

Abstract
The growth hormone receptor was the first cytokine receptor to be cloned and crystallized, and provides a valuable exemplar for activation of its cognate kinase, JAK2. We review progress in understanding its activation mechanism, in particular the molecular movements made by this constitutively dimerized receptor in response to ligand binding, and how these lead to a separation of JAK-binding Box1 motifs. Such a separation leads to removal of the pseudokinase inhibitory domain from the kinase domain of a partner JAK2 bound to the receptor, and vice versa, leading to apposition of the kinase domains and transactivation. This may be a general mechanism for class I cytokine receptor action.
Keywords: receptor dimer, conformational change, transmembrane helix, FRET, Box1, molecular dynamics
It has been two decades since the cloning of Janus kinases JAKs 1, 2, and 3, and of tyk2,1,2 and there are now over 5000 Pubmed citations for JAK and more than 7000 citations for their key transcription factor targets, the seven signal transducers and activators of transcription (STATs). Yet, despite their widespread importance in biology and medicine, the molecular mechanism used by cytokine receptors to activate the JAKs has remained an enigma. There are over 30 class I cytokine receptors identified which activate JAK1, JAK2, JAK3, or tyk2 tyrosine kinases,3 and likewise at least 12 class II cytokine receptors lacking the classic WSxWS motif in their extracellular domains.4 The JAKs bind to these receptors via their N-terminal 4.1, Ezrin, Radixin, Moesin (FERM) domain (and probably elsewhere) to a conserved membrane proximal proline rich sequence (Box1). The crystal structures of the ligand binding extracellular domains has been published for several class I cytokine receptors, and careful analysis of the role of the conserved FNIII domains has identified key roles for particular hydrophobic as well as ionic residues in the ligand binding interaction. Conversely, the crystal structure of the kinase domain of JAK2 was elucidated in 20065 and more recently, that of the pseudokinase domain.6 Given all this knowledge, it is curious that there was no mechanism for the JAK activation process which accounts for the key feature of its pseudokinase domain (which presumably locks the kinase domain, and must be removed for activation), or for the evidence that a number of these receptors exist as a dimer held together by their single transmembrane domain, which eliminates ligand-dependent receptor dimerization as a possible activation mechanism to bring the kinase domains together for trans-activation.
We have approached this problem through an investigation of the means used by one of the simplest of the class I cytokine receptors, the growth hormone (GH) receptor, to activate JAK2. This receptor was the first such receptor to be cloned7 and the first to have elucidated its extracellular domain crystal structure determined in complex with the ligand, GH.8 A series of landmark publications from Genentech formed the basis of the textbook view that the role of the bivalent ligand was to initiate receptor dimerization, and this led to activation of the associated JAK2, which was identified as the relevant kinase in 1993.9 This model was then extended to all class I and II cytokine receptors, and was concordant with the prevailing view of ligand dependent activation of the receptor tyrosine kinases such as the EGF receptor by dimerization, although this did not apply to insulin or IGF-1 receptors, which exist as ligand-independent dimers.
Apart from the crystal structure of the GH(GHR2) extracellular domain, and the 2:1 stoichiometry of binding to the extracellular domain, the main support for the induced dimerization model was set out in Fuh et al.10 which showed that three of three bivalent monoclonal antibodies to the extracellular domain of the receptor were capable of activating a hybrid receptor expressed in a FDC-P1 cell line, while monovalent fragments of these could not dimerize and activate this receptor. The hybrid receptor consisted of the extracellular domain of the human GH receptor fused to the N-terminal fibronectin domain of the G-CSF receptor. Hence, not only was G-CSF rather than GH receptor signaling actually being measured, but the additional 3 fibronectin domains may promote association of the extracellular domains themselves. A second line of evidence for induced dimerization was the bell shaped dose response curve for hGH with this hybrid receptor, which was taken as evidence that a second hGH would occupy the second receptor at high concentration (over 2 µg/mL), preventing effective signaling. It should be noted that Yang et al.11 have shown a C-N-terminal fusion hGH dimer is actually capable of signaling, although with substantially lower potency, so that it is possible to fit 2 hormone molecules into the active complex. A third line of evidence proposed to support receptor dimerization was that disrupting binding of the second receptor by mutation of Gly120 (G120) to a bulky side-chain resulted in antagonism.12
Subsequently, we created FDC-P1 and Ba/F3 cell lines expressing full-length GH receptor, which responded appropriately to low concentrations of hGH. These were challenged with our panel of monoclonal antibodies to the extracellular domain of the GH receptor, and surprisingly none of the 14 antibodies were able to activate the full-length receptor in FDC-P1 cells, although 8 could do so with the hybrid receptor expressing cells.13 Nine acted as an antagonist with these cells. One antibody (mAb 263) was able to convincingly act as an agonist in the more sensitive Ba/F3 line expressing full-length GH receptor, and the epitope of this antibody was subsequently mapped by PCR mediated mutagenesis and expression in yeast.14 We concluded from these studies that the activation process must be quite selective, and involve a specific conformational change rather than simple receptor dimerization. Could it involve a conformational change within a constitutive dimer?
