in the different tissues and growth conditions.
Despite the unanimously recognized role of plant walls as cellular structures sensing and responding to stimuli [21]–[24] and despite the economic significance of alfalfa, to our knowledge no study is yet available on the expression analysis of key wall biosynthetic genes (as CesAs) in response to different abiotic stresses in M. sativa. We here provide such a study and identify the main trends characterizing the response to abiotic stresses in alfalfa stems.
Materials and Methods
Plant growth and abiotic stress treatments
Medicago sativa L. seeds, variety Giulia (Italy), were inoculated with a peat-based inoculant (HiStick, Becker Underwood) according to the manufacturer’s instructions. Five seeds were sown per pot in 1 L containers filled with soil (50% topsoil, 25% potting soil, 25% sand). After 4 weeks of cultivation under controlled greenhouse conditions (photoperiod of 13 h light/11 h darkness, minimum temperature of 20°C, maximum 27°C), plants were moved to incubators (programmed to provide exactly the same light/dark cycles) for moderate cold and heat stress treatments, while those subjected to moderate salt stress were left in the greenhouse. For the cold stress condition, plants were grown at a constant temperature of 5°C; for the heat stress condition, they were grown at 28°C/32°C (night/day); for the salt stress treatment, plants were supplemented with 100 mM NaCl. A total of 3 biological replicates (each consisting of a pool of 15 plants) were used per treatment. For each time point studied (0- 24- 72- 96 h) a control group was always kept (i.e. plants grown without any treatment for 24- 72- 96 h), for appropriate comparisons.
RNA extraction and cDNA synthesis
Sampled tissues (roots, leaves and whole stems) were ground to a fine powder in liquid nitrogen, using a mortar and a pestle. One hundred mg of finely ground sample were weighed on a balance and total RNA was extracted using the RNeasy Plant Mini Kit with the on-column DNase I treatment (Qiagen). The integrity of the extracted RNA was checked with an Agilent Bioanalyzer (all the RINs were >8) and the purity/concentration measured using a NanoDrop ND-1000 spectrophotometer (A260/280 and A260/230 ratios between 1.9 and 2.2). Subsequently, 1 µg of extracted RNA was retro-transcribed using the Superscript II cDNA Synthesis kit (Invitrogen), according to the manufacturer’s instructions.
Identification of CesA genes from alfalfa
To identify and amplify putative CesA genes from M. sativa, initial data mining was performed on M. truncatula [25], a closely related species for which the genome is available. A total of nine putative M. truncatula cellulose synthase proteins (hereafter indicated MtCESAs) were identified. BLASTp searches were performed against non-redundant protein databases of Arabidopsis thaliana and Populus trichocarpa from the National Centre for Biotechnology [26] to check the percentage of identity of the identified sequences. To amplify the orthologous genes from alfalfa, primers were designed on the identified MtCesAs genes (listed in Table S1). Three full-length CesA genes from alfalfa were identified and designated MsCesA3, MsCesA4 and MsCesA7-A [GenBank: KJ398155, KJ398156, KJ398157; Fig. S1], on the basis of their phylogenetic kinship, while partial sequences were obtained for the other MsCesAs (Figs. S1 and S2). The phylogenetic tree was built by aligning the amino acid regions of CESAs from M. truncatula, M. sativa, P. trichocarpa and A. thaliana encompassing the U1–U4 regions, the QXXRW motif and the HVR2 region, which allows class discrimination [27], using MUSCLE [28]. Phylogeny was analyzed using PhyML [29]. The maximum-likelihood phylogenetic tree was rendered using TreeDyn [30]. Microarray data for M. truncatula CesAs were retrieved at [31] and electronic fluorescent pictographic (eFP) representations at [32].
Quantitative real-time PCR and statistical analysis
For quantitative real-time PCR analysis, 10 ng cDNA were used as template. The cDNA was amplified using the MESA GREEN qPCR MasterMix Plus.
Low ROX (Eurogentec) on a ViiA 7 Real-Time PCR System (Applied Biosystems) in a final volume of 25 µl.
The reactions were performed in technical triplicates and repeated on the above-described 3 biological independent replicates. The PCR conditions consisted of an initial denaturation at 95° for 10 min, followed by 45 cycles of denaturation at 95° for 15 sec, annealing/extension at 60° for 60 sec.
A dissociation kinetics analysis was performed at the end of the experiment to check the specificity of the annealing.
Ten candidate reference genes were analyzed, namely actin, tubulin, ubiquitin-conjugating protein 13 (UBC13), cyclophilin (cyclo), elongation initiation factor 4A (eif4A), elongation initiation factor 5A (eif5A), translation initiation factor IIA (TFIIA), glyceraldehyde-3P dehydrogenase (GAPDH), actin-depolymerizing protein (ADF1), poly(A) binding protein 4 (PAB4). Their stability was evaluated using NormFinder [33] and geNormPLUS [2], two of the most commonly used software, which rank candidate reference genes on the basis of their stability. The software geNormPLUS performs a pairwise comparison and computes the M-value, i.e. the variation of a gene compared to all the remaining candidates, while NormFinder computes first the intra-group and subsequently the inter-group expression variability of a candidate reference gene [33]–. NormFinder calculates both a single best gene (best gene) and an optimal gene pair (best pair); the best pair might display compensating expression in the different experimental groups. The candidate reference genes primers for actin, tubulin, GAPDH were designed using the sequences from M. truncatula [GenBank: XM_003621971, XM_003603622, XM_003595990]. The primers for the other reference genes were designed using the sequences of the candidate housekeeping genes reported by Yang et al. [15], which show an average RPKM-normalized value higher than 10 and the lowest coefficient of variation identified with RNAseq. The list of primers used to perform RT-qPCR analyses is shown in Table S2. The RT-qPCR primers for CAD and CslD4 (Table S2) were designed on the sequences from M. truncatula genes (probesets Mtr.8985.1.S1_at and Mtr.45005.1.S1_at, respectively) [19], while those for SuSy and PAL (Table S2) were designed on the reported sequences from alfalfa (probeset Msa.2902.1.S1_at) [19] and [GenBank: CAA41169]. Primers were designed using Primer3Plus [35] and analyzed with OligoAnalyzer 3.1 [36]. The primers size was 20 bp, the amplicon sizes were between 70–150 bp (Table S2), the %GC was between 40–60% and Tm 60°C. The primers used were not intron spanning. Primer efficiencies were tested and are reported in Table S2. All the amplicons were verified by sequencing on an Applied Biosystems 3500 Genetic Analyser using the BigDye Terminator v3.1 Cycle Sequencing and the BigDye XTerminator Purification kits, according to the manufacturer’s instructions.
The results relative to the expression of the target genes were analyzed using the software qBasePLUS version 2.5 (Biogazelle, [37]) and normalized taking into account the most stable reference genes (as indicated in the text). The expression levels of the genes detected in the different tissues and conditions analyzed are here expressed as “Normalized relative expression”. A one-way ANOVA (with Tukey’s HSD post-hoc test) was performed on the log2 transformed calibrated normalized relative quantities (CNRQs), using IBM SPSS Statistics (version 19), after having checked the normal distribution of the data with a Kolmogorov-Smirnov test.
