

Abstract
We report the case of a young, never-smoker woman with Li–Fraumeni syndrome and advanced lung adenocarcinoma refractory to multiple lines of conventional chemotherapy and negative for actionable alterations by routine testing. Comprehensive genomic profiling by clinical-grade next generation sequencing was performed on 3320 exons of 184 cancer-related genes and 37 introns of 14 genes frequently rearranged in cancer. The tumor was found to harbor both EGFR L858R and ERBB2 S310F alterations and also tested positive for a known TP53 germline mutation. The presence of the EGFR mutation was further validated by direct sequencing. Based on these results, a dual EGFR/ERBB2 inhibitor, afatinib, was chosen for treatment. The patient achieved a rapid, complete, and durable response to afatinib monotherapy, both clinically and radiographically. The treatment was very well tolerated. This unique case raises practical questions as to the challenges of molecular testing and highlights the potential association of p53 mutations with concurrent EGFR and ERBB2 aberrations. As this case powerfully illustrates, the combination of broad genomic profiling and targeted therapy guided by mutational analysis offers the possibility of precision management of refractory advanced adenocarcinoma in the background of neoplastic syndromes.
Keywords: EGFR, ERBB2, Li–Fraumeni syndrome, afatinib, genomic profiling, lung cancer, next generation sequencing
Introduction
Recently, significant advances have been made in our understanding of the molecular abnormalities contributing to lung cancer pathogenesis. In a large majority of never smoking, lung adenocarcinoma patients, dominant oncogenic alterations are found in epidermal growth factor receptor (EGFR), ALK, ROS, human epidermal growth factor receptor 2 (ERBB2 or HER2), and other genes. KRAS mutations occur in about 20–25% of lung adenocarcinoma patients, mostly in smokers. The majority of “driver” oncogenic alterations are mutually exclusive except for the frequent co-occurrence of PI3K mutations with other mutational events, such as EGFR or KRAS mutations.1 ERBB2 mutations, occurring in about 1–10% of lung adenocarcinomas, encompass both exon 20 insertions and extracellular domain mutations, including the ERBB2 S310F alteration identified in our patient.2,3 The ERBB2 S310F mutation is a recently discovered mutation identified by a comprehensive genetic study.4 Up until now 10 cancer cases have been reported in the literature, 3 in lung, 2 in large intestine, 1 in stomach, 1 in urinary tract, 2 in breast, and 1 in ovarian.2,4-9
We report a fascinating case of a young, non-smoker patient with Li–Fraumeni syndrome and advanced, chemotherapy-refractory, EGFR and ERBB2 double mutant lung adenocarcinoma with a rapid, complete, and durable response to afatinib monotherapy.
Clinical Case Report
The patient is a 46-y-old, never smoker, Hispanic woman with Li–Fraumeni syndrome (LFS) who was diagnosed with recurrent, metastatic lung adenocarcinoma in September 2010. She initially presented for medical attention in 2005 when she was found to be PPD-positive and completed a course of isoniazid treatment. A chest X-ray indicated a small right middle lobe lesion, which was followed for two years with interval growth. Two years later some growth in this lesion was noted. A fused positron emission tomography/computed topography (PET/CT) scan revealed three FDG-avid lesions: a 2.7 × 2.2 cm cavitary right middle lobe nodule suggesting a pulmonary malignancy, a left breast lesion, and a lesion in the left gluteal area. She underwent a right middle lobe lobectomy and nodal dissection in November 2007, which yielded a TTF1-positive, T1aN0M0, 1.6 cm lung tumor with moderately differentiated adenosquamous carcinoma histology (Fig. 1). At that time no further therapy post-operation for her lung cancer was recommended. In January 2008, she underwent a left breast lumpectomy and sentinel node biopsy, which revealed a T1cN0M0 ER+/PR+/ERBB2+ invasive ductal breast carcinoma. She received 4 cycles of cyclophosphamide/docetaxel, followed by adjuvant tamoxifen therapy and one year of trastuzumab therapy. The gluteal lesion, a biopsy proven Schwannoma, was initially observed and ultimately resected in February 2010. She was noted to have a strong family history of early onset cancers: her sister and her aunt had breast cancer in their 40s, her cousin had a brain tumor at a young age, and her mother had leukemia. Given her multiple malignancies and strong family history of early onset breast cancer, brain cancer, and leukemia, genetic testing was performed, revealing a TP53 germline mutation in the patient and two of her children. BRCA1 and BRCA2 testing was negative. Given these results and the presence of complex adnexal cysts on imaging studies, she underwent prophylactic bilateral total mastectomies with breast reconstructive surgeries and total abdominal hysterectomy-bilateral salpingo-oophorectomy. Surgical pathology was unremarkable except an atypical leiomyoma.