The first evidence for the existence of a constitutive receptor dimer (predimer) came with our study of binding of the B2036 antagonist (essentially a high site 1 affinity G120R) to membrane preparations from HEK293 cells expressing GH receptor.15 By Scatchard analysis the number of receptors available for binding to this antagonist ligand was doubled when MAb5 was present. MAb5 is able to bind to the extracellular receptor-receptor interaction domain (site 3, the “dimerization domain”) and prevent dimer formation. Hence if the receptors were present as dimers before hormone addition, one would predict that MAb5 would, by blocking dimerization, free an equal number of receptors for binding of this site 1-specific G120R ligand. If the receptors were monomeric before hormone addition, the MAb5 should be without effect on receptor number, since it does not compete at the ligand binding site. We found that the MAb5 did double the binding sites for the G120R site 1 antagonist, supporting the presence of a constitutive receptor dimer in the membrane. Further, PEGylated B2036 bound well to the isolated extracellular domain (GHBP), but poorly to the cell surface receptor, likely because the PEGylation was interfering with binding to the second receptor in a constitutive dimer.
Our prediction of a constitutive dimer was shortly confirmed by Strous’s group using co-immunoprecipitation (CoIP) of truncated receptors of differing size, with the additional finding that the extracellular domain was not needed for its formation since its removal with proteinase K treatment did not influence dimer extent in intact cells.16 Strous’s group also reported that receptor dimer formation occurred in the endoplasmic reticulum, and was not affected by the addition of hormone to receptor-expressing cells. We then published CoIP studies to show that constitutive dimers were evident when the cytoplasmic sequence was truncated just below the Box2 sequence at residue 360.17 Similarly, using a range of truncated receptor constructs with appropriate placement of FRET or BRET reporters, we were able to conclude that the transmembrane helix with 6 additional C-terminal residues was sufficient to drive dimer formation. Importantly, as the reporters were successively placed closer to the cytoplasmic membrane surface, both FRET and BRET ratios increased markedly, indicating proximity of the JAK2 binding Box1 sequences. Notably, deletion of the Box1 motif by alanine substitution did not change the FRET or BRET ratio, indicating that receptor dimer was not a consequence of JAK2 association. Placement of the FRET reporters at the receptor C-terminus resulted in a low FRET value, indicating wide separation. Contrary to the ligand-induced dimerization model, addition of hGH ligand to cell membrane preparations from these receptor overexpressing cells did not change the FRET or BRET ratios with the reporter group attached to the N- or C-terminus of the receptor.17
The finding that the transmembrane helix was primarily responsible for formation of the constitutive GH receptor dimer was concordant with studies from Langosch/Klingmuller18 and Lodish19 groups. These groups used the bacterial ToxR/ToxCAT assay of TM helix association and immunofluorescence co-patching respectively to show that the homologous EPO receptor is constitutively dimerized through its TM helix domains by a leucine zipper-like motif. The ToxR assay, together with cysteine substitution and crosslinking was subsequently used to show that the TPO receptor also exists as a ligand independent dimer through TM helix interaction in the cell membrane.20 Likewise, the homologous prolactin receptor exists as a homodimer in ligand-independent manner, even at physiological levels of expression, and this association appears to be mediated by the TM helices.21,22 Clearly then, for this class of homodimerizing cytokine receptors, signal transmission into the cell must involve a conformational change or a subunit arrangement.
We sought a defined conformational change by resolving the crystal structure of the unliganded extracellular domain of the GH receptor at 2.7 Å,17 and compared this to the structure of receptor 1 in the 2:1 liganded receptor dimer elucidated by de Vos et al.8 There were no major differences, although there were minor changes in residue position within the ligand binding sites (around Trp104 and Trp169), and an alteration of the F’G’ loop position in the lower receptor domain. The latter was shown to regulate receptor signaling by a novel Src/ERK pathway by mutagenesis,23 but did not influence JAK2 activation. No alterations in the position of residues involved in the extracellular receptor-receptor “dimerization domain” (loop 144–148) were evident, despite finding this in the 1:1 complex with the G120R antagonist.24 There was evident a small rotation of around 8° between upper and lower cytokine receptor domains in the z-plane, which suggested a role for subunit rotation in the activation process. Indeed, when comparing the location of the two receptor subunits in the 2:1 complex, what is most evident is the elevation of receptor 1 and the rotation of receptor subunits by around 30° from the midline as a result of the asymmetric placement of the binding sites on the hormone. Support for a rigid body subunit rearrangement was obtained by Poger and Mark,25 who used molecular dynamics to show that when the hormone is removed from the 2:1 complex in silico, the second receptor subunit rotates around 45° counterclockwise with respect to the first.
