

Abstract
The phosphorylation state of heptapeptide repeats within the C-terminal domain (CTD) of the largest subunit of RNA Polymerase II (PolII) controls the transcription cycle and is maintained by the competing action of kinases and phosphatases. Rtr1 was recently proposed to be the enzyme responsible for the transition of PolII into the elongation and termination phases of transcription by removing the phosphate marker on Serine 5, but this attribution was questioned by the apparent lack of enzymatic activity. Here we demonstrate that Rtr1 is a phosphatase of new structure that is auto-inhibited by its own C-terminus. The enzymatic activity of the protein in vitro is functionally important in vivo as well: a single amino acid mutation that reduces activity leads to the same phenotype in vivo as deletion of the protein-coding gene from yeast. Surprisingly, Rtr1 dephosphorylates not only Serine 5 on the CTD, but also the newly described anti-termination Tyrosine 1 marker, supporting the hypothesis that Rtr1 and its homologs promote the transition from transcription to termination.
Introduction
The phosphorylation state of the C-terminal domain of RNA Polymerase II (PolII) Rpb1 subunit controls transcription1. The CTD consists of a highly conserved heptapeptide (Y1S2P3T4S5P6S7) repeated between 26 times in Saccharomyces cerevisiae and 52 times in humans. Ser2 and Ser5 are reversibly phosphorylated, while the prolines are subject to cis-trans isomerization facilitated by isomerases such as Ess12–6. In addition, the Tyr1, Thr4 and Ser7 residues can also be phosphorylated, although the impact and scope of these modifications is less well understood7–9. The dynamic combination of post-translational modifications constitutes a ‘CTD code’ which helps recruit or activate various factors to the polymerase during the transcription cycle10–12.
High levels of phosphorylation of Ser5 (Ser5P) on the CTD occur at or near the promoter and help recruit mRNA capping and transcription elongation factors13–15. This modification can also act as a signal for the snoRNA/snRNA termination pathway via the Nrd1-Nab3-Sen1 complex in yeasts16. Ser5P is progressively dephosphorylated as the polymerase progresses into the elongation and termination phases of transcription. In contrast, Ser2 phosphorylation (Ser2P) levels are low at the start of transcription and increase as the polymerase moves along a gene, where this modification signals the recruitment and/or activation of transcription termination factors17–19.
Multiple Ser2/5 kinases and phosphatases have been identified1, but the identity of the phosphatase responsible for the critical transition from Ser5P to Ser2P during transcriptional elongation remains unclear. Yeast Rtr1, a highly conserved protein in all eukaryotes (Figure S1), was recently proposed to be the Ser5P phosphatase responsible for this transition20, a hypothesis further supported by the independent observation that its human orthologue (RPAP2) has phosphatase activity with identical selectivity profile: active on Ser5P, but not upon Ser2P nor Ser7P20,21. However, this attribution was negated by the lack of in vitro phosphatase activity in Kluyveromyces lactis Rtr1, whose crystal structure also failed to reveal a canonical active site observed in other phosphatases22. It was proposed that the phosphatase activity detected for Rtr1 might arise from the co-purification of an E. coli phosphatase enzyme, although it would appear unlikely that the accidental presence of a recombinant protein from bacterial sources would yield an enzyme that selectively dephosphorylates a substrate without an equivalent in bacteria.
Here we resolve this controversy by reporting that Rtr1 is active as a phosphatase and that its enzymatic activity is functional: mutation in a single absolutely conserved residue that significantly reduces catalytic activity in vitro also abolishes its function in vivo. We further show that Rtr1 can target and dephosphorylate PolII CTD repeats carrying both Ser5P and the newly described anti-termination Tyr1 phosphorylation marker, providing additional evidence that Rtr1 is the phosphatase that promotes the transition from initiation to the elongation and termination phases of transcription.
Results
Rtr1 is a phosphatase
We independently determined the crystal structure of the K. lactis Rtr1 (KlRtr1) NTD (amino acids 1-156, Table 1), which is nearly identical to the previously determined structure22 (Cα RMSD = 0.35Å) (Figure 1A). Purification of the full-length KlRtr1 protein using standard protocols (Figure 1B, upper flow) resulted in preparations that lacked activity when assayed against both phosphorylated GST-CTD (data not shown) and the acid phosphatase substrate 6,8-difluoro-4-methylumbelliferyl phosphate (DiFMUP) (Figure 1C), a classical phosphatase substrate. However, a closer examination of purification protocols in light of reports that some phosphatases are inhibited by very low concentrations of divalent metal ions23, prompted us to consider the possibility that activity was abolished by an inhibitory metal. Thus, we re-purified KlRtr1 with just one additional step: washing the protein with EDTA prior to the final gel filtration step (Figure 1B, lower flow). This EDTA-treated protein exhibited robust activity against the phosphatase substrate (Figure 1C) and the GST-CTD (see below). Careful quantitation of the protein samples used in the assays by various methods, including SDS-PAGE, demonstrates that equal amounts of proteins were used, suggesting that activity is intrinsic to the EDTA treated sample (Figure S2A).
Table 1.
Crystal data collection and refinement statistics
| KlRtr1 SeMet | KlRtr1 NTD nativea | ScRtr1 full-length | |
|---|---|---|---|
| Data collection | |||
| Space group | P212121 | P212121 | P213 |
| Cell dimensions | |||
| a, b, c (Å) | 45.39, 103.35, 105.39 | 43.27, 88.92, 102.49 | 170.75, 170.75, 170.75 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 |
| Resolution (Å) | 50–3.19 (3.30–3.19)b | 50–2.06 (2.10–2.06) | 50–4.00 (4.07–4.00) |
| Rsym or Rmerge | 0.097 (0.620) | 0.069 (0.612) | 0.101 (0.540) |
| I/σI | 25.0 (3.9) | 36.2 (3.1) | 24.4 (3.9) |
| Completeness (%) | 99.9 (100.0) | 99.9 (100.0) | 100.0 (100.0) |
| Redundancy | 13.4 (12.1) | 5.6 (4.8) | 11.0 (11.2) |
| Refinement | |||
| Resolution (Å) | 44.44–2.10 | ||
| No. reflections | 22581 | ||
| Rwork/Rfree | 0.202/0.253 | ||
| No. atoms | |||
| Protein | 2,355 | ||
| Ligand/ion | 2 | ||
| Water | 137 | ||
| B-factors | |||
| Protein | 46.3 | ||
| Ligand/ion | 35.7 | ||
| Water | 64.7 | ||
| R.m.s. deviations | |||
| Bond lengths (Å) | 0.018 | ||
| Bond angles (°) | 2.015 | ||
Crystals were grown from an enzymatically active protein preparation. Final structure refinement was executed on an active protein sample.
