

Abstract
IMPORTANCE
Whole-genome sequencing (WGS) is increasingly applied in clinical medicine and is expected to uncover clinically significant findings regardless of sequencing indication.
OBJECTIVES
To examine coverage and concordance of clinically relevant genetic variation provided by WGS technologies; to quantitate inherited disease risk and pharmacogenomic findings in WGS data and resources required for their discovery and interpretation; and to evaluate clinical action prompted by WGS findings.
DESIGN, SETTING, AND PARTICIPANTS
An exploratory study of 12 adult participants recruited at Stanford University Medical Center who underwent WGS between November 2011 and March 2012. A multidisciplinary team reviewed all potentially reportable genetic findings. Five physicians proposed initial clinical follow-up based on the genetic findings.
MAIN OUTCOMES AND MEASURES
Genome coverage and sequencing platform concordance in different categories of genetic disease risk, person-hours spent curating candidate disease-risk variants, interpretation agreement between trained curators and disease genetics databases, burden of inherited disease risk and pharmacogenomic findings, and burden and interrater agreement of proposed clinical follow-up.
RESULTS
Depending on sequencing platform, 10% to 19% of inherited disease genes were not covered to accepted standards for single nucleotide variant discovery. Genotype concordance was high for previously described single nucleotide genetic variants (99%-100%) but low for small insertion/deletion variants (53%-59%). Curation of 90 to 127 genetic variants in each participant required a median of 54 minutes (range, 5-223 minutes) per genetic variant, resulted in moderate classification agreement between professionals (Gross κ, 0.52; 95%CI, 0.40-0.64), and reclassified 69%of genetic variants cataloged as disease causing in mutation databases to variants of uncertain or lesser significance. Two to 6 personal disease-risk findings were discovered in each participant, including 1 frameshift deletion in the BRCA1 gene implicated in hereditary breast and ovarian cancer. Physician review of sequencing findings prompted consideration of a median of 1 to 3 initial diagnostic tests and referrals per participant, with fair interrater agreement about the suitability of WGS findings for clinical follow-up (Fleiss κ, 0.24; P < 001).
CONCLUSIONS AND RELEVANCE
In this exploratory study of 12 volunteer adults, the use of WGS was associated with incomplete coverage of inherited disease genes, low reproducibility of detection of genetic variation with the highest potential clinical effects, and uncertainty about clinically reportable findings. In certain cases, WGS will identify clinically actionable genetic variants warranting early medical intervention. These issues should be considered when determining the role of WGS in clinical medicine.
As technical barriers to human DNA sequencing decrease and the cost of whole-genome sequencing (WGS) approaches$1000,WGS and protein-coding genome sequencing (whole-exome sequencing [WES]) are increasingly used in clinical medicine. Both WGS/WES can successfully aid clinical diagnosis,1-3 reveal the genetic basis of rare familial diseases,4-6 and explicate novel disease biology.7,8 Regardless of context, even in apparently healthy individuals, WGS/WES are expected to uncover genetic findings of potential clinical importance.9-11 However, comprehensive clinical interpretation and reporting of clinically significant findings are seldom performed. As WGS/WES are applied more broadly, questions have been raised about the duty for discovery, interpretation, and reporting of clinical findings. Recently published recommendations define genetic variant types in a minimum list of inherited disease genes that are suggested to be subject to discovery, reporting, and clinical follow-up regardless of the primary indication for sequencing, patient preference, or patient age.12 Despite this, the technical sensitivity and reproducibility of clinical genetic findings using WGS and the clinical opportunities and costs associated with discovery and reporting of these and other clinical findings in WGS data remain undefined.
We previously described a framework for interpretation of WGS data in clinical context involving individuals13 and families.14 The Stanford Genomic Medicine Application Pilot Project was designed to explore clinically interpretable sequence coverage and genetic variant reproducibility using current sequencing technology, the burden of clinically reportable inherited disease risk and pharmacogenomic findings in WGS according to current genetic knowledge, the resources required for discovery and interpretation of these findings, and the burden and costs of initial clinical follow-up prompted by WGS findings (a glossary of genetic terms are provided in Box 1).
Box 1. Genetics Glossary.
Allele: Alternative form of a gene or DNA sequence. Variations in clinical traits and phenotypes are allelic if they arise from the same gene sequence or locus and are nonallelic if they arise from different gene sequences of different loci.
Annotation: Adding pertinent information such as gene coded for, amino acid sequence, or other commentary to the database entry of raw sequence of DNA bases.
Calls: Assignment of a nucleotide base (A, T, G, C) at specific positions in the genome during genotyping or sequencing.
Curate/curation: The process of manual review of the literature, databases, and other data sources to collect evidence related to a specific genetic variant, with the ultimate goal of assessing its potential pathogenicity.
Frame shift variant: Any mutation that disrupts the normal sequence of triplets causing a new sequence to be created that codes for different amino acids. Frame shift mutations are usually caused by an insertion/deletion of DNA and typically eventually produce a premature stop codon.
Haplotype: The combination of linked marker alleles (may be polymorphisms or mutations) for a given region of DNA on a single chromosome.
Locus (plural loci): The physical site on a chromosome occupied by a particular gene or other identifiable DNA sequence characteristic.
Sequencing coverage/depth of coverage: The number of independent times a location in the genome has been sequenced.
Stopgain: A DNA sequence variant in which one base is substituted for another, resulting in the formation of a premature stop codon. Synonymous with the term “nonsense” variant.
Stoploss: A DNA sequence variant is one in which one base is substituted for another, resulting in the loss of the normal stop codon.
Structural variant: DNA sequence variants that involve large segments of DNA(at least1000base pairs), including copy number variants, inversions, and translocations.
Truncating variant: A DNA sequence variant that results in the formation of a premature stop codon and therefore a truncated protein.
Whole-exome sequencing: A method for determining the precise order of bases in a DNA molecule in the exome, which represents the entire protein-coding portion of the genome.
Whole-genome sequencing: A method for determining the precise order of bases in the genome, which is the sum total of all genetic material of a cell or an organism.
Zygosity: the characterization of an individual’s hereditary traits in terms of gene pairing in the zygote from which it developed.
Sources: Glossary of genomics terms. JAMA. 2013;309(14):1533-1535 and Human Genome Project Information Archive. http://web.ornl.gov/sci/techresources/Human_Genome/glossary.shtml. Accessed February 20, 2014.
Methods
Setting and Study Population
Unrelated adults who expressed interest in WGS during the pilot phase of our clinical genomic service at Stanford Hospital and Clinics were recruited between November 2011 and March 2012. None of the study participants had manifest inherited disease. Study participants received genetic counseling at the time of sample collection and during disclosure of results with the primary physician. This study was approved by Stanford University’s institutional review board, and all participants gave informed written consent.