Hierarchical clustering was generated with Cluster 3.0 [38] and visualized with Java TreeView [39], available at [40].
Results
Stability of putative reference genes in different tissues of M. sativa subjected to abiotic stresses
Analyses with geNormPLUS were performed to rank the expression stability of the 10 candidate reference genes in the tissues and conditions analyzed (Fig. 1). According to geNormPLUS, TFIIA ranks among the most stable genes in roots, leaves and stems, but interestingly this gene is not among the most stably expressed when all the tissues are grouped together (Fig. 1). However NormFinder ranks TFIIA among the 4 most stable genes when all the tissues are taken into account (Table 1). The gene eIF4A is very stable in leaves and stems, while it is among the least stable in roots (Fig. 1). The most stably expressed genes when all the tissues are grouped together are ADF1 and PAB4 (Fig. 1). Actin is among the least stable genes in all the conditions tested (Fig. 1). These results show that care should be taken when choosing candidate reference genes for expression analysis in different plant tissues, as stable genes in a specific tissue might not be suitable for normalization of expression data in another one. In the literature, several studies have shown the importance of determining the stability of the reference genes in the different plant tissues, in order to use the most reliable ones in the condition examined e.g. [11], [20]. The stability data obtained with geNormPLUS have been compared to the rankings generated via the other widely used software, i.e. NormFinder. Ranking lists were generated for each single tissue under all the conditions tested, for all the tissues together under each single condition (which can be of particular interest when a specific abiotic stress is studied), as well as for all the tissues and conditions together (Table 1). From the rankings it is possible to confirm the expression stability of TFIIA in the single tissues under the different stress treatments; in particular the pairs ADF1/TFIIA and eIF4a/TFIIA are confirmed as the most suitable genes for normalization in roots and stems respectively (Table 1). PAB4 ranks always among the 5 most stable genes when all the tissues are analyzed together in each of the conditions tested, while the high stability of eIF5A and PAB4 is confirmed when all the tissues and conditions are taken into consideration (Table 1). Both geNormPLUS and NormFinder show how unsuitable tubulin and actin are for data normalization in our experimental system: this is quite important, as these genes, although suitable for normalization in some instances [41]–[42], might not be ideal in others [8], [20], [43]. The best gene pairs identified by NormFinder are different in some tissues from those identified by geNormPLUS, a finding which has already been reported in other studies e.g. [20] and might be due to the different ranking methodology used by the two softwares: normalization in the leaves requires TFIIA and eIF4A according to geNormPLUS, while NormFinder suggests ADF1 and PAB4, which in the geNormPLUS ranking are the 4th and 7th least stable gene, respectively (Fig. 1). Similarly, if all the tissues and stresses are considered, the best gene pair is GAPDH/PAB4 according to NormFinder, however geNormPLUS ranks GAPDH as the most unstable gene in this configuration (Fig. 1).
Figure 1. Candidate reference genes in alfalfa.

Ranking of ten candidate reference genes in different tissues of M. sativa according to the parameter M calculated by geNormPLUS. Increasing stability of the candidate genes is determined by a decrease in the M value.
Table 1. Ranking of candidate reference genes according to NormFinder.
| Leaves (all treatments) | Roots(all treatments) | Stems(all treatments) | Control(all tissues) | Cold(all tissues) | Heat(all tissues) | Salt(all tissues) | All tissues and treatments | |||||||||
| Gene | Stability | Gene | Stability | Gene | Stability | Gene | Stability | Gene | Stability | Gene | Stability | Gene | Stability | Gene | Stability | |
| Act | 0.150 | Act | 0.251 | Act | 0.306 | Act | 0.117 | Tub | 0.148 | Tub | 0.280 | ADF1 | 0.154 | Tub | 0.168 | |
| Tub | 0.126 | Tub | 0.211 | ADF1 | 0.153 | TFIIA | 0.109 | eIF5A | 0.125 | Act | 0.202 | Act | 0.105 | Act | 0.133 | |
| eIF5A | 0.103 | Cyclo | 0.139 | Tub | 0.143 | Tub | 0.108 | Act | 0.106 | eIF5A | 0.118 | Cyclo | 0.080 | UBC13 | 0.084 | |
| GAPDH | 0.091 | UBC13 | 0.138 | Cyclo | 0.128 | eIF5A | 0.104 | TFIIA | 0.093 | Cyclo | 0.116 | Tub | 0.068 | eIF4A | 0.076 | |
| UBC13 | 0.087 | eIF5A | 0.110 | UBC13 | 0.127 | GAPDH | 0.059 | GAPDH | 0.081 | UBC13 | 0.112 | eIF5A | 0.073 | Cyclo | 0.076 | |
| Cyclo | 0.086 | PAB4 | 0.102 | eIF5A | 0.102 | Cyclo | 0.053 | ADF1 | 0.064 | GAPDH | 0.109 | TFIIA | 0.050 | ADF1 | 0.074 | |
| ADF1 | 0.072 | eIF4A | 0.094 | PAB4 | 0.081 | eIF4A | 0.053 | eIF4A | 0.054 | PAB4 | 0.085 | GAPDH | 0.049 | TFIIA | 0.067 | |
| eIF4A | 0.056 | GAPDH | 0.089 | eIF4A | 0.067 | UBC13 | 0.051 | UBC13 | 0.051 | eIF4A | 0.080 | PAB4 | 0.046 | eIF5A | 0.058 | |
| TFIIA | 0.051 | TFIIA | 0.071 | GAPDH | 0.062 | ADF1 | 0.040 | PAB4 | 0.047 | ADF1 | 0.074 | UBC13 | 0.041 | GAPDH | 0.055 | |
| Best gene | PAB4 | 0.043 | ADF1 | 0.036 | TFIIA | 0.046 | PAB4 | 0.033 | Cyclo | 0.041 | TFIIA | 0.040 | eIF4A | 0.029 | PAB4 | 0.050 |
| Best pair | ADF1/PAB4 | 0.033 | ADF1/TFIIA | 0.040 | TFIIA/eIF4A | 0.040 | Cyclo/eIF4A | 0.019 | Cyclo/PAB4 | 0.033 | Cyclo/GAPDH | 0.042 | GAPDH/UBC13 | 0.020 | GAPDH/PAB4 | 0.010 |
The best gene and the best combination of genes are shown. The analysis has been carried out to find the most stable reference genes in the different tissues under all the treatments tested, in all the tissues under different treatments and when all the tissues and treatments studied are grouped together. Abbreviations here used: Act (actin), Tub (tubulin), GAPDH, Cyclo (cyclophilin).
Nevertheless, taking into account the rankings of NormFinder and geNormPLUS, it emerges that for expression studies in alfalfa tissues (and possibly in other legume crops) actin and tubulin are not ideal, whereas a suitable panel of reference genes should include eIF4A, PAB4, ADF1 and TFIIA, as they rank among the most stable genes according to the two softwares. This result can be of particular interest when studying gene expression in different plant tissues subjected to a specific treatment: if a tissue-maximization strategy is selected in the experimental design, it is helpful to know a priori which panel of candidates to include for stability test.