Values in parentheses indicate highest-resolution shell.
Figure 1. Rtr1 is an active phosphatase.

a) Crystal structure of KlRtr1 NTD (left, cyan). The dotted line outlines a flexible loop unseen in the electron density maps. The previously reported crystal structure KlRtr1’s NTD (PDB 4FC8) is shown on the right (yellow); an overlay of both structures is in the center.
b) Purification scheme adopted in this study to obtain active recombinant Rtr1; the determining step in the purification is highlighted in red.
c) Steady state phosphatase assays (n = 3) (with DiFMUP substrate) performed using KlRtr1 proteins (10μM) obtained from both purification schemes (+EDTA in black, −EDTA in red), and the E66Q mutant (blue).
d) Inhibition experiments (n = 3) performed using 10μM KlRtr1 and 1mM DiFMUP against two traditional competitive phosphatase inhibitors (vanadate in blue, BGP in red).
e) Sequence alignment of Rtr1 (numbering based on K. lactis) across yeasts and vertebrates highlighting the strictly conserved Glu66 residue (asterisked).
f) Fitness of yeast cells lacking either Rtr1 and/or its paralog Rtr2 at 37°C. A single activity disrupting mutant (E66A) was integrated into the chromosomal Rtr1 locus with a URA3 marker for selection. The phenotypes observed with the single mutant are comparable to those observed when the Rtr1 protein is deleted.
Since a simple treatment with EDTA yielded an active protein, we performed the assay in the presence of Mg, Ni and Ca (Figure S2B) but none of these metals had significant effects on the activity of the protein. However, the enzymatic activity is sensitive to specific phosphatase inhibitors. The classical competitive phosphatase inhibitor orthovanadate inhibited KlRtr1 with an inhibition constant Ki of approximately 0.8μM (Figure 1D), while the inhibitor β-glycerophosphate (BGP) had little effect at similar concentrations. Exhaustive efforts to soak or co-crystallize a wide range of known phosphatase inhibitors and/or peptides to obtain an enzyme:substrate complex were unsuccessful, perhaps due in part to its poor kinetics (see below, Table 2).
Table 2.
Steady state kinetic parameters of Rtr1 assayed against the synthetic substrate DiFMUP.
| Rtr1 construct | Km (μM) | Kcat (s−1) |
|---|---|---|
| Wild-type KlRtr1 FL | 587 ± 70 | 0.0010 ± 0.00003 |
| Wild-type KlRtr1 NTD | 694 ± 74 | 0.0015 ± 0.00005 |
| KlRtr1 CTR | Not detectable | Not detectable |
| KlRtr1 FL E66Q | Not detectable | Not detectable |
| KlRtr1 FL E197Q | 529 ± 45 | 0.0016 ± 0.00004 |
| hRPAP2 NTD | 1668 ± 205 | 0.0005 ± 0.00003 |
Mass spectrometry did not reveal the presence of any other significant co-purified protein in our preparations, but very low levels of contamination cannot be ruled out, leaving open the possibility that phosphatase activity could arise from an E. coli protein22. To demonstrate that catalytic activity resides in Rtr1, we introduced point mutants within highly conserved residues (Figure S1). Mutations of the conserved Cys residues (C73/78/111) resulted in insoluble, most likely unfolded proteins, which were obviously inactive, consistent with the critical role of zinc coordination in protein folding. More revealing, a conservative mutation of the strictly conserved Glu66 (Figure 1E) to Gln produced soluble folded protein with significantly reduced activity against DiFMUP (Figure 1B). The loss of activity following a structurally conservative Glu-Gln substitution, suggests that enzymatic activity originates with Rtr1 and not from co-purified contaminants.
In order to completely rule out a co-purifying E. coli phosphatase, we expressed and purified Rtr1 as a GST fusion protein from insect cells using a baculovirus expression system and assayed its activity against DiFMUP. We observe activity comparable to that of protein purified from bacteria, indicating that activity stems directly from Rtr1 and not an E. coli contaminant (Figure S2B).
To investigate whether the phosphatase activity of Rtr1 is functionally important in cells, we introduced these mutants into yeast in vivo. While Rtr1 is not an essential gene, cells lacking Rtr1 (Figure 1F, rtr1Δ) grow poorly at 37°C24. This temperature sensitivity phenotype is further strengthened when Rtr2 is also deleted (Figure 1F, rtr1Δ/rtr2Δ). Strikingly, cells in which the chromosomal copy of Rtr1 is replaced with an Rtr1 E66A mutant behave similarly to cells lacking Rtr1. In an rtr2Δ background, the E66A mutant also displays increased temperature sensitivity similar to rtr1Δ/rtr2Δ (Figure 1F, Rtr1-E66A/rtr2Δ). Western blot analysis of protein levels show that the E66A mutant is similarly expressed as the wild-type (Figure S2D). Thus, abrogating the enzymatic activity of the protein by a single amino acid change leads to a growth defect comparable to that observed with the deletion of the protein in yeast.
We conclude that Rtr1 is an active phosphatase, which is unrelated structurally to other known such enzymes, and that the function of the protein in vivo is directly related to its enzymatic activity.
The phosphatase activity resides within the conserved N-terminal domain and is regulated by the C-terminal region
Rtr1 is a highly conserved protein in all eukaryotes (Figure S1), even if it is a nonessential gene in yeasts24. Based on the crystal structure and the sequence alignment, yeast Rtr1’s can be divided into a highly conserved N-terminal domain (NTD) of approximately 150 residues and a less conserved region in its C-terminus, referred to as the C-terminal region (CTR) (Figure 2A). The metazoan Rtr1 protein (RPAP2) tends to be much larger proteins of approximately 600 residues, with the only significant conservation found within the NTD.
Figure 2. The N-terminal domain of Rtr1 is the functional phosphatase domain.