WGS Generation
Genomic DNA was isolated from whole blood. For all study participants, genomic DNA was sequenced at Illumina Inc (Methods section in the Supplement).15 Confirmatory sequencing of genomic DNA from 9 of the study participants was performed by Complete Genomics Inc16 to evaluate reproducibility of sequence data between commonly used sequencing platforms.
The analytical workflow for clinical WGS interpretation is presented in Figure 1. Illumina sequence reads were processed as described in the Methods section in the Supplement by comparison to major allele reference genomes for the European ancestry from Utah or Han Chinese from Beijing and Japanese from Tokyo HapMap population groups, depending on self-reported ethnicity.14 Complete Genomics Inc sequence data were processed as described.16 In all cross-platform sequence comparisons Illumina data were used as reference.
Figure 1. Overview of Whole-Genome Sequence Analysis Work flow for the Genomic Medicine Application Pilot Project (GMAPP).
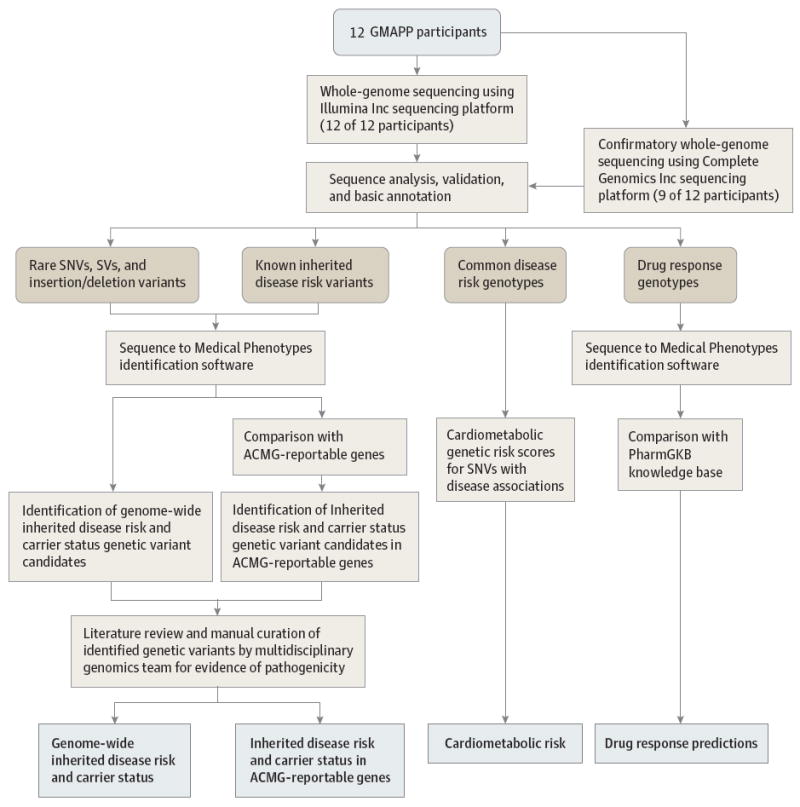
Rare genetic variants were defined as alleles with frequency <0.01 in an ethnically matched population genetic survey. Inherited disease risk candidate identification and genetic drug response prediction are outlined in the Supplement. Abbreviations: ACMG, American College of Medical Genetics and Genomics; GMAPP, Genomic Medicine Application Pilot Project; SNV, single nucleotide variant; SV, structural variant.
In addition to genetic variants from an ethnically matched reference sequence, genotypes for 72 383 male and 72 316 female genetic variants cataloged in the Human Gene Mutation Database (HGMD)17 that reside within inherited disease genes cataloged by the ClinVar database,18 118 genetic variants with replicated associations with diabetes mellitus type 2 and coronary artery disease risk in white and East Asian populations,19-22 and 555 genetic variants with clinical drug response associations (Pharm GKB database download January 6, 2013)23 were specifically called in WGS data from all participants (Methods section in the Supplement).
Inherited Disease–Risk Assessment
The workflow for inherited disease risk and carrier status evaluation and a sample of a medical genomics report are described in the Methods section in the Supplement. Basic annotation of genetic variants were found in 2725 male and 2716 female inherited disease genes, bioinformatic prioritization of medical genetic findings were performed using Sequence to Medical Phenotypes, a phenotype identification software program (http://ashleylab.stanford.edu/tools/tools_synthetic/stmp), which prioritizes previously reported and novel genetic variants in inherited disease genes based on allele frequency, functional class, and consensus evidence for evolutionary constraint or conservation and predicted pathogenicity. Genetic variants in 56 genes that have been recommended by the American College of Medical Genetics and Genomics [ACMG] for pathogenic variant discovery, review, and reporting WGS/WES (ACMG-reportable genes) were also identified.12
Variants identified by the phenotype identification software program for curation were reviewed blind to patient phenotype by a multidisciplinary genomics team composed of 3 genetic counselors (M.E.G., K.E.O., and C.C.), 3 physician-informaticists (F.E.D., E.A.A., and M.T.W.), and 1 molecular pathologist (J.D.M.). This curation classified candidate variants with respect to variant- and gene-level evidence for pathogenicity and characteristics of the clinical phenotype for reporting. One of 9 summary pathogenicity classification statements (Table 1S in the Supplement) was generated for each genetic variant. Using these pathogenicity classifications, the genomics team members determined the suitability for reporting and assigned a reporting category (Box 2) adapted from the ACMG standards to interpret and report genetic sequence variations.24 At least 2 team members reviewed all variant classifications. For participants sequenced on both platforms, only variants with concordant genotype calls were curated and reported. We estimated upstream costs of sequencing and curation at $10 000 based on Illumina Clinical WGS service cost in December 2012 and at $50 per hour for curation based on local salary rates for licensed genetic counselors and commercial contracting rates.
Box 2. Reporting Criteria for Inherited Disease Risk Variantsa.
Reported disease-associated mutations (adapted from ACMG category 1: “Sequence variation is previously reported and is a recognized cause of the disorder”)b
Mutations convincingly segregating with inherited disease in 1 or more families and are absent or observed very infrequently in ethnically matched population controls.
Mutations observed in multiple unrelated probands with inherited disease and are absent or observed very infrequently in ethnically matched population controls.
Mutations observed in single probands with inherited disease, with functional evidence of pathogenicity and are absent or observed very infrequently in ethnically matched population controls.