Optimal number of reference genes for normalization in M. sativa tissues using geNormPLUS ranking
In order to calculate the appropriate number of reference genes for data normalization in alfalfa, we used geNormPLUS to compute the pairwise variation (Vn/Vn+1) between two consecutive normalization factors (NFn and NFn+1). The analysis shows that for accurate normalization in roots, stems and leaves, 2 reference genes are required: the addition of a third gene is indeed not necessary, as the V value relative to 2 reference genes is already below the cut-off threshold of 0.15 (Fig. 2). However, if all the tissues are grouped together, the number of genes required for accurate data normalization increases to 3, since the V value relative to 2 genes is above the cut-off threshold (0.159) (Fig. 2).
Figure 2. Determination of the appropriate number of reference genes for data normalization in M. sativa tissues under abiotic stress conditions, as computed by geNormPLUS.

The pairwise variation (Vn/Vn+1) was calculated between the normalization factors NFn and NFn+1. The recommended cut-off threshold of 0.15 was kept in the present study.
Validation of the selected reference genes in different tissues
The validity of the candidate reference genes identified via the geNormPLUS and NormFinder analyses was tested in the different tissues and conditions by studying the expression profiles of a stress-associated kinase orthologous to MtSK1 [GenBank: XP_003592980] [44]. This gene is a member of the SnRK group of plant kinases and was shown to be induced upon wounding in cultured tissues [44].
Since SnRKs are involved in stress response in plants e.g. [45], we decided to use this gene both to validate the identified reference genes in the different conditions and to study its expression profile in response to different abiotic stresses in alfalfa tissues. It was assumed that the experimental treatment would not alter the expression of the reference genes, but would instead affect the expression of the stress-associated kinase. The data were analyzed with qBASEPLUS and normalized using ADF1/.
TFIIA and eIF4A/TFIIA for the roots and stems respectively, since these candidates were selected by both geNormPLUS and NormFinder, then a comparison of normalization strategies was performed for the leaves (Figs. 3 and 4), since the two softwares chose different candidates (namely ADF1/PAB4 by NormFinder and TFIIA/eIF4A by geNormPLUS; Fig. 1 and Table 1). As can be seen in Fig. 3, the stresses which triggered the most significant changes were cold and heat: in all the tissues examined, a significant decrease in expression could indeed be observed during cold stress treatment, while heat stress induced expression, where the highest increase was present in roots. Salt stress, on the other hand, did not appreciably change the expression of the stress-associated kinase, apart from a mild increase at 24 and 72h in the stems (Fig. 3). This result was unexpected, as it was previously shown that the expression of the ortholog from M. truncatula increased in the leaves after salt stress treatment [44], however it should be noted that the analysis was here performed on another species and that fluctuations in expression were observed in control condition over the different time-points (Fig. 3). These fluctuations contribute to make the expression changes not significant.
Figure 3. MsSK1 expression in alfalfa tissues under abiotic stresses.

Expression profiles of MsSK1 in the different tissues under abiotic stresses (yellow dotted frame is control; blue dotted frame is cold stress; red dotted frame is heat stress; green dotted frame is salt stress). The Y-axis indicates NRE (Normalized Relative Expression of MtSK1). Data were normalized using ADF1/TFIIA and eIF4A/TFIIA for the roots and stems respectively and TFIIA/eIF4A for the leaves. Means sharing a letter are not significantly different at α = 0.05.
Figure 4. Comparison of NormFinder and geNormPLUS normalization methods.

Comparison of MsSK1 expression profiles in leaves when normalization is performed using ADF1/PAB4 (according to NormFinder), or TFIIA/eIF4A (according to geNormPLUS).
In order to compare the normalization strategies using the gene pairs recommended by NormFinder and geNormPLUS, we chose to perform a test on the leaves, since for the roots and the stems the two softwares agreed on the best gene pairs (Fig. 1 and Table 1). As can be seen in Fig. 4, the expression trend in response to the different stresses did not change: different normalized relative expression values could be observed for a same time point between the NormFinder and geNormPLUS normalization (Fig. 4). In particular, higher error bars could be observed at some time points (e.g. 24 h heat, 72 h heat, 96 h heat) for the expression values obtained with NormFinder-based normalization (Fig. 4): this is most likely a reflection of the intrinsic computing differences of the two algorithms. However the Student’s t-test did not show statistically significant differences between the magnitude changes calculated by NormFinder and geNormPLUS (not shown).
Identification and phylogenetic analysis of CesAs from M. truncatula and M. sativa
In silico analysis of M. truncatula genome led to the identification of 9 putative CesA genes (Table 2). On the basis of the amino acid sequence identity with the orthologs from A. thaliana and poplar, a nomenclature is here proposed (Table 2) which follows the one recently proposed for Populus [46]. M. truncatula CESAs are between 981 and 1098 amino acids long and show from 6 to 8 transmembrane domains (TMDs; Table S3) according to the parameters of TMHMM [47]. The CESAs showing 6 TMDs actually display the occurrence of 2 additional potential TMDs, which however do not reach the critical threshold of the software (not shown). Therefore, since the CESAs so far described typically show the occurrence of 8 TMDs, the alfalfa proteins might as well all share the same feature. All the proteins show the occurrence of the signature motif typical of processive glycosyltransferases from family 2 (GT2s), i.e. D, D, DxD, QxxRW; MtCESA6-F, however, shows amino acid substitutions in the conserved motif (Fig. S3). The genes also have the zinc-finger domain (CxxC)4 (Fig. S3). Other genes with amino acid substitutions in the processive GT2s motif have been classified as CESAs (i.e. in Cicer aretinum and Phaseolus vulgaris) [GenBank: XP_004499618.1, ESW20735.1](Fig. S3), moreover phylogenetic and blast analyses both classify MtCESA6-F as a putative CESA and assign it to the primary CESAs clade (Fig. 5). Therefore this gene was assigned to the CesA6 branch and retained for expression analysis.
Table 2. Proposed nomenclature for the CesA genes from M. truncatula based on amino acid identities with the orthologous proteins from A. thaliana and P. trichocarpa.