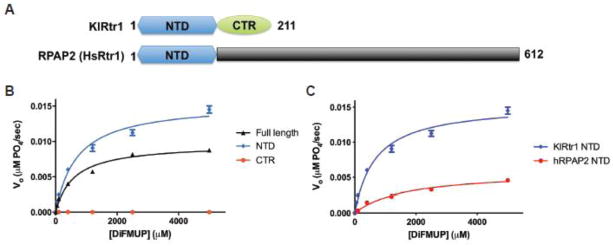
a) Domain breakdown of Rtr1 in yeasts and vertebrates. Yeast Rtr1 proteins are smaller proteins with conserved N-terminal (NTD) and C-terminal domains (CTR). Vertebrate Rtr1 (RPAP2) proteins are typically larger with the only conservation to yeast Rtr1 proteins found within the NTD.
b) Steady state phosphatase assays (n = 3) of KlRtr1 against DiFMUP for full-length (black), NTD (blue), and CTR (red) (10μM protein, all constructs). The full-length curve is derived from the dataset used in Fig. 1C.
c) Steady state phosphatase assays (n = 3) of hRPAP2 NTD (red) (10μM) against DiFMUP. KlRtr1 NTD (from Fig. 2B) is shown as a comparison.
Given that the crystal structure of the most conserved domain revealed no putative active site, we considered the possibility that activity could originate at the interface between the two domains. However, the CTR exhibited no activity above background, while both NTD and full protein had robust activity and similar affinities for the substrate (Table 2). Interestingly, the NTD exhibited a nearly 30% increase in Vmax/Kcat relative to the full protein (Figure 2B), suggesting that the CTR regulates enzymatic activity via a non-competitive mechanism. To confirm the activity of the NTD is conserved in yeasts and animals, we also purified and assayed human RPAP2’s NTD (residues 1-189) against DiFMUP. We observe activity from RPAP2 NTD against DiFMUP, although albeit weaker compared to the K. lactis NTD (Figure 2C). Thus, these data demonstrate that the NTD of Rtr1 represents the active phosphatase domain conserved from yeast to human.
Based on the noncompetitive inhibition of the NTD by the CTR, we asked if the previously observed inhibition resulting from purification is alleviated by the deletion of the CTR. Indeed, the NTD construct purified using either method of Figure 1B yielded active protein with similar kinetic profiles (Figure S2E). Thus, Rtr1’s NTD is an evolutionarily conserved phosphatase domain, albeit inefficient.
Auto-inhibition confines the active site
We were unable to crystallize the complete K. lactis Rtr1 proteins, and NMR analysis on S. cerevisiae Rtr1 yields spectra of mixed quality. Peaks from the NTD are well dispersed, as expected for a domain with a well-defined fold, while peaks originating from the CTR cluster within a narrow spectral region (8–8.5 ppm) and are more intense, suggesting that this region of the protein is only partially structured (Figure S3A and B). Unambiguous NOEs were nonetheless observed between residues belonging to the CTR and the NTD (Figure 3A, left), indicating an interaction between these two regions. Significantly, the residues contacted by the CTR occur near the zinc finger on the “back” face of the protein (Figure 3A, right), on the opposite side of the invariant Glu66 residue that drastically reduces activity (Figure 1C, E and Figure 3A, right). In several partially refined NMR structures of the complete ScRtr1 protein, the CTR can assume positions on the back face of the NTD (Figure S4). However, since kinetic data from Figure 2B suggest that the CTR is a noncompetitive regulator of NTD activity, the active site of Rtr1 cannot reside on this back face of the protein.
Figure 3. Yeast Rtr1 proteins are auto-inhibited by the C-terminal region.

a) NOE connectivities between residues in the CTR of ScRtr1 to residues in the NTD (left panel). The equivalent residues within the NTD that the CTR makes contacts with are highlighted in orange on the crystal structure of the K. lactis protein, and indicated in numbers. The invariant Glu66 residue is highlighted in red.
b) Sequence alignment of Rtr1 (numbering based on K. lactis) across yeasts and vertebrates, focusing on the extreme C-terminus of the protein. Glu197 is asterisked.
c) Steady state phosphatase assay (n = 3) against DiFMUP of KlRtr1 E197Q (red) compared with full-length protein (black) and with the NTD phosphatase domain (blue). Curves for full-length and NTD are derived from Fig. 1C and Fig. 2B respectively.
d) Cartoon illustration of the regulation of Rtr1 activity by its C-terminus. Zinc is shown as a grey sphere and the structurally dynamic loop is shown as a dashed line. Glu66 is denoted by a red line on the same face as the active site forming loop. The CTR weakly interacts with the NTD in the absence of a metal, as shown by the dashed lines; among interacting residues is the critical Glu197 necessary for regulation.
Sequence alignment of Rtr1 (Figure 3B) reveals an invariant (in yeasts) glutamate (KlRtr1 Glu197) near the extreme C-terminus of the enzyme, as well as a number of highly conserved residues both N- and C-terminal to the glutamate. Two of these residues in the CTR (S. cerevisiae W204/L205; equivalent K. lactis M189/L190) displayed NOEs residues found in the NTD (Figure 3A, left). Given E197’s invariance and relative proximity to residues that were structurally connected to the NTD, we hypothesized that this conserved residue of the protein regulates enzyme activity. When Glu197 was conservatively mutated to Gln, we observed an increase in Kcat relative to the wild type full-length enzyme. Comparison of the kinetic parameters of this mutant with the NTD construct shows a similar Vmax, suggesting abrogation of auto-inhibition by mutation of the single conserved glutamate (Figure 3C, Table 2). We collected NOESY spectra of the equivalent mutant in our ScRtr1 construct, but observed no differences, suggesting that this glutamate regulates activity in a manner that does not disrupt the NTD-CTR interface (data not shown).
We also crystallized and solved the structure of ScRtr1, although to lower resolution compared to KlRtr1’s NTD (~4Å) (Table 1). The structure revealed no additional electron density for the CTR aside from a single helix. While the NTD structure is nearly identical to the K. lactis protein, we observed very clear features of a loop (residues ~75-100) in the low-resolution map of ScRtr1 (Figure S5A, left), not seen in the crystal structure of the KlRtr1 NTD. Examination of backbone dynamics conducted by NMR relaxation methods confirmed that the loop is flexible (Figure S3B). It is only stabilized in the crystal by a packing interaction with a neighboring molecule (Figure S5A) and forms a V-shaped crevasse with helices 4 (including Glu66) and 5, potentially forming a structurally dynamic active site. While the sequence is poorly conserved, the length of this loop appears to always range between 20–30 amino acids across species.
