

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is one of the most aggressive and deadly malignancies. Recently, the deubiquitinating protease USP9X has been shown to behave as an oncogene in a number of neoplasms, including those of breast, brain, colon, esophagus and lung, as well as KRAS wild-type PDAC. However, other studies suggest that USP9X may function as a tumor-suppressor in a murine PDAC model when USP9X expression is depleted during early pancreatic development. To address the conflicting findings surrounding the role of USP9X in PDAC, we examined the effects of knocking down USP9X in five human PDAC cell lines (BxPC3, Capan1, CD18, Hs766T, and S2-013). We demonstrate that knocking down USP9X in each of the PDAC cell lines reduces their anchorage-dependent growth. Using an inducible shRNA system to knock down USP9X in both BxPC3 and Capan1 cells, we also determined that USP9X is necessary for the anchorage-independent growth. In addition, knockdown of USP9X alters the cell cycle profile of BxPC3 cells and increases their invasive capacity. Finally, we show that an inhibitor of deubiquitinating proteases, WP1130, induces significant cytotoxicity in each of the five PDAC cell lines tested. Overall, our work and the work of others indicate that the function and role of USP9X is highly context-dependent. Although USP9X may function as a tumor-suppressor during the establishment of PDAC, data presented here argue that USP9X promotes cell growth in advanced PDAC cells when PDAC is typically diagnosed. Hence, USP9X may be a promising therapeutic target for the treatment of advanced PDAC.
Keywords: USP9X, deubiquitinating protease, pancreatic ductal adenocarcinoma, WP1130, KRAS, oncogene, tumor suppressor gene
Introduction
Pancreatic cancer is one of the most aggressive malignancies, and although it is the tenth most commonly diagnosed cancer in the United States, it is the fourth most common cause of cancer deaths. The vast majority of pancreatic cancers are pancreatic ductal adenocarcinomas (PDAC). For several decades, the 5-y survival of patients with PDAC has remained largely unchanged at ~6%, with a median survival of under a year. Remarkable progress has been made during the last decade toward identifying and understanding the complex signaling pathways that contribute to the initiation and progression of PDAC. Despite our improved understanding, the majority of cases are diagnosed at advanced stages, which have proven extremely difficult to treat. Thus, it is crucial to continue efforts toward unraveling the molecular mechanisms that support and drive this cancer if progress is to be made in improving treatment of this highly deadly disease.
Ubiquitin specific peptidase 9X (USP9X), a deubiquitinating protease, has recently emerged as a potential driver of growth and self-renewal in neoplastic cells. Analogous to phosphatases in kinase signaling pathways, USP9X has been shown to remove ubiquitin moieties, which principally direct target proteins toward proteosomal degradation. Because of its ability to modify a multitude of target proteins, the function of USP9X is likely to be highly context-dependent. Several reports have portrayed USP9X as an oncogene. USP9X stabilizes the pro-survival protein MCL1 in follicular and diffuse large B-cell lymphomas.1 USP9X also stabilizes SMURF1, an E3-ligase shown to affect diverse cellular processes, including BMP signaling and cell migration in the context of breast cancer cells.2 Knockdown of USP9X in the context of colorectal carcinoma leads to increased activity of pro-apoptotic pathways and increased sensitivity to the cytotoxic effects of 5-fluorouracil.3 In the context of prostate cancer, USP9X stabilizes the oncogenic transcription factor ERG, and disruption of USP9X by the deubiquitinating protease inhibitor, WP1130, inhibited prostate tumor cell growth in mice.4 Moreover, knockdown of USP9X in medulloblastoma and glioblastoma cells dramatically impairs their growth.5 Consistent with findings in other neoplasms, knockdown of USP9X in a KRAS wild-type PDAC cell line tumor xenograft model led to a significant decrease (~50%) in tumor volume, suggesting that USP9X is an oncogene in the context of established PDAC cells.1
In strong contrast, other studies suggest that USP9X may function as a tumor-suppressor in PDAC. It was recently reported that USP9X behaves as a tumor-suppressor in the context of murine PDAC.6,7 Both studies used the Sleeping Beauty transposon system, and observed that interfering with USP9X expression during pancreatic development (E8.5) in the context of mutated KRAS is associated with a more rapid onset of PDAC in this mouse model. Additionally, Pérez-Mancera and coworkers reported that a large proportion of advanced human pancreatic cancer samples had reduced overall levels of USP9X compared with normal pancreatic tissue7 and that knockdown of USP9X in mouse PDAC cell lines did not affect monolayer growth in a short-term study (over a 4 d period), but increased the growth of PDAC cells in suspension. The conflicting conclusions surrounding USP9X function as either a tumor-suppressor6,7 or an oncogene1 may point to a more complex role for USP9X in PDAC. In this regard, USP9X may parallel the behavior of TGF-β observed in some cancers, where TGF-β behaves a tumor-suppressor during the early stages of disease, but as an oncogene during later stages of some cancers.8-10 Thus, it is important to establish the role of USP9X in the context of PDAC, as therapeutic manipulation of USP9X function, either positively or negatively, may be an effective means to treat PDAC.
In this study, we closely examined whether USP9X functions primarily as an oncogene or as a tumor-suppressor in five different human PDAC cell lines by examining the longer-term effects of reducing the levels of USP9X in PDAC cells. Here, we report that knocking down USP9X by shRNA significantly reduces the monolayer growth of five different human PDAC cell lines, one that expresses wild-type KRAS and four that express mutant KRAS. Interestingly, our studies demonstrate that the effects of knocking down USP9X on monolayer growth of PDAC cells become evident only after 4 d. To confirm and extend these findings, we engineered PDAC cells for inducible knockdown of USP9X. We demonstrate that inducible knockdown of USP9X levels leads to a reduction in both monolayer and soft-agar growth of PDAC cells. We also demonstrate that inducible knockdown of USP9X leads to an increase in the G1 cell cycle compartment, and an increase in the invasive capacity of PDAC cells. We extended these findings and determined that the deubiquitinating protease inhibitor WP1130 impairs the growth of multiple PDAC cell lines. We conclude that the effects of USP9X are highly dependent upon cellular context. In the case of PDAC, USP9X may function primarily as a tumor-suppressor during the early stages of PDAC, especially in a mouse model, but promotes tumor cell growth later in the progression of human PDAC.