What could be the role of relative subunit rotation? We have proposed that it is necessary for the locking of the “dimerization domains” in the lower cytokine receptor (FNIII) module,17 since mutations in this domain block signaling without influencing hormone binding. This is evident both in the case of Laron dwarfism26 and from our in vitro mutagenesis of these residues.27 A network of hydrogen bonds and salt bridges stabilize this “site 3” interaction in the presence of bound hormone in solution28 and in vivo,27 but no interaction is evident in the absence of hormone in solution by physical methods.29 We also found that blockade of these “dimerization domains” with an epitope mapped mAb also blocks signal generation with minimal effect on ligand binding.30
We were able to mimic locking of the dimerization domain by replacing the extracellular domain with a Jun–Jun zipper at approximately the position of the dimerization domain, and this resulted in constitutive activation.31 The jun zipper approach was then used to reveal the steric requirements for receptor activation, using retroviral infection to avoid clonal artifacts and overexpression.32 Through the use of these zippers we could eliminate compensatory movements of the extracellular domain, and hold the receptor transmembrane and cytoplasmic domain tightly in a chosen configuration. First, we placed the zipper successively closer to the cell surface in 4-residue increments. The closer the zipper came to the cell surface, the stronger was the activation. Using the construct with the leucine zipper closest to the cell surface, we then inserted 1–4 alanine residues between the zipper and the N-terminus of the TM helices to assess the effect of rotation of the TM helix on signaling. These experiments showed that there was a preferred orientation for activation, suggesting a rotation of the TM helix was involved in activation.
A key element in understanding the receptor activation mechanism stemmed from the use of monomeric FRET reporters placed just below Box1 (i.e., 37 residues below Box1).32 These constructs could bind JAK2 normally and were able to activate JAK2 on addition of ligand. To our surprise we found that every case of constitutive activation was associated with increased separation of the FRET reporters, contrary to the conventional view that ligand binding induced proximity of the Box1 sequences. We considered the possibility that the reduced FRET efficiency was a result of fluorophore rotation, but found that insertion of a flexible linker between the receptor and the fluorophores did not change the extent of reduced FRET seen with receptor activation. The inverse relationship between cell proliferation and FRET efficiency could be extended to other means of altering the extent of constitutive activation. In particular, removing the acidic residues (EED) which capped the TM helix, and replacing them with non-repulsive alanine residues in the Jun zipper chimera enhanced cell proliferation, and resulted in an even lower FRET efficiency. Moreover, if we co-transfected full-length wt receptor (containing EED) with a mutant receptor possessing a charge reversal of EED to KKR, we observed constitutive activation, again associated with a decreased FRET efficiency. It appears that these acidic residues act as a gating mechanism for receptor activation, where the electrostatic repulsion must be overcome by ligand-induced proximity of the upper TM helices. Similarly, we had found that alanine substitution of the first lysine of the submembrane SKQQRIK sequence resulted in increased receptor activation (similar to what was reported for the TPO receptor33) and again found a corresponding decrease in FRET efficiency with our reporters. Could we show this decrease with ligand binding to a receptor with intact extracellular domain, and the same FRET reporter placement? Using appropriately low transfection levels in HEK293 cells, hGH binding indeed resulted in a substantial decrement in FRET efficiency. Moreover, we could block this separation movement by preincubation with the G120R hGH antagonist which does not engage the second receptor, hence prevents the extracellular dimerization domains locking together. Importantly, when we introduced the D170H mutation26 into this dimerization domain, which results in Laron dwarfism, we observed no change in the FRET efficiency on addition of GH ligand.