To investigate the role of this loop in enzymatic activity, we introduced a series of internal deletions in this loop in our KlRtr1 NTD construct to assay for activity. Assays of NTDΔ90–99 show a near 40% drop in Vmax; an even larger deletion (NTDΔ85–99) shows a more significant decrease in Vmax (near 70%) (Figure S5B). Gel filtration of these constructs show elution volumes similar to that of the wild type NTD, suggesting that deletion of this loop did not affect the folding of this domain (Figure S5C), and that the abrogation of activity is solely attributed to the loss of the loop. Altogether these data show that the loop plays a role in enzymatic activity, although the structural basis is unclear.
To summarize (Figure 3D), the non-competitive nature of the regulation of the NTD suggests that the CTR does not mask the active site. The data suggest instead that the active site is located within the front side of this protein near this loop and the Glu66 residue.
Rtr1 targets both Ser5P and Tyr1P for dephosphorylation
In order to establish the specificity of purified KlRtr1 protein on the CTD, we carried out phosphatase assays using GST-CTD phosphorylated with purified TFIIH (Figure 4A). KlRtr1 preferentially dephosphorylates Ser5P but with little effect on Ser2P and Ser7P (Figure 4B). Quantitation of the signals from the blots clearly shows robust dephosphorylation of Ser5P with increasing enzyme (Figure 4C).
Figure 4. Rtr1 is a dual specificity phosphatase that acts both on Ser5 and Tyr1.

a) TFIIH efficiently phosphorylates the GST-CTD. With the exception of the anti-Ser7P antibody, all antibodies used recognize only the phosphorylated form of the CTD.
b) GST-CTD phosphorylated with either purified TFIIH (left) or TFIIH/Abl kinase (right) were used as substrates for KlRtr1. Reaction products were separated by SDS-PAGE and then western blots were then probed with antibodies against Ser2, Ser5, Ser7, and Tyr1. Two exposures for Tyr1 are shown for clarity.
c) Quantitation of phospho-signals from the Ser2P, Ser5P, Ser7P, and Tyr1P blots (n = 3) on GST-CTD modified by TFIIH or TFIIH plus Abl kinase.
Recent work identified Tyr1 phosphorylation within the S. cerevisiae CTD as an anti-termination marker9. Namely, chromatin immunoprecipitation profiles of Tyr1P show an enrichment of the phospho-marker during the elongation phase of transcription, in agreement with its putative role in preventing the premature recruitment of transcription termination factors. Given the role of Rtr1 in dephosphorylating Ser5P during transcription to generate the predominant Ser2P form observed in late phases of transcription, we asked whether Rtr1 would dephosphorylate Tyr1P as well.
The kinase activity of TFIIH is only weakly active on tyrosines in vitro (Figure 4A, top panel; Figure 4B, lower left panels). Thus, we used Abl kinase and TFIIH together to effectively phosphorylate both serines and tyrosines on the CTD in vitro25. We then assayed the modified polypeptide with an anti-Tyr1P antibody (Chromotek) in parallel with the other phospho-specific antibodies. As shown in Figure 4A, Rtr1 dephosphorylates Tyr1P as well: the signal for the Tyr1-specific antibody decreases with increasing amounts of Rtr1. The presence of the Tyr1P marker does not disrupt the ability of the enzyme to dephosphorylate Ser5P (Figure 4A and B), since quantitation shows that dephosphorylation is comparable for Tyr1 and Ser5, but the signals for Ser2P and Ser7P remain constant after Rtr1 treatment. In addition to highlighting a new and unexpected phosphatase specificity, this result suggests that Rtr1 does not recognize and bind to the CTD like the well-characterized CTD-interacting domain proteins, which are repelled by the presence of the Tyr1P marker9.
Finally, we tested Rtr1’s ability to dephosphorylate synthetic CTD peptides phosphorylated only on either Tyr1P, Ser5P, and a disphosphorylated Ser2/5P, but strangely, Rtr1 displayed no activity against these phosphorylated peptides both in the presence and absence of Ess15,26 (data not shown). These data are in contrast with the data on the long form of the CTD (Fig. 4), and raise the possibility of a CTD code embedded in the heterogeneously phosphorylated GST-CTD substrate which Rtr1 can recognize and dephosphorylate.
Discussion
The interplay of kinases and phosphatases that act upon the C-terminal domain (CTD) of RNA PolII regulates and times the synthesis and biogenesis of cellular RNAs. However, the identity of the critical transition phosphatase that removes the Ser5P marker and shift the polymerase to the elongation and termination mode remains to be firmly established. Rtr1 (RPAP2 in vertebrates), a highly conserved protein in all eukaryotes, was proposed to be such a phosphatase in two independent studies showing that Rtr1 in both yeasts and vertebrates can specifically dephosphorylate the CTD Ser5P20,27, but this conclusion was negated by the report that a highly purified, crystallized K. lactis Rtr1 was inactive22. We demonstrate here that Rtr1 is a phosphatase of new structure and attribute previous results on the lack of enzymatic activity to the absence of an identifiable step in the purification protocol, which resulted in an inactive protein. We further show that the phosphatase activity of Rtr1 is functionally important. Mutation of the absolutely conserved Glu66 to Gln reduces catalytic activity significantly and leads to the same phenotype in vivo observed for mutations of the zinc coordinating Cys residues, which generate an unfolded, obviously misfunctional protein.
The use of EDTA during purification was understandably overlooked because Rtr1 requires a single structural zinc ion to maintain its fold. However, Rtr1 maintains its hold on the structural zinc ion, once expressed, even in the presence of high concentrations of chelating agents, and in our hands required no additional zinc to be added to the growth media, or purification solutions22. While we were able to obtain crystals of ScRtr1 full-length that diffracted to 4Å, we did not identify an obvious contaminating metal, likely due to the low resolution of the maps.