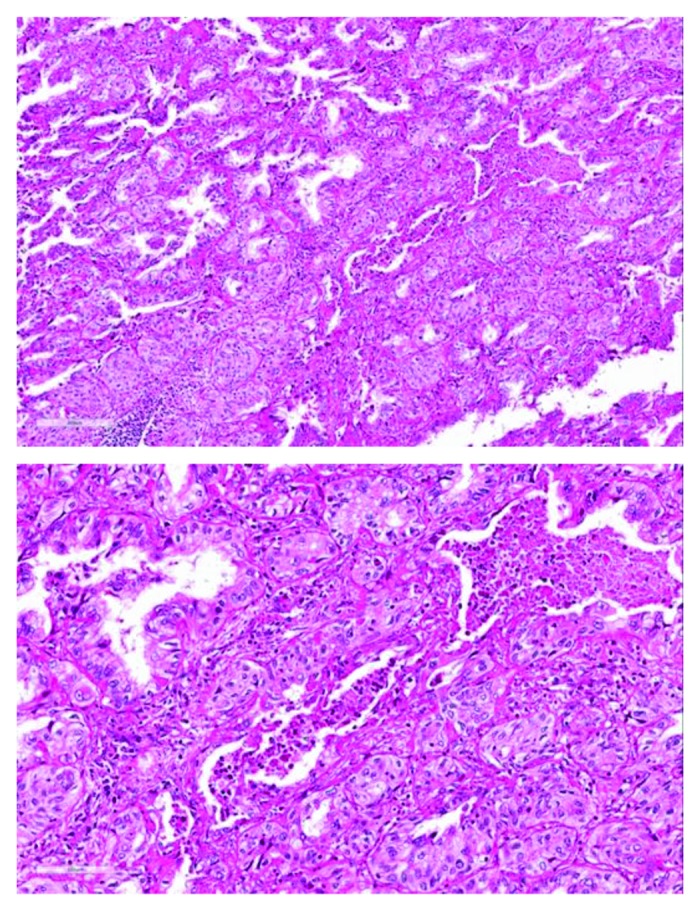
Figure 1. H&E (hematoxylin and eosin) staining of a lung resection specimen shows predominantly adenocarcinoma histology with areas of squamous differentiation.
She was followed expectantly and yearly scans were negative until three years later when in September 2010 she was noted to have a large right pleural effusion and pleural nodularity. Cytology from the pleural effusion confirmed lung adenocarcinoma and pleurodesis was successfully performed. Direct sequencing for EGFR and KRAS and 2p23 break-apart FISH for ALK of a cell block from a pleural fluid specimen was negative. At the time of original diagnosis, mutation study of the surgical specimen from the primary specimen was not initially done, because it was not a routine practice at the time. She was treated with carboplatin and pemetrexed combination chemotherapy for six cycles with a near-complete response. Maintenance pemetrexed was administered for eight months thereafter until disease progression in November 2011. She opted to participate in a clinical trial randomizing patients to docetaxel with or without a PI3 kinase inhibitor and received 4 cycles of docetaxel monotherapy. She again progressed with worsening pleural-based nodules and was switched to gemcitabine chemotherapy. Her disease remained stable for 6 mo before clinical and radiographic progression with worsening pleural-based masses, parenchymal lung nodules, hilar, subcarinal, and supraclavicular adenopathy and progressive fatigue, dyspnea, and chest discomfort in December 2012.
The perceived lack of effective standard therapies in the face of the patient’s still excellent performance status and multiple prior lines of therapy, prompted broader genomic profiling for the presence of other actionable genomic alterations, including RET fusion.
A formalin-fixed paraffin-embedded (FFPE) tumor specimen was tested for comprehensive genomic profiling using clinical next generation sequencing by Foundation Medicine. Hybrid capture of 3320 exons in 182 cancer-related genes and 37 introns from 14 genes frequently rearranged in cancer was performed for >50 ng of DNA and sequenced to a median coverage depth of 120×.5 Analysis of the right middle lobe resection specimen confirmed the known germline TP53 gene mutation, G245S (estimated 59% of reads); and demonstrated the presence of both an EGFR L858R mutation (estimated 27% of reads) and an ERBB2 extracellular domain S310F mutation (estimated 25% of reads). In light of the dual EGFR and ERBB2 mutations, instead of pursuing erlotinib therapy, in January 2013 the patient was enrolled into an ongoing compassionate use program of afatinib, a dual inhibitor of EGFR and ERBB2.