In order to further validate our surprising finding, we took two approaches: (1) cysteine crosslinking between receptor TM helices and their juxtamembrane segments and (2) molecular dynamics modeling of the TM helices in lipid membranes. For the crosslinking experiments we individually replaced with cysteine every residue from the top of the linker that joins the lower cytokine receptor domain to the TM domain (i.e., residue Leu251) to the bottom of the TM domain (Ser288), transiently transfected these constructs in COS-1 cells, then added either the crosslinking agents MTS or o-phenanthroline, and ran solubilized extracts on non-reducing gels. When plotted on to a helix wheel projection of the TM sequence, it was apparent that one side of each helix was in contact, which allowed us to define the relative orientation of the two helices in the basal state. This not only provided further evidence for constitutive dimerization, but also allowed us to identify the basal state in molecular dynamics simulations of the two helices as they approached in lipid membranes. However, we could not observe a difference in helix orientation with hormone addition in the crosslinking experiments, most likely because most of the receptor was trapped within the cells in vesicles, and because the crosslinkers could not penetrate into the cell. Nevertheless, we did find that residues of the EED sequence just above the TM helix became more crosslinked with hormone addition. We also observed spontaneous crosslinking of the introduced cysteines in the upper helix and juxtamembrane sequence down to Ile270, and found that this was associated with activation of STAT5 when the full-length receptor was studied. We take this as supporting the finding that increased receptor proximity in the upper TM/juxtamembrane sequence results in receptor activation.
To understand the helix dynamics we undertook in silico molecular dynamics in DPPC membranes in collaboration with M Doxastakis at the University of Houston. To obtain the free energy profile as a function of distance (PMF) between two transmembrane domains (TMDs), exhaustive Monte Carlo simulations using 128 replica pairs were performed for a range of separations between the centers of mass of the helices as they approach. The fact this profile has a deep minimum at a separation of 0.7–0.8 nm supports the finding that the two TMDs interact strongly. The simulations showed that as the two helices approach, they first interact at their C-termini as a result of repulsive interactions at the N-termini. Subsequently, the TMDs follow a path that maximizes interactions between the Phe residues and Thr–Thr hydrogen bonding. This initially leads an essentially parallel dimer (State 1) corresponding to the crosslink pattern seen in the absence of hormone (see Fig. 1). Closer approach of the helices (as could result from hormone binding) leads to a close-packed structure (State 2) with increased tilting and rotation, resulting in a left handed cross over dimer with increased separation at the C-terminus. Both State 1 and 2 lie within the identified energy minimum, indicating a relatively facile State 1–State 2 transition. The GHR TMDs form the active left hand crossover dimer by rotating Phe276 and 283 out of the interface. Importantly, when the inactive State 1 is compared with the left handed crossover form, separation between the C-termini has increased substantially. This parallels our finding that activity correlates with higher separation of the C-termini as monitored by FRET. Moreover, in the proposed active State 2 form the N-terminal Trp267 sits favorably at the upper membrane interface, facing outward to favor association, while the C-terminal Lys289 is in an unfavorable configuration for helix interaction. Thus the model can also explain how alanine substitution of Lys289 promotes the left handed dimer form, correlating with the increased activation seen on mutation of Lys289 to a smaller alanine residue. It is important to note that these helix movements are essentially sequence independent, so could be applied to other class I receptors.
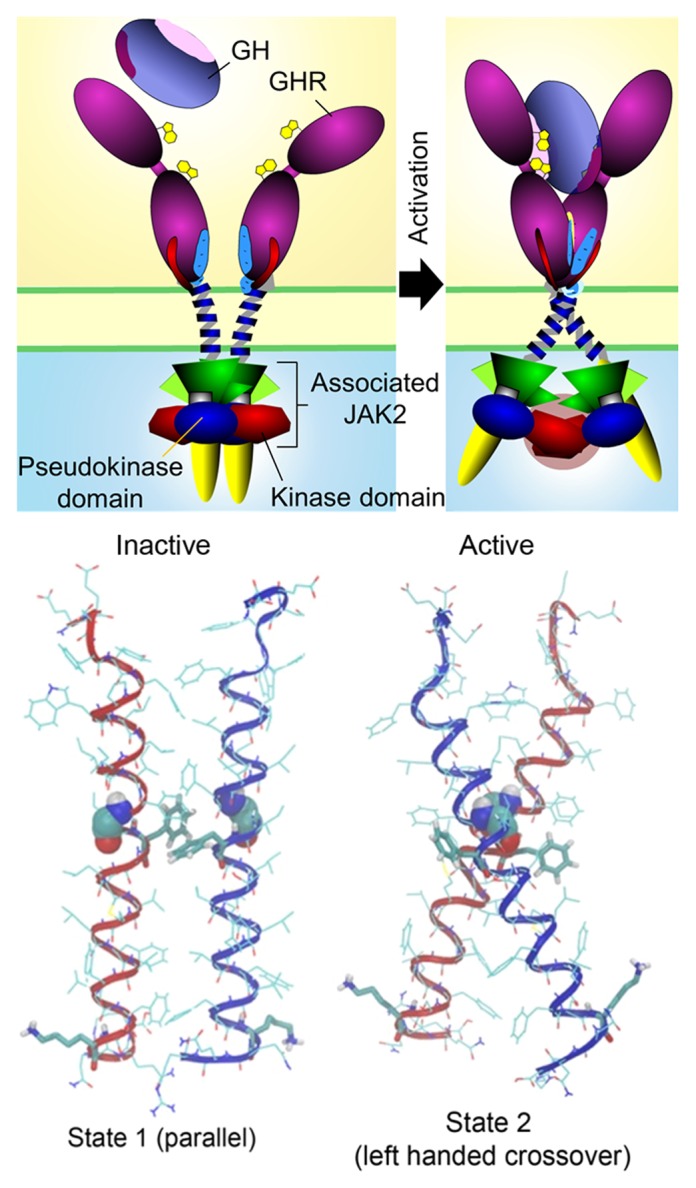
Figure 1. Receptor/JAK2 activation process. Cartoon of basal state (State 1) and of active state (State 2) with helix alignments for these states derived by modeling shown below each cartoon. In State 1 the pseudokinase domains inhibit their partner JAK2 kinase domains, while in the active state the pseudokinase domains are removed as a result of tilting of the receptor helices, bringing the kinase domains into proximity for trans-activation. Reproduced from Brooks et al.32 with permission.