Rtr1 displays linear enzyme kinetics over a one hour time course as assayed by both fluorescence (observation of product), or malachite green (observation of phosphate release) and is selectively inhibited by a classical phosphatase inhibitor. However, it is an inefficient enzyme by comparison with other Ser5P phosphatases, such as Ssu72 and Scp128,29. Even when compared to the Ser2P phosphatase Fcp130, the slowest known CTD phosphatase, Rtr1 is nearly 400 times slower, at about 1×10−3 s−1 in vitro against DiFMUP. The poor turnover rate of Rtr1 is likely a reflection of its structure, which lacks a well-defined pocket or groove to serve as an active site. We provide this conjecture also as an explanation for our inability to crystallize an enzyme-inhibitor complex despite exhaustive attempts.
Kinetic measurements revealed a noncompetitive auto-inhibitory function for the partially conserved CTR of Rtr1, mediated by a conserved glutamate located near the C-terminus of the protein. Mutation of this single residue alleviated the partial inhibition of Rtr1. While suppression of enzymatic activity is only ~30%, it would seem that Rtr1 was naturally evolved to be a kinetically slow enzyme. In our in vitro characterization, we cannot rule out that Rtr1’s inhibition by a divalent metal is not reflective of a native state where a regulatory mechanism is in place to activate the protein by removing the inhibitory metal. Both auto-inhibition by the C-terminus and the regulatory loop found in the NTD provide attractive candidates for regulation during transcription by an as yet unidentified protein partners.
We also observe that Rtr1 is a dual specificity phosphatase, which acts not only on Ser5P but on Tyr1P, a new anti-termination marker9. The activity towards Tyr1P in vitro is specific: similar levels of dephosphorylation were observed for both Tyr1P and Ser5P, while levels of Ser2P and Ser7P were not affected at all (Fig. 4B). Interestingly, ChIP data show that the levels of Rtr1 do not decrease to background until the end of transcription20, in coincidence with the decline of the Tyr1P marker9, but additional work will be needed to address the in vivo effect Rtr1 has on Tyr1P.
Rtr1 is active on long CTD repeats, but it displays no activity towards synthetic CTD phosphopeptide mimics. However, these substrates differ considerably from the GST-CTD, which is heterogeneously phosphorylated across its entire length, while synthetic peptides have a narrowly defined phospho-pattern. Our contrasting data lead us to speculate that Rtr1 recognizes an unidentified CTD code, which can be found on a heavily modified substrate, a hypothesis supported by studies on RPAP2 that show that it can bind to Ser7P peptides21 and studies on Rtr1 that show interaction with both the Ser5 and Ser2 forms of PolII32. The elongation phase of transcription in which Rtr1 is active is also the phase of transcription where the CTD is most heavily modified, with Ser2/5 and Tyr1 as known markers, and additional potential marks at Thr4 and Ser7. Overlapping phospho-marks between neighboring repeats can also specify a recruitment signal for CTD interacting proteins33. Additional work will be needed to systematically identify the CTD substrate recognized and dephosphorylated by Rtr1.
Based on our observations, and previous work we propose that Rtr1 is recruited to PolII during import and assembly of the polymerase34 in a form where enzymatic activity is limited. Since Rtr1 does not dephosphoryate peptides carrying only isolated Tyr1 and Ser5 phospho-marks, Rtr1 remains inactive during transcriptional initiation, capping and promoter clearance (Figure 5 top). As the polymerase shifts fully into processive elongation and additional phospho-markers are deposited along the CTD, Rtr1 begins to recognize a specific but unknown ‘CTD code’ and begins to remove the Ser5P markers, progressively setting the polymerase into the transcription termination mode (Figure 5 bottom). Additional factors could also stimulate the activity of Rtr1 during the transition phase either by direct binding or post translational modifications.
Figure 5.

A model for Rtr1’s role in the transcription cycle. Phosphorylation of Ser5 on the CTD at the start of transcription facilitates the recruitment of the mRNA capping complexes (top). As the polymerase moves into elongation and termination modes, the CTD is highly phosphorylated by multiple kinases, including an as of yet identified Tyr1 kinase. Rtr1 becomes active against Ser5P during the elongation phase (bottom) of transcription due to the recognition of an as-of-yet determined CTD code. The Tyr1 marker is heavily phosphorylated during elongation and is also a target for dephosphorylation by Rtr1.
In conclusion, the data presented here demonstrate that Rtr1 is a phosphatase of novel structure that removes the Tyr1P and Ser5P markers from the PolII CTD, albeit inefficiently. Thus, it is the phosphatase responsible for the transition to the elongation and termination phase of transcription. Future work will be needed to elucidate its catalytic mechanism, which may be distinct from that of known phosphatases to which Rtr1 bears no structural homology.
Materials and methods
Protein expression and purification
Saccharomyces cerevisiae (Sc) and Kluoverymyces lactis (Kl) Rtr1 proteins, and human RPAP2 (1-189) were cloned into a modified pET-28a (Novagen) vector with a Protein G B1 domain (GB1) fused to the N-terminus to facilitate expression. Plasmids encoding the gene were transformed into Rosetta DE3 E. coli, shaken at 37°C until induction with IPTG and expressed overnight at 18°C. Cells were harvested the next morning and resuspended in lysis buffer (50mM HEPES pH7.5, 200mM NaCl, 30mM imidazole, 5mM βME), lysed by sonication and cleared by high-speed centrifugation. Lysate was applied to a HisTrap column (GE Healthcare) equilibrated in lysis buffer. Bound protein was eluted from the column by a linear gradient against elution buffer (lysis buffer + 500mM imidazole). Protein-containing fractions were pooled and placed into dialysis buffer (20mM HEPES pH7.5, 100mM NaCl, 5mM βME); TEV protease was incubated overnight to remove the His-GB1 tag.
Dialyzed material was collected and re-applied to a HisTrap column equilibrated in dialysis buffer to remove the tag, TEV protease, and any uncleaved protein. Prior to applying the protein to the gel filtration column, 10mM EDTA was added to the protein to remove any residual divalent metals that may have co-purified with the protein. After incubation with EDTA, the protein was applied to a Superdex 75 (GE Healthcare) equilibrated in storage buffer (dialysis buffer but 5mM DTT substituted the βME). The protein eluted at a volume consistent with a monomer. Protein-containing fractions were concentrated to 10–20mg/mL (as determined by both Bradford assay and absorbance at 280nm) and flash frozen using liquid nitrogen. For the KlRtr1 NTD construct, the protein was only concentrated to ~2mg/mL, due to more limited solubility before storage.