Results
Stable knockdown of USP9X reduces the growth of PDAC cells
There is conflicting evidence regarding the role of USP9X in PDAC. Knockdown of USP9X in wild-type KRAS expressing BxPC3 cells reduces their tumorigenicity,1 whereas depletion of USP9X in a mutant KRAS mouse model of PDAC reduces the latency of tumor formation.6,7 To help resolve this discrepancy, we initially transduced BxPC3 and mutant KRAS Capan1 PDAC cells11 with lentiviral vectors that constitutively express an shRNA directed against USP9X transcripts (Table S1). Three shRNA sequences directed against USP9X were tested. As a control, a previously described12 non-specific Scrambled shRNA was transduced into the cells. Western blot analysis demonstrated approximately 60% and ~50% reduction in both the cytoplasmic and nuclear pools of USP9X in BxPC3 and Capan1 PDAC cells three days after transduction (Fig. S1A). Strikingly, after 6 d, the size of cell colonies was markedly reduced in cells transduced with the USP9X shRNA constructs in both BxPC3 and Capan1 cells (Fig. S1B). Results of MTT assays corroborated our microscopy observations that reduced USP9X levels dramatically impaired cell growth (Fig. S1C). These observations were extended to three additional PDAC cell lines (CD18, Hs766T, and S2–013). Similar to results with BxPC3 cells and Capan1 cells, reduction in USP9X levels slowed monolayer growth of these three PDAC cell lines (Fig. S2).
Inducible depletion of USP9X reduces the anchorage-dependent and anchorage-independent growth of PDAC cells
Although stable knockdown of USP9X demonstrates a requirement of USP9X for PDAC cell growth, further characterization of the role of USP9X is best done using cells engineered for inducible knockdown of USP9X. In this regard, repeated transduction of PDAC cells may contribute to experimental variability secondary to transduction efficiency. Additionally, long-term culture of cells (multiple passages) in which factors are constitutively expressed or depleted may introduce selective pressure. For example, stable expression of the transcription factor SOX2 in neoplastic cells enriches a subpopulation with enhanced growth,13-15 whereas rapid induction of SOX2 levels via an inducible system leads to dramatic decreases in the growth of some cells.16,17 Therefore, we engineered BxPC3 and Capan1 cells with a stably integrated, Dox-inducible USP9X shRNA vector. More specifically, we employed a lentiviral vector that constitutively expresses the reverse tet-transactivator, as well as introduces an USP9X shRNA construct with an associated red-fluorescent protein reporter, under the control of a tet-responsive element (Fig. 1A), which was used to produce iKD-USP9X-BxPC3 and iKD-USP9X-Capan1 cell lines, as described in the Materials and Methods. Importantly, induction of shRNA expression in both iKD-USP9X-BxPC3 (Fig. 1B) and iKD-USP9X-Capan1 (Fig. 2A) reduced the overall levels of USP9X to a degree similar to our stable knockdown studies (Fig. S1). This reduction in USP9X levels was observed as early as 2 d after the addition of Dox (data not shown).

Figure 1. Knockdown of USP9X in BxPC3 cells using a Dox inducible lentiviral system. (A) Schematic of the vectors used to inducibly knockdown USP9X. A constitutively active UBC promoter drives expression of a reverse-tet transactivator (rtTA) and also allows for the selection of transduced cells via puromycin. This vector also contains a tet-responsive element that allows for the inducible expression of an RFP marker and a USP9X shRNA sequence. (B) Western blot analysis of USP9X levels in iKD-USP9X-BxPC3 cells grown in the absence or presence of Dox (1 µg/mL) for 6 d. USP9X levels were normalized against GAPDH loading controls, and relative levels are indicated in parentheses. (C) MTT assay examining the growth of iKD-USP9X-BxPC3 cells over time in the absence or presence of Dox (1 µg/mL). The experiment was repeated three times. Data points were averaged and normalized to the day 2 time point, which was set to one. Error bars represent standard deviation, and significant differences between cultures grown with and without Dox are indicated. (D) Representative photomicrographs of iKD-USP9X-BxPC3 cells grown in the absence and presence of Dox for 6 d. (E) Soft-agar growth of iKD-USP9X-BxPC3 cells grown in the absence or presence of Dox. iKD-USP9X-BxPC3 cells grown in the absence of Dox were placed into soft-agar culture conditions, as described in the Materials and Methods. Dox-induced cells were treated with 1 µg/mL Dox where indicated. A scorer, unaware of sample designation, counted the number of colonies observed in the indicated number of high-powered fields (n). Counts were averaged and graphed. Error bars represent standard deviation. The Student t test was used to analyze statistical significance. Growth under anchorage-dependent and anchorage-independent conditions was repeated and similar results were obtained.

Figure 2. Knockdown of USP9X in Capan1 pancreatic cancer cells. (A) Western blot analysis of USP9X levels in iKD-USP9X-Capan1 cells grown in the absence or presence of Dox (1 µg/mL) for 6 d. USP9X levels were normalized against GAPDH loading controls, and relative levels are indicated in the parentheses. (B) MTT assay examining the growth of iKD-USP9X-Capan1 cells over time in the absence or presence of Dox (1 µg/mL). The experiment was repeated three times. Data points were averaged and normalized to the day 2 time point, which was set to one. Error bars represent standard deviation, and significant differences between cultures grown with or without Dox are indicated. (C) Representative photomicrographs of iKD-USP9X-Capan1 cells grown in the absence or presence of Dox for 6 d. (D) Soft-agar growth of iKD-USP9X-Capan1 cells grown in the absence or presence of Dox. iKD-USP9X-Capan1 cells grown in the absence of Dox were placed into soft-agar culture conditions, as described in the Materials and Methods. Dox-induced cells were treated with 1 µg/mL Dox where indicated. A scorer, unaware of sample designation, counted the number of colonies observed in the indicated number of high-powered fields (n). Counts were averaged and graphed. Error bars represent standard deviation. The Student t test was used to analyze statistical significance. Growth under anchorage-dependent and anchorage-independent conditions was repeated and similar results were obtained.
As was observed with stable transduction of BxPC3 cells, reduction of USP9X levels in iKD-USP9X-BxPC3 cells led to reduced cell growth in monolayer. Importantly, differences in growth properties were subtle 4 d after knockdown (similar to a prior report7), but became evident 6 d after the USP9X shRNA was induced (Fig. 1C). This reduction in overall cell number was likely due to reduced cellular proliferation, as colony sizes were smaller in USP9X-deficient cells when compared with their uninduced counterparts (Fig. 1D). These observations were extended by examining anchorage-independent growth of iKD-USP9X-BxPC3 cells, because anchorage-independent growth has been shown to correlate with the tumorigenic potential of neoplastic cells.18 For these studies, the anchorage-independent growth of iKD-USP9X-BxPC3 cells was correlated with endogenous levels or depleted levels of USP9X. For this purpose, cells were cultured in soft-agar, in the absence or presence of Dox for ~1 wk in serum-free growth factor supplemented medium. USP9X levels were reduced to a similar extent in anchorage-independent conditions as compared with monolayer growth conditions (see below; Fig. S4). Reduced levels of USP9X impaired anchorage-independent growth of iKD-USP9X-BxPC3 cells (Fig. 1E). Together, these data suggest that USP9X is important for both anchorage-dependent and anchorage-independent growth of BxPC3 PDAC cells.
iKD-USP9X-Capan1 cells exhibited reduced cell proliferation following USP9X knockdown, similar to their BxPC3 counterparts (Fig. 2). Specifically, knockdown of USP9X did not produce a clear difference at the day 2 or the day 4 time points; however, a significant reduction in growth was observed when USP9X had been knocked down for 6 d (Fig. 2B). As in the case of the iKD-USP9X-BxPC3 cells, iKD-USP9X-Capan1 colonies were smaller following USP9X knockdown (Fig. 2C). Importantly, examination of iKD-USP9X-Capan1 cells grown in anchorage-independent conditions also demonstrated that reduction of USP9X significantly diminished suspension growth (Fig. 2D). Taken together, these data suggest that, in the context of PDAC cells BxPC3 and Capan1, USP9X is necessary to maintain their proliferative capacity both in monolayer and in soft-agar.