| Populus/Medicago %identity | P. trichocarpa | M. truncatula | A. thaliana | Arabidopsis/Medicago %identity |
| 87 | PtiCesA1-A estExt_fgenesh4_pm.C_LG_XVIII0125 | MtCesA1 Medtr3g107520 | AtCesA1/RSW1 AT4G32410 | 84 |
| 87 | PtiCesA1-B fgenesh4_pg.C_LG_VI001789 | MtCesA1 Medtr3g107520 | AtCesA1/RSW1 AT4G32410 | 84 |
| 83 | PtiCesA3-A eugene3.00060479 | MtCesA3 Medtr3g030040 | AtCesA3/CEV1 AT5G05170 | 85 |
| 84 | PtiCesA3-B eugene3.00160483 | MtCesA3 Medtr3g030040 | AtCesA3/CEV1 AT5G05170 | 85 |
| 88 | PtiCesA3-C estExt_fgenesh4_pg.C_LG_IX0979 | MtCesA3 Medtr3g030040 | AtCesA3/CEV1 AT5G05170 | 85 |
| 88 | PtiCesA3-D estExt_Genewise1_v1.C_LG_I1792 | MtCesA3 Medtr3g030040 | AtCesA3/CEV1 AT5G05170 | 85 |
| 84 | PtiCesA4 eugene3.00002636 | MtCesA4 Medtr2g035780 | AtCesA4/IRX5 AT5G44030 | 77 |
| 87 | PtiCesA6-B estExt_fgenesh4_pg.C_LG_VII0650 | MtCesA6-B Medtr8g092590 | AtCesA6/IXR2/PRC1 AT5G64740 | 82 |
| 81 | PtiCesA6-C estExt_fgenesh4_pg.C_LG_V1107 | MtCesA6-C Medtr1g098550 | AtCesA6/IXR2/PRC1 AT5G64740 | 74 |
| 68 | PtiCesA6-F fgenesh4_pg.C_scaffold_133000012 | MtCesA6-F Medtr3g007770 | AtCesA6/IXR2/PRC1 AT5G64740 | 62 |
| 87 | PtiCesA7-A estExt_Genewise1_v1.C_LG_VI2188 | MtCesA7-A Medtr4g130510 | AtCesA7/IRX3 AT5G17420 | 85 |
| 63 | PtiCesA7-B gw1.XVIII.3152.1 | MtCesA7-B Medtr8g063270 | AtCesA7/IRX3 AT5G17420 | 64 |
| 80 | PtiCesA8-A gw1.XI.3218.1 | MtCesA8 Medtr8g086600 | AtCesA8/IRX1 AT4G18780 | 76 |
| 77 | PtiCesA8-B eugene3.00040363 | MtCesA8 Medtr8g086600 | AtCesA8/IRX1 AT4G18780 | 76 |
Loci are as reported in the Phytozome web portal [25].
Figure 5. Phylogenetic relationships of CESAs from M. truncatula, M. sativa, P. trichocarpa and A. thaliana by maximum-likelihood analysis.

Bootstrap = 100. Numbers indicate percentage of branch support values. The scale bar indicates an evolutionary distance of 0.2 amino acid substitutions per positions. Mesotaenium caldariorum CESA1 [GenBank: AAM83096] was used as outgroup to root the tree. The branch of secondary CESAs is indicated in blue. Arrows point to the three full-length CESAs identified in M. sativa.
Phylogenetic analysis showed the occurrence of 6 CESA clades with proteins involved in primary and secondary cell wall biosynthesis (Fig. 5). MtCESA1, MtCESA3, MtCESA6-B, MtCESA6-C, MtCESA6-F belong to the primary cell wall clade, while MtCESA4, MtCESA7-A, MtCESA7-B and MtCESA8 belong to the secondary cell wall clade (Fig. 5). Although MtCESA7-B and MtCESA6-F show the lowest % identity (Table 2), the phylogenetic tree classifies them as representatives of the CESA6 and CESA7 clade respectively (Fig. 5). The branches relative to these genes correspond to higher evolutionary distance (Fig. 5), a finding, which might indicate different roles with respect to their paralogs. Nevertheless the branch support values for the CESA6-E/F and CESA7-A/B clades are high (98 and 100%, respectively; Fig. 5).
The phylogenetic analysis shows, as expected, that orthologous genes from different species are more related than homologs from the same species [48]. Some of the identified CESAs are represented by different genes in M. truncatula. Three orthologs of AtCESA6 are present: these genes might display specific roles in primary cell wall biosynthesis, but it is possible that they participate in secondary cell wall biosynthesis too, since poplar CESA6-E and CESA6-F were shown to be part of one of the two types of complexes found in differentiating xylem [49]. The CESA6 members group together with the A. thaliana CESA2, CESA5, CESA9: this finding reflects their possible interchangeability in the primary CESA complex [50]–[51]. In the secondary cell wall clade, the occurrence of 2 AtCESA7 orthologs is observed. This is especially interesting if one considers that the presence of 2 CesA7 and CesA8 genes is a reported feature for woody angiosperms such as poplar, where the biosynthesis of wood represents an important process. Further functional characterizations are necessary to unveil the role of the 2 CESA7 in M. truncatula. However, in the light of the specialization and promiscuity that the different CESAs display, e.g. mucilage or seed coat biosynthesis [52]–[54], involvement in both primary and secondary cell wall biosynthesis or formation of mixed complexes [51],[55]–[56], it is plausible to hypothesize that these 2 proteins co-participate in the assembly of secondary wall complexes and/or possess specific functions in cell wall biosynthesis. The tissue-specific expression of M. truncatula CesAs obtained from publicly available microarray data [31] confirmed the annotation of the genes into the primary and secondary clades: as can be seen from Fig. 6, the primary CesAs show a homogeneous expression in roots, leaves and stems, while the secondary display a higher expression in the stems. Notably the primary CesAs MtCesA3 and MtCesA1 show a high level of expression in the stems (Fig. 6 and Fig. S4), a finding which suggests a role for these genes in alfalfa stem cell wall biosynthesis.
Figure 6. Radar plots of M. truncatula CesAs obtained plotting the microarray data retrieved at [31].
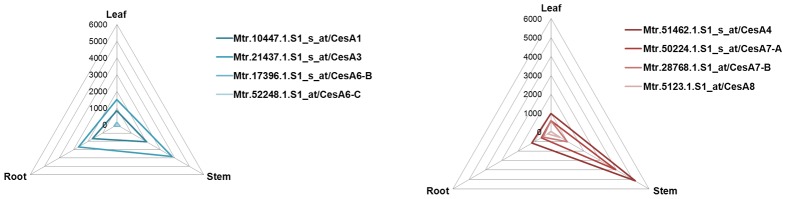
Three full-length CesA sequences from M. sativa have been here obtained [GenBank: KJ398155, KJ398156, KJ398157; Fig. S1]; the phylogenetic analysis classifies them as MsCESA3, MsCESA4 and MsCESA7-A (Fig. 5). Partial sequences have been obtained for the other CesAs of M. sativa (Figs. S1 and S2).
Cell wall-related genes from M. sativa show two main trends in response to abiotic stresses
Variations in the expression pattern in response to abiotic stresses can be observed among the different cell wall-related genes. From the Heat Map visualization, it is possible to discern two main groups: a heat/salt-induced and a cold/heat-repressed group of cell wall genes (Fig. 7). Salt/heat-induced genes are represented by the primary CesAs MsCesA1, MsCesA3, MsCesA6-B (with a Pearson correlation coefficient of 0.883) and to this group CAD belongs too (although with a lower correlation coefficient of 0.690). CslD4 and PAL are also assigned to this group, although they cluster in a different branch, as their trend is less sharp than the one observed for primary CesAs (Fig. 7). The cold/heat-repressed group is represented by the secondary CesAs, together with SuSy (correlation coefficient of 0.91 for the cluster SuSy, MsCesA4 and MsCesA7-A, and of 0.93 for MsCesA4 and MsCesA7-B). The hierarchical clustering assigns to this group also MsCesA6-C and MsCesA6-F (Fig. 7). The statistical analyses carried out on the RT-qPCR data (Fig. S5) reveal that the changes in expression for MsCesA1 and MsCesA6-F are statistically not significant; however their expression patterns can be interpreted as an overall trend which enables their classification in the heat/salt-induced and cold/heat-repressed group, respectively (Fig. 7).