For expression in insect cells, KlRtr1 was expressed as a GST fusion protein in Hi5 monolayer insect cells and immobilized using glutathione affinity chromatography (GE Healthcare) using GST lysis buffer containing 40mM HEPES pH7.5, 250mM NaCl, 2mM EDTA, 2mM DTT. After extensive column washing, protein was eluted from the column using GST lysis buffer supplemented with 10mM reduced glutathione. The GST tag was cleaved with TEV protease and the protein was placed into dialysis buffer. Uncleaved protein and free GST tag was then removed by another pass over glutathione beads, and then concentrated and loaded onto a Superdex 75 (GE Healthcare) equilibrated in storage buffer. Eluted protein was then concentrated and flash frozen in liquid nitrogen for future use.
Mutagenesis
Point mutants were generated using the QuikChange kit (Stratagene). Internal deletions were generated by using overlap extension PCR. All mutants were verified by sequencing. Expression and purification of the mutants were done exactly as for the wild type protein.
NMR sample preparation and experiments
NMR samples were prepared by growing Rosetta DE3 transformed with ScRtr1 in M9 minimal media supplemented with 0.5 g/L 15NH4Cl, 2 g/L 13C-glucose and 0–100% D2O (Sigma-Aldrich) as needed. Selective methyl labeling of ILV residues was done as described35. Samples were purified as described above, but the final storage buffer was different (20mM Bistris pH6.5, 50mM NaCl, 2mM DTT).
NMR spectra were recorded at 298K on Bruker Avance 600 and Avance 800 spectrometers equipped with triple-resonance cryoprobes and pulse field gradients. Data were processed with NMRpipe36 and analyzed with CCPNMR37. Rtr1 backbone assignments were obtained using TROSY, trHNCA, trHN(CO)CA, trHN(CO)CACB, trHNCACB, trHNCO and trHN(CA)CO spectra on a 15N, 13C, 2H labeled protein in 90% H2O, 10%D2O. Methyl assignments of ILEs, LEUs and VALs were obtained using (H)CC(CO)NH and H(CC)(CO)NH spectra recorded on perdeuterated Rtr1 retaining 1H, 13C labels at the ILV methyl positions.
Crystallization, data collection, structure determination and refinement
Initial crystals of KlRtr1 were obtained by adding trypsin (Sigma) to full-length protein at a ratio of 1:10000 w/w and incubating for 30 minutes at room temperature prior to setting up drops. Crystals were obtained by hanging drop by mixing one volume of sample with an equal volume of precipitant (0.1M Bicine pH8.5, 10–20% MPD) at 4°C. Crystals appeared in ~2 days and matured to final size after a week. Crystals were cryoprotected by mother liquor supplemented with 30% MPD, flash frozen in liquid nitrogen and harvested for data collection.
All datasets were collected at the Advanced Light Source at the Lawrence Berkeley National Laboratory at beam lines BL5.0.1, 5.0.2 and 5.0.3. Datasets were indexed, integrated, and scaled with the HKL2000 package38. Initial phases were determined from a SAD dataset using a single SeMet derivatized crystal. The SAD dataset was collected at BL5.0.1 at a wavelength of 0.98Å and phases were determined by the Solve/Resolve program 39. Initial model building and refinement were done by Coot and CNS40,41.
Using the initial model as a template, we re-crystallized KlRtr1 NTD in similar conditions and obtained phases using the SeMet model as a molecular replacement solution. Final model building and refinement were done on this construct using Coot and Refmac5 as part of the CCP4 package42. The final model had 97.8% of all residues in the favored region of the Ramachandran plot, and 2.2% in the allowed region.
Crystals of full length ScRtr1 were obtained using one volume of reductively methylated protein 43 with one volume of precipitant (0.04M Bicine pH8.5, 150mM LiCl, 20mM hexammine cobalt(III) chloride, 22–30% MPD). Crystals were cryoprotected with mother liquor +35% MPD and flash frozen in liquid nitrogen prior to data collection. The best crystals diffracted to 4Å at the Advanced Light Source. Phases were obtained by molecular replacement using our KlRtr1 NTD structure as a search model.
Phosphatase assays
Steady state kinetic assays were performed with varying concentrations of DiFMUP (Life Technologies) and 10μM KlRtr1 (various constructs as described in the text) in 50mM MES, pH 5.5 at 30°C. Monitoring of product formation was observed by either extracting aliquots of the phosphatase reaction at fixed time points and quenching with Biomol Green reagent (Enzo Sciences), or by continuous observation of fluorescence emission spectra at a wavelength of 450nm. Product formation/phosphate release was determined by comparison against a standard curve of either DiFMU or phosphate. All experiments were done in triplicate, and data were analyzed and fit to the Michaelis-Menten equation using GraphPad Prism.
IC50 experiments were performed using sodium orthovanadate (New England Biolabs) and β-glycerophosphate (Sigma-Aldrich) as inhibitors. Data were plotted and analyzed using GraphPad Prism. Inhibition constants (Ki) were determined by converting IC50 values using the Cheng-Prusoff equation.
In vivo experiments
All yeast strains used in this study were derived from BY4741. To create Rtr1 mutant strains, a RTR1 fragment (−266 to +678) was amplified from BY4741 yeast genomic DNA and cloned into pBS1539 to create RTR1-TAP44. Mutant plasmids were created by site directed mutagenesis. Yeast strains were created by transforming BY4741 with a PCR product amplified from wild type or mutant RTR1-TAP plasmids and selecting transformants on complete synthetic media lacking uracyl. Expression of the mutant constructs was confirmed by western blotting. For the growth assays, 5-fold serial dilutions of yeast were spotted on YPD or YPD + 2% form amide as previously described24.
GST-CTD phosphatase reactions
GST-CTD phosphatase reactions were performed as previously described20. Briefly, purified recombinant GST-CTD was phosphorylated in vitro by TFIIH and/or Abl kinase in the presence of ATP. Unreacted ATP was removed by gel filtration. For each phosphatase reaction, approximately 5pmol of modified GST-CTD was used as a substrate in the presence of increasing concentrations of KlRtr1 as indicated. Reactions were quenched with 2X SDS-PAGE loading buffer followed by western blot analysis using the CTD phosphorylation antibodies described above.