After two weeks of afatinib monotherapy at 40 mg a day orally, her fatigue, right-sided chest pain and dyspnea improved dramatically, and all her symptoms resolved completely after 2 mo. Repeat fused PET-CT scans revealed complete anatomic and metabolic response with minimal residual FDG avidity in the right pleura consistent with prior talc pleurodesis (Fig. 2). Twelve months afterwards, complete response was maintained, both clinically and by repeated imaging. She has been tolerating afatinib very well with minimal adverse effects, such as easily manageable grade 1 rash and grade 1 diarrhea.

Figure 2. (A) PET/CT (positron emission tomography/computed tomography) scan prior to afatinib treatment. (B) Follow-up PET/CT scan after 2 and (C) 10 mo of afatinib treatment with complete anatomic and metabolic response.
Discussion
This case describes a never-smoker, LFS patient, whose recurrent, metastatic lung adenocarcinoma harbored activating and sensitizing EGFR and ERBB2 mutations and who achieved a complete response to a dual EGFR and ERBB2 inhibitor. This case brings up a number of relevant clinical, diagnostic, and scientific considerations.
It would be highly interesting to learn whether the EGFR and ERBB2 mutations within our patient co-exist in a single tumor clone or represent two independent clones and further testing to address this will be pursued in the future if feasible, such as through the use of mutation-specific antibodies. We were able to assess allele frequencies of these mutations from the clinical test sample and very similar allele frequencies of the EGFR and ERBB2 mutations do strongly suggest their co-occurrence within the same clone of tumor cells. Further testing in such cases could be contemplated at the time of progression as well to better address which mutation/pathway is key in driving drug resistance. Only about 1% of lung cancers carry dual driver mutations at the time of diagnosis.10 However, this frequency may be underestimated since in advanced lung cancer patients usual testing methods, such as direct sequencing, might experience reduced sensitivity as the proportion of tumor DNA in a typical specimen frequently is below 20%.11
Our patient carries a germ-line TP53 alteration, predisposing her to multiple malignancies, such as Schwannoma, lung, and breast adenocarcinomas, and atypical leiomyoma. TP53 is a tumor-suppressing protein, whose function is frequently lost in cancers through TP53 gene missense mutations. TP53 mutations have been found to co-occur with EGFR mutations in lung cancer patients as noted in 38% of Japanese lung cancer patients with EGFR mutations and reported in 34% of lung cancer patients in the international project of The Clinical Lung Cancer Genome Project (CLGGP) and Network Genomic Medicine (NGM) on genomics-based classification.12,13 Arguably, TP53 loss may predispose to the synchronous development of oncogenic EGFR and ERBB2 alterations through impaired DNA damage response, as these oncogenic events rarely co-occur otherwise. Very intriguingly, a recent case report of a patient with LFS and breast and lung cancers reported similar dual mutations of EGFR and ERBB2 and a response to lapatinib therapy.14 The relationship between TP53 and dual EGFR/ERBB2 mutations may be causative or associative, but is currently undefined and may be investigated through LFS tumor registries. Interestingly, TP53 mutant tumors with EGFR mutation did worse than TP53 wild-type tumors with EGFR mutation.13 Further exploration of the molecular characteristics of TP53 mutation-driven oncogenic pathways may reveal additional diagnostic and therapeutic insights into LFS tumors.
The patient’s L858R EGFR gene mutation had originally not been found from the pleural fluid cell block during direct EGFR sequencing, a standardized molecular test. The specimen contained tumor cells admixed with non-malignant cells, which coupled with a highly cellular inflammatory background, likely affected sensitivity and produced a false negative test result. Later on, the presence of the L858R mutation was confirmed through standard sequencing on the patient’s original primary specimen, which was a more cellular resection specimen from 2007. This situation demonstrates that careful communication between treating physicians and pathology testing facilities is necessary to ascertain that the most clinically relevant and technically best specimens are used for testing. Although ERBB2 mutations have long been known to occur in lung cancer and are viewed as actionable, widespread ERBB2 testing is not performed.15 In our patient’s case, standard molecular testing would not have revealed the presence of the concurrent ERBB2 mutation. Such cases might suggest that future standard molecular testing may need to encompass ERBB2 alterations. Genomic profiling by a comprehensive clinical next generation sequencing assay as performed in this case, uncovers actionable genomic alterations in a high proportion of patients, thus allowing optimal selection of molecularly targeted therapies for improved patient outcomes.16 A recent study demonstrated the feasibility of comprehensive genomic profiling based on clinical grade next generation sequencing in describing alterations in cytologic samples, including fine-needle aspirations (FNAs) of pulmonary neoplasms. For pancreatic neoplasms, there was perfect (100%) concordance between the genomic profiles of FNAs and all available matched tumor resections.17
Clinically and radiographically, this patient responded remarkably to afatinib, a second-generation tyrosine kinase inhibitor (TKI) that irreversibly inhibits the EGFR and ERBB2/HER2 kinases.18,19 Although this patient’s response may predominantly rely upon inhibition of one oncogenic pathway, this case highly suggests both pathways were key targets. Tumors harboring the common activating EGFR alterations, L858R base substitutions, and exon 19 deletions, are highly responsive to EGFR tyrosine kinase inhibitor therapy with response rates of 60–80%. Clinically, the responsiveness of ERBB2-mutated lung adenocarcinomas has not been rigorously evaluated heretofore mainly because of lack of routine testing. However, a prospective “basket trial” of neratinib, a pan-ERBB2 inhibitor in patients with ERBB2 alterations largely independent of site of origin was recently commenced (NCT01953926). Preclinical reports show ERBB2 alterations responding to anti-ERBB2 therapy, including the irreversible HER2 inhibitors afatinib and dacomitinib.20
Afatinib is a potent small-molecule irreversible pan ErbB inhibitor (EGFR, IC50 0.5 nM and ERBB2, IC50 14 nM).19 Complete response to afatinib has been reported in lung cancer with EGFR mutations (deletion 19 or L858R).21 In comparison to first line doublet chemotherapy, afatinib improved progression-free survival in patients with EGFR-activating mutations, leading to its recent FDA approval. Afatinib is also a potent ERBB2 kinase inhibitor.22 Because of afatinib’s additional activity against HER2, it is actively investigated for breast cancer and for other EGFR and HER2 driven cancers.18,23-25 Afatinib monotherapy demonstrated clinical activity in ERBB2-positive breast cancer, including trastuzumab-resistant breast cancer.24
Afatinib is not only active against EGFR mutations targeted by first generation TKIs like erlotinib or gefitinib, but also appears to have some preclinical activity against EGFR mutations not sensitive to these standard therapies, such as EGFR T790M and exon 20 insertions.22 Accumulating evidence has demonstrated cross-talk between HER family members in the development of acquired resistance to EGFR-TKI therapy.26,27 HER2 overexpression also can confer resistance to EGFR-TKIs in cell line models and the HER2 gene was found to be amplified in a significant fraction of both murine and human tumors with acquired resistance to erlotinib. As noted earlier, re-biopsy of such a unique tumor upon progression could provide further clues as to the contribution of the two synchronous oncogenic pathways to acquired resistance.
To our knowledge, our report of a LFS patient with advanced lung adenocarcinoma containing EGFR L858R, ERBB2 S310F, and TP53 germ-line mutations, demonstrating a complete and durable response to afatinib, is the first case of its kind. The patient responded to afatinib completely after failure of multiple lines of chemotherapy. This case well exemplifies the emerging power of molecular testing and targeted therapeutics for the management of refractory advanced adenocarcinoma.
Disclosure of Potential Conflicts of Interest
S.M.A. and V.A.M. are employees of and have equity interest in Foundation Medicine, Inc. B.H. receives clinical research grant support from Boehringer-Ingelheim Pharmaceuticals. For the remaining authors, no conflicts of interest were declared.
Glossary
Abbreviations:
- LFS
Li–Fraumeni syndrome
- PET
positron emission tomography
- CT
computed tomography
- FDG
fluorodeoxyglucose
- SUV
standardized uptake value
- FFPE
formalin fixed paraffin embedded
- FNA
fine needle aspiration
- H&E stain
hematoxylin and eosin
References
- 1.Pao W, Hutchinson KE. Chipping away at the lung cancer genome. Nat Med. 2012;18:349–51. doi: 10.1038/nm.2697. [DOI] [PubMed] [Google Scholar]
- 2.Han SW, Kim HP, Shin JY, Jeong EG, Lee WC, Lee KH, Won JK, Kim TY, Oh DY, Im SA, et al. Targeted sequencing of cancer-related genes in colorectal cancer using next-generation sequencing. PLoS One. 2013;8:e64271. doi: 10.1371/journal.pone.0064271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stephens P, Hunter C, Bignell G, Edkins S, Davies H, Teague J, Stevens C, O’Meara S, Smith R, Parker A, et al. Lung cancer: intragenic ERBB2 kinase mutations in tumours. Nature. 2004;431:525–6. doi: 10.1038/431525b. [DOI] [PubMed] [Google Scholar]
- 4.Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, Sougnez C, Greulich H, Muzny DM, Morgan MB, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–75. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bell D, Berchuck A, Birrer M, Chien J, Cramer D, Dao F, Dhir R, DiSaia P, Gabra H, Glenn P, et al. Cancer Genome Atlas Research Network Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–15. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seo JS, Ju YS, Lee WC, Shin JY, Lee JK, Bleazard T, Lee J, Jung YJ, Kim JO, Shin JY, et al. The transcriptional landscape and mutational profile of lung adenocarcinoma. Genome Res. 2012;22:2109–19. doi: 10.1101/gr.145144.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, Turashvili G, Ding J, Tse K, Haffari G, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012;486:395–9. doi: 10.1038/nature10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang K, Kan J, Yuen ST, Shi ST, Chu KM, Law S, Chan TL, Kan Z, Chan AS, Tsui WY, et al. Exome sequencing identifies frequent mutation of ARID1A in molecular subtypes of gastric cancer. Nat Genet. 2011;43:1219–23. doi: 10.1038/ng.982. [DOI] [PubMed] [Google Scholar]
- 9.Ali SM, Alpaugh RK, Downing SR, Stephens PJ, Yu JQ, Wu H, Buell JK, Miller VA, Lipson D, Palmer GA, et al. Response of an ERBB2-Mutated Inflammatory Breast Carcinoma to Human Epidermal Growth Factor Receptor 2-Targeted Therapy. J Clin Oncol. 2014 doi: 10.1200/JCO.2013.49.0599. Forthcoming. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson BE, Kris MG, Berry LD, Kwiatkowski DJ, Iafrate A J, Varella-Garcia M, Wistuba I I, Franklin W A, Ladanyi M, Su P F, et al. A multicenter effort to identify driver mutations and employ targeted therapy in patients with lung adenocarcinomas: The Lung Cancer Mutation Consortium (LCMC) J Clin Oncol. 2013;31:abstr 8019. [Google Scholar]
- 11.Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, Schnall-Levin M, White J, Sanford EM, An P, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31:1023–31. doi: 10.1038/nbt.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kosaka T, Yatabe Y, Endoh H, Kuwano H, Takahashi T, Mitsudomi T. Mutations of the epidermal growth factor receptor gene in lung cancer: biological and clinical implications. Cancer Res. 2004;64:8919–23. doi: 10.1158/0008-5472.CAN-04-2818. [DOI] [PubMed] [Google Scholar]
- 13.Clinical Lung Cancer Genome Project (CLCGP) Network Genomic Medicine (NGM) A genomics-based classification of human lung tumors. Sci Transl Med. 2013;5:ra153. doi: 10.1126/scitranslmed.3006802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Serra V, Vivancos A, Puente XS, Felip E, Silberschmidt D, Caratù G, Parra JL, De Mattos-Arruda L, Grueso J, Hernández-Losa J, et al. Clinical response to a lapatinib-based therapy for a Li-Fraumeni syndrome patient with a novel HER2V659E mutation. Cancer Discov. 2013;3:1238–44. doi: 10.1158/2159-8290.CD-13-0132. [DOI] [PubMed] [Google Scholar]
- 15.Mazières J, Peters S, Lepage B, Cortot AB, Barlesi F, Beau-Faller M, Besse B, Blons H, Mansuet-Lupo A, Urban T, et al. Lung cancer that harbors an HER2 mutation: epidemiologic characteristics and therapeutic perspectives. J Clin Oncol. 2013;31:1997–2003. doi: 10.1200/JCO.2012.45.6095. [DOI] [PubMed] [Google Scholar]
- 16.Ali SM, Palma NA, Wang K, Ross J S, Stephens P J, Yelensky R, Palmer G A, Lipson D, Miller VA, et al. Clinical next generation sequencing (NGS) to reveal high frequency of alterations to guide targeted therapy in lung cancer patients. J Clin Oncol. 2013;31:abstr 8020. [Google Scholar]
- 17.Young G, Wang K, He J, Otto G, Hawryluk M, Zwirco Z, Brennan T, Nahas M, Donahue A, Yelensky R, et al. Clinical next-generation sequencing successfully applied to fine-needle aspirations of pulmonary and pancreatic neoplasms. Cancer Cytopathol. 2013;121:688–94. doi: 10.1002/cncy.21338. [DOI] [PubMed] [Google Scholar]
- 18.Minkovsky N, Berezov A. BIBW-2992, a dual receptor tyrosine kinase inhibitor for the treatment of solid tumors. Curr Opin Investig Drugs. 2008;9:1336–46. [PubMed] [Google Scholar]
- 19.Li D, Ambrogio L, Shimamura T, Kubo S, Takahashi M, Chirieac LR, Padera RF, Shapiro GI, Baum A, Himmelsbach F, et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene. 2008;27:4702–11. doi: 10.1038/onc.2008.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Engelman JA, Zejnullahu K, Gale CM, Lifshits E, Gonzales AJ, Shimamura T, Zhao F, Vincent PW, Naumov GN, Bradner JE, et al. PF00299804, an irreversible pan-ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res. 2007;67:11924–32. doi: 10.1158/0008-5472.CAN-07-1885. [DOI] [PubMed] [Google Scholar]
- 21.Yang JC, Shih JY, Su WC, Hsia TC, Tsai CM, Ou SH, Yu CJ, Chang GC, Ho CL, Sequist LV, et al. Afatinib for patients with lung adenocarcinoma and epidermal growth factor receptor mutations (LUX-Lung 2): a phase 2 trial. Lancet Oncol. 2012;13:539–48. doi: 10.1016/S1470-2045(12)70086-4. [DOI] [PubMed] [Google Scholar]
- 22.Bean J, Riely GJ, Balak M, Marks JL, Ladanyi M, Miller VA, Pao W. Acquired resistance to epidermal growth factor receptor kinase inhibitors associated with a novel T854A mutation in a patient with EGFR-mutant lung adenocarcinoma. Clin Cancer Res. 2008;14:7519–25. doi: 10.1158/1078-0432.CCR-08-0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miller VA, Hirsh V, Cadranel J, Chen YM, Park K, Kim SW, Zhou C, Su WC, Wang M, Sun Y, et al. Afatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-Lung 1): a phase 2b/3 randomised trial. Lancet Oncol. 2012;13:528–38. doi: 10.1016/S1470-2045(12)70087-6. [DOI] [PubMed] [Google Scholar]
- 24.Lin NU, Winer EP, Wheatley D, Carey LA, Houston S, Mendelson D, Munster P, Frakes L, Kelly S, Garcia AA, et al. A phase II study of afatinib (BIBW 2992), an irreversible ErbB family blocker, in patients with HER2-positive metastatic breast cancer progressing after trastuzumab. Breast Cancer Res Treat. 2012;133:1057–65. doi: 10.1007/s10549-012-2003-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Spicer JF, Rudman SM. EGFR inhibitors in non-small cell lung cancer (NSCLC): the emerging role of the dual irreversible EGFR/HER2 inhibitor BIBW 2992. Target Oncol. 2010;5:245–55. doi: 10.1007/s11523-010-0140-y. [DOI] [PubMed] [Google Scholar]
- 26.Regales L, Gong Y, Shen R, de Stanchina E, Vivanco I, Goel A, Koutcher JA, Spassova M, Ouerfelli O, Mellinghoff IK, et al. Dual targeting of EGFR can overcome a major drug resistance mutation in mouse models of EGFR mutant lung cancer. J Clin Invest. 2009;119:3000–10. doi: 10.1172/JCI38746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takezawa K, Pirazzoli V, Arcila ME, Nebhan CA, Song X, de Stanchina E, Ohashi K, Janjigian YY, Spitzler PJ, Melnick MA, et al. HER2 amplification: a potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFRT790M mutation. Cancer Discov. 2012;2:922–33. doi: 10.1158/2159-8290.CD-12-0108. [DOI] [PMC free article] [PubMed] [Google Scholar]