Figure 7. Heat Map representation of the data in Fig. S5 showing the hierarchical clustering of cell wall-related genes in response to the abiotic stresses at the different time points in alfalfa stems.

The data collected refer to 3 independent biological replicates, each consisting of a pool of 15 plants. For each stress treatment a control group was always kept for the 24-48 h-96 h time points, for appropriate comparisons. The group clustering was generated with Cluster 3.0 [38] and visualized with Java TreeView [39], as described in Material and Methods.
A more detailed analysis of MsCesAs expression profiles shows mild but significant change for MsCesA3, with respect to the control, in response to salt stress after 96 h (Fig. S5; Table S4). MsCesA6-B displays an increase in expression at late stages of heat and salt application, which reaches a maximum after 96 h of treatment (Fig. S5; Table S4). MsCesA6-C shows a noteworthy decrease after 24 and 72 h of cold stress treatment (Fig. S5; Table S4).
MsCesA4, MsCesA7-A and MsCesA7-B show a trend towards decrease in expression already after 24 h of heat stress, while CesA8 responds later, 72 h after the application of the stress (Fig. S5; Table S5). The correlation analysis of the wall-related genes performed with qBasePLUS revealed a strong correlation between MsCesA4 and MsCesA7-A in all the conditions tested (Fig. S6). This is not surprising, since these two genes belong both to the secondary CESAs clade, they are necessary for secondary cell wall biosynthesis together with CESA8 [57] and have been shown to interact in Arabidopsis [56].
Discussion
The use of RT-qPCR for gene expression studies is a tool of unanimously recognized value, even in the current scientific era marked by the next generation sequencing revolution. Its utility is indeed unquestionable and necessary for validation of results massively produced via high-throughput methods.
For relative gene expression studies using RT-qPCR, the selection of suitable reference genes is a factor of paramount importance. Several studies in the literature have already undertaken the analysis of a set of candidate reference genes for normalization strategies in different plant species and conditions. Lists of stable genes are already available for relative RT-qPCR studies in plant tissues; however it is important to check their suitability in the experimental set-up adopted.
We have here identified and validated the use of reference genes for expression studies in alfalfa plants under different abiotic stresses. Two well-known and widely-used softwares, geNormPLUS [2] and NormFinder [33], have been chosen to rank the stability of the selected genes and we show that for some tissues, the best gene pairs identified differed between the 2 methods (Fig. 1 and Table 1). However, we were able to identify and propose a set of reference genes, ranked among the most stable by both softwares, namely eIF4A, PAB4, ADF1 and TFIIA. These genes can therefore be included in a panel of candidates to be tested for RT-qPCR studies in alfalfa and, potentially, in other leguminous plants.
For the validation phase, we have used as a model gene a plant kinase, SK1, known for its susceptibility to stresses [44] and we show that the response pattern is similar in the different tissues, where cold and heat stress cause the most pronounced responses, namely reduction and increase of expression, respectively (Fig. 3).
We have subsequently extended our RT-qPCR study to cell wall biosynthetic genes in stems, since our efforts are currently devoted towards understanding the regulation of cell wall biosynthesis dynamics in stems of alfalfa plants. In particular we here show the expression of nine putative CesAs, belonging to both primary and secondary wall clades (Fig. 5), together with other wall-related genes (Fig. 7). Although several reports in the literature have shown a link between cell wall biosynthesis/modification and abiotic stresses [58]–[61], a detailed investigation of cell wall gene expression changes in response to different abiotic stresses is lacking.
The main finding of our investigation is the elucidation of the wall-related gene dynamism in alfalfa plants subjected to abiotic stresses. The hierarchical clustering analysis identified two main trends in response to abiotic stresses: a salt/heat-induced and a cold/heat-repressed group of genes. Interestingly, a gene known to be involved in lignin biosynthesis, CAD, grouped together with the primary CesAs MsCesA1, MsCesA3 and MsCesA6-B (Fig. 7): this indicates that these genes, although not strictly related, show a common response mechanism to abiotic stress. In this respect it should be noted that induction of a peroxidase, triggering in its turn an increase in lignin and suberin deposition, has been reported in tomato plants exposed to salt stress [62] and that tomato plants under salt stress show an increased number of lignified cells [63]. In addition to this, a link between miRNAs, abiotic stresses and lignification has been unveiled in A. thaliana, as miR397b, a miRNA targeting a laccase (and consequently affecting lignification), was shown to be up-regulated in response to salt stress [64]–[65]. PAL and CslD4 also clustered with the CAD-primary CesAs group, although with a lower correlation: both genes display a heat and salt-stress responsive trend at later stages of treatment (Fig. 7; Fig. S5; Table S6). Cellulose synthase-like genes belong to the CESA superfamily and several members involved in wall glycan biosynthesis have been identified [66]. Many members of the Csl group of genes have not yet been functionally characterized, however representatives of the CslD clade are required for tip-growing cells [67] and CslD1 and CslD4 have been shown to affect cellulose biosynthesis in pollen tubes [68]. Moreover, another member of the CslD clade, CslD5, was shown to be required for osmotic stress tolerance in A. thaliana [60]. The results shown by the hierarchical clustering (Fig. 7) suggest that PAL and CslD4 might be involved in cell wall remodeling in response to heat stress in alfalfa stems in a pathway likely involving increased lignin biosynthesis and cellulose deposition to strengthen the wall under the adverse condition. Heat stress triggers substantial modifications in plants: changes in ultrastructural anatomy and cell wall polysaccharide composition have been observed in coffee leaves subjected to heat stress, with an increase in monolignol content [69].
The susceptibility of primary CesAs to exogenous stresses is a known feature: the A. thaliana cev1/CesA3 mutant shows constitutive expression of stress responsive genes, together with an increased resistance to fungal attack [70].
The second group of genes identified by the hierarchical clustering is represented by the secondary CesAs together with SuSy, MsCesA6-C and MsCesA6-F (Fig. 5). MsCesA6-C and MsCesA6-F belong to the primary CesAs and it is interesting that these genes cluster with secondary CesAs. This might indicate that, as already discussed for CAD, a similar response mechanism exists between these genes and the secondary CesAs, or it can indirectly show that they are more functionally related to secondary CesAs. This needs verification, however the presence of multiple CesA6 genes in alfalfa might indicate overlapping and/or distinct roles in cell wall biosynthesis.
The second group of genes identified by the hierarchical clustering shows down-regulation in response to cold and heat stress. Heat stress is known to cause a decreased expression of SuSy in pollen grains (accompanied by a decrease in the expression of other wall-related genes and vacuolar invertases; [71]) and in chickpea leaves [72]. The RT-qPCR analysis performed on alfalfa stems suggests that the decrease observed in secondary CesAs expression upon cold and heat stress treatment might be related to an impaired fueling of UDP-glucose by SuSy, which, despite not strictly required for cellulose synthesis [73]–[74], might contribute to increase the rate of synthesis, by concentrating the substrate [74].