Supplementary Material
Highlights.
Rtr1 is a new phosphatase unrelated structurally to known such enzymes
Rtr1’s N-terminal domain represents a novel fold for phosphatase function.
Disruption of in vitro activity display gene deletion phenotypes in vivo.
The C-terminal region of Rtr1 inhibits phosphatase activity.
Rtr1 selectively dephosphorylates both Tyr1 and Ser5 on the polymerase II CTD.
Acknowledgments
The authors would like to thank the staff at the Advanced Light Source (Berkeley, CA) for assistance in data collection. We would also like to thank the Stoddard laboratory at the Fred Hutchinson Cancer Research Center for providing access to their home X-ray source for initial crystal screening. We thank Dr. Ning Zheng for his advice and guidance in solving the crystal structure of KlRtr1 NTD. Chromotek generously allowed us early access to the anti-Tyr1P antibody. TFIIH was kindly provided by Tsuyoshi Imasaki and Yuichiro Takagi. The rtr1Δ/rtr2Δ was kindly provided by Kevin Morano.
This project was supported by a grant from NIH-NIGMS RO1 064440 to G.V. and R01 GM099714 to A.L.M. P.H. was partially supported by the Training in Molecular Biophysics training grant (T32GM008268).
Footnotes
Accession numbers
Atomic coordinates and structure factors of active KlRtr1 NTD have been deposited in the Protein Data Bank with the accession code 4M30.
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hsin J-P, Manley JL. The RNA polymerase II CTD coordinates transcription and RNA processing. Genes Dev. 2012;26:2119–2137. doi: 10.1101/gad.200303.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kubicek K, Cerna H, Holub P, Pasulka J, Hrossova D, Loehr F, Hofr C, Vanacova S, Stefl R. Serine phosphorylation and proline isomerization in RNAP II CTD control recruitment of Nrd1. Genes Dev. 2012;26:1891–1896. doi: 10.1101/gad.192781.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meinhart A, Kamenski T, Hoeppner S, Baumli S, Cramer P. A structural perspective of CTD function. Genes Dev. 2005;19:1401–1415. doi: 10.1101/gad.1318105. [DOI] [PubMed] [Google Scholar]
- 4.Morris DP, Phatnani HP, Greenleaf AL. Phospho-carboxyl-terminal domain binding and the role of a prolyl isomerase in pre-mRNA 3′-End formation. J Biol Chem. 1999;274:31583–31587. doi: 10.1074/jbc.274.44.31583. [DOI] [PubMed] [Google Scholar]
- 5.Xiang K, Nagaike T, Xiang S, Kilic T, Beh MM, Manley JL, Tong L. Crystal structure of the human symplekin-Ssu72-CTD phosphopeptide complex. Nature. 2010;467:729–733. doi: 10.1038/nature09391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang M, Wang XJ, Chen X, Bowman ME, Luo Y, Noel JP, Ellington AD, Etzkorn FA, Zhang Y. Structural and kinetic analysis of prolyl-isomerization/phosphorylation cross-talk in the CTD code. ACS Chem Biol. 2012;7:1462–1470. doi: 10.1021/cb3000887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chapman RD, Heidemann M, Albert TK, Mailhammer R, Flatley A, Meisterernst M, Kremmer E, Eick D. Transcribing RNA polymerase II is phosphorylated at CTD residue serine-7. Science. 2007;318:1780–1782. doi: 10.1126/science.1145977. [DOI] [PubMed] [Google Scholar]
- 8.Hsin JP, Sheth A, Manley JL. RNAP II CTD Phosphorylated on Threonine-4 Is Required for Histone mRNA 3′ End Processing. Science. 2011;334:683–686. doi: 10.1126/science.1206034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mayer A, Heidemann M, Lidschreiber M, Schreieck A, Sun M, Hintermair C, Kremmer E, Eick D, Cramer P. CTD tyrosine phosphorylation impairs termination factor recruitment to RNA polymerase II. Science. 2012;336:1723–1725. doi: 10.1126/science.1219651. [DOI] [PubMed] [Google Scholar]
- 10.Buratowski S. The CTD code. Nat Struct Biol. 2003;10:679–680. doi: 10.1038/nsb0903-679. [DOI] [PubMed] [Google Scholar]
- 11.Buratowski S. Progression through the RNA polymerase II CTD cycle. Mol Cell. 2009;36:541–546. doi: 10.1016/j.molcel.2009.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schwer B, Shuman S. Deciphering the RNA Polymerase II CTD Code in Fission Yeast. Molecular Cell. 2011 doi: 10.1016/j.molcel.2011.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Komarnitsky P, Cho EJ, Buratowski S. Different phosphorylated forms of RNA polymerase II and associated mRNA processing factors during transcription. Genes Dev. 2000;14:2452–2460. doi: 10.1101/gad.824700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghosh A, Shuman S, Lima CD. Structural insights to how mammalian capping enzyme reads the CTD code. Mol Cell. 2011;43:299–310. doi: 10.1016/j.molcel.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mayer A, Lidschreiber M, Siebert M, Leike K, Söding J, Cramer P. Uniform transitions of the general RNA polymerase II transcription complex. Nat Struct Mol Biol. 2010;17:1272–1278. doi: 10.1038/nsmb.1903. [DOI] [PubMed] [Google Scholar]
- 16.Vasiljeva L, Kim M, Mutschler H, Buratowski S, Meinhart A. The Nrd1-Nab3-Sen1 termination complex interacts with the Ser5-phosphorylated RNA polymerase II C-terminal domain. Nat Struct Mol Biol. 2008;15:795–804. doi: 10.1038/nsmb.1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ahn SH, Kim M, Buratowski S. Phosphorylation of Serine 2 within the RNA Polymerase II C-Terminal Domain Couples Transcription and 3′ End Processing. Molecular Cell. 2004;13:67–76. doi: 10.1016/s1097-2765(03)00492-1. [DOI] [PubMed] [Google Scholar]
- 18.Gu B, Eick D, Bensaude O. CTD serine-2 plays a critical role in splicing and termination factor recruitment to RNA polymerase II in vivo. Nucleic Acids Res. 2013;41:1591–1603. doi: 10.1093/nar/gks1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lunde BM, Reichow SL, Kim M, Suh H, Leeper TC, Yang F, Mutschler H, Buratowski S, Meinhart A, Varani G. Cooperative interaction of transcription termination factors with the RNA polymerase II C-terminal domain. Nat Struct Mol Biol. 2010;17:1195–1201. doi: 10.1038/nsmb.1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mosley AL, Pattenden SG, Carey M, Venkatesh S, Gilmore JM, Florens L, Workman JL, Washburn MP. Rtr1 is a CTD phosphatase that regulates RNA polymerase II during the transition from serine 5 to serine 2 phosphorylation. Mol Cell. 2009;34:168–178. doi: 10.1016/j.molcel.2009.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Egloff S, Zaborowska J, Laitem C, Kiss T, Murphy S. Ser7 phosphorylation of the CTD recruits the RPAP2 Ser5 phosphatase to snRNA genes. Mol Cell. 2012;45:111–122. doi: 10.1016/j.molcel.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xiang K, Manley JL, Tong L. The yeast regulator of transcription protein Rtr1 lacks an active site and phosphatase activity. Nat Commun. 2012;3:946. doi: 10.1038/ncomms1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wilson M, Hogstrand C, Maret W. Picomolar concentrations of free zinc(II) ions regulate receptor protein-tyrosine phosphatase β activity. J Biol Chem. 2012;287:9322–9326. doi: 10.1074/jbc.C111.320796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gibney PA, Fries T, Bailer SM, Morano KA. Rtr1 is the Saccharomyces cerevisiae homolog of a novel family of RNA polymerase II-binding proteins. Eukaryotic Cell. 2008;7:938–948. doi: 10.1128/EC.00042-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baskaran R, Chiang GG, Mysliwiec T, Kruh GD, Wang JY. Tyrosine phosphorylation of RNA polymerase II carboxyl-terminal domain by the Abl-related gene product. J Biol Chem. 1997;272:18905–18909. doi: 10.1074/jbc.272.30.18905. [DOI] [PubMed] [Google Scholar]
- 26.Werner-Allen JW, Lee CJ, Liu P, Nicely NI, Wang S, Greenleaf AL, Zhou P. cis-Proline-mediated Ser(P)5 Dephosphorylation by the RNA Polymerase II C-terminal Domain Phosphatase Ssu72. J Biol Chem. 2011;286:5717–5726. doi: 10.1074/jbc.M110.197129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Egloff S, Zaborowska J, Laitem C, Kiss T, Murphy S. Ser7 Phosphorylation of the CTD Recruits the RPAP2 Ser5 Phosphatase to snRNA Genes. Molecular Cell. doi: 10.1016/j.molcel.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Y, Kim Y, Genoud N, Gao J, Kelly JW, Pfaff SL, Gill GN, Dixon JE, Noel JP. Determinants for dephosphorylation of the RNA polymerase II C-terminal domain by Scp1. Mol Cell. 2006;24:759–770. doi: 10.1016/j.molcel.2006.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Y, Zhang M, Zhang Y. Crystal structure of Ssu72, an essential eukaryotic phosphatase specific for the C-terminal domain of RNA polymerase II, in complex with a transition state analogue. Biochem J. 2011;434:435–444. doi: 10.1042/BJ20101471. [DOI] [PubMed] [Google Scholar]
- 30.Hausmann S, Shuman S. Characterization of the CTD phosphatase Fcp1 from fission yeast. Preferential dephosphorylation of serine 2 versus serine 5. J Biol Chem. 2002;277:21213–21220. doi: 10.1074/jbc.M202056200. [DOI] [PubMed] [Google Scholar]
- 31.Rodriguez CR, Cho E-J, Keogh M-C, Moore CL, Greenleaf AL, Buratowski S. Kin28, the TFIIH-Associated Carboxy-Terminal Domain Kinase, Facilitates the Recruitment of mRNA Processing Machinery to RNA Polymerase II. Mol Cell Biol. 2000;20:104–112. doi: 10.1128/mcb.20.1.104-112.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mosley AL, Hunter GO, Sardiu ME, Smolle M, Workman JL, Florens L, Washburn MP. Quantitative Proteomics Demonstrates That the RNA Polymerase II Subunits Rpb4 and Rpb7 Dissociate during Transcriptional Elongation. Mol Cell Proteomics. 2013;12:1530–1538. doi: 10.1074/mcp.M112.024034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Egloff S, Szczepaniak SA, Dienstbier M, Taylor A, Knight S, Murphy S. The Integrator Complex Recognizes a New Double Mark on the RNA Polymerase II Carboxyl-terminal Domain. Journal of Biological Chemistry. 2010;285:20564–20569. doi: 10.1074/jbc.M110.132530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Forget D, Lacombe A-A, Cloutier P, Lavallée-Adam M, Blanchette M, Coulombe B. Nuclear import of RNA polymerase II is coupled with nucleocytoplasmic shuttling of the RNA polymerase II-associated protein 2. Nucl Acids Res. 2013 doi: 10.1093/nar/gkt455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rosen MK, Gardner KH, Willis RC, Parris WE, Pawson T, Kay LE. Selective methyl group protonation of perdeuterated proteins. J Mol Biol. 1996;263:627–636. doi: 10.1006/jmbi.1996.0603. [DOI] [PubMed] [Google Scholar]
- 36.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 37.Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinas M, Ulrich EL, Markley JL, Ionides J, Laue ED. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins. 2005;59:687–696. doi: 10.1002/prot.20449. [DOI] [PubMed] [Google Scholar]
- 38.Otwinowski Z, Minor W. [20] Processing of X-ray diffraction data collected in oscillation mode. Methods in Enzymology. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 39.Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Acta Crystallogr D Biol Crystallogr. 1999;55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 41.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 42.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Walter TS, Meier C, Assenberg R, Au KF, Ren J, Verma A, Nettleship JE, Owens RJ, Stuart DI, Grimes JM. Lysine Methylation as a Routine Rescue Strategy for Protein Crystallization. Structure. 2006;14:1617–1622. doi: 10.1016/j.str.2006.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Puig O, Caspary F, Rigaut G, Rutz B, Bouveret E, Bragado-Nilsson E, Wilm M, Séraphin B. The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods. 2001;24:218–229. doi: 10.1006/meth.2001.1183. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.