Rare, expected pathogenic variants (adapted from ACMG category 2: “Sequence variation is previously unreported and is of the type which is expected to cause the disorder”)
Rare or novel nonsense, stop loss, splice-disrupting, or frameshift insertion/deletion variants affecting the majority of protein coding transcripts of genes associated with inherited disease.
Genetic variants of uncertain significance (adapted from ACMG category 3: “Sequence variation is previously unreported and is of the type which may or may not be causative of the disorder”)
Rare or novel missense and non-frameshifting insertion/deletion variants with evolutionary conservation or biochemical prediction support for predicted pathogenicity, and variants of all types previously reported as disease causing inwhich primary literature reports of pathogenicity provide conflicting or incomplete evidence of causality. These variants must occur in inherited disease genes in which there is evidence that variants of the type discovered in study participants are disease causing.
Abbreviations: ACMG, American College of Medical Genetics and Genomics.
aACMG categories as described by Richards et al.24
bFrequency is defined by an allele frequency consistent with mode of inheritance and population prevalence of disease.
Estimation of the Population Prevalence of Genetic Variants in ACMG-Reportable Genes
To assess the prevalence of rare, potentially pathogenic genetic findings in ACMG reportable genes in a large cohort selected without respect to phenotype, we estimated the number of candidate reportable findings in a new analysis of genetic variant data from 1092 participants sequenced as part of the 1000 Genomes Project.25 The genetic variants that were predicted to cause loss of function or that were described as “disease- causing mutations” by the HGMD among the 56 ACMG reportable genes were extracted from existing variant calls (http://www.1000genomes.org) for prevalence estimates.
Genetic Risk of Cardiometabolic Disease
Using methods described by Knowles et al,26 percentile genetic risk scores were generated for diabetes mellitus type 2 and coronary artery disease using genotypes and odds ratios for 118commonsingle nucleotide variants with replicated disease associations (Tables S3-S6 in the Supplement).19-22
Genetic Drug Response Predictions
Genotype and haplotype data at loci with reported clinical drug-response predictions were intersected with annotations cataloged in the PharmGKB knowledge base.23 Thirty drug-response associations with level of evidence of 1B or higher (replicated associations, implemented ina major health system or pharmacogenomics research network site, or endorsed in a medical society guideline) were reported, as well as drug dosing and administration schedules recommended by the Clinical Pharmacogenomics Implementation Consortium guidelines for simvastatin, warfarin, clopidogrel, thiopurines, and codeine.27-31
Concordance in Inherited Disease-Risk Classification
Eighteen randomly chosen genetic variants prioritized for curation were blindly reclassified by 2 or more members of the genomics team. All raters had prior experience with WGS variant interpretation in research and clinical contexts. Interrater agreement in assigning genetic variant summary classifications and suitability for reporting was calculated using the Gross κ statistic as described in R version 2.11.32
Burden and Cost of Clinical Follow-up
Three academic primary care physicians and 2 academic medical geneticists separately reviewed all 12 complete personal medical genomics reports and proposed clinical follow-up based on the genetic findings alone. Two of the primary care physicians had no previous experience with genomic medicine or genetics training. One primary care physician had formal research genetics training. None of the reviewing physicians were involved in primary data generation, analysis, review of variants, or generation of the reports. The financial cost of proposed clinical follow-up was estimated using the Centers for Medicare & Medicaid Services calendar-year 2013 national physician fee schedule for nonfacility billing and used the 2013 Clinical Laboratory Fee Schedule national midpoint( Healthcare Common Procedure Coding System [Table S7 in the Supplement]). Costs for a high-complexity established patient-physician encounter and a 120-minute follow-up genetic counseling encounter were added to all participant-specific costs to estimate the total cost. Interrater agreement of suitability of genetic findings for clinical follow-up was calculated using the Fleiss κ for multiple raters33 in R version 2.11. Throughout, a P value of less than .01 was considered statistically significant.
Results
Study Participants and WGS
The characteristics of the study participants and a summary of WGS are presented in Table 1. Five participants were white, 7were East Asian, and 7werewomen.Depending on sequencing platform, a median of 10% (Illumina Inc; range, 5%-34%) to 19% (Complete Genomics Inc; range, 18%-21%) of genes associated with inherited disease and a median of 9% (Illumina Inc; range, 2%-27%) to 17% (Complete Genomics Inc; range, 17%-19%) of ACMG-reportable genes were not covered at a minimum threshold for genetic variant discovery (Figure 2 and Figures 1S 2S in the Supplement).We were able to call genotypes with 99.9% confidence for 99% to 100% of previously reported genetic variants in inherited disease genes, genetic variants used to assess genetic risk of complex cardiometabolic phenotypes, and genetic variants with clinical drug response associations.
Table 1.
Study Participants and Sequencing Summary
| Characteristics | Median (Range) |
|---|---|
| Participant characteristics | |
| Age, y | 53 (42-85) |
| Women, No./Total | 7/12 |
| Self-reported ethnicity, No./Total | |
| White | 5/12 |
| East Asian | 7/12 |
| Sequencing coverage and depth | |
| Illumina Inc, No. of patients | 12 |
| Haploid coverage depth | 50 (38-62) |
| Genome covered, % | 95.0 (94.8-95.6) |
| Complete Genomics Inc, No. of participants | 9 |
| Haploid coverage depth | 61 (61-62) |
| Genome covered, % | 96.6 (96.3-96.9) |
| Single nucleotide variants | |
| All | 2 403 504 (2 313 092-2 508 838) |
| Novel | 39 582 (22 861-65 818) |
| Exonic | 15 312 (14 933-16 394) |
| Stoploss or stopgain | 74 (66-82) |
| Splice disrupting | 419 (368-511) |
| Missense | 5447 (5280-5800) |
| Small insertion/deletion variants | |
| All | 583 273 (558 693-676 634) |
| Novel | 31 653 (27 350-73 287) |
| Exonic | 355 (231-389) |
| Frameshift | 91 (63-101) |
| Structural variants | |
| All | 2790 (2220-2900) |
| Novel | 88 (77-123) |
| Exonic | 120 (111-130) |
Figure 2. Missing Coverage of 56 Genes the ACMG Recommends for Pathogenic Variant Discovery, Review, and Reporting in WGS.