How could a separation of the TM helices at their C-termini result in activation of JAK2? To understand this, we must review what is known of the structure of JAK2. As with other JAK family members, this kinase possesses a modified FERM domain at its N-terminus (JH7–JH5) which is used for binding to the receptor via the receptor Box1/Box2 motifs. This is followed by an apparently non-functional SH2 domain (JH4–JH3) which links to a pseudokinase domain (JH2) followed by the kinase domain (JH1) at the C-terminus. The crystal structures of the kinase5 and pseudokinase6 domains have been resolved, and the former has been of great utility in the design of potential therapeutic agents. It has recently been established that the pseudokinase domain does function as a serine/threonine kinase acting to regulate JH1 kinase activity, although the significance of this is not yet clear.34 There are many other regulatory tyrosine phosphorylation sites in JAK2 which have been identified by mass spectrometry technology, but again there is no overall understanding of how these interact and function.35 In relation to a kinase activation mechanism based on increased separation of the Box1 sequences, we postulated that this movement provides a means to remove the inhibitory pseudokinase domain from the kinase domain.32 What is relevant here is the finding of Lodish group36 that in order for oncogenic V617F JAK2 to manifest its constitutive activity, the EPO receptor must be present, i.e., the JAK2 must be bound to complimentary EPOR subunits within a preformed receptor dimer. If, as believed, this mutation prevents the pseudokinase domain from inhibiting the kinase domain, why does it need the receptor present to manifest such activity? We reasoned that the pseudokinase domain of one JAK2 could be inhibiting the kinase domain of the other JAK2 and vice versa. Pulling them apart would allow the kinase domains to come into proximity, facilitating trans activation and JAK2 action (see Fig. 1).
To support this hypothesis we took four approaches. First, we monitored movements of the kinase and pseudokinase domains by FRET using receptors which were constitutively activated by co-transfecting receptor constructs with the wild type juxtamembrane EED sequence and with KKR replacing this, so that charge attraction at this point led to receptor activation. The baseline controls were EED receptor alone, or the KKR substituted receptor alone. FRET reporters were placed at the C-terminus of JAK2 to monitor movement of the catalytic domains, or C-terminal to the SH2 domain at Asn533, in place of the pseudokinase domain. This showed that receptor activation resulted in an increased FRET for the C-terminal kinase domain placement of reporters, but a decrease in FRET for the pseudokinase reporter placement at Asn533, as predicted by our model.
A second approach was to swap the kinase and pseudokinase domains, then to co-transfect this construct with wild-type JAK2. If our model is correct, this should place the kinase domains together in the tetrameric receptor/JAK2 assembly, resulting in constitutive activation of the JAK2, in a receptor-dependent manner. This is indeed what we found, provided the JAK2 level was expressed at low levels to avoid auto-activation.
A third approach was to quantify interactions between two pseudokinase-kinase (PK-K) domains, or these domains separately, using both Alpha screen and single molecule fluorescence technologies. These experiments showed that the PK-K pair can associate in trans with another such pair, as predicted by our model.