Depletion of USP9X alters the cell cycle distribution of PDAC cells and increases their invasive properties
To further investigate the roles of USP9X in PDAC, we examined how the knockdown of USP9X influenced several other properties that are relevant to the behavior of tumor cells. For these studies, we used iKD-USP9X-BxPC3 cells because ~70% these cells (data not shown) express cell surface markers (CD44+, CD24+, ESA+) shown to be associated with PDAC tumor-initiating cells.19 By contrast, far fewer (<10%) iKD-USP9X-Capan1 cells express all three markers. Initially, we examined how the knockdown of USP9X influenced the cell cycle profile of iKD-USP9X-BxPC3 cells (Fig. 3). USP9X knockdown cells exhibited a small, but statistically significant, increase in the G1 and a decrease in the S phase compartments (Fig. 3). These data, along with our microscopic observations (Fig. 1) are consistent with USP9X functioning to support the growth of PDAC cells.
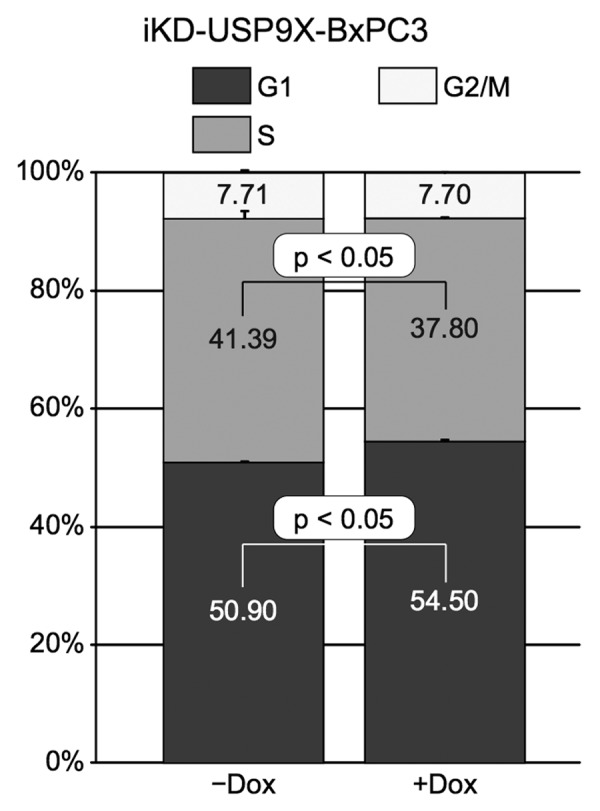
Figure 3. Effects of knocking down USP9X on cell cycle distribution. Cell cycle analysis of iKD-USP9X-BxPC3 cells cultured in the absence or presence of Dox (1 µg/ml) for 4 d. The Telford Reagent46 and FACS analysis (UNMC Cell Analysis Core Facility) were used to determine cell cycle distribution. The Student t test was used to determine P values (n = 2), and error bars represent standard deviations. This experiment was repeated with similar results.
An important property of tumor cells is their migratory and invasive behavior. To assess the effect of knocking down USP9X on cell migration, iKD-USP9X-BxPC3 cells were grown in the absence or presence of Dox for 2 d. Next, the cells were subcultured into serum-free medium and placed in the upper chamber of an uncoated, porous BD Biocoat Invasion chamber, as described in the Materials and Methods. After 24 h, migration toward serum-containing medium in the lower chamber was assessed. Knockdown of USP9X had no significant impact on the ability of cells to migrate through a porous membrane (Figs. 4A). To confirm this finding, we performed a wound-healing assay, and observed similar results: knockdown of USP9X had no observable effect on the migratory behavior of iKD-USP9X-BxPC3 cells (Fig. S3). Next, we examined the effect of knocking down USP9X on the invasive behavior of these cells. For this purpose, Matrigel coated membranes were used, as described in the Materials and Methods. Remarkably, knockdown of USP9X led to a substantial enhancement of the invasion activity of iKD-USP9X-BxPC3 cells (Fig. 4B). Together, these data suggest that USP9X is not essential for the free migration of iKD-USP9X-BxPC3 cells, but reduction of USP9X levels enhances their ability to invade through a Matrigel biomatrix.

Figure 4. Migration and invasion by iKD-USP9X-BxPC3 cells following knockdown of USP9X. (A) A migration assay was performed as described in the Materials and Methods. iKD-USP9X-BxPC3 cells were grown in the absence or presence (1 µg/mL) of Dox for 48 h and then transferred onto uncoated porous membranes in serum-free medium. Cells were allowed to migrate through the membrane, toward serum-containing medium, for 24 h, before being fixed, stained and counted. The values presented are averages of cells in 14 random, 6.25 mm2 fields. Error bars represent standard error of the mean. The Student t test was used to evaluate statistical significance. This experiment was repeated two additional times with similar results. (B) Similarly, invasion assays were performed as described in the Materials and Methods. Membranes coated with Matrigel were used along with uncoated membranes. Stained cells were counted in 18 random, 6.25 mm2 fields. The bar graph depicts the percentage of invasion (average number of cells per field observed and standard error of the mean for the invasion membranes divided by the average number of cells per field observed on the uncoated membranes). Error bars represent conversion of standard error of the mean. The Student t test was used to test average counts per field for significant difference. This experiment was repeated two additional times, with similar results.