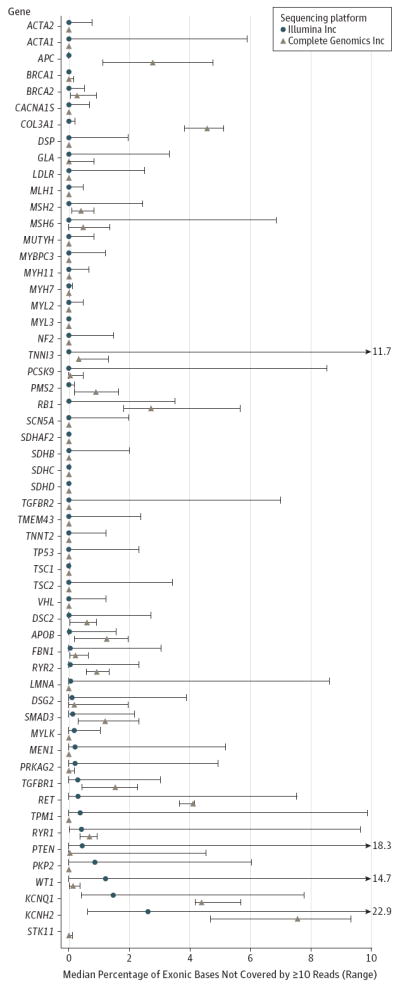
The percentage of exonic bases of genes for pathogenic variant discovery and reporting in genome and exome sequencing that were not covered by 10 or more reads. ACMG indicates American College of Medical Genetics and Genomics; WGS, whole-genome sequencing.
The genotype concordance between sequencing platforms was high for common genetic variants (99% overall, Figure S3 in the Supplement). Similarly, genotype concordance was high for single nucleotide variants overall, in protein coding regions of the genome, and among candidate variants for inherited disease risk (Figure 3). However, genotype concordance for small insertion/deletion variants was moderate overall (median, 57%; range, 53%-59%) and in protein coding regions of the genome (median, 66%; range, 64%-70%) but was substantially lower among genetic variants that were candidates for inherited disease risk (median, 33%; range, 10%- 75%).
Figure 3. Genotype Concordance Between Whole-Genome Sequencing Platforms in 12 Participants Sequenced in the Genomic Medicine Application Pilot Project.
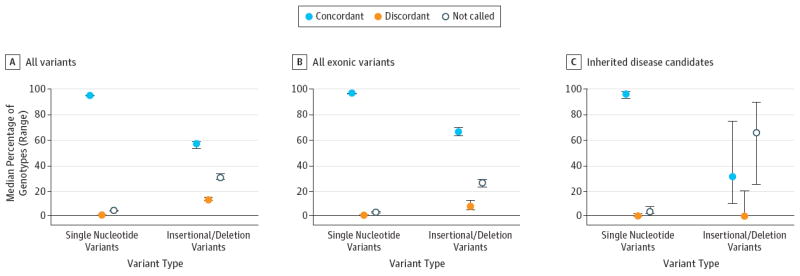
A, Genotype concordance for all variant positions. B, Genotype concordance for variants in protein-coding regions. C, Genotype concordance for candidate inherited disease risk variants. Concordant refers to sequence variants with the same alleles and zygosity (ie, homozygous calls in both platforms) called by both platforms; discordant, refers to sequence variants called by both platforms but with different alleles or zygosity; and not called, sequence variants identified using the Illumina platform for which no variant genotype call was made by the Complete Genomics platform.
Resource Needs for WGS Interpretation
For each participant, the phenotype software program identified 89 to 125 novel and rare single nucleotide variants or insertion/ deletion variants and 0 to 4 large structural variants (90-127 total genetic variants) that were candidates for curation with respect to personal risk and carrier status for inherited disease (Table 2). Review of evidence for pathogenicity, including time required for literature search (finding literature cataloged in mutation databases and performing independent PubMed and Google searches for the genetic variant and gene) and secondary review, required a median of 54 minutes (range, 5-223minutes) per genetic variant. We estimated that the median cost for sequencing and variant interpretation was $ 14 815 (range, $14 050-$15 715), plus the costs of computing infrastructure and data storage.
Table 2.
Inherited Disease Risk and Carrier Status Findings in Whole-Genome Sequence Data
| No. of Variants per Participant, Median (Range) | |
|---|---|
| Candidate variants identified for manual curation | 108 (90-127) |
| Candidate previously reported or potentially pathogenic variants in ACMG-reportable genes | 3 (1-7) |
| Reportable variants associated with personal inherited disease riska | 5 (2-6) |
| Reported disease-associated mutations | 0 (0-2) |
| Rare, expected pathogenic variants | 0 (0-1) |
| Genetic variants of uncertain significance | 3 (1-6) |
| Reportable variants with implications for carrier status for inherited diseasea | 13 (8-18) |
| Reported disease-associated mutations | 2 (0-4) |
| Rare, expected pathogenic variants | 2 (1-4) |
| Genetic variants of uncertain significance | 9 (4-12) |
Abbreviation: ACMG, American College of Medical Genetics and Genomics.
Reportable according to reporting criteria specified in Box 2 and classification criteria (eTable 1 in Supplement).
Interrater Agreement in Inherited Disease Gene Variant Classification
Independent classification of 18 randomly chosen genetic variants with potential personal risk or carrier status implications for inherited disease (Tables S8-S9 in the Supplement) by 6 genomics professionals resulted in moderate interrater agreement about genetic variant pathogenicity (Gross κ, 0.52; 95%CI, 0.40-0.64) and close interrater agreement about suitability for reporting (Gross κ, 0.83; 95% CI, 0.73-0.93).
In the cohort, we found 574total genetic variants that were cataloged in HGMD and prioritized for curation by the genomics team (Table 3). Among these genetic variants, we identified 1 variant classified as a disease-causing mutation according to the HGMD and 18 of other classifications that were the most common allele in an ethnically matched population. The genomics team reclassified 8 of 68 genetic variants (11.8%) that HGMD lists as “disease causing mutations” to be “very likely pathogenic” and reclassified 47of 68 (69%) as variants of uncertain or lesser significance.
Table 3.