Finally, we docked the crystal structures of the kinase and pseudokinase domains in silico in the predicted orientation. For this purpose we used HADDOCK to reach two optimum minimum energy solutions. The docked structures predict a close complementary interaction between the opposing kinase and pseudokinase domains. Importantly, the structure shows proximity between the activation loop and V617 in the pseudokinase domain which, when mutated, results in constitutive activation and oncogenesis because of impaired kinase inhibition. To make a model of the complete complex, covalently linked pseudokinase and kinase domains of one JAK2 were then placed with another PK-K pair in alternative orientations, yielding two arrangements which were consistent with our experimental results. These arrangements are also consistent with the recent publication of Varghese et al.37 which shows the PK-K domains are linear in solution, and appear to bind in trans.
An animation based on the entire JAK2 activation mechanism is shown at http://web-services.imb.uq.edu.au/waters/hgh.html. We anticipate that this model applies to other class I cytokine receptors such as the EPO receptor. Indeed, we have created chimeras with EPOR ECD fused to the GH receptor TMD and cytoplasmic domain and find the EPOR agonist peptide dimer EMP1 activates GHR signaling, whereas the antagonist peptide dimer EMP-33 does not. Evidently the receptor activation mechanism does not have a specific sequence requirement for the TM helix, but rather the movement we observe is a function of helix dynamics in a lipid membrane contingent on close apposition of the N-terminal end of the paired helices. We anticipate this concept will be valuable in designing therapeutic modulators of class 1 cytokine function.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by NHMRC (Australia) grants 511120, 1002893, 1025082, and 1020317 to M.J.W.
Glossary
Abbreviations:
- GH
growth hormone
- G-CSF
granulocyte-colony stimulating factor
- FRET
fluorescence resonance energy transfer
- BRET
bioluminescence resonance energy transfer
- TM
transmembrane
- FNIII
fibronectin 3
References
- 1.Wilks AF, Harpur AG, Kurban RR, Ralph SJ, Zürcher G, Ziemiecki A. Two novel protein-tyrosine kinases, each with a second phosphotransferase-related catalytic domain, define a new class of protein kinase. Mol Cell Biol. 1991;11:2057–65. doi: 10.1128/mcb.11.4.2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ihle JN. STATs: signal transducers and activators of transcription. Cell. 1996;84:331–4. doi: 10.1016/S0092-8674(00)81277-5. [DOI] [PubMed] [Google Scholar]
- 3.Liongue C, Ward AC. Evolution of Class I cytokine receptors. BMC Evol Biol. 2007;7:120. doi: 10.1186/1471-2148-7-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Langer JA, Cutrone EC, Kotenko S. The Class II cytokine receptor (CRF2) family: overview and patterns of receptor-ligand interactions. Cytokine Growth Factor Rev. 2004;15:33–48. doi: 10.1016/j.cytogfr.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 5.Lucet IS, Fantino E, Styles M, Bamert R, Patel O, Broughton SE, Walter M, Burns CJ, Treutlein H, Wilks AF, et al. The structural basis of Janus kinase 2 inhibition by a potent and specific pan-Janus kinase inhibitor. Blood. 2006;107:176–83. doi: 10.1182/blood-2005-06-2413. [DOI] [PubMed] [Google Scholar]
- 6.Bandaranayake RM, Ungureanu D, Shan Y, Shaw DE, Silvennoinen O, Hubbard SR. Crystal structures of the JAK2 pseudokinase domain and the pathogenic mutant V617F. Nat Struct Mol Biol. 2012;19:754–9. doi: 10.1038/nsmb.2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leung DW, Spencer SA, Cachianes G, Hammonds RG, Collins C, Henzel WJ, Barnard R, Waters MJ, Wood WI. Growth hormone receptor and serum binding protein: purification, cloning and expression. Nature. 1987;330:537–43. doi: 10.1038/330537a0. [DOI] [PubMed] [Google Scholar]
- 8.de Vos AM, Ultsch M, Kossiakoff AA. Human growth hormone and extracellular domain of its receptor: crystal structure of the complex. Science. 1992;255:306–12. doi: 10.1126/science.1549776. [DOI] [PubMed] [Google Scholar]
- 9.Argetsinger LS, Campbell GS, Yang X, Witthuhn BA, Silvennoinen O, Ihle JN, Carter-Su C. Identification of JAK2 as a growth hormone receptor-associated tyrosine kinase. Cell. 1993;74:237–44. doi: 10.1016/0092-8674(93)90415-M. [DOI] [PubMed] [Google Scholar]
- 10.Fuh G, Cunningham BC, Fukunaga R, Nagata S, Goeddel DV, Wells JA. Rational design of potent antagonists to the human growth hormone receptor. Science. 1992;256:1677–80. doi: 10.1126/science.256.5064.1677. [DOI] [PubMed] [Google Scholar]
- 11.Yang N, Langenheim JF, Wang X, Jiang J, Chen WY, Frank SJ. Activation of growth hormone receptors by growth hormone and growth hormone antagonist dimers: insights into receptor triggering. Mol Endocrinol. 2008;22:978–88. doi: 10.1210/me.2007-0424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Waters MJ, Brooks AJ. Growth hormone receptor: structure function relationships. Horm Res Paediatr. 2011;76(Suppl 1):12–6. doi: 10.1159/000329138. [DOI] [PubMed] [Google Scholar]
- 13.Rowlinson SW, Behncken SN, Rowland JE, Clarkson RW, Strasburger CJ, Wu Z, Baumbach W, Waters MJ. Activation of chimeric and full-length growth hormone receptors by growth hormone receptor monoclonal antibodies. A specific conformational change may be required for full-length receptor signaling. J Biol Chem. 1998;273:5307–14. doi: 10.1074/jbc.273.9.5307. [DOI] [PubMed] [Google Scholar]
- 14.Wan Y, Zheng YZ, Harris JM, Brown R, Waters MJ. Epitope map for a growth hormone receptor agonist monoclonal antibody, MAb 263. Mol Endocrinol. 2003;17:2240–50. doi: 10.1210/me.2003-0162. [DOI] [PubMed] [Google Scholar]
- 15.Ross RJ, Leung KC, Maamra M, Bennett W, Doyle N, Waters MJ, Ho KK. Binding and functional studies with the growth hormone receptor antagonist, B2036-PEG (pegvisomant), reveal effects of pegylation and evidence that it binds to a receptor dimer. J Clin Endocrinol Metab. 2001;86:1716–23. doi: 10.1210/jcem.86.4.7403. [DOI] [PubMed] [Google Scholar]
- 16.Gent J, van Kerkhof P, Roza M, Bu G, Strous GJ. Ligand-independent growth hormone receptor dimerization occurs in the endoplasmic reticulum and is required for ubiquitin system-dependent endocytosis. Proc Natl Acad Sci U S A. 2002;99:9858–63. doi: 10.1073/pnas.152294299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brown RJ, Adams JJ, Pelekanos RA, Wan Y, McKinstry WJ, Palethorpe K, Seeber RM, Monks TA, Eidne KA, Parker MW, et al. Model for growth hormone receptor activation based on subunit rotation within a receptor dimer. Nat Struct Mol Biol. 2005;12:814–21. doi: 10.1038/nsmb977. [DOI] [PubMed] [Google Scholar]
- 18.Kubatzky KF, Ruan W, Gurezka R, Cohen J, Ketteler R, Watowich SS, Neumann D, Langosch D, Klingmüller U. Self assembly of the transmembrane domain promotes signal transduction through the erythropoietin receptor. Curr Biol. 2001;11:110–5. doi: 10.1016/S0960-9822(01)00018-5. [DOI] [PubMed] [Google Scholar]
- 19.Constantinescu SN, Keren T, Socolovsky M, Nam H, Henis YI, Lodish HF. Ligand-independent oligomerization of cell-surface erythropoietin receptor is mediated by the transmembrane domain. Proc Natl Acad Sci U S A. 2001;98:4379–84. doi: 10.1073/pnas.081069198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matthews EE, Thévenin D, Rogers JM, Gotow L, Lira PD, Reiter LA, Brissette WH, Engelman DM. Thrombopoietin receptor activation: transmembrane helix dimerization, rotation, and allosteric modulation. FASEB J. 2011;25:2234–44. doi: 10.1096/fj.10-178673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gadd SL, Clevenger CV. Ligand-independent dimerization of the human prolactin receptor isoforms: functional implications. Mol Endocrinol. 2006;20:2734–46. doi: 10.1210/me.2006-0114. [DOI] [PubMed] [Google Scholar]
- 22.Qazi AM, Tsai-Morris CH, Dufau ML. Ligand-independent homo- and heterodimerization of human prolactin receptor variants: inhibitory action of the short forms by heterodimerization. Mol Endocrinol. 2006;20:1912–23. doi: 10.1210/me.2005-0291. [DOI] [PubMed] [Google Scholar]
- 23.Rowlinson SW, Yoshizato H, Barclay JL, Brooks AJ, Behncken SN, Kerr LM, Millard K, Palethorpe K, Nielsen K, Clyde-Smith J, et al. An agonist-induced conformational change in the growth hormone receptor determines the choice of signalling pathway. Nat Cell Biol. 2008;10:740–7. doi: 10.1038/ncb1737. [DOI] [PubMed] [Google Scholar]
- 24.Clackson T, Ultsch MH, Wells JA, de Vos AM. Structural and functional analysis of the 1:1 growth hormone:receptor complex reveals the molecular basis for receptor affinity. J Mol Biol. 1998;277:1111–28. doi: 10.1006/jmbi.1998.1669. [DOI] [PubMed] [Google Scholar]
- 25.Poger D, Mark AE. Turning the growth hormone receptor on: evidence that hormone binding induces subunit rotation. Proteins. 2010;78:1163–74. doi: 10.1002/prot.22636. [DOI] [PubMed] [Google Scholar]
- 26.Brooks AJ, Waters MJ. The growth hormone receptor: mechanism of activation and clinical implications. Nat Rev Endocrinol. 2010;6:515–25. doi: 10.1038/nrendo.2010.123. [DOI] [PubMed] [Google Scholar]
- 27.Chen C, Brinkworth R, Waters MJ. The role of receptor dimerization domain residues in growth hormone signaling. J Biol Chem. 1997;272:5133–40. doi: 10.1074/jbc.272.8.5133. [DOI] [PubMed] [Google Scholar]
- 28.Bernat B, Pal G, Sun M, Kossiakoff AA. Determination of the energetics governing the regulatory step in growth hormone-induced receptor homodimerization. Proc Natl Acad Sci U S A. 2003;100:952–7. doi: 10.1073/pnas.0235023100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cunningham BC, Ultsch M, De Vos AM, Mulkerrin MG, Clauser KR, Wells JA. Dimerization of the extracellular domain of the human growth hormone receptor by a single hormone molecule. Science. 1991;254:821–5. doi: 10.1126/science.1948064. [DOI] [PubMed] [Google Scholar]
- 30.Jiang J, Wan Y, Wang X, Xu J, Harris JM, Lobie PE, Zhang Y, Zinn KR, Waters MJ, Frank SJ. Inhibitory GH receptor extracellular domain monoclonal antibodies: three-dimensional epitope mapping. Endocrinology. 2011;152:4777–88. doi: 10.1210/en.2011-1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Behncken SN, Billestrup N, Brown R, Amstrup J, Conway-Campbell B, Waters MJ. Growth hormone (GH)-independent dimerization of GH receptor by a leucine zipper results in constitutive activation. J Biol Chem. 2000;275:17000–7. doi: 10.1074/jbc.275.22.17000. [DOI] [PubMed] [Google Scholar]
- 32.Brooks AJ, Dai W, O’Mara ML, Abankwa D, Chhabra Y, Pelekanos RA, Gardon O, Tunny KA, Blucher KM, Morton CJ, et al. Mechanism of activation of protein kinase JAK2 by the growth hormone receptor. Science. 2014;344:1249783. doi: 10.1126/science.1249783. [DOI] [PubMed] [Google Scholar]
- 33.Staerk J, Lacout C, Sato T, Smith SO, Vainchenker W, Constantinescu SN. An amphipathic motif at the transmembrane-cytoplasmic junction prevents autonomous activation of the thrombopoietin receptor. Blood. 2006;107:1864–71. doi: 10.1182/blood-2005-06-2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ungureanu D, Wu J, Pekkala T, Niranjan Y, Young C, Jensen ON, Xu CF, Neubert TA, Skoda RC, Hubbard SR, et al. The pseudokinase domain of JAK2 is a dual-specificity protein kinase that negatively regulates cytokine signaling. Nat Struct Mol Biol. 2011;18:971–6. doi: 10.1038/nsmb.2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Argetsinger LS, Stuckey JA, Robertson SA, Koleva RI, Cline JM, Marto JA, Myers MG, Jr., Carter-Su C. Tyrosines 868, 966, and 972 in the kinase domain of JAK2 are autophosphorylated and required for maximal JAK2 kinase activity. Mol Endocrinol. 2010;24:1062–76. doi: 10.1210/me.2009-0355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lu X, Huang LJ, Lodish HF. Dimerization by a cytokine receptor is necessary for constitutive activation of JAK2V617F. J Biol Chem. 2008;283:5258–66. doi: 10.1074/jbc.M707125200. [DOI] [PubMed] [Google Scholar]
- 37.Varghese LN, Ungureanu D, Liau NP, Young SN, Laktyushin A, Hammaren H, Lucet IS, Nicola NA, Silvennoinen O, Babon JJ, et al. Mechanistic insights into activation and SOCS3-mediated inhibition of myeloproliferative neoplasm-associated JAK2 mutants from biochemical and structural analyses. Biochem J. 2014;458:395–405. doi: 10.1042/BJ20131516. [DOI] [PMC free article] [PubMed] [Google Scholar]