Conclusions
The present work constitutes a useful guide for the identification of appropriate reference genes in expression studies on alfalfa, which can be extended to other legume crops for analysis. Through analyses using NormFinder and geNormPLUS, we have identified a set of suitable candidates, which can be included in a panel of reference genes to be tested for differential expression analysis.
The results concerning CesAs and a few other wall-related genes confirm an active role played by the cell wall in response to exogenous stimuli and constitute a step forward in delineating the complex pathways fine-tuning the response of plants to abiotic stresses.
Supporting Information
Nucleotide sequences of MsCesAs. Sequence details of the CesAs identified in alfalfa.
(DOC)
Alignment of alfalfa partial CesA sequences with M. truncatula CesAs. Alignment of the CesAs from alfalfa with the respective orthologs from M. truncatula.
(DOC)
Sequence details of MtCESA6-F. Alignment of MtCESA6-F with CesAs from C. aretinum [GenBank: XP_004499618.1] and P. vulgaris [GenBank: ESW20735.1] showing the amino acid substitutions in the processive GT2s motif (bold and underlined). The zinc-finger domain (CxxC)4 is highlighted in yellow.
(DOC)
Electronic Fluorescence Pictographic (eFP) representations of M. truncatula CesA1, CesA3, CesA6-B, CesA6-C, CesA4, CesA7-A, CesA7-B, CesA8.
(TIF)
Gene expression profiles of cell wall-related genes in stems of alfalfa plants subjected to abiotic stress. Data were normalized using eif4A/TFIIA. Means sharing a letter are not significantly different at α = 0.05. NRE indicates Normalized Relative Expression.
(TIF)
MsCesA7-A and MsCesA4 relationship. Correlation between MsCesA7-A and MsCesA4 in stems under abiotic stress conditions. Pearson (log) r = 0.962; Spearman (log) r = 0.961.
(TIF)
List of primers used to amplify MsCesAs. Name of the primers, with the respective sequences, used to amplify the CesAs from M. sativa.
(DOC)
List of primers used for the RT-qPCR study. Name of the primers used for the RT-qPCR study, with the respective sequences. Details concerning the amplicons details (length, Tm), PCR efficiencies and regression coefficients are included.
(DOC)
CESAs from M. truncatula. Details concerning number of predicted transmembrane helices (TMHs, according to [47]) and the length of the putative CESAs from M. truncatula.
(DOC)
Normalized Relative Expression for primary CesAs. Normalized Relative Expression values ± standard deviation and significance (Sig.) for the primary CesAs. Data were normalized using eif4A/TFIIA.
(DOC)
Normalized Relative Expression for secondary CesAs. Normalized Relative Expression values ± standard deviation and significance (Sig.) for the secondary CesAs. Data were normalized using eif4A/TFIIA.
(DOC)
Normalized Relative Expression for for CAD, CslD4, PAL and SuSy. Normalized Relative Expression values ± standard deviation and significance (Sig.) for CAD, CslD4, PAL and SuSy. Data were normalized using eif4A/TFIIA.
(DOC)
Acknowledgments
The authors wish to thank Dr. Lucien Hoffmann for critical reading of the manuscript. Laurent Solinhac is acknowledged for technical assistance.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper.
Funding Statement
Financial support was obtained through the Fonds National de la Recherche Luxembourg (FNR) Project CANCAN C13/SR/5774202 and through internal funding sources. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Schmittgen TD, Lee EJ, Jiang J, Sarkar A, Yang L, et al. (2008) Real-time PCR quantification of precursor and mature microRNA. Methods 44: 31–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, et al. (2002) Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 3: RESEARCH0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, et al. (2009) The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem 55: 611–622. [DOI] [PubMed] [Google Scholar]
- 4. Prasch CM, Sonnewald U (2013) Simultaneous application of heat, drought, and virus to Arabidopsis plants reveals significant shifts in signaling networks. Plant Physiol 162: 1849–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sultan SE (2000) Phenotypic plasticity for plant development, function and life history. Trends Plant Sci 5: 537–542. [DOI] [PubMed] [Google Scholar]
- 6. Sultan SE (2003) Phenotypic plasticity in plants: a case study in ecological development. Evol Dev 5: 25–33. [DOI] [PubMed] [Google Scholar]
- 7. Puijalon S, Bornette G (2006) Phenotypic plasticity and mechanical stress: biomass partitioning and clonal growth of an aquatic plant species. Am J Bot 93: 1090–1099. [DOI] [PubMed] [Google Scholar]
- 8. Nicot N, Hausman JF, Hoffmann L, Evers D (2005) Housekeeping gene selection for real-time RT-PCR normalization in potato during biotic and abiotic stress. J Exp Bot 56: 2907–2914. [DOI] [PubMed] [Google Scholar]
- 9. Wan H, Zhao Z, Qian C, Sui Y, Malik AA, et al. (2010) Selection of appropriate reference genes for gene expression studies by quantitative real-time polymerase chain reaction in cucumber. Anal Biochem 399: 257–261. [DOI] [PubMed] [Google Scholar]
- 10. Gu C, Chen S, Liu Z, Shan H, Luo H, et al. (2011) Reference gene selection for quantitative real-time PCR in Chrysanthemum subjected to biotic and abiotic stress. Mol Biotechnol 49: 192–197. [DOI] [PubMed] [Google Scholar]
- 11. Le DT, Aldrich DL, Valliyodan B, Watanabe Y, Ha CV, et al. (2012) Evaluation of candidate reference genes for normalization of quantitative RT-PCR in soybean tissues under various abiotic stress conditions. PLoS One 7: e46487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Selim M, Legay S, Berkelmann-Löhnertz B, Langen G, Kogel KH, et al. (2012) Identification of suitable reference genes for real-time RT-PCR normalization in the grapevine-downy mildew pathosystem. Plant Cell Rep 31: 205–216. [DOI] [PubMed] [Google Scholar]
- 13. Zhu J, Zhang L, Li W, Han S, Yang W, et al. (2013) Reference gene selection for quantitative real-time PCR normalization in Caragana intermedia under different abiotic stress conditions. PLoS One 8: e53196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kakar K, Wandrey M, Czechowski T, Gaertner T, Scheible WR, et al. (2008) A community resource for high-throughput quantitative RT-PCR analysis of transcription factor gene expression in Medicago truncatula . Plant Methods 4: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yang SS, Tu ZJ, Cheung F, Xu WW, Lamb JF, et al. (2011) Using RNA-Seq for gene identification, polymorphism detection and transcript profiling in two alfalfa genotypes with divergent cell wall composition in stems. BMC Genomics 12: 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pini F, Frascella A, Santopolo L, Bazzicalupo M, Biondi EG, et al. (2012) Exploring the plant-associated bacterial communities in Medicago sativa L. BMC Microbiol. 12: 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tesfaye M, Yang SS, Lamb JFS, Jung H-JG, Samac DA, et al. (2009) Medicago truncatula as a model for dicot cell wall development. Bioenergy Res 2: 59–76. [Google Scholar]
- 18. Young ND, Debellé F, Oldroyd GE, Geurts R, Cannon SB, et al. (2011) The Medicago genome provides insight into the evolution of rhizobial symbioses. Nature 480: 520–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang SS, Xu WW, Tesfaye M, Lamb JF, Jung HJ, et al. (2010) Transcript profiling of two alfalfa genotypes with contrasting cell wall composition in stems using a cross-species platform: optimizing analysis by masking biased probes. BMC Genomics 11: 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Huis R, Hawkins S, Neutelings G (2010) Selection of reference genes for quantitative gene expression normalization in flax (Linum usitatissimum L.). BMC Plant Biol 10: 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Humphrey TV, Bonetta DT, Goring DR (2007) Sentinels at the wall: cell wall receptors and sensors. New Phytol 176: 7–21. [DOI] [PubMed] [Google Scholar]
- 22. Seifert GJ, Blaukopf C (2010) Irritable walls: the plant extracellular matrix and signaling. Plant Physiol 153: 467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hamann T, Denness L (2011) Cell wall integrity maintenance in plants: lessons to be learned from yeast? Plant Signal Behav 6: 1706–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hamann T (2012) Plant cell wall integrity maintenance as an essential component of biotic stress response mechanisms. Front Plant Sci 3: 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Phytozome website. Available: www.phytozome.com. Accessed 2014 July 10.