Agreement Between Human Gene Mutation Database and Multidisciplinary Genomics Team Classification for 574 Unique, Previously Described, Inherited Disease Risk Candidates Found in 12 Participants by Whole-Genome Sequencing
| HGMD Genomics Teama | No. (%) of Total Variants for Each HGMD Classification | Total | |||||
|---|---|---|---|---|---|---|---|
| Disease-Causing Mutation | Likely Disease Causing Mutation | Disease Associated Functional Polymorphism | Disease Associated Polymorphism | Functional Polymorphism | Frame Shift or Truncating Variantb | ||
| Not associated with an inherited disease | 1 (1.4) | 44 (67.8) | 111 (84.1) | 176 (75.9) | 64 (88.9) | 0 | 396 |
| Likely benign, strong evidence | 1 (1.4) | 0 | 5 (3.8) | 19 (8.2) | 1 (1.4) | 5 (100.0) | 31 |
| Some evidence suggesting benign | 17 (25.0) | 10 (15.4) | 4 (3.0) | 17 (7.3) | 3 (4.2) | 0 | 51 |
| Uncertain significance | 29 (42.6) | 9 (13.8) | 8 (6.1) | 17 (7.3) | 1 (1.4) | 0 | 64 |
| Some evidence suggesting pathogenic | 4 (5.9) | 1 (1.5) | 2 (1.5) | 2 (0.9) | 3 (4.2) | 0 | 12 |
| Likely pathogenic | 8 (11.8) | 0 | 0 | 1 (0.4) | 0 | 0 | 9 |
| Very likely pathogenic | 8 (11.8) | 1 (1.5) | 2 (1.5) | 0 | 0 | 0 | 11 |
| Total | 68 | 65 | 132 | 232 | 72 | 5 | 574 |
Abbreviations: HGMD, Human Gene Mutation Database.
All variants cataloged in HGMD are, by definition, previously reported. Thus, the categories from Table S1 (in the Supplement) “Novel, predicted benign,” “Novel, predicted damaging by one program”, and “Novel, predicted damaging by two or more programs” do not apply and are not displayed.
Polymorphic or rare variant reported in the literature (eg, detected in the process of whole-genome/exome screening) that is predicted to truncate or otherwise alter the gene product but with no disease association reported as yet.
Burden of Inherited Disease Gene Variants
According to classification criteria (Table S1 in the Supplement), the reporting criteria (Box 2), and curation by the genomics team, 11%to 25%of genetic variants identified by the phenotype identification software program for curation were reportable. The genomics team classified 26 of 68 variants (38%) that were described in HGMD as “disease-causing mutations” as reportable; 9 of these were reported as variants of uncertain significance. Overall, 2 to 6 genetic variants with potential personal disease-risk implications were reported to each participant, including 1 to 5 associated with diseases with reported adult onset. Eight to 18 genetic variants were found in each participant with potential carrier status implications (Table 2).
Burden of Clinically Significant Findings in ACMG Reportable Genes
Among the ACMG-reportable genes, we found 12 to 20 genetic variants per participant that met our criteria for curation. This set included 1 or more previously reported disease associated genetic variants in all 12 participants and at least 1 rare, expected pathogenic genetic variant in 9 participants. Seven participants harbored novel, expected pathogenic variants in these genes that could not be replicated by the second sequencing platform or that were found at a frequency indicating systematic sequencing artifact or common polymorphism. After exclusion of these genetic variants, 1 to 7 genetic variants remained for curation per individual (Table 2), including 1 or more cataloged as disease causing in 5 and likely disease causing in 9 participants, according to the HGMD classification (Table S10 in the Supplement). To assess the generalizability of these findings in a larger cohort ascertained without respect to clinical phenotype, we estimated the population prevalence of mutations cataloged as “disease causing” in the HGMD in 1092 participants who underwent WGS and WES as part of the 1000Genomes Project.25 In this cohort, 373 of 1092 study participants (34.2%) harbored a very rare (allele frequency, <0.5%) genetic variant classified by the HGMD as disease causing, and 40 (3.7%) harbored a very rare genetic variant predicted to cause loss of ACGM-reportable gene function (Table S11 in the Supplement).
After the genomics team’s curation of genetic variants that had been discovered in the 12 participants in ACMG reportable genes, we determined that 1 inherited disease-risk variant was very likely pathogenic, an open-reading-frame-shifting deletion of 19 nucleotides inBRCA1 (rs80359876; NCBI Entrez Gene 672) with strong evidence to support risk of hereditary breast and ovarian cancer.34-36 This finding, which was confirmed via capillary sequencing in a commercial clinical genetic testing laboratory, was discovered ina white woman who did not meet family history referral criteria. Evaluation by an independent clinical team led the participant to undergo prophylactic bilateral salpingooophorectomy and intensified imaging– based breast cancer screening per the National Comprehensive Cancer Network guidelines for carriers of pathogenic BRCA1 mutations.37 Four additional participants carried genetic variants cataloged as disease causing in the HGMD that the genomics team had reclassified as reportable variants of uncertain significance.
Genetic Drug–Response Assessment
According to Clinical Pharmacogenomics Implementation Consortium guidelines, 11 individuals carried 1ormoregenetic variants with an associated clinical guideline for change in drug dosing or administration (Table S12 in the Supplement); each individual carried 1 to 3 such variants. Three to 10 additional genetic variants were found in each individual with drug-response associations supported by PharmGKB evidence level 1B or higher (Figure S4 in the Supplement).
Clinical Follow-up Proposed by Medical Professionals
Review of medical genomics reports by 3 primary care physicians prompted consideration of a median of 1 to 3 initial follow- up referrals and diagnostic tests per participant. The median estimated total costs per person, which includes a $340 high complexity established patient visit and a 120-minute genetic counseling session for all participants, were $351 to $776.
An independent review of reports by 2 medical geneticists prompted consideration of a median of 3 initial follow-up tests and referrals at median estimated total costs per person of $626 to $773 (Table S13 in the Supplement). Interrater agreement about suitability for follow-up of WGS findings was fair for all reported variants (Fleiss κ, 0.24; P < .001) and variants with potential personal inherited disease risk implications (κ, 0.22; P<.001), but only slight for variants with potential inherited carrier status implications (κ, 0.08; P = .007) and poor for cardiometabolic disease risk scores (κ, −0.03; P = .62).
Discussion
Whole-genome sequencing and WES are increasingly used in clinical medicine and have the potential not only to answer specific diagnostic questions but also to uncover clinically important genetic findings unrelated to the primary indication for sequencing. Whole-exome sequencing has been the primary sequencing modality in clinical genetics and in surveys of reportable genetic findings in unselected participants.38,39 Whole-genome sequencing is expected to provide superior coverage of certain genomic regions, including intronic and other noncoding regions associated with inherited disease, noncoding pharmacogenomic alleles, and complex disease-risk alleles.