Expression of USP9X and one of its target proteins, ITCH, in transformed pancreatic ductal cells
A prior study demonstrated that loss of USP9X in a murine mutant KRAS model accelerated the formation of PDAC.7 That study also reported that loss of USP9X led to a reduction in the expression of ITCH, an E3-ubiquitin ligase involved in a wide-array of cellular processes. ITCH auto-ubiquitinates itself leading to its degradation; however, USP9X can stabilize ITCH by deubiquitinating it.20 These findings led us to examine how in vitro transformation of human primary pancreatic ductal cells influences the expression of USP9X and ITCH. For this purpose, we compared the expression of both proteins in primary human pancreatic ductal cells immortalized by hTERT (hTERT-HPNE cells) as well as in their transformed counterparts. More specifically, we compared the expression of USP9X and ITCH in hTERT-HPNE cells and hTERT-HPNE cells that ectopically express different combinations of E6/E7, SV40 small t-antigen, and mutant KRAS. Previous studies have shown that full transformation to tumorigenic cells requires ectopic expression of E6/E7, SV40 small t-antigen, and mutant RAS.21 Western blot analysis of extracts prepared from hTERT-HPNE cells and hTERT-HPNE cells transformed by E6/E7, SV40 small t-antigen, and mutant KRAS, determined that there were no significant changes in the expression of USP9X and ITCH (Fig. 5). Moreover, similar expression of both proteins was observed in hTERT-HPNE cells that ectopically express E6/E7, E6/E7, and SV40 small t-antigen, or E6/E7 and mutant KRAS. The findings for hTERT-HPNE E6/E7 cells are particularly interesting, because immortalization of keratinocytes with HPV E6/E7 has been shown to upregulate USP9X activity.22 Thus, in the in vitro model of pancreatic cell transformation, USP9X and ITCH protein levels do not appear to change significantly.
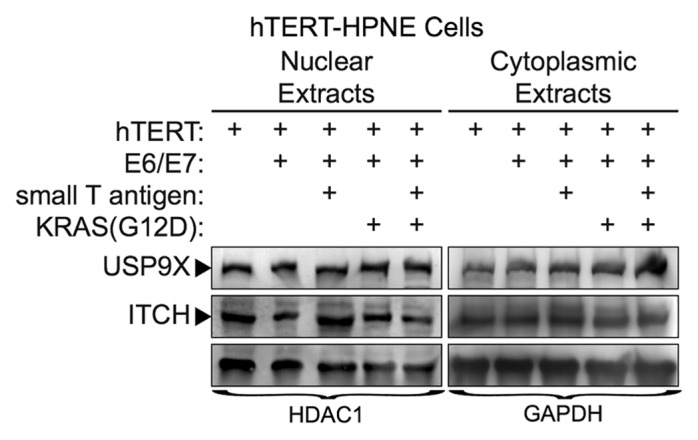
Figure 5. USP9X and ITCH expression in transformed, nestin expressing human pancreatic cell lines. Cell lines were immortalized by expression of the indicated proteins (e.g., hTERT, E6/E7, small T-antigen, KRAS-G12D), as described previously.40 Expression of USP9X and ITCH were examined by western blot analysis. HDAC1 and GAPDH served as loading controls.
The relationship between USP9X and ITCH was examined more closely in protein extracts prepared from cells in which USP9X was knocked down. We observed a reduction in ITCH primarily in nuclear extracts when USP9X was knocked down in iKD-USP9X-BxPC3 cells grown in suspension (Fig. S4). In contrast, we did not observe a significant reduction in ITCH expression in either nuclear or cytoplasmic extracts prepared from iKD-USP9X-BxPC3 cells grown in monolayer.
A partially selective deubiquitinating protease inhibitor strongly reduces the growth of PDAC cells
Lastly, we examined whether the small molecule inhibitor, WP1130, could perturb the growth of human PDAC cells, as our data supports the role of USP9X as a growth promoter. WP1130 is a partially selective deubiquitinating protease inhibitor that directly inhibits the enzymatic activity of USP9X, as well as the activities of USP5, USP14, and UCH37.23,24 As discussed below, WP1130 has been shown to impair the growth of a number of neoplastic cell types, including chronic myelogenous leukemia24 and mantle cell lymphoma,25 without inducing significant toxicity in mice. To examine whether WP1130 could impair PDAC cell growth, five human PDAC cell lines (BxPC3, Capan1, CD18, Hs766T, and S2-013) were grown with various concentrations of WP1130 for a total of 72 h. After 48 h, increased cellular toxicity was observed by microscopy, which corresponded to increased concentrations of WP1130 (Fig. S5). Importantly, WP1130 impaired the growth of each of the PDAC cell lines at the highest concentration of WP1130 tested (5 µM), although the effects on Hs766T cells were more modest (Fig. 6). Together, these data demonstrate that WP1130 impairs the growth of five PDAC cell lines in vitro, and suggest that USP9X and/or other deubiquitinating enzymes are potential therapeutic targets for the treatment of PDAC.

Figure 6. Effects of USP9X inhibitor, WP1130, on in vitro PDAC cell growth. BxPC3, Capan1, CD18, Hs766T, and S2-013, PDAC cells were cultured in the indicated concentrations of WP1130 for 72 h. All conditions tested included the same concentration of the DMSO vehicle. Cell viability was assessed by MTT assay, as described in the Materials and Methods. Measurements from triplicate cultures (n = 3) were averaged and plotted. Error bars represent standard deviations, and the Student t test was used to determine significance. This experiment was repeated and similar results were obtained.
Discussion
The deubiquitinating enzyme USP9X has been shown to participate in large list of biological pathways and processes. The roles of USP9X are likely to be highly context-dependent, because of the broad diversity of its targets. Mounting evidence suggests that USP9X behaves principally as an oncogene in the context of many neoplasms. Studies have demonstrated that USP9X levels correlate with tumor cell growth and staging in a number of cancers including lung,26 breast,27 cervical,22 chronic myelogenous leukemia,24 colon,28 esophageal carcinoma,29 brain,5 and to a limited extent, PDAC.1 The data presented in this report supports the role of USP9X as a growth promoter in the context of PDAC. Specifically, we demonstrate that USP9X is required for the monolayer growth of five PDAC cell lines. Use of inducible knockdown of USP9X in two PDAC cell lines, one with wild-type KRAS and one with mutant KRAS, indicates that knockdown of USP9X also inhibits their anchorage-independent growth. Interestingly, we demonstrate that the knockdown of USP9X does not affect the migratory behavior of iKD-USP9X-BxPC3 cells, but does enhance their ability to invade through a biomatrix. We also demonstrate that an in vitro model of pancreatic cell transformation, which utilizes HPNE cells and their transformed counterparts, does not alter the relative levels of USP9X, nor one of its target, ITCH. Additionally, we determined that the ability of USP9X to act upon ITCH is dependent upon growth conditions. Knockdown of USP9X decreases the levels of ITCH when the cells are grown in suspension and principally in the nucleus. Lastly, we determined that an inhibitor of deubiquitinating enzymes, WP1130, substantially reduces the growth of five PDAC tumor cell lines.
Roles of USP9X in PDAC cells are context-dependent
Recently, it was reported that USP9X behaves as a tumor-suppressor in a murine model of PDAC in which the Sleeping Beauty transposon interfered with USP9X expression early in development.7 Although USP9X may play an important role in the prevention of PDAC generation, our data lead to the conclusion that, for established PDAC tumor cells, USP9X promotes cell growth. Importantly, the observation that USP9X may function as a tumor-suppressor or as a promoter of cell growth under different contexts may be analogous to the role of TGF-β. During the early development of many cancers, TGF-β behaves as a tumor-suppressor, but during the progression of some cancers, including breast cancer, TGF-β signaling behaves as an oncogene, e.g., by promoting metastasis.8-10
The differing observations and conclusions reached in this report and prior studies supporting USP9X as a tumor-suppressor in PDAC may be due, in part, to differences in experimental design. Notably, the study by Pérez-Mancera and coworkers did not observe a decrease in monolayer growth in a short-term study that did not go beyond 4 d.7 Our studies demonstrate that the effects upon cell growth following knockdown of USP9X do not become evident until after day 4. We observed a reduction in growth by day 6 in each of the five pancreatic cell lines studied, including our engineered USP9X inducible knockdown pancreatic tumor cells. Currently, it is unclear why the growth inhibitory effects of knocking down USP9X only become evident after 4 d. However, the delay in growth inhibition was not due to a long delay in the knockdown of USP9X. We observed decreases in USP9X as early as 2 d following induction of USP9X shRNA. We suspect that the delay in growth reduction is the result of subtle disturbances in multiple pathways, which eventually culminate in growth inhibition after several cell cycles.
Another discrepancy between our data and the findings of Pérez-Mancera and coworkers is the effects on growth under anchorage-independent conditions.7 We observed a reduction in anchorage-independent growth when USP9X was reduced in iKD-USP9X-BxPC3 and iKD-USP9X-Capan1 cells, whereas Pérez-Mancera et al. reported an increase in anchorage-independent growth when USP9X was knocked down in other PDAC cell lines. The reasons for the differing results are unclear. It is possible that the method of knocking down USP9X contributes to the different outcomes: USP9X was knocked down by inducible expression of shRNA directed against USP9X in our study vs. knockdown by stable expression of USP9X shRNA by Pérez-Mancera et al. Other differences exist in the experimental systems, including the culture medium used in the anchorage-independent growth studies. Although both studies examined anchorage-independent growth in soft-agar, our study was performed in serum-free, stem cell medium, supplemented with growth factors as reported by others,30 whereas Pérez-Mancera et al. appear to have used serum-containing medium.
The most important difference between our work and that of others is the assignment of the overall impact of loss of USP9X on pancreatic tumor cells. The studies conducted in a mouse model indicate that interfering with USP9X expression in the context of mutant KRAS can accelerate PDAC formation, which points to USP9X as a tumor-suppressor.6,7 In contrast, our studies indicate that knocking down USP9X in five different pancreatic tumor cell lines leads to a significant growth inhibition. A likely explanation for the difference in conclusions is the endpoint of these studies. Specifically, studies conducted in mice point to an important tumor-suppressor role of USP9X during the early stages of PDAC, whereas our studies indicate that for cells isolated from advanced pancreatic tumors USP9X promotes cell growth, at least in vitro. Thus, our studies suggest that USP9X expression has a more sinister side. USP9X expression may facilitate growth during the later stages of PDAC. Interestingly, USP9X may help limit the spread of these tumor cells, which, again, points to the context-dependent effects of USP9X in this cancer.
The role of UPS9X in cellular function is likely to be pliable due to the vast diversity of biological processes influenced by USP9X. For example, USP9X has been shown to stabilize MCL11 and β-catenin,31,32 moderators of cell viability and proliferation, which would support the role of USP9X as an oncogene in the appropriate context. Interestingly, USP9X has been shown here (Fig. 4) and elsewhere to affect cell motility and invasion.33 Reduction of USP9X levels was previously shown to reduce levels of EFA6, a promoter of de novo tight junction assembly.33 Conversely, USP9X has been shown to stabilize SMURF1, a protein that is required for breast cancer cell motility.2 It is also noteworthy that USP9X forms protein complexes with SOX2 in multiple cell types,5,34 a transcription factors whose expression increases during the progression of PDAC.35 The complexity of assigning a specific role to USP9X in the context of PDAC is further compounded by recent studies indicating that there may be three subtypes of PDAC, as defined by transcriptional profiles.36 Importantly, these subtypes differ in their clinical outcomes and therapeutic responses, and similar differences may exist between our experimental model and the murine models used previously. However, examination of the expression of USP9X and several USP9X targets at the RNA level do not indicate obvious differences in the three PDAC subtypes.
USP9X as a potential target for improvement in the treatment of PDAC
Defining the roles of USP9X during PDAC development and progression may have important implications for the treatment of PDAC. In this regard, if USP9X behaves as a tumor-suppressor in PDAC, implementation of small molecule factors to increase USP9X expression, such as DNA methylase inhibitors,7 may be beneficial. In contrast, if the function of USP9X changes to that of a growth promoter in advanced disease, then deubiquitinating protease inhibitors, such as WP1130, would be more appropriate. However, our data suggest that inhibiting USP9X may reduce tumor growth, but enhance tumor cell dispersal and, as a result, promote further metastasis. In that case, inhibiting USP9X may extend the overall survival of patients with PDAC, but not provide a cure. Given the short survival of PDAC patients, this may be acceptable until more effective therapies are developed in the future.
Numerous reports have recently suggested that impairing the function of USP9X, and more broadly deubiquitinating proteases, is likely to be beneficial in the treatment of other types of cancer. In this regard, USP9X inhibition has been shown to enhance the effects of SAHA and 5-FU,3 and USP9X increases radio-resistance by stabilizing MCL1.37 Moreover, small molecule inhibitors of ubiquitin specific peptidases have been developed, including WP113023 and HBX41108.38 WP1130 has been shown to impair the growth of chronic myelogenous leukemia cells,24 mantle cell lymphoma cells,25 as well as prostate cancer cells,4 and we have demonstrated that WP1130 impairs the growth of both medulloblastoma and glioblastoma cells (Cox and Rizzino, unpublished data), which require USP9X.5 Because our data argue that USP9X is a growth promoter in the context of established PDAC cells, we examined the effects of WP1130 and determined that it impairs the growth of 5 human PDAC cell lines. It is unclear whether this effect is mediated solely through USP9X or through a combination of deubiquitinating proteases, and currently, there are no USP9X-specific inhibitors.
In summary, USP9X, as well as other deubiquitinating proteases, participate in an astounding diversity of biological processes within cells. Undoubtedly, these proteins influence the growth and activity of neoplasms as well. Because of the differences observed in our report and those reported in murine PDAC models, further studies are warranted to more carefully understand the role of USP9X in the context of PDAC. Taken further, USP9X as well as other deubiquitinating proteases may be valuable targets for the development of novel therapeutic modalities to improve clinical outcomes for patients with PDAC.
Materials and Methods
Cell culture conditions
BxPC3, Capan1, CD18, Hs766T, S2-013, iKD-USP9X-BxPC3, and iKD-USP9X-Capan1 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM, Invitrogen) supplemented with 10% fetal bovine serum (FBS, HyClone). Stock cultures of hTERT-HPNE cells and their counterparts that ectopically express E6/E7, SV40 small t-antigen and/or mutant KRAS21,39 were cultured in Medium D (1 volume M3 base [InCell Corp.], 3 volumes glucose-free DMEM, 5% FBS, 5.5 mmol/L glucose, 10 ng/mL epidermal growth factor, and 50 μg/mL gentamicin), as described previously.40 BxPC3, Capan1, CD18, Hs766T, and S2-013 were obtained from M.A. Hollingsworth as cryopreserved cells. hTERT-HPNE cells and their transformed derivatives were obtained from M.M. Ouellette as cryopreserved cells. All cells were used within 6 mo after being recovered from cryopreserved stocks. WP1130 was obtained from Selleck Chemicals (S2243). Cells were maintained at 37 °C in a moist atmosphere of 95% air and 5% CO2. For suspension cultures (ITCH expression), cells were seeded on top of a layer of 0.5% noble agar in DMEM + 10% FBS. MTT assays were used to assess relative cell growth, as described previously.41,42
Lentivirus production and transduction
Lentiviruses used to knockdown USP9X were prepared as described previously.43 Vectors to produce lentiviruses for constitutive (USP9X shRNA no. 1–3) or doxycycline-inducible (Inducible USP9X shRNA) expression of shRNAs were obtained from Open Biosystems. A previously validated non-targeting shRNA (Scrambled) was used as a negative control in knockdown experiments.12 Additional shRNA information is provided in Table S1. The procedure used to transduce cells was described previously.43 Briefly, 24 h after being subcultured, the pancreatic tumor cells were refed with fresh medium containing lentivirus. After 24 h of lentivirus exposure, untransduced cells were eliminated by selection for 48 h in medium containing 5 µg/mL puromycin. iKD-USP9X-BxPC3 cells were expanded as a sub-population of surviving BxPC3 cells. iKD-USP9X-Capan1 cells were produced in the same fashion. USP9X shRNA was induced by culturing cells in doxycycline (Dox, Sigma-Aldrich), for the times and concentrations indicated.
Soft-agar growth
For soft-agar growth, culture dishes were first coated with a hard-agar layer (culture medium + 0.5% noble agar). Counted cells were diluted in medium containing 0.3% noble agar and added on top of the hard-agar layer. Once this soft layer solidified, culture medium and heat-labile factors were added atop the soft-agar layer. Every other day, additional media was added. Dox (1 µg/mL), where indicated, was included in all layers. Culture medium used for soft-agar growth consists of DMEM-F12 (Gibco, 12500-062), 20 µg/mL Insulin (Sigma-Aldrich, I5500), 0.4% BSA (Gibco, 15260037), N2 supplement (100X stock, Gibco, 17502048), B27 supplement (50X stock, Gibco, 17504044), 20 ng/mL EGF (Fisher, 50-813-058), and 10 ng/mL bFGF (Fisher, 50-398-429), as described by others.30
Western blot analysis
An NE-PER kit (Thermo-Scientific) was used to isolate nuclear and cytoplasmic extracts for western blot analysis, as described previously.44 Western blotting was performed as previously described.45 Blots were normalized to indicated loading controls and quantified, as described previously.34 Primary and secondary antibodies are listed in Table S2.
Migration/invasion assays
Migration assays were performed using uncoated, control BD Biocoat Invasion chambers with 8.0 µm pore size PET membrane inserts in 24-well plates (BD Bioscience, 354578). Invasion assays were conducted using BD Matrigel Invasion chambers (354480) according to the manufacturers’ guidelines. The outer chamber of each well was filled with 750 µL DMEM containing 10% FBS. A total of 3 × 104 iKD-USP9X-BxPC3 cells in 500 µL serum-free DMEM-F12 in the absence or presence of Dox (1 µg/mL, 48 h) were loaded into each inner chamber. After 24 h of incubation, the insert was removed and rinsed in 1× PBS. Cotton swabs were used to remove cells from the serum-free side of the membrane. The membranes were fixed in methanol, stained using 1% toluidine blue, and washed three times in PBS. Membranes were dried, removed using a scalpel, and mounted on a slide. Stained cells were counted in the indicated number of random fields (6.25 mm2) using a grid in the microscope eyepiece. Duplicate samples were used to increase the area from which random fields were chosen.
In vitro wound-healing assay
For in vitro wound-healing studies, iKD-USP9X-BxPC3 cells grown in the absence or presence of Dox (1 µg/mL, 3 d) were seeded in 6-well plates at 1 × 106 per well. Twenty-four hours later, wounds were created by scraping the cells with a sterile pipette tip. The cells were photographed with a Canon Rebel XTi camera at 10×, at the time points indicated after wounding. The sizes of the wound at each time point were determined using the rectangle tool in Photoshop.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
Fred and Pamela Buffett Cancer Center and the Nebraska Department of Health (2013-25). Core Facilities of the Cancer Center are supported in part by the National Cancer Institute (CA74771).
Acknowledgments
Michelle Desler is thanked for technical support.
Glossary
Abbreviations:
- Dox
doxycycline
- PDAC
pancreatic ductal adenocarcinoma
- USP9X
ubiquitin specific peptidase 9X
References
- 1.Schwickart M, Huang X, Lill JR, Liu J, Ferrando R, French DM, Maecker H, O’Rourke K, Bazan F, Eastham-Anderson J, et al. Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature. 2010;463:103–7. doi: 10.1038/nature08646. [DOI] [PubMed] [Google Scholar]
- 2.Xie Y, Avello M, Schirle M, McWhinnie E, Feng Y, Bric-Furlong E, Wilson C, Nathans R, Zhang J, Kirschner MW, et al. Deubiquitinase FAM/USP9X interacts with the E3 ubiquitin ligase SMURF1 protein and protects it from ligase activity-dependent self-degradation. J Biol Chem. 2013;288:2976–85. doi: 10.1074/jbc.M112.430066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harris DR, Mims A, Bunz F. Genetic disruption of USP9X sensitizes colorectal cancer cells to 5-fluorouracil. Cancer Biol Ther. 2012;13:1319–24. doi: 10.4161/cbt.21792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang S, Kollipara RK, Srivastava N, Li R, Ravindranathan P, Hernandez E, Freeman E, Humphries CG, Kapur P, Lotan Y, et al. Ablation of the oncogenic transcription factor ERG by deubiquitinase inhibition in prostate cancer. Proc Natl Acad Sci U S A. 2014;111:4251–6. doi: 10.1073/pnas.1322198111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cox JL, Wilder PJ, Gilmore JM, Wuebben EL, Washburn MP, Rizzino A. The SOX2-interactome in brain cancer cells identifies the requirement of MSI2 and USP9X for the growth of brain tumor cells. PLoS One. 2013;8:e62857. doi: 10.1371/journal.pone.0062857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mann KM, Ward JM, Yew CCK, Kovochich A, Dawson DW, Black MA, Brett BT, Sheetz TE, Dupuy AJ, Chang DK, et al. Australian Pancreatic Cancer Genome Initiative Sleeping Beauty mutagenesis reveals cooperating mutations and pathways in pancreatic adenocarcinoma. Proc Natl Acad Sci U S A. 2012;109:5934–41. doi: 10.1073/pnas.1202490109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pérez-Mancera PA, Rust AG, van der Weyden L, Kristiansen G, Li A, Sarver AL, Silverstein KAT, Grützmann R, Aust D, Rümmele P, et al. Australian Pancreatic Cancer Genome Initiative The deubiquitinase USP9X suppresses pancreatic ductal adenocarcinoma. Nature. 2012;486:266–70. doi: 10.1038/nature11114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dalal BI, Keown PA, Greenberg AH. Immunocytochemical localization of secreted transforming growth factor-beta 1 to the advancing edges of primary tumors and to lymph node metastases of human mammary carcinoma. Am J Pathol. 1993;143:381–9. [PMC free article] [PubMed] [Google Scholar]
- 9.Siegel PM, Shu W, Cardiff RD, Muller WJ, Massagué J. Transforming growth factor beta signaling impairs Neu-induced mammary tumorigenesis while promoting pulmonary metastasis. Proc Natl Acad Sci U S A. 2003;100:8430–5. doi: 10.1073/pnas.0932636100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tian F, Byfield SD, Parks WT, Stuelten CH, Nemani D, Zhang YE, Roberts AB. Smad-binding defective mutant of transforming growth factor beta type I receptor enhances tumorigenesis but suppresses metastasis of breast cancer cell lines. Cancer Res. 2004;64:4523–30. doi: 10.1158/0008-5472.CAN-04-0030. [DOI] [PubMed] [Google Scholar]
- 11.Kita K, Saito S, Morioka CY, Watanabe A. Growth inhibition of human pancreatic cancer cell lines by anti-sense oligonucleotides specific to mutated K-ras genes. Int J Cancer. 1999;80:553–8. doi: 10.1002/(SICI)1097-0215(19990209)80:4<553::AID-IJC12>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 12.Wiebe MS, Traktman P. Poxviral B1 kinase overcomes barrier to autointegration factor, a host defense against virus replication. Cell Host Microbe. 2007;1:187–97. doi: 10.1016/j.chom.2007.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen Y, Shi L, Zhang L, Li R, Liang J, Yu W, Sun L, Yang X, Wang Y, Zhang Y, et al. The molecular mechanism governing the oncogenic potential of SOX2 in breast cancer. J Biol Chem. 2008;283:17969–78. doi: 10.1074/jbc.M802917200. [DOI] [PubMed] [Google Scholar]
- 14.Jia X, Li X, Xu Y, Zhang S, Mou W, Liu Y, Liu Y, Lv D, Liu C-H, Tan X, et al. SOX2 promotes tumorigenesis and increases the anti-apoptotic property of human prostate cancer cell. J Mol Cell Biol. 2011;3:230–8. doi: 10.1093/jmcb/mjr002. [DOI] [PubMed] [Google Scholar]
- 15.Nakatsugawa M, Takahashi A, Hirohashi Y, Torigoe T, Inoda S, Murase M, Asanuma H, Tamura Y, Morita R, Michifuri Y, et al. SOX2 is overexpressed in stem-like cells of human lung adenocarcinoma and augments the tumorigenicity. Lab Invest. 2011;91:1796–804. doi: 10.1038/labinvest.2011.140. [DOI] [PubMed] [Google Scholar]
- 16.Kopp JL, Ormsbee BD, Desler M, Rizzino A. Small increases in the level of Sox2 trigger the differentiation of mouse embryonic stem cells. Stem Cells. 2008;26:903–11. doi: 10.1634/stemcells.2007-0951. [DOI] [PubMed] [Google Scholar]
- 17.Cox JL, Wilder PJ, Desler M, Rizzino A. Elevating SOX2 levels deleteriously affects the growth of medulloblastoma and glioblastoma cells. PLoS One. 2012;7:e44087. doi: 10.1371/journal.pone.0044087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carney DN, Gazdar AF, Minna JD. Positive correlation between histological tumor involvement and generation of tumor cell colonies in agarose in specimens taken directly from patients with small-cell carcinoma of the lung. Cancer Res. 1980;40:1820–3. [PubMed] [Google Scholar]
- 19.Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, Wicha M, Clarke MF, Simeone DM. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67:1030–7. doi: 10.1158/0008-5472.CAN-06-2030. [DOI] [PubMed] [Google Scholar]
- 20.Mouchantaf R, Azakir BA, McPherson PS, Millard SM, Wood SA, Angers A. The ubiquitin ligase itch is auto-ubiquitylated in vivo and in vitro but is protected from degradation by interacting with the deubiquitylating enzyme FAM/USP9X. J Biol Chem. 2006;281:38738–47. doi: 10.1074/jbc.M605959200. [DOI] [PubMed] [Google Scholar]
- 21.Campbell PM, Groehler AL, Lee KM, Ouellette MM, Khazak V, Der CJ. K-Ras promotes growth transformation and invasion of immortalized human pancreatic cells by Raf and phosphatidylinositol 3-kinase signaling. Cancer Res. 2007;67:2098–106. doi: 10.1158/0008-5472.CAN-06-3752. [DOI] [PubMed] [Google Scholar]
- 22.Rolén U, Kobzeva V, Gasparjan N, Ovaa H, Winberg G, Kisseljov F, Masucci MG. Activity profiling of deubiquitinating enzymes in cervical carcinoma biopsies and cell lines. Mol Carcinog. 2006;45:260–9. doi: 10.1002/mc.20177. [DOI] [PubMed] [Google Scholar]
- 23.Kapuria V, Peterson LF, Fang D, Bornmann WG, Talpaz M, Donato NJ. Deubiquitinase inhibition by small-molecule WP1130 triggers aggresome formation and tumor cell apoptosis. Cancer Res. 2010;70:9265–76. doi: 10.1158/0008-5472.CAN-10-1530. [DOI] [PubMed] [Google Scholar]
- 24.Sun H, Kapuria V, Peterson LF, Fang D, Bornmann WG, Bartholomeusz G, Talpaz M, Donato NJ. Bcr-Abl ubiquitination and Usp9x inhibition block kinase signaling and promote CML cell apoptosis. Blood. 2011;117:3151–62. doi: 10.1182/blood-2010-03-276477. [DOI] [PubMed] [Google Scholar]
- 25.Pham LV, Tamayo AT, Li C, Bornmann W, Priebe W, Ford RJ. Degrasyn potentiates the antitumor effects of bortezomib in mantle cell lymphoma cells in vitro and in vivo: therapeutic implications. Mol Cancer Ther. 2010;9:2026–36. doi: 10.1158/1535-7163.MCT-10-0238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dasgupta S, Jang JS, Shao C, Mukhopadhyay ND, Sokhi UK, Das SK, Brait M, Talbot C, Yung RC, Begum S, et al. SH3GL2 is frequently deleted in non-small cell lung cancer and downregulates tumor growth by modulating EGFR signaling. J Mol Med (Berl) 2013;91:381–93. doi: 10.1007/s00109-012-0955-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deng S, Zhou H, Xiong R, Lu Y, Yan D, Xing T, Dong L, Tang E, Yang H. Over-expression of genes and proteins of ubiquitin specific peptidases (USPs) and proteasome subunits (PSs) in breast cancer tissue observed by the methods of RFDD-PCR and proteomics. Breast Cancer Res Treat. 2007;104:21–30. doi: 10.1007/s10549-006-9393-7. [DOI] [PubMed] [Google Scholar]
- 28.Peddaboina C, Jupiter D, Fletcher S, Yap JL, Rai A, Tobin RP, Jiang W, Rascoe P, Rogers MKN, Smythe WR, et al. The downregulation of Mcl-1 via USP9X inhibition sensitizes solid tumors to Bcl-xl inhibition. BMC Cancer. 2012;12:541. doi: 10.1186/1471-2407-12-541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peng J, Hu Q, Liu W, He X, Cui L, Chen X, Yang M, Liu H, Wei W, Liu S, et al. USP9X expression correlates with tumor progression and poor prognosis in esophageal squamous cell carcinoma. Diagn Pathol. 2013;8:177. doi: 10.1186/1746-1596-8-177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Herreros-Villanueva M, Zhang J-S, Koenig A, Abel EV, Smyrk TC, Bamlet WR, de Narvajas AA-M, Gomez TS, Simeone DM, Bujanda L, et al. SOX2 promotes dedifferentiation and imparts stem cell-like features to pancreatic cancer cells. Oncogenesis. 2013;2:e61. doi: 10.1038/oncsis.2013.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murray RZ, Jolly LA, Wood SA. The FAM deubiquitylating enzyme localizes to multiple points of protein trafficking in epithelia, where it associates with E-cadherin and beta-catenin. Mol Biol Cell. 2004;15:1591–9. doi: 10.1091/mbc.E03-08-0630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Taya S, Yamamoto T, Kanai-Azuma M, Wood SA, Kaibuchi K. The deubiquitinating enzyme Fam interacts with and stabilizes beta-catenin. Genes Cells. 1999;4:757–67. doi: 10.1046/j.1365-2443.1999.00297.x. [DOI] [PubMed] [Google Scholar]
- 33.Théard D, Labarrade F, Partisani M, Milanini J, Sakagami H, Fon EA, Wood SA, Franco M, Luton F. USP9x-mediated deubiquitination of EFA6 regulates de novo tight junction assembly. EMBO J. 2010;29:1499–509. doi: 10.1038/emboj.2010.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gao Z, Cox JL, Gilmore JM, Ormsbee BD, Mallanna SK, Washburn MP, Rizzino A. Determination of protein interactome of transcription factor Sox2 in embryonic stem cells engineered for inducible expression of four reprogramming factors. J Biol Chem. 2012;287:11384–97. doi: 10.1074/jbc.M111.320143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sanada Y, Yoshida K, Ohara M, Oeda M, Konishi K, Tsutani Y. Histopathologic evaluation of stepwise progression of pancreatic carcinoma with immunohistochemical analysis of gastric epithelial transcription factor SOX2: comparison of expression patterns between invasive components and cancerous or nonneoplastic intraductal components. Pancreas. 2006;32:164–70. doi: 10.1097/01.mpa.0000202947.80117.a0. [DOI] [PubMed] [Google Scholar]
- 36.Collisson EA, Sadanandam A, Olson P, Gibb WJ, Truitt M, Gu S, Cooc J, Weinkle J, Kim GE, Jakkula L, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med. 2011;17:500–3. doi: 10.1038/nm.2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Trivigno D, Essmann F, Huber SM, Rudner J. Deubiquitinase USP9x confers radioresistance through stabilization of Mcl-1. Neoplasia. 2012;14:893–904. doi: 10.1593/neo.12598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Colland F, Formstecher E, Jacq X, Reverdy C, Planquette C, Conrath S, Trouplin V, Bianchi J, Aushev VN, Camonis J, et al. Small-molecule inhibitor of USP7/HAUSP ubiquitin protease stabilizes and activates p53 in cells. Mol Cancer Ther. 2009;8:2286–95. doi: 10.1158/1535-7163.MCT-09-0097. [DOI] [PubMed] [Google Scholar]
- 39.Lee KM, Nguyen C, Ulrich AB, Pour PM, Ouellette MM. Immortalization with telomerase of the Nestin-positive cells of the human pancreas. Biochem Biophys Res Commun. 2003;301:1038–44. doi: 10.1016/S0006-291X(03)00086-X. [DOI] [PubMed] [Google Scholar]
- 40.Lee J, Jang K-T, Ki C-S, Lim T, Park YS, Lim HY, Choi D-W, Kang WK, Park K, Park JO. Impact of epidermal growth factor receptor (EGFR) kinase mutations, EGFR gene amplifications, and KRAS mutations on survival of pancreatic adenocarcinoma. Cancer. 2007;109:1561–9. doi: 10.1002/cncr.22559. [DOI] [PubMed] [Google Scholar]
- 41.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 42.Hansen MB, Nielsen SE, Berg K. Re-examination and further development of a precise and rapid dye method for measuring cell growth/cell kill. J Immunol Methods. 1989;119:203–10. doi: 10.1016/0022-1759(89)90397-9. [DOI] [PubMed] [Google Scholar]
- 43.Cox JL, Mallanna SK, Ormsbee BD, Desler M, Wiebe MS, Rizzino A. Banf1 is required to maintain the self-renewal of both mouse and human embryonic stem cells. J Cell Sci. 2011;124:2654–65. doi: 10.1242/jcs.083238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mallanna SK, Ormsbee BD, Iacovino M, Gilmore JM, Cox JL, Kyba M, Washburn MP, Rizzino A. Proteomic analysis of Sox2-associated proteins during early stages of mouse embryonic stem cell differentiation identifies Sox21 as a novel regulator of stem cell fate. Stem Cells. 2010;28:1715–27. doi: 10.1002/stem.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mallanna SK, Rizzino A. Emerging roles of microRNAs in the control of embryonic stem cells and the generation of induced pluripotent stem cells. Dev Biol. 2010;344:16–25. doi: 10.1016/j.ydbio.2010.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Telford WG, King LE, Fraker PJ. Evaluation of glucocorticoid-induced DNA fragmentation in mouse thymocytes by flow cytometry. Cell Prolif. 1991;24:447–59. doi: 10.1111/j.1365-2184.1991.tb01173.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.