- 26.National Center for Biotechnology Information (NCBI) website. Available: http://www.ncbi.nlm.nih.gov/. Accessed 2014 July 10.
- 27. Carroll A, Specht CD (2011) Understanding plant cellulose synthases through a comprehensive investigation of the cellulose synthase family sequences. Front Plant Sci 2: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32: 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Guindon S, Gascuel O (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52: 696–704. [DOI] [PubMed] [Google Scholar]
- 30. Chevenet F, Brun C, Bañuls AL, Jacq B, Christen R (2006) TreeDyn: towards dynamic graphics and annotations for analyses of trees. BMC Bioinformatics 7: 439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Medicago truncatula Gene Expression Atlas website. Available: http://mtgea.noble.org/v3/. Accessed 2014 July 10.
- 32. Medicago electronic fluorescent pictographic (eFP) representations website. Available: http://bar.utoronto.ca/efpmedicago/cgi-bin/efpWeb.cgi. Accessed 2014 July 10.
- 33. Andersen CL, Jensen JL, Ørntoft TF (2004) Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res 64: 5245–5250. [DOI] [PubMed] [Google Scholar]
- 34. Sørby LA, Andersen SN, Bukholm IR, Jacobsen MB (2010) Evaluation of suitable reference genes for normalization of real-time reverse transcription PCR analysis in colon cancer. J Exp Clin Cancer Res 29: 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Untergasser A, Nijveen H, Rao X, Bisseling T, Geurts R, et al. (2007) Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res 35: W71–W74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.OligoAnalyzer 3.1 website. Available: http://eu.idtdna.com/analyzer/Applications/OligoAnalyzer/. Accessed 2014 July 10.
- 37. Hellemans J, Mortier G, De Paepe A, Speleman F, Vandesompele J (2007) qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol 8: R19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Eisen MB, Spellman PT, Brown PO, Botstein D (1998) Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A 95: 14863–14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Saldanha AJ (2004) Java Treeview-extensible visualization of microarray data. Bioinformatics 20: 3246–3248. [DOI] [PubMed] [Google Scholar]
- 40.Java TreeView website. Available: http://jtreeview.sourceforge.net/. Accessed 2014 July 10.
- 41. Jian B, Liu B, Bi YR, Hou WS, Wu CX, et al. (2008) Validation of internal control for gene expression study in soybean by quantitative real-time PCR. BMC Mol Biol 9: 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. González-Verdejo CI, Die JV, Nadal S, Jiménez-Marín A, Moreno MT, et al. (2008) Selection of housekeeping genes for normalization by real-time RT-PCR: analysis of Or-MYB1 gene expression in Orobanche ramosa development. Anal Biochem 379: 176–181. [DOI] [PubMed] [Google Scholar]
- 43. Guénin S, Mauriat M, Pelloux J, Van Wuytswinkel O, Bellini C, et al. (2009) Normalization of qRT-PCR data: the necessity of adopting a systematic, experimental conditions-specific, validation of references. J Exp Bot 60: 487–493. [DOI] [PubMed] [Google Scholar]
- 44. Nolan KE, Saeed NA, Rose RJ (2006) The stress kinase gene MtSK1 in Medicago truncatula with particular reference to somatic embryogenesis. Plant Cell Rep 25: 711–722. [DOI] [PubMed] [Google Scholar]
- 45. Coello P, Hey SJ, Halford NG (2011) The sucrose non-fermenting-1-related (SnRK) family of protein kinases: potential for manipulation to improve stress tolerance and increase yield. J Exp Bot 62: 883–893. [DOI] [PubMed] [Google Scholar]
- 46. Kumar M, Thammannagowda S, Bulone V, Chiang V, Han KH, et al. (2009) An update on the nomenclature for the cellulose synthase genes in Populus . Trends Plant Sci 14: 248–254. [DOI] [PubMed] [Google Scholar]
- 47.TMHMM website. Available: http://www.cbs.dtu.dk/services/TMHMM/. Accessed 2014 July 10.