However, our initial clinical experience with this technology illustrates several challenges to clinical adoption of WGS. First, our results suggest that although analytical validity of WGS is improving, technical challenges to sensitive and accurate assessment of individual genetic variation remain. Depending on sequencing platform, between 10% and 19% of inherited disease genes, including 9% to 17% that are ACMG reportable genes,12 were not consistently covered at a read depth that was sufficient for a comprehensive survey of genetic variants. We observed high overall genotype concordance between sequencing platforms, and single nucleotide variant concordance was high among candidate-inherited disease- risk variants. However, although overall concordance between insertion/deletion variants was higher than previously reported,40 fewer than one-third of insertion/deletion variants in inherited disease genes were confirmed by the second sequencing platform. This finding suggests that genetic variants of a type that are quite likely to be pathogenic are more often inconsistently identified. Other investigators have made similar observations about potential loss-of-function mutations.9,41 This may be particularly pronounced in individuals with low prior probability of inherited disease or when no clear diagnostic end point is pursued. Thus, although WGS is increasingly inexpensive and rapid, there is a persistent need for technical confirmation of potentially significant findings and supplementation with other genetic assays to achieve clinical grade sensitivity and specificity.
Human resource needs for full clinical interpretation of WGS data remain considerable, and much uncertainty remains in classification of potentially pathogenic genetic variants. Discovery and evaluation of genome-wide reportable genetic findings, even after stringent bioinformatic filtering, required curation (literature review and secondary assessment of pathogenicity) of approximately 100 findings in each participant. This included a median of 3 genetic findings per participant that required curation per published recommendations for incidental findings in WES/WGS.12 In 4 of 12 participants, mutations reported as disease causing in mutation databases among ACMG-reportable genes were found and determined by the genomics team to be of uncertain or lesser significance; a similar frequency of such findings was discovered in more than 1000 individuals selected without respect to phenotype. Curation required nearly an hour of time per genetic variant in our study and resulted in reclassification of 69% of genetic variants cataloged in mutation databases as disease causing mutations to variants of uncertain or lesser significance. This finding is similar to previous reports of high rates of classification discordance between existing disease mutation databases and independent raters, and highlights the persistent need for recuration of putatively pathogenic genetic variants prior to clinical action.38,42,43
Our analysis of classification concordance among genomics professionals revealed very good interrater agreement about suitability for reporting but less good interrater agreement of genetic variant classification. This finding indicates classification uncertainty even when published evidence is concurrently reviewed by trained raters and specific criteria for variant pathogenicity are provided. Although general guidelines exist for interpretation of sequence variants,24 little consensus exists beyond expert opinion for distinguishing clearly pathogenic from less certainly pathogenic variation, leaving curators, laboratories, and clinicians to rely on often subjective assessments of genetic variant pathogenicity. Until uniformly accepted guidelines and generally accessible and reliable data sources exist for clinical interpretation of genetic variants, pathogenicity assessment will likely continue to involve substantial uncertainty.
Following curation, 2 to 6 genetic variants with potential personal inherited disease risk implications were identified in each participant. The majority of these genetic variants were associated with reportedly adult-onset disease. Most were previously unreported but of similar type to previously reported pathogenic variants, eg, a novel protein-truncating mutation in the gene POLG2 (HGNC 9180), a gene associated with progressive external ophthalmoplegia.44 Prior estimates, extrapolated from the prevalence of mutations associated with familial cancer, proposed that the population prevalence of well-established inherited disease risk findings in the ACMG-reportable genes may be very low (1%).12,39 Recent estimates based on WES data in individuals largely unselected with respect to clinical phenotype suggest a prevalence of such findings of approximately 3% in 52 of these 56 genes.38
In our study, which in contrast to these prior studies involved return of actionable results to the treating physician and participant and clinical action where appropriate, we determined that 1 participant harbored such a genetic variant. This unanticipated finding of germline risk for hereditary breast and ovarian cancer prompted risk-reducing surgery and intensified cancer screening in the absence of a family or personal history of breast or ovarian cancer. The practical burden of reportable genetic findings from WGS/WES is likely to vary substantially with institutional sequencing expertise, reliance on pathogenicity classifications in mutation databases, and access to and methods for evaluation of published evidence.
The prognostic meaning of reportable genetic discoveries in a patient population with a low prior probability of inherited disease remains unclear and will only be determined by iterative clinical evaluation. Our assessment of the burden and estimated costs of initial clinical follow-up proposed by 5 physicians revealed estimated costs of generally less than$1000 per person. Interrater agreement about suitability of follow- up for genetic variants was at best fair, with the highest agreement for WGS findings with potential personal inherited disease risk implications, and lowest agreement for cardiometabolic disease-risk scores. The burden and costs of this initial and subsequent clinical care are likely to depend greatly on individual practice patterns in the face of uncertain findings and the results of initial clinical evaluation.
Our study has limitations. Substantial challenges exist to clinical validation of these tools because of the vast number of decision nodes in such pipelines and lack of clear gold standards for genome interpretation. The study did not capture clinical evaluations performed by primary care physicians and consultants downstream of initial clinical tests and referrals, such as imaging and risk-reducing surgery recommended by an initial referral to an oncologist. The costs of initial clinical follow-up may be higher than our estimates in some clinical contexts.
Conclusions
In this exploratory study of 12 volunteer adults, the use of WGS was associated with incomplete coverage of inherited disease genes, low reproducibility of genetic variation with the highest potential clinical effects, and uncertainty about clinically reportable WGS findings. Despite this, in certain cases WGS will identify clinically actionable genetic variants warranting early medical intervention. These issues should be considered when determining the role of WGS in clinical medicine.
Supplementary Material
Acknowledgments
Funding/Support: Supported in part by grants NHLBI T32 HL094274-01A2 (Dr Dewey), NHGRI P50 HG003389-05 (Ms Ormond), NIGMS R24 GM61374 (Drs Klein, Whirl-Carrillo, and Altman), NIH R01 GM079719 9 (Dr Butte), NHGRI U01 HG004267-03 (Dr Snyder), and Director’s New Innovator Award DP2 OD004613, RO1 HL113006, UO1 HG007436, XO1 HL115206, and HL094274 from the National Institutes of Health and by a grant from the Breetwor Family Foundation (Dr Ashley) and a grant from the LeDucq Foundation (Dr Quertermous).
Role of the Sponsor: The sponsors had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Footnotes
Author Contributions: Dr Dewey had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. Drs Dewey and Pan and Ms Grove contributed equally to the manuscript. Drs Ashley and Quertermous contributed equally to the manuscript and study.
Study concept and design: Dewey, Grove, Bernstein, Ormond, Ioannidis, Altman, Snyder, Ashley, Quertermous.
Acquisition of data: Dewey, Grove, Bernstein, Chaib, Kingham, Ford, Boxer, Yeung, Assimes, Snyder, Ashley, Quertermous.
Analysis and interpretation of data: Dewey, Grove, Pan, Goldstein, Merker, Goldfeder, Enns, David, Pakdaman, Ormond, Caleshu, Klein, Whirl-Carrillo, Sakamoto, Wheeler, Butte, Boxer, Ioannidis, Assimes, Snyder, Ashley, Quertermous.
Drafting of the manuscript: Dewey, Klein, Butte, Ford, Ashley.
Critical revision of the manuscript for important intellectual content: Dewey, Grove, Pan, Goldstein, Bernstein, Chaib, Merker, Goldfeder, Enns, David, Pakdaman, Ormond, Caleshu, Kingham, Klein, Whirl-Carrillo, Sakamoto, Wheeler, Ford, Boxer, Ioannidis, Yeung, Altman, Assimes, Snyder, Ashley, Quertermous.
Statistical analysis: Dewey, Pan, Goldstein, Ioannidis, Ashley.
Obtained funding: Snyder, Ashley, Quertermous.
Administrative, technical, and material support: Grove, Chaib, Merker, Caleshu, Klein, Ford, Boxer, Snyder, Quertermous.
Study supervision: Bernstein, Butte, Ford, Yeung, Altman, Assimes, Snyder, Ashley, Quertermous.
Additional Contributions: We thank the study participants for their involvement. We also thank Amin Zia, PhD, from the Stanford University Department of Genetics for commentary regarding structural variant identification and James R. Priest, MD, from the Stanford University Division of Pediatric Cardiology for helpful commentary on the manuscript. Neither investigator was compensated for their contribution.
Conflict of Interest Disclosures: All authors have completed and submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Dr Dewey reported that he is a stockholder and member of the scientific advisory board of Personalis Inc, a privately held genome interpretation company, and receives royalties for patented technology related to genome sequencing.Ms Grove reported that she has received speaker’s fees from Illumina Inc. Dr Klein reported that that she is a member of the scientific advisory board for Coriell Inc. Mss Caleshu and Ormond and Dr Wheeler reported that they receive royalties for patented technology related to genome sequencing. Drs Butte, Altman, and Snyder and Mr Ashley are founders, stockholders, and members of the scientific advisory board of Personalis Inc and receive royalties for patents related to genome sequencing. Dr Butte reported that he is a stockholder and member of the scientific advisory board of NuMedii Inc; consultant to Lilly, Regeneron, Johnson & Johnson, Roche, Geisinger, Verinata, Pfizer, and Samsung; has received speaker’s fees from Pfizer, Lilly, Siemens, Bristol-Myers Squibb, and Genentech; and holds stock in Carmenta, Eceos, Assay Depot, and Genstruct/Selva. Dr Snyder reported that he is a member of the scientific advisory board and stockholder of Genapsys Inc. Dr Quertermous reported that he is a member of the scientific advisory board of Aviir Inc. No other disclosures were reported.
Supplemental content at jama.com
References
- 1.Choi M, Scholl UI, Ji W. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci U S A. 2009;106(45):19096–19101. doi: 10.1073/pnas.0910672106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Worthey EA, Mayer AN, Syverson GD, et al. Making a definitive diagnosis: successful clinical application of whole exome sequencing in a child with intractable inflammatory bowel disease. Genet Med. 2011;13(3):255–262. doi: 10.1097/GIM.0b013e3182088158. [DOI] [PubMed] [Google Scholar]
- 3.Gahl WA, Markello TC, Toro C. The National Institutes of Health Undiagnosed Diseases Program: insights into rare diseases. Genet Med. 2012;14(1):51–59. doi: 10.1038/gim.0b013e318232a005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ng SB, Buckingham KJ, Lee C, et al. Exome sequencing identifies the cause of a mendelian disorder. Nat Genet. 2010;42(1):30–35. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Musunuru K, Pirruccello JP, Do R, et al. Exome sequencing, ANGPTL3 mutations, and familial combined hypolipidemia. N Engl J Med. 2010;363(23):2220–2227. doi: 10.1056/NEJMoa1002926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bamshad MJ, Ng SB, Bigham AW, et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet. 2011;12(11):745–755. doi: 10.1038/nrg3031. [DOI] [PubMed] [Google Scholar]
- 7.Lupski JR, Reid JG, Gonzaga-Jauregui C, et al. Whole-genome sequencing in a patient with Charcot-Marie-Tooth neuropathy. N Engl J Med. 2010;362(13):1181–1191. doi: 10.1056/NEJMoa0908094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnson JO, Mandrioli J, Benatar M, et al. ITALSGEN Consortium. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron. 2010;68(5):857–864. doi: 10.1016/j.neuron.2010.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.MacArthur DG, Balasubramanian S, Frankish A, et al. 1000 Genomes Project Consortium. A systematic survey of loss-of-function variants in human protein-coding genes. Science. 2012;335(6070):823–828. doi: 10.1126/science.1215040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xue Y, Chen Y, Ayub Q, et al. 1000 Genomes Project Consortium. Deleterious- and disease-allele prevalence in healthy individuals: insights from current predictions, mutation databases, and population-scale resequencing. Am J Hum Genet. 2012;91(6):1022–1032. doi: 10.1016/j.ajhg.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ball MP, Thakuria JV, Zaranek AW, et al. A public resource facilitating clinical use of genomes. Proc Natl Acad Sci U S A. 2012;109(30):11920–11927. doi: 10.1073/pnas.1201904109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Green RC, Berg JS, Grody WW, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 2013;15(7):565–574. doi: 10.1038/gim.2013.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ashley EA, Butte AJ, Wheeler MT, et al. Clinical assessment incorporating a personal genome. Lancet. 2010;375(9725):1525–1535. doi: 10.1016/S0140-6736(10)60452-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dewey FE, Chen R, Cordero SP, et al. Phased whole-genome genetic risk in a family quartet using a major allele reference sequence. PLoS Genet. 2011;7(9):e1002280. doi: 10.1371/journal.pgen.1002280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bentley DR, Balasubramanian S, Swerdlow HP, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456(7218):53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Drmanac R, Sparks AB, Callow MJ, et al. Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science. 2010;327(5961):78–81. doi: 10.1126/science.1181498. [DOI] [PubMed] [Google Scholar]
- 17.Stenson PD, Mort M, Ball EV, et al. The Human Gene Mutation Database: 2008 update. Genome Med. 2009;1(1):13. doi: 10.1186/gm13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Riggs ER, Wain KE, Riethmaier D, et al. Hum Mutat; Towards a universal clinical genomics database: the 2012 International Standards for Cytogenomic Arrays Consortium meeting; 2013. pp. 915–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deloukas P, Kanoni S, Willenborg C, et al. CARDIoGRAMplusC4D Consortium; DIAGRAM Consortium; CARDIOGENICS Consortium; MuTHER Consortium; Wellcome Trust Case Control Consortium. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet. 2013;45(1):25–33. doi: 10.1038/ng.2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu X, Wang L, Chen S, et al. Coronary Artery Disease Genome-Wide Replication and Meta-analysis (CARDIoGRAM) Consortium. Genome-wide association study in Han Chinese identifies four new susceptibility loci for coronary artery disease. Nat Genet. 2012;44(8):890–894. doi: 10.1038/ng.2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morris AP, Voight BF, Teslovich TM, et al. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet. 2012;44(9):981–990. doi: 10.1038/ng.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cho YS, Chen CH, Hu C, et al. Meta-analysis of genome-wide association studies identifies eight new loci for type 2 diabetes in east Asians. Nat Genet. 2012;44(1):67–72. doi: 10.1038/ng.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Whirl-Carrillo M, McDonagh EM, Hebert JM, et al. Pharmacogenomics knowledge for personalized medicine. Clin Pharmacol Ther. 2012;92(4):414–417. doi: 10.1038/clpt.2012.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Richards CS, Bale S, Bellissimo DB, et al. Molecular Subcommittee of the ACMG Laboratory Quality Assurance Committee. ACMG recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007. Genet Med. 2008;10(4):294–300. doi: 10.1097/GIM.0b013e31816b5cae. [DOI] [PubMed] [Google Scholar]
- 25.Abecasis GR, Auton A, Brooks LD, et al. 1000 Genomes Project Consortium. An integrated map of genetic variation from 1092 human genomes. Nature. 2012;491(7422):56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Knowles JW, Assimes TL, Kiernan M, et al. Randomized trial of personal genomics for preventive cardiology: design and challenges. Circ Cardiovasc Genet. 2012;5(3):368–376. doi: 10.1161/CIRCGENETICS.112.962746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Relling MV, Gardner EE, Sandborn WJ, et al. Clinical pharmacogenetics implementation consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing: 2013 update. Clin Pharmacol Ther. 2013;93(4):324–325. doi: 10.1038/clpt.2013.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wilke RA, Ramsey LB, Johnson SG, et al. Clinical Pharmacogenomics Implementation Consortium (CPIC). The clinical pharmacogenomics implementation consortium: CPIC guideline for SLCO1B1 and simvastatin-induced myopathy. Clin Pharmacol Ther. 2012;92(1):112–117. doi: 10.1038/clpt.2012.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Crews KR, Gaedigk A, Dunnenberger HM, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guidelines for codeine therapy in the context of cytochrome P450 2D6 (CYP2D6) genotype. Clin Pharmacol Ther. 2012;91(2):321–326. doi: 10.1038/clpt.2011.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnson JA, Gong L, Whirl-Carrillo M, et al. Clinical Pharmacogenetics Implementation Consortium. Clinical Pharmacogenetics Implementation Consortium Guidelines for CYP2C9 and VKORC1 genotypes and warfarin dosing. Clin Pharmacol Ther. 2011;90(4):625–629. doi: 10.1038/clpt.2011.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scott SA, Sangkuhl K, Gardner EE, et al. Clinical Pharmacogenetics Implementation Consortium. Clinical Pharmacogenetics Implementation Consortium guidelines for cytochrome P450-2C19 (CYP2C19) genotype and clopidogrel therapy. Clin Pharmacol Ther. 2011;90(2):328–332. doi: 10.1038/clpt.2011.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gross ST. The kappa coefficient of agreement for multiple observers when the number of subjects is small. Biometrics. 1986;42(4):883–893. [PubMed] [Google Scholar]
- 33.Fleiss JL. Measuring nominal scale agreement among many raters. Psychol Bull. 1971;76(5):378–382. [Google Scholar]
- 34.Couch FJ, DeShano ML, Blackwood MA, et al. BRCA1 mutations in women attending clinics that evaluate the risk of breast cancer. N Engl J Med. 1997;336(20):1409–1415. doi: 10.1056/NEJM199705153362002. [DOI] [PubMed] [Google Scholar]
- 35.Baudi F, Quaresima B, Grandinetti C, et al. Evidence of a founder mutation of BRCA1 in a highly homogeneous population from southern Italy with breast/ovarian cancer. Hum Mutat. 2001;18(2):163–164. doi: 10.1002/humu.1167. [DOI] [PubMed] [Google Scholar]
- 36.Quaresima B, Romeo F, Faniello MC, et al. BRCA1 5083del19 mutant allele selectively up-regulates periostin expression in vitro and in vivo. Clin Cancer Res. 2008;14(21):6797–6803. doi: 10.1158/1078-0432.CCR-07-5208. [DOI] [PubMed] [Google Scholar]
- 37.Daly MB, Axilbund JE, Buys S, et al. National Comprehensive Cancer Network. Genetic/familial high-risk assessment: breast and ovarian. J Natl Compr Canc Netw. 2010;8(5):562–594. doi: 10.6004/jnccn.2010.0043. [DOI] [PubMed] [Google Scholar]
- 38.Dorschner MO, Amendola LM, Turner EH, et al. National Heart, Lung, and Blood Institute Grand Opportunity Exome Sequencing Project. Actionable, pathogenic incidental findings in 1,000 participants’ exomes. Am J Hum Genet. 2013;93(4):631–640. doi: 10.1016/j.ajhg.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Johnston JJ, Rubinstein WS, Facio FM, et al. Secondary variants in individuals undergoing exome sequencing: screening of 572 individuals identifies high-penetrance mutations in cancer-susceptibility genes. Am J Hum Genet. 2012;91(1):97–108. doi: 10.1016/j.ajhg.2012.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lam HY, Clark MJ, Chen R, et al. Performance comparison of whole-genome sequencing platforms. Nat Biotechnol. 2012;30(1):78–82. doi: 10.1038/nbt.2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.MacArthur DG, Tyler-Smith C. Loss-of-function variants in the genomes of healthy humans. Hum Mol Genet. 2010;19(R2):R125–R130. doi: 10.1093/hmg/ddq365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Berg JS, Adams M, Nassar N, et al. An informatics approach to analyzing the incidentalome. Genet Med. 2013;15(1):36–44. doi: 10.1038/gim.2012.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bell CJ, Dinwiddie DL, Miller NA, et al. Carrier testing for severe childhood recessive diseases by next-generation sequencing. Sci Transl Med. 2011;3(65):65ra64. doi: 10.1126/scitranslmed.3001756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hudson G, Chinnery PF. Mitochondrial DNA polymerase-gamma and human disease. Hum Mol Genet. 2006;15(spec No 2):R244–R252. doi: 10.1093/hmg/ddl233. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.