- 48. Handakumbura PP, Matos DA, Osmont KS, Harrington MJ, Heo K, et al. (2013) Perturbation of Brachypodium distachyon CELLULOSE SYNTHASE A4 or 7 results in abnormal cell walls. BMC Plant Biol 13: 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Song D, Shen J, Li L (2010) Characterization of cellulose synthase complexes in Populus xylem differentiation. New Phytol 187: 777–790. [DOI] [PubMed] [Google Scholar]
- 50. Persson S, Paredez A, Carroll A, Palsdottir H, Doblin M, et al. (2007) Genetic evidence for three unique components in primary cell-wall cellulose synthase complexes in Arabidopsis . Proc Natl Acad Sci U S A 104: 15566–15571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Betancur L, Singh B, Rapp RA, Wendel JF, Marks MD, et al. (2010) Phylogenetically distinct cellulose synthase genes support secondary wall thickening in Arabidopsis shoot trichomes and cotton fiber. J Integr Plant Biol 52: 205–220. [DOI] [PubMed] [Google Scholar]
- 52. Stork J, Harris D, Griffiths J, Williams B, Beisson F, et al. (2010) CELLULOSE SYNTHASE9 serves a nonredundant role in secondary cell wall synthesis in Arabidopsis epidermal testa cells. Plant Physiol 153: 580–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sullivan S, Ralet MC, Berger A, Diatloff E, Bischoff V, et al. (2011) CESA5 is required for the synthesis of cellulose with a role in structuring the adherent mucilage of Arabidopsis seeds. Plant Physiol 156: 1725–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mendu V, Griffiths JS, Persson S, Stork J, Downie AB, et al. (2011) Subfunctionalization of cellulose synthases in seed coat epidermal cells mediates secondary radial wall synthesis and mucilage attachment. Plant Physiol 157: 441–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Carroll A, Mansoori N, Li S, Lei L, Vernhettes S, et al. (2012) Complexes with mixed primary and secondary cellulose synthases are functional in Arabidopsis plants. Plant Physiol 160: 726–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Li S, Lei L, Gu Y (2013) Functional analysis of complexes with mixed primary and secondary cellulose synthases. Plant Signal Behav 8: e23179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Taylor NG, Howells RM, Huttly AK, Vickers K, Turner SR (2003) Interactions among three distinct CesA proteins essential for cellulose synthesis. Proc Natl Acad Sci U S A 100: 1450–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pilling E, Höfte H (2003) Feedback from the wall. Curr Opin Plant Biol 6: 611–616. [DOI] [PubMed] [Google Scholar]
- 59. Moura JC, Bonine CA, de Oliveira FVJ, Dornelas MC, Mazzafera P (2010) Abiotic and biotic stresses and changes in the lignin content and composition in plants. J Integr Plant Biol 52: 360–376. [DOI] [PubMed] [Google Scholar]
- 60. Zhu J, Lee BH, Dellinger M, Cui X, Zhang C, et al. (2010) A cellulose synthase-like protein is required for osmotic stress tolerance in Arabidopsis . Plant J 63: 128–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wolf S, Mravec J, Greiner S, Mouille G, Höfte H (2012) Plant cell wall homeostasis is mediated by brassinosteroid feedback signaling. Curr Biol 22: 1732–1737. [DOI] [PubMed] [Google Scholar]
- 62. Quiroga M, Guerrero C, Botella MA, Barceló A, Amaya I, et al. (2000) A tomato peroxidase involved in the synthesis of lignin and suberin. Plant Physiol 122: 1119–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sánchez-AguayoI, Rodríguez-Galán JM, García R, Torreblanca J, Pardo JM (2004) Salt stress enhances xylem development and expression of S-adenosyl-L-methionine synthase in lignifying tissues of tomato plants. Planta 220: 278–285. [DOI] [PubMed] [Google Scholar]
- 64. Sunkar R, Zhu JK (2004) Novel and stress-regulated microRNAs and other small RNAs from Arabidopsis . Plant Cell 16: 2001–2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Martin RC, Liu PP, Goloviznina NA, Nonogaki H (2010) microRNA, seeds, and Darwin?: diverse function of miRNA in seed biology and plant responses to stress. J Exp Bot 61: 2229–2234. [DOI] [PubMed] [Google Scholar]
- 66. Yin Y, Huang J, Xu Y (2009) The cellulose synthase superfamily in fully sequenced plants and algae. BMC Plant Biol 9: 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bernal AJ, Yoo CM, Mutwil M, Jensen JK, Hou G, et al. (2008) Functional analysis of the cellulose synthase-like genes CSLD1, CSLD2, and CSLD4 in tip-growing Arabidopsis cells. Plant Physiol 148: 1238–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wang W, Wang L, Chen C, Xiong G, Tan XY, et al. (2011) Arabidopsis CSLD1 and CSLD4 are required for cellulose deposition and normal growth of pollen tubes. J Exp Bot 62: 5161–5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lima RB, dos Santos TB, Vieira LG, Ferrarese Mde L, Ferrarese-Filho O, et al. (2013) Heat stress causes alterations in the cell-wall polymers and anatomy of coffee leaves (Coffea arabica L.). Carbohydr Polym 93: 135–143. [DOI] [PubMed] [Google Scholar]
- 70. Ellis C, Karafyllidis I, Wasternack C, Turner JG (2002) The Arabidopsis mutant cev1 links cell wall signaling to jasmonate and ethylene responses. Plant Cell 14: 1557–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bita CE, Gerats T (2013) Plant tolerance to high temperature in a changing environment: scientific fundamentals and production of heat stress-tolerant crops. Front Plant Sci 4: 273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kaushal N, Awasthi R, Gupta K, Gaur P, Siddique KHM, et al. (2013) Heat-stress-induced reproductive failures in chickpea (Cicer arietinum) are associated with impaired sucrose metabolism in leaves and anthers. Funct Plant Biol 40: 1334–1349. [DOI] [PubMed] [Google Scholar]
- 73. Barratt DHP, Derbyshire P, Findlay K, Pike M, Wellner N, et al. (2009) Normal growth of Arabidopsis requires cytosolic invertase but not sucrose synthase. Proc Natl Acad Sci U S A 106: 13124–13129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Carpita NC (2011) Update on mechanisms of plant cell wall biosynthesis: how plants make cellulose and other (1->4)-β-D-glycans. Plant Physiol 155: 171–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Nucleotide sequences of MsCesAs. Sequence details of the CesAs identified in alfalfa.
(DOC)
Alignment of alfalfa partial CesA sequences with M. truncatula CesAs. Alignment of the CesAs from alfalfa with the respective orthologs from M. truncatula.
(DOC)
Sequence details of MtCESA6-F. Alignment of MtCESA6-F with CesAs from C. aretinum [GenBank: XP_004499618.1] and P. vulgaris [GenBank: ESW20735.1] showing the amino acid substitutions in the processive GT2s motif (bold and underlined). The zinc-finger domain (CxxC)4 is highlighted in yellow.
(DOC)
Electronic Fluorescence Pictographic (eFP) representations of M. truncatula CesA1, CesA3, CesA6-B, CesA6-C, CesA4, CesA7-A, CesA7-B, CesA8.
(TIF)
Gene expression profiles of cell wall-related genes in stems of alfalfa plants subjected to abiotic stress. Data were normalized using eif4A/TFIIA. Means sharing a letter are not significantly different at α = 0.05. NRE indicates Normalized Relative Expression.
(TIF)
MsCesA7-A and MsCesA4 relationship. Correlation between MsCesA7-A and MsCesA4 in stems under abiotic stress conditions. Pearson (log) r = 0.962; Spearman (log) r = 0.961.
(TIF)
List of primers used to amplify MsCesAs. Name of the primers, with the respective sequences, used to amplify the CesAs from M. sativa.
(DOC)
List of primers used for the RT-qPCR study. Name of the primers used for the RT-qPCR study, with the respective sequences. Details concerning the amplicons details (length, Tm), PCR efficiencies and regression coefficients are included.
(DOC)
CESAs from M. truncatula. Details concerning number of predicted transmembrane helices (TMHs, according to [47]) and the length of the putative CESAs from M. truncatula.
(DOC)
Normalized Relative Expression for primary CesAs. Normalized Relative Expression values ± standard deviation and significance (Sig.) for the primary CesAs. Data were normalized using eif4A/TFIIA.
(DOC)
Normalized Relative Expression for secondary CesAs. Normalized Relative Expression values ± standard deviation and significance (Sig.) for the secondary CesAs. Data were normalized using eif4A/TFIIA.
(DOC)
Normalized Relative Expression for for CAD, CslD4, PAL and SuSy. Normalized Relative Expression values ± standard deviation and significance (Sig.) for CAD, CslD4, PAL and SuSy. Data were normalized using eif4A/TFIIA.
(DOC)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper.