

Abstract
Background
The PCR technique and its variations have been increasingly used in the clinical laboratory and recent advances in this field generated new higher resolution techniques based on nucleic acid denaturation dynamics. The principle of these new molecular tools is based on the comparison of melting profiles, after denaturation of a DNA double strand. Until now, the secondary structure of single-stranded nucleic acids has not been exploited to develop identification systems based on PCR. To test the potential of single-strand RNA denaturation as a new alternative to detect specific nucleic acid variations, sequences from viruses of the Totiviridae family were compared using a new in silico melting curve approach. This family comprises double-stranded RNA virus, with a genome constituted by two ORFs, ORF1 and ORF2, which encodes the capsid/RNA binding proteins and an RNA-dependent RNA polymerase (RdRp), respectively.
Results
A phylogenetic tree based on RdRp amino acid sequences was constructed, and eight monophyletic groups were defined. Alignments of RdRp RNA sequences from each group were screened to identify RNA regions with conserved secondary structure. One region in the second half of ORF2 was identified and individually modeled using the RNAfold tool. Afterwards, each DNA or RNA sequence was denatured in silico using the softwares MELTSIM and RNAheat that generate melting curves considering the denaturation of a double stranded DNA and single stranded RNA, respectively. The same groups identified in the RdRp phylogenetic tree were retrieved by a clustering analysis of the melting curves data obtained from RNAheat. Moreover, the same approach was used to successfully discriminate different variants of Trichomonas vaginalis virus, which was not possible by the visual comparison of the double stranded melting curves generated by MELTSIM.
Conclusion
In silico analysis indicate that ssRNA melting curves are more informative than dsDNA melting curves. Furthermore, conserved RNA structures may be determined from analysis of individuals that are phylogenetically related, and these regions may be used to support the reconstitution of their phylogenetic groups. These findings are a robust basis for the development of in vitro systems to ssRNA melting curves detection.
Electronic supplementary material
The online version of this article (doi:10.1186/1471-2105-15-243) contains supplementary material, which is available to authorized users.
Keywords: RNA secondary structure; Infectious Myonecrosis Virus; high resolution melting curve; Virus detection; IHHNV, WSSV, Trichomonas
Background
Despite the emergence of new techniques to nucleic acids investigation such as next generation sequence and array chips, the Polymerase Chain Reaction (PCR) and its variations still prevail in clinical laboratories. The use of PCR has grown increasingly in different applications ranging from microorganisms detection to diagnosis of complex genetic diseases [1–3]. The simple implementation and the possibility of post-PCR analysis automation make PCR a great tool for high throughput analysis [3]. Since its introduction with LifeCycler®, the post PCR low resolution melting analysis using SYBR® Green I dye is the method used to confirm the reaction specificity or to detect primer dimer formation and other non-specific products [4]. Some years later, the High Resolution Melting Analysis (HRMA) became possible with the advent of new intercalating dyes that could be used in high concentrations without compromising the PCR efficiency [5]. The HRMA technique allows fast high throughput analysis of PCR products and reinvigorated the use of DNA melting for a wide range of applications, including SNP genotyping and DNA mapping [6–9], gene scanning [10, 11], heterozygosity screening [12], species identification [13, 14] and many others.
The secondary structure formed by a particular nucleic acid molecule influences their DNA melting profile. Many bioinformatic tools designed to predict melting curves of nucleic acids are available [15–17]. Softwares that predict melting curves can efficiently validate regions with different denaturation profiles and these regions can be exploited to differentiate similar sequences and to define targets to post-PCR tests [18]. Until now, studies that attempt to develop molecular tools based on melting curves are restricted to denaturation of double-stranded DNA (dsDNA) molecules. Reports of secondary structures formed by a single nucleic acid strand, particularly single strand RNA (ssRNA), are focused in the determination of viral or viroid genome structures [19–22], noncoding RNAs (ncRNAs) and small interfering RNAs (iRNAs) [23–26].
Using carefully calculated thermodynamic parameters, algorithms can be used to predict the secondary structure of a RNA strand [27–33]. One of the most cited online servers that provide tools to work with RNA structures is the Vienna RNA Package [29]. Among the tools provided, RNAfold calculates the minimum free energy and predicts an optimal secondary structure using McCaskill’s algorithm [30]. Vienna RNA Package also provides the unique tool to assess ssRNA melting curves, the RNAheat software, which reads RNA sequences and calculates their specific heat in a determined temperature range, from the partition function by numeric differentiation [31–33].
The identification of RNA secondary structures is particularly interesting when viral genomes are analyzed. Previous studies demonstrated that conserved stem loops, extensive long-range interactions and small palindromic stem–loops generate structures that are generally associated with viral packing capacity and regulate viral replication [19, 21, 34]. However, such processes and mechanisms are not fully understood in Totiviridae family. Viruses of this family infect protozoa, fungi, insects and shrimps and some of these organisms have medical, zootechnical and agricultural importance [35–38]. Totiviridae members have monopartite double strand RNA (dsRNA) genomes organized in two ORFs. ORF1 encodes a capsid protein (CP) and ORF2 encodes an RNA-dependent RNA polymerase (RdRp) that is highly conserved among the family species [39].
In the present study we propose that the information extracted from a melting curve of a single stranded RNA molecule allows more precise detection of nucleotide variations than the traditional HRMA. To demonstrate our hypothesis, two softwares, RNAheat and MELTSIM, were used to generate melting curves of nucleic acid sequences from Totiviridae viruses. Melting curves generated were used to reconstruct groups determined by a traditional phylogenetic analysis, based on RdRp sequence alignment. Subsequently, ssRNA and dsDNA melting curves were compared for its potential to discriminate Trichomonas vaginalis virus isolates. Our results indicate that the information obtained by ssRNA denaturation may be used as a support to the development of more accurate methods to detect differences in nucleic acid sequences.
Results and discussion
Phylogenetic analysis of Totiviridae family
RNA-dependent RNA polymerases sequences are conserved within members of the families Totiviridae and Chrysoviridae [40]. Hence they were used to estimate the phylogenetic relationships among these viruses. Twenty eight RdRp aminoacid sequences referenced inTable 1 and two sequences referenced in Table 2 were aligned, and their phylogenetic relationships are shown in Figure 1A. Eight monophyletic groups can be defined in the obtained dendogram, and they were named following Liu et al. classification [40]. The groups IMNV-like, which comprises viruses that infect arthropods, GLV-like and ScV-like matched with previously described inferences [40]. Four new groups were retrieved: MoV-like that comprising viruses that infect plants and fungi, TVV-like and LRV-like that comprises virus that infect human protozoan parasites, and GaRV-like comprising fungus viruses. To ensure the efficiency of the analysis, relationships between TVV-like, LRV-like and GLV-like groups and their integrants were determined using the sequences referenced in Table 2 in a second phylogenetic analysis showed in Figure 1B. All observed groups are in agreement with the classification proposed by International Committee on Taxonomy of Viruses (ICTV) [41]. GLV-like comprises viruses of the genus Giardiavirus and ScV-like comprises viruses of the genus Totivirus. The genus Victorivirus includes two gropus, MoV-like and GaRV-like. The genera Leishmaniavirus and Trichomonasvirus include groups LRV-like and TVV-like respectively. IMNV-like group appears as less derived group near to GLV-like and does is not classified by ICTV. The Zygosaccharomyces bailii virus (ZbV-Z) and two other related viruses isolated from plants and fungus clustered together to form a ZbV-Z-like clade, on a basal branch of the phylogenetic tree (Figure 1A). Indeed, this group was formerly referred as a primitive, less derived group, distantly related to Totiviruses, and includes virions with distinct genomic organization from this family. A new family Amalgamaviridae has been proposed to accommodate these three viruses [40].
Table 1.
Totiviridae aminoacid sequences used in this study grouped according to phylogenetic analysis
| Virus name | Accession No. | Abbreviation |
|---|---|---|
| MoV-like | ||
| Beauveria bassiana RNA virus 1 | CCC42235 | BeauV |
| Tolypocladium cylindrosporum virus 1 | YP_004089630 | TcV-1 |
| Botryotinia fuckeliana totivirus 1 | YP_001109580 | BotryV |
| Helminthosporium victoriae virus 190S | NP_619670 | HvV-190S |
| Chalara elegans RNA virus 1 | YP_024728 | ChalElV |
| Helicobasidium mompa totivirus 1-17 | NP_898833 | HmV1-17 |
| Magnaporthe oryzae virus 1 | YP_122352.1 | MoV-1 |
| IMNV-like | ||
| Infectious myonecrosis virus | AHY18670.1 | IMNV |
| Tianjin totivirus | AFE02920.1 | TianV |
| Omono river virus | BAJ21511.1 | ORV |
| Drosophila melanogaster totivirus SW-2009a | YP_003289293.1 | DmV-SW-2009a |
| Armigeres subalbatus virus SaX06-AK20 | YP_003934934.1 | AsV-SaX06-AK20 |
| GLV-like | ||
| Piscine myocarditis virus AL V-708 | YP_004581250.1 | PMV-AL V-708 |
| Giardia canis virus from China (2883–5981)* | DQ238861.1 | GCV |
| Giardia lamblia virus (2880–5978)* | NC_003555.1 | GLV2 |
| ZbV-Z like | ||
| Blueberry latent virus isolate AR (936–3332)* | HM029248.1 | BLV |
| Southern tomato virus isolate Mexico-1(1039–3327)* | EF442780.1 | STV |
| Zygosaccharomyces bailii virus Z | NP_624325.1 | ZbV-Z |
| ScV-like | ||
| Ustilago maydis virus H1 (735–6002)* | NC_003823.1 | UmV-H1 |
| Saccharomyces cerevisiae virus L-BC (La) | NP_042581.1 | ScV L-BC |
| Saccharomyces cerevisiae virus L-A | NP_620495.1 | ScV L-A |
| Black raspberry virus F | YP_001497151.1 | BRV-F |
| Tuber aestivum virus 1 | ADQ54106.1 | TaV-1 |
| GaRV-like | ||
| Sphaeropsis sapinea RNA virus 2 | AAD11603.1 | SphaeroV |
| Coniothyrium minitans RNA virus | YP_392467.1 | CmRV |
| Epichloe festucae virus 1 | CAK02788.1 | EpiFesV |
| Gremmeniella abietina RNA virus L2 | AAT48885.1 | GaRV-L2 |
| Other sequence | ||
| Eimeria brunetti RNA virus 1 | NP_108651 | EbRV-1 |
*Accession numbers correspond to nucleotide sequences of complete genomes. Numbers in brackets correspond to first and last nucleotides of RdRp coding sequences.
Table 2.
Aminoacid sequences of Trichomonasvirus , Leishmaniavirus and Giardiavirus used in this study grouped according to phylogenetic analysis
| Virus name | Accession no. | Abbreviation |
|---|---|---|
| TVV4 | ||
| Trichomonas vaginalis virus 4 strain TVV4-1 | AED99796.1 | TVV4-1 |
| Trichomonas vaginalis virus 4 strain TVV4-OC3 | AED99794.1 | TVV4-OC3 |
| Trichomonas vaginalis virus 4 strain TVV4-OC5 | AED99798.1 | TVV4-OC5 |
| TVV3 | ||
| Trichomonas vaginalis virus 3 strain TVV3-UR1 | AED99800.1 | TVV3-UR1 |
| Trichomonas vaginalis virus 3 strain TVV3-OC5 | AED99804.1 | TVV3-OC5 |
| Trichomonas vaginalis virus 3 strain TVV3-OC3 | AED99802.1 | TVV3-OC3 |
| Trichomonas vaginalis virus 3 | NP_659390.1 | Trichomonasvirus_3 |
| TVV2 | ||
| Trichomonas vaginalis virus 2 strain TVV2-OC3 | AED99808.1 | TVV2-OC3 |
| Trichomonas vaginalis virus 2 strain TVV2-UR1 | AED99806.1 | TVV2-UR1 |
| Trichomonas vaginalis virus 2 strain TVV2-OC5 | AED99810.1 | TVV2-OC5 |
| Trichomonas vaginalis virus II | NP_624323.2 | Trichomonasvirus_II |
| Trichomonas vaginalis virus 2 isolate C76 | AET81014.1 | TVV2-C76 |
| Trichomonas vaginalis virus 2 isolate C351 | AET81016.1 | TVV2-C351 |
| TVV1 | ||
| Trichomonas vaginalis virus | NP_620730.2 | Trichomonasvirus_I |
| Trichomonas vaginalis virus 1 isolate C344 | AET81012.1 | TVV1-C344 |
| Trichomonas vaginalis virus 1 strain TVV1-UH9 | AED99814.1 | TVV1-UH9 |
| Trichomonas vaginalis virus 1 strain TVV1-OC4 | AED99818.1 | TVV1-OC4 |
| Trichomonas vaginalis virus 1 strain TVV1-OC3 | AED99816.1 | TVV1-OC3 |
| Trichomonas vaginalis virus 1 strain TVV1-UR1 | AED99812.1 | TVV1-UR1 |
| Trichomonas vaginalis virus 1 strain TVV1-OC5 | AED99820.1 | TVV1-OC5 |
| LRV-like | ||
| Leishmania RNA virus 2 - 1 | NP_043465.1 | LRV 2-1 |
| Leishmania RNA virus 1 - 4 | NP_619653.1 | LRV 1-4 |
| Leishmania RNA virus 1 - 1 | NP_041191.1 | LRV 1-1 |
| GLV-like | ||
| Giardia canis virus from China | ABB36743.1 | GCV |
| Giardia lamblia virus | AAM77694.1 | GLV1 |
| Giardia lamblia virus | NP_620070.1 | GLV2 |
Figure 1.
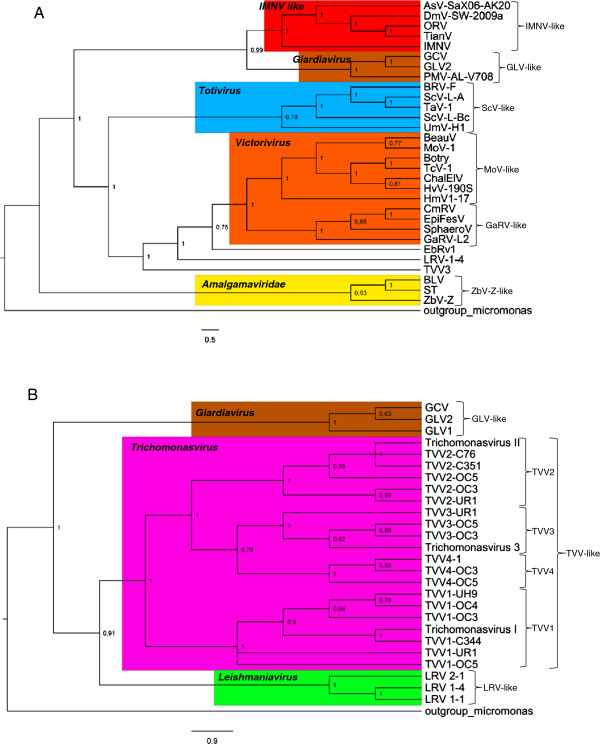
Phylogenetic relationships between Totiviridae family members. Trees were calculated from an alignment of RdRp aminoacid sequences from representative members of the Totiviridae family, using Bayesian inference. The IDs of the sequences in trees A and B are shown in Tables 1 and 2. The numbers in branch nodes indicate posterior probabilities. The right curly brackets indicate the groups identified in this study, named in accordance with Liu et al. [40] and de colors represent the genera in according to ICTV.
Identification of conserved RNA secondary structures and melting curves generation
In HRMA, nucleotide variations between two PCR products are detected comparing their melting curves. Although this approach has been successfully used to identify sequence polymorphisms [6–8], and to discriminate bacterial strains and viruses variants [13, 14, 42], it can be rather inconclusive in some cases. High-resolution instruments and expensive dyes are required to detect punctual mutations or in situations where is necessary to detect multiple mutations in a same sequence [43, 44]. Considering that ssRNA melting curve is closed related to the secondary structure assumed by a ssRNA molecule, we decided to investigate if a melting curve of a ssRNA is more informative than a melting curve generated from a dsDNA. For this, RNA sequences from Totiviridae viruses coding for RdRp proteins were inspected in order to identify regions with conserved secondary structures. Conserved regions were selected to avoid major structural variation between the sequences. Initially, RNA sequences referenced in Tables 3 and 4 were screened but conserved RNA structures common to all sequences were not found. Interestingly, the alignment of the sequences from each group individually revealed regions with high probability (>90%) to form conserved RNA structures in groups IMNV-like, GaRV-like and GLV-like. Members of the MoV-like group showed conserved regions only when analyzed in subgroups, BotryV, TcV-1 and HvV-190S showed regions with conserved RNA structure in the second half of ORF2 when taken together. The same was observed to MoV-like members BeauV and MoV-1 when analyzed individually (data not shown). The groups ZbV-Z-like, ScV-like, TVV-like and LRV-like do not show RNA conserved regions with high RNAz score, nevertheless, one conserved region of each group could be selected manually from alignments (data not show). It is clear that the similarity between sequences increases the chance of finding regions with conserved RNA structure. In agreement with phylogenetic trees showed in Figure 1, individuals that share a secondary RNA structure belongs to groups with shorter branches.
Table 3.
Totiviridae nucleotide sequences used in this study grouped according to phylogenetic analysis
| Virus name | Acession code (GI) | Abbreviation |
|---|---|---|
| MoV-like | ||
| Beauveria bassiana RNA virus 1 (2672–5176) | 345108726 | BeauV |
| Tolypocladium cylindrosporum virus 1 (2604–5126) | 315573168 | TcV-1 |
| Botryotinia fuckeliana totivirus 1 (2631–5147) | 134141995 | BotryV |
| Helminthosporium victoriae virus 190S (2605–5112) | 124484600 | HvV-190S |
| Chalara elegans RNA virus 1 (2619–4067) | 48696977 | ChalElV |
| Helicobasidium mompa totivirus 1–17 (2563–5100) | 33867950 | HmV1-17 |
| Magnaporthe oryzae virus 1 (2818–5316) | 54193767 | MoV-1 |
| IMNV-like | ||
| Penaeid shrimp infectious myonecrosis virus (5241–7490) | 459680256 | IMNV |
| Tianjin totivirus (5319–7535) | 380715048 | TianV |
| Omono river virus (5202–7535) | 307933349 | ORV |
| Drosophila melanogaster totivirus SW - 2009a (4841–6706) | 268053723 | DmV-SW-2009a |
| Armigeres subalbatus virus SaX06-AK20 (5145–7430) | 309259994 | AsV-SaX06-AK20 |
| GLV-like | ||
| Piscine myocarditis virus AL V-708 (3114–5294) | 336042307 | PMV-AL V-708 |
| Giardia canis virus from China (2883–5981) | 78217291 | GCV |
| Giardia lamblia virus (2880–5978) | 20143439 | GLV2 |
| ZbV-Z Like | ||
| Blueberry latent virus isolate AR (936–3332) | 308097100 | BLV |
| Southern tomato virus isolate Mexico-1(1039–3327) | 133776995 | STV |
| Zygosaccharomyces bailii virus Z (1106–3037) | 20889374 | ZbV-Z |
| ScV-like | ||
| Ustilago maydis virus H1 | 20564172 | UmV-H1 |
| Saccharomyces cerevisiae virus L-BC (La) (1970–4561) | 9627980 | ScV L-Bc |
| Saccharomyces cerevisiae virus L-A (2351–4546) | 20428567 | ScV L-A |
| Black raspberry virus F (2565–5009) | 157939583 | BRV-F |
| Tuber aestivum virus 1 (2169–4556) | 312233874 | TaV-1 |
| GaRV-like | ||
| Sphaeropsis sapinea RNA virus 2 (2658–5135) | 3808226 | SphaeroV |
| Coniothyrium minitans RNA virus (2386–4875) | 78762702 | CmRV |
| Epichloe festucae virus 1 (2568–5051) | 94536498 | EpiFesV |
| Gremmeniella abietina RNA virus L2 (2599–5076) | 49036574 | GaRV-L2 |
| Other sequence | ||
| Eimeria brunetti RNA virus 1(2667–5321) | NP_108651 | EbRV-1 |
Accession codes correspond to nucleotide sequences of complete genomes. Numbers in brackets correspond to first and last nucleotides of RdRp coding sequences.
Table 4.
Nucleotide sequences of Trichomonasvirus , Giardiavirus and Leishmaniavirus used in this study grouped according to phylogenetic analysis
| Virus name | Acession code (GI) | Abbreviation |
|---|---|---|
| TVV4 | ||
| Trichomonas vaginalis virus 4 strain TVV4 -1 (2534–4782) | 332015871 | TVV4-1 |
| Trichomonas vaginalis virus 4 strain TVV4-OC3 (2535–4783) | 332015868 | TVV4-OC3 |
| Trichomonas vaginalis virus 4 strain TVV4-OC5 (2534–4782) | 332015874 | TVV4-OC5 |
| TVV3 | ||
| Trichomonas vaginalis virus 3 strain TVV3-UR1 (2448–4693) | 332015877 | TVV3-UR1 |
| Trichomonas vaginalis virus 3 strain TVV3-OC5 (2445–4690) | 332015883 | TVV3-OC5 |
| Trichomonas vaginalis virus 3 strain TVV3-OC3 (2449–4694) | 332015880 | TVV3-OC3 |
| Trichomonas vaginalis virus 3 (2645–4690) | 21450040 | Trichomonasvirus_3 |
| TVV2 | ||
| Trichomonas vaginalis virus 2 strain TVV2-OC3 (2380–4607) | 332015889 | TVV2-OC3 |
| Trichomonas vaginalis virus 2 strain TVV2-UR1 (2379–4606) | 332015886 | TVV2-UR1 |
| Trichomonas vaginalis virus 2 strain TVV2-OC5 (2378–4605) | 332015892 | TVV2-OC5 |
| Trichomonas vaginalis virus II (2302–4605) | 20889358 | Trichomonasvirus_II |
| Trichomonas vaginalis virus 2 isolate C76 (2317–4620) | 357529890 | TVV2-C76 |
| Trichomonas vaginalis virus 2 isolate C351 (2314–4617) | 357529893 | TVV2-C351 |
| TVV1 | ||
| Trichomonas vaginalis virus (2308–4578) | 20564174 | Trichomonasvirus_I |
| Trichomonas vaginalis virus 1 isolate C344 (2316–4578) | 357529887 | TVV1-C344 |
| Trichomonas vaginalis virus 1 strain TVV1-UH9 (2353–4615) | 332015898 | TVV1-UH9 |
| Trichomonas vaginalis virus 1 strain TVV1-OC4 (2353–4615) | 332015904 | TVV1-OC4 |
| Trichomonas vaginalis virus 1 strain TVV1-OC3 (2355–4617) | 332015901 | TVV1-OC3 |
| Trichomonas vaginalis virus 1 strain TVV1-UR1 (2354–4616) | 332015895 | TVV1-UR1 |
| Trichomonas vaginalis virus 1 strain TVV1-OC5 (2351–4613) | 332015907 | TVV1-OC5 |
| LRV-like | ||
| Leishmania RNA virus 2–1 (2858–5191) | 9628596 | LRV 2-1 |
| Leishmania RNA virus 1–4 (2605–5241) | 20153346 | LRV 1-4 |
| Leishmania RNA virus 1–1 (2612–5236) | 9626920 | LRV 1-1 |
| GLV-like | ||
| Giardia canis virus from China (2883–5981) | 78217291 | GCV |
| Giardia lamblia virus (2880–5978) | 20143439 | GLV2 |
| Giardia lamblia virus (2880–5978) | 21780360 | GLV1 |
Accession codes correspond to nucleotide sequences of complete genomes. Numbers in brackets correspond to first and last nucleotides of RdRp coding sequences.
RNA secondary structures of the conserved regions found in groups IMNV-like, GLV-like and GaRV-like were predicted using the software RNAfold. RNA fragments that show conserved RNA secondary structures in IMNV-like group are indicated in Figure 2 column A. The respective models generated from each sequence are showed in Figure 2 column B. These sequences were also used to perform a in silico melting curve analysis using softwares RNAheat and MELTSIM, in order to obtain ssRNA melting curves (Figure 2 column C) and dsDNA melting curves (Figure 2 column D). The results of the same analysis from groups GLV-like and GaRV-like are showed in Additional file 1: Figure S1 and Additional file 2: Figure S2 respectively. Is interesting to observe that, in all cases, ssRNA melting curve presents higher variation than the profile generated by denaturation of dsDNA. This variation is possibly due to the presence of "bubbles" or “hairpins” formed as result of regions that not have perfect base pair complementarities. These regions may comprise several small pieces that present different melting temperatures. When dsDNA is used, the melting curve variation is generated only due to differences in the number of hydrogen bonds between the strands, which can be caused by nucleotide mispairing. This subtle variation in dsDNA melting curve can be detected only using more sensitive and expensive methods. Denaturation profile generated by ssRNA, as a result of the loss of its secondary structure, reflects more intense variations in nucleotide sequence unambiguously. These variations are more pronounced if the number of paired regions interspersed with non complementary regions is high. This can be easily observed when comparing the graphs of columns C and D in Figure 2. Is possible to distinguish five profiles in column C visually, but is not possible to do it comparing profiles that are in column D.
Figure 2.
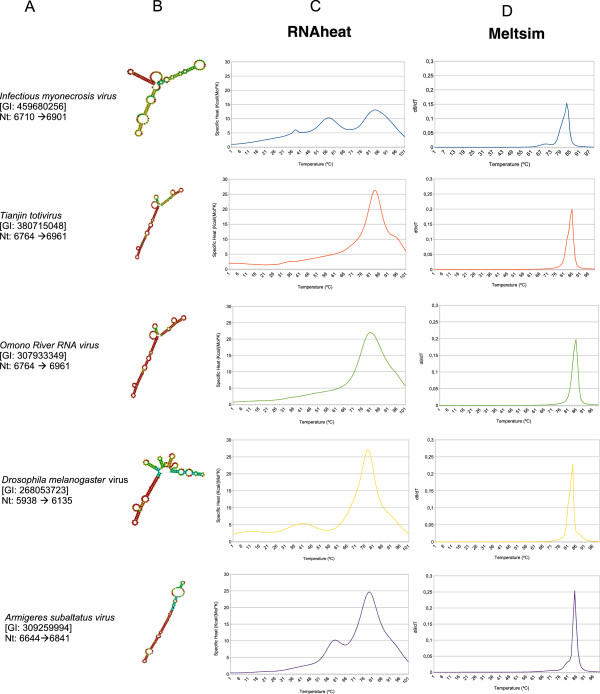
Regions with conserved RNA secondary structures and their respective melting curves. This figure corresponds to analysis of IMNV-like group sequences. (A) Indication of regions with conserved secondary structure inside RdRp coding sequences, identified using RNAz. (B) Minimum free energy models calculated using RNAfold corresponding to each conserved region identified by RNAz. Structures are colored according to base-pairing probabilities. Red color denotes the high probability and purple denotes low probability of a given base is paired or not. For unpaired regions the color denotes the probability of being unpaired. (C and D) Melting curves calculated from conserved regions using softwares RNAheat and MELTSIM respectively.
Clustering groups using melting curves
To confirm that information extracted from a ssRNA melting curve is more detailed than its correspondent dsDNA melting curve, clustering analyses were performed using melting curves from each ssRNA and dsDNA sequences of groups IMNV-like, GaRV-like and GLV-like. The curves were compared per group and clustered using R [45]. The results are represented as dendograms in Figure 3 and in Additional file 3: Figure S3. The relationships between individuals are determined exclusively by the similarity between the melting curves generated by the programs RNAheat and MELTSIM. The groups obtained from R analyses (Figure 3) were compared to groups obtained in phylogenetic analysis. It was surprising that the IMNV-like and GaRV-like groups could be perfectly reconstructed by the clustering of the RNAheat melting curves data, but not by the clustering based on MELTSIM melting curves. In these two cases, the analysis using ssRNA melting curves showed more resolution than the analysis using dsDNA melting curve. In other words, these results strongly indicate that ssRNA melting curve is a good source of information about the nucleic acid sequence. Additional tests using the conserved sequences manually selected from the other groups confirm that the resolution of dendrograms generated from RNAheat curves is never less than the resolution of dendograms generated from MELTSIM curves (data not show).
Figure 3.
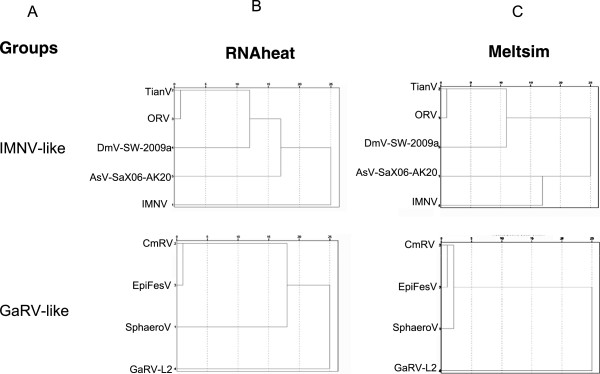
Cluster analysis and dendograms of groups IMNV-like and GaRV-like. Melting curves generated for each conserved RNA sequence in a same group were compared and clustered using a statistical inference. The proximity between individuals of groups indicated in the column (A) is due exclusively to the similarity between the melting curves generated in silico. Columns (B) and (C) shows the dendograms calculated from curves generated by RNAheat and MELSTSIM for the members of each group.
It is already known that the formation of secondary structures in DNA can exerts significant influence in the molecule functions during DNA replication, transcription or translation. These secondary structures may vary within the molecule or when DNA is transcribed to RNA in according to cellular context involved. Considering this fact, is perfectly plausible that a given nucleic acid sequence may suffer different selective pressures in according with variations of it conformation in different stages of its “life cyle”. In single stranded RNA viruses, the secondary structure of RNA could be selected by a large number of factors acting at the same time, including the compactation of the genetic material into capsid. Therefore, we opted to eliminate any noise that could compromise the analysis of RNA conserved secondary structures and ensure that natural selection would be acting mainly on the structure detected by RNAz. Due to this fact the Totiviridae family seems a perfect model. During all replication steps the genetic material of Totiviridae remains as RNA and the formation of RNA secondary structures occur only when RNA is being replicated. This factor can be decisive for the perfect reconstruction of phylogenetic groups comparing secondary structures of RNA.
Potential of single strand melting curve to pathogen identification
Whereas the analysis of single strand denaturation enables a higher resolution to phylogenetic groups reconstruction, it is expected to be also more efficient in distinguishing individuals within the same group. To confirm this, a phylogenetic analysis of sequences from a large number of members of TVV-like group was performed (Figure 1B). The analysis of different variants of Trichomonas vaginalis virus, revealed four five sub-groups called TVV2, TVV3, TVV4 and TVV1, all belonging to the group TVV-like and to genus Trichomonasvirus. The sub-group TVV1 was selected to generate melting curves in silico. An alignment of RdRp RNA sequences was used for RNAz screening. This analysis revealed one region with conserved RNA structure shared by all viruses of this group in the second half or RdRp RNA sequence. Then, two regions were selected, a non-conserved region and the conserved region detected by RNAz (Figure 4A and 4B, respectively). These regions were used to generate melting curves using RNAheat and MELTSIM (Figure 4C and 4D respectively). It was clear that the melting curves generated from ssRNA are more informative than the curves generated by denaturation of dsDNA. Observing the curves generated by RNAheat in both sets of melting curves is possible to differentiate seven Trichomonasvirus variants. The discrimination of each virus is more difficult if the graphs generated by MELTSIM, because the variation in the melting curves occurs in a restricted temperature range.
Figure 4.
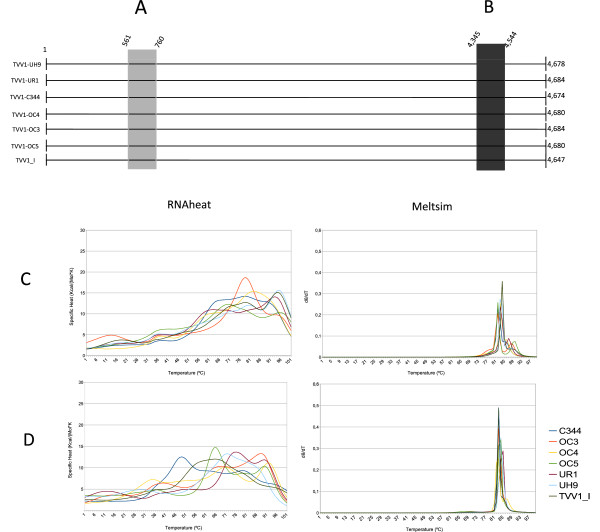
Differentiating members of the subgroup TVV1 using in silico melting curves. A region with variable RNA secondary structure (A) and a region with conserved RNA structure (B) were obtained from the alignment of ORF2 of all members of the group using the software RNAz. The curves were generated by RNAheat and MELTSIM to conserved regions (C) and variable regions (D). Each denaturation curve is marked with a different color: Dark blue lines to TVV1-C344; orange lines to TVV1-OC3; yellow lines to TVV1-OC4; green lines to TVV1-OC5; dark red lines to TVV1-UR1; light blue lines to TVV1-UH9 and dark green lines to TVV1_I.
Conclusions
The results presented here are a strong indication that the ssRNA melting curves are more informative than dsDNA melting curves. In addition, they demonstrate that common RNA conserved regions may be determined from analysis of individuals that are phylogenetically related, and that these regions may be used to support the reconstitution of their phylogenetic groups. These findings are a robust basis for the development of in vitro systems to ssRNA melting curves detection.
Methods
Data acquisition and phylogenetic analysis
The nucleotide and amino acid sequences from Totiviridae viruses were retrieved from public repositories such as GenBank [http://www.ncbi.nlm.nih.gov] and UniProt [http://www.uniprot.org]. Sequences were aligned using TCOFFEE and MCOFFEE algorithms [46] using default parameters, and manually edited using Jalview v. 2.8 [47]. ORFs and protein conserved domains identification were performed using ORF finder and NCBI conserved domain database (CDD), respectively. The RdRP sequence from Micromonas pusilla virus (Reoviridae family; Accession number YP654545) was used as outgroup due to its higher proximity and similarity to the family Totiviridae. The RdRP sequences were then aligned at the amino acid level, using the program MAFFT v. 6.85 [48, 49] with the L-INS-I parameter, gap opening penalty 1.53 and offset value 0.1, guided by the structural alignment from protein family pfam02123, present in the Conserved Domains Database (CDD) [50]. Then, they were re-aligned using the program Muscle [51]. Afterwards, the best-fit amino acid substitution model was estimated using ProtTest v.3.2 [52] and the dendograms were calculated based on a Bayesian analysis using MrBayes 3.1.2 [53, 54] and BEAST v.1.8 [55]. All indels and non-informative sites (missing data) in the alignment were treated as partial deletion, with a cutoff of 75%, to avoid potentially ambiguous regions in topologies. The Bayesian inferences were conducted using three independent runs, with fixed LG or WAG model, gamma distributed rates among sites and fixed amino acid frequencies. Each Markov Chain was initiated with a random tree and run for 106 generations, sampled every 100 generations, and a consensus tree was estimated by using a burning in of 1,000,000 trees. The convergence of the simultaneous runs was assessed using the Tracer tool v. 1.5 [56], in order to evaluate the statistic support and robustness of the bayesian analysis. The trees generated by the programs were edited in the program FigTree [56].
RNA secondary structure prediction
Conserved RNA secondary structures were detected from TCOFFEE multiple alignments of RdRp RNA sequences (Tables 3 and 4), using the RNAz software, provided by Vienna RNA Web Services [57]. This tool detects a consensus secondary structure for an alignment based on thermodynamic stability and structural conservation. A normalized measure of thermodynamic stability is computed by comparing the minimal free energy (MFE) of a native sequence to the MFEs of a large number of random sequences of the same length and base composition. Then, a z-score is calculated from the relation z = (m -μ )/σ, where μ and σ are the mean and standard deviations, respectively, of the MFEs of the random samples [58]. Negative z-scores indicate that a sequence is more stable than expected by chance. The structural conservation is predicted using the RNAalifold approach [59]. The secondary structures were then calculated using the sequences selected from the RNAz output using RNAfold software provided by Vienna RNA Package [57]. RNAfold reads RNA sequences, calculates their MFE structure and free energy. The -p option was used to compute the partition function (PF) and base pairing probability matrix, as well as the overall free energy of the thermodynamic ensemble. RNAfold produces PostScript files with plots of the resulting secondary structure graph and a dot plot of the base pairing matrix. Default parameters were used to generate the interactive RNA structure plots.
Melting curve analysis
The dsDNA melting curves were estimated using the MELTSIM software, which generates derivative profiles. In the model used by this software, proposed by Blake et al. [15], the loop entropy has been appended in a one-dimensional Ising lattice [60–62]. By default, the program starts the simulation at 60°C (T1), increasing the temperature in every 0.050 degrees, until it reaches 100°C (T2). The single strand RNA melting curves were estimated using the RNAheat software [31]. This program reads RNA sequences and calculate their specific heat in a predetermined temperature range, from a partition function by numeric differentiation that describes the statistical properties of a system in thermodynamic equilibrium. The temperature dependence of the partition function gives information about the secondary structure melting behavior. The overwhelming majority of configurations are in the unfolded state and the high temperature ensemble is unfolded. According to reference point proposed by McCaskill [30] for the entropy of zero for an unfolded chain, the partition function must decrease toward one at high temperature and the specific heat reflects the occurrence of any structural transitions as the temperature increases. The result is written as a list of temperature degrees in °C versus specific heat in Kcal/(Mol * K) [31]. The results calculated from 0 to 100°C were plotted using R [45].
Statistical and grouping analysis
Based on the melting denaturation scores, the melting curves were clustered using a hierarchical cluster analysis, using R [45]. This technique was used to identify the mutually exclusive groups that could be obtained based in the sample, considering only the similarities or differences between them. In this procedure, dendograms with the clusters were identified using the single linkage (nearest neighbor) method with the measure of Euclidean distance squared. This algorithm takes the two objects with the smallest distance and clusters them in the first group. Then, it takes the next object with the smallest distance and this third object is clustered with the first group, being included in the group a new group with two objects is obtained. This process keeps going until all objects are allocated to a group. The nucleotide sequences from the identified regions with conserved secondary structures were aligned in MEGA5 [63] using the MUSCLE algorithm. Each alignment was used in a neighbor-joining grouping analysis, using Maximum-composite likelihood distance and 500 bootstrap replications. The obtained dendograms were visually compared to the ones from hierarchical cluster analyses, based on the single and double strand DNA melting denaturation cores.
Availability of supporting data
All supporting data are included in Additional files.
Electronic supplementary material
Additional file 1: Figure S1: Regions with conserved RNA secondary structures identified in GLV-like group and their respective melting curves. (A) Regions with secondary structures identified using RNAz software, from the alignment of ORF2 RNA sequences of GLV-like group members. (B) Secondary structure calculated using RNAfold, corresponding to each conserved region identified by RNAz. (C) Melting curves calculated from the conserved region, using the software RNAheat which considers ssRNA denaturation. (D) Melting curves calculated from the conserved region, using the software MELTSIM which considers dsDNA denaturation. (PPT 270 KB)
Additional file 2: Figure S2: Regions with conserved RNA secondary structures identified in GaRV-like group and their respective melting curves. (A) Regions with secondary structures identified using RNAz software, from the alignment of ORF2 RNA sequences of GaRV-like group members. (B) Secondary structure calculated using RNAfold, corresponding to each conserved region identified by RNAz. (C) Melting curves calculated from the conserved region, using the software RNAheat which considers ssRNA denaturation. (D) Melting curves calculated from the conserved region, using the software MELTSIM which considers dsDNA denaturation. (PPT 302 KB)
Additional file 3: Figure S3: Cluster analysis and dendogram of GLV-like group. The curves generated for each sequence were compared and clustered using a statistical inference. The proximity between individuals of groups indicated in the column (A) is due exclusively to the similarity between the melting curves generated in silico. Columns (B) and (C) shows the dendograms calculated from the curves generated by the programs RNAheat and MELSTSIM for the members of GLV group. (PPT 355 KB)
Ackonwledgements
The authors wish to thank Coordenação de Aperfeiçoamento Pessoal de Nível Superior (CAPES) and Fundação Apoio à Pesquisa do RN (FAPERN) for financial support. R. V. M. Almeida and M.D. A. Dantas have master’s scholarship from (CAPES). The authors are also grateful to two anonymous reviewers for valuable comments and suggestions that helped to improve the manuscript.
Abbreviations
- PCR
Polymerase chain reaction
- ORF
Open reading frame
- RdRP
RNA-dependent RNA polymerase
- dsDNA
Double-stranded DNA
- ssRNA
Single-stranded RNA
- HRMA
High resolution melting analysis
- SNP
Single nucleotide polymorphism
- ncRNA
Non-coding RNA
- siRNA
small interfering RNA
- CP
Capsid protein
- CDD
Conserved domain database
- ML
Maximum likelihood
- MP
Maximum parsimony
- TBR
Tree bissection reconnection
- NNI
Nearest neighbor interchange
- MFE
Minimum free energy
- PF
Partition function.
Footnotes
Competing interests
There are no financial or non-financial competing interests regarding the publication of this work.
Authors’ contributions
DCFL conceived the idea, RACO made in silico experiments and figures; RACO and MDAD collected and organized the sequence database; RVMA and JPMSL performed the pylogenetic analysis; FNC performed the cluster statistical analysis; DCFL and JPMSL discussed the results; DCFL, JPMSL, RACO and RVMA wrote the manuscript. All authors read and approved the final manuscript.
Contributor Information
Raffael AC Oliveira, Email: raffael.azevedo@gmail.com.
Ricardo VM Almeida, Email: ricardovictorm@gmail.com.
Márcia DA Dantas, Email: dani_araujod@yahoo.com.br.
Felipe N Castro, Email: castrofn@gmail.com.
João Paulo MS Lima, Email: jpmslima@gmail.com.
Daniel CF Lanza, Email: danielclanza@gmail.com.
References
- 1.Katsanis SH, Katsanis N. Molecular genetic testing and the future of clinical genomics. Nat Rev Genet. 2013;14:415–426. doi: 10.1038/nrg3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Espy MJ, Uhl JR, Sloan LM, Buckwalter SP, Jones MF, Vetter EA, Yao JD, Wengenack NL, Rosenblatt JE, Cockerill FR, 3rd, Smith TF. Real-time PCR in clinical microbiology: applications for routine laboratory testing. Clin Microbiol Rev. 2006;19:165–256. doi: 10.1128/CMR.19.1.165-256.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Munro SB, Kuypers J, Jerome KR. Comparison of a multiplex real-time PCR assay with a multiplex Luminex assay for influenza virus detection. J Clin Microbiol. 2013;51:1124–1129. doi: 10.1128/JCM.03113-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wittwer CT, Herrmann MG, Moss AA, Rasmussen RP. Continuous fluorescence monitoring of rapid cycle DNA amplification. Biotechniques. 1997;22:130–131. doi: 10.2144/97221bi01. [DOI] [PubMed] [Google Scholar]
- 5.Wittwer CT, Reed GH, Gundry CN, Vandersteen JG, Pryor RJ. High-resolution genotyping by amplicon melting analysis using LCGreen. Clin Chem. 2003;49:853–860. doi: 10.1373/49.6.853. [DOI] [PubMed] [Google Scholar]
- 6.Liew M, Pryor R, Palais R, Meadows C, Erali M, Lyon E, Wittwer C. Genotyping of single-nucleotide polymorphisms by high-resolution melting of small amplicons. Clin Chem. 2004;50:1156–1164. doi: 10.1373/clinchem.2004.032136. [DOI] [PubMed] [Google Scholar]
- 7.Rouleau E, Lefol C, Bourdon V, Coulet F, Noguchi T, Soubrier F, Bieche I, Olschwang S, Sobol H, Lidereau R. qPCR HRM, a new approach to screen simultaneously point mutations and large rearrangements—Application to MLH1 germline mutations in Lynch Syndrome. Hum Mutat. 2009;30:867–875. doi: 10.1002/humu.20947. [DOI] [PubMed] [Google Scholar]
- 8.Nguyen-Dumont T, Le Calvez-Kelm F, Forey N, McKay-Chopin S, Garritano S, Gioia-Patricola L, De Silva D, Weigel R, Sangrajrang S, Lesueur F, Tavtigian SV, Breast Cancer Family Registries (BCFR); Kathleen Cuningham Foundation Consortium for Research into Familial Breast Cancer (kConFab) Description and validation of high-throughput simultaneous genotyping and mutation scanning by high-resolution melting curve analysis. Hum Mutat. 2009;30:884–890. doi: 10.1002/humu.20949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith BL, Lu CP, Alvarado Bremer JR. High-resolution melting analysis (HRMA): a highly sensitive inexpensive genotyping alternative for population studies. Mol Ecol Resour. 2010;10:193–196. doi: 10.1111/j.1755-0998.2009.02726.x. [DOI] [PubMed] [Google Scholar]
- 10.Erali M, Wittwer CT. High resolution melting analysis for gene scanning. Methods. 2010;50:250–261. doi: 10.1016/j.ymeth.2010.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Montgomery J, Wittwer CT, Kent JO, Zhou L. Scanning the cystic fibrosis transmembrane conductance regulator gene using high-resolution DNA melting analysis. Clin Chem. 2007;53:1891–1898. doi: 10.1373/clinchem.2007.092361. [DOI] [PubMed] [Google Scholar]
- 12.Gundry CN, Dobrowolski SF, Martin YR, Robbins TC, Nay LM, Boyd N, Coyne T, Wall MD, Wittwer CT, Teng DH. Base-pair neutral homozygotes can be discriminated by calibrated high-resolution melting of small amplicons. Nucleic Acids Res. 2008;36:3401–3408. doi: 10.1093/nar/gkn204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cousins MM, Swan D, Magaret CA, Hoover DR, Eshleman SH. Analysis of HIV using a high resolution melting (HRM) diversity assay: automation of HRM data analysis enhances the utility of the assay for analysis of HIV incidence. PLoS One. 2012;7:e51359. doi: 10.1371/journal.pone.0051359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Porcellato D, Grønnevik H, Rudi K, Narvhus J, Skeie SB. Rapid lactic acid bacteria identification in dairy products by high-resolution melt analysis of DGGE bands. Lett Appl Microbiol. 2012;54:344–351. doi: 10.1111/j.1472-765X.2012.03210.x. [DOI] [PubMed] [Google Scholar]
- 15.Blake RD, Bizzaro JW, Blake JD, Day GR, Delcourt SG, Knowles J, Marx KA, SantaLucia J., Jr Statistical Mechanical Simulation of Polymeric DNA Melting with MELTSIM. Bioinformatics. 1999;15:370–375. doi: 10.1093/bioinformatics/15.5.370. [DOI] [PubMed] [Google Scholar]
- 16.Dwight Z, Palais R, Wittwer CT. uMELT: prediction of high-resolution melting curves and dynamic melting profiles of PCR products in a rich web application. Bioinformatics. 2011;27(7):1019–1020. doi: 10.1093/bioinformatics/btr065. [DOI] [PubMed] [Google Scholar]
- 17.Dwight ZL, Palais R, Wittwer CT. uAnalyze: web-based high-resolution DNA melting analysis with comparison to thermodynamic predictions. IEEE/ACM Trans Comput Biol Bioinform. 2012;9:1805–1811. doi: 10.1109/TCBB.2012.112. [DOI] [PubMed] [Google Scholar]
- 18.Rasmussen JP, Saint CP, Monis PT. Use of DNA melting simulation software for in silico diagnostic assay design: targeting regions with complex melting curves and confirmation by real-time PCR using intercalating dyes. BMC Bioinformatics. 2007;8:107. doi: 10.1186/1471-2105-8-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lok AS, Akarca U, Greene S. Mutations in the pre-core region of hepatitis B virus serve to enhance the stability of the secondary structure of the pre-genome encapsidation signal. Proc Natl Acad Sci U S A. 1994;91:4077–4081. doi: 10.1073/pnas.91.9.4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Flores R, Serra P, Minoia S, Di Serio F, Navarro B. Viroids: from genotype to phenotype just relying on RNA sequence and structural motifs. Front Microbiol. 2012;3:217. doi: 10.3389/fmicb.2012.00217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lusvarghi S, Sztuba-Solinska J, Purzycka KJ, Pauly GT, Rausch JW, Grice SF. The HIV-2 Rev-response element: determining secondary structure and defining folding intermediates. Nucleic Acids Res. 2013;41(13):6637–6649. doi: 10.1093/nar/gkt353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bergeron É, Chakrabarti AK, Bird BH, Dodd KA, McMullan LK, Spiropoulou CF, Nichol ST, Albariño CG. Reverse genetics recovery of Lujo virus and role of virus RNA secondary structures in efficient virus growth. J Virol. 2012;86(19):10759–10765. doi: 10.1128/JVI.01144-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tan X, Lu ZJ, Gao G, Xu Q, Hu L, Fellmann C, Li MZ, Qu H, Lowe SW, Hannon GJ, Elledge SJ. Tiling genomes of pathogenic viruses identifies potent antiviral shRNAs and reveals a role for secondary structure in shRNA efficacy. Proc Natl Acad Sci U S A. 2012;109:869–874. doi: 10.1073/pnas.1119873109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mercer TR, Mattick JS. Structure and function of long noncoding RNAs in epigenetic regulation. Nat Struct Mol Biol. 2013;20:300–307. doi: 10.1038/nsmb.2480. [DOI] [PubMed] [Google Scholar]
- 25.Shao Y, Chan CY, Maliyekkel A, Lawrence CE, Roninson IB, Ding Y. Effect of target secondary structure on RNAi efficiency. RNA. 2007;13(10):1631–1640. doi: 10.1261/rna.546207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Esteban DJ, Upton C, Bartow-McKenney C, Buller RML, Chen NG, Schriewer J, Lefkowitz EJ, Wang C. Expression of a non-coding RNA in ectromelia virus is required for normal plaque formation. Virus Genes. 2013;48:38–47. doi: 10.1007/s11262-013-0983-2. [DOI] [PubMed] [Google Scholar]
- 27.Zuker M, Mathews DH, Turner DH. Algorithms and Thermodynamics for RNA Secondary Structure Prediction: A Practical Guide. In: Barciszewski J, Clark BFC, editors. RNA Biochemistry and Biotechnology. Poznan, Poland: NATO ASI Series, Kluwer Academic Publishers; 1999. [Google Scholar]
- 28.Bellaousov S, Reuter JS, Seetin MG, Mathews DH. RNAstructure: Web servers for RNA secondary structure prediction and analysis. Nucleic Acids Res. 2013;41(Web Server issue):W471–W474. doi: 10.1093/nar/gkt290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lorenz R, Bernhart SH, Siederdissen CH, Tafer H, Flamm C, Stadler PF, Hofacker IL. ViennaRNA Package 2.0. Algorithms Mol Biol. 2011;6:26. doi: 10.1186/1748-7188-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCaskill JS. The equilibrium partition function and base pair binding probabilities for RNA secondary structures. Biopolymers. 1999;29:1105–1119. doi: 10.1002/bip.360290621. [DOI] [PubMed] [Google Scholar]
- 31.Hofacker IL, Fontana W, Stadler PF, Bonhoeffer S, Tacker M, Schuster P. Fast Folding and Comparison of RNA Secondary Structures. Monatshefte f Chemie. 1994;125:167–188. doi: 10.1007/BF00818163. [DOI] [Google Scholar]
- 32.Mathews DH, Disney MD, Matthew D, Childs JL, Schroeder SJ, Susan J, Zuker M, Turner DH. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc Natl Acad Sci U S A. 2004;101:7287–7292. doi: 10.1073/pnas.0401799101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Turner DH, Mathews DH. NNDB: The nearest neighbor parameter database for predicting stability of nucleic acid secondary structure. Nucleic Acids Res. 2009;38:280–282. doi: 10.1093/nar/gkp892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tycowski KT, Shu MD, Borah S, Shi M, Steitz JA. Conservation of a triple-helix-forming RNA stability element in noncoding and genomic RNAs of diverse viruses. Cell Rep. 2012;2(1):26–32. doi: 10.1016/j.celrep.2012.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bessarab IN, Nakajima R, Liu HW, Tai JH. Identification and characterization of a type III Trichomonas vaginalis virus in the protozoan pathogen Trichomonas vaginalis. Arch Virol. 2011;156:285–294. doi: 10.1007/s00705-010-0858-y. [DOI] [PubMed] [Google Scholar]
- 36.Ghabrial SA, Nibert ML. Victorivirus, a new genus of fungal viruses in the family Totiviridae. Arch Virol. 2009;154:373–379. doi: 10.1007/s00705-008-0272-x. [DOI] [PubMed] [Google Scholar]
- 37.Zhai Y, Attoui H, Mohd Jaafar F, Wang HQ, Cao YX, Fan SP, Sun YX, Liu LD, Mertens PP, Meng WS, Wang D, Liang G. Isolation and full-length sequence analysis of Armigeres subalbatus totivirus, the first totivirus isolate from mosquitoes representing a proposed novel genus (Artivirus) of the family Totiviridae. J Gen Virol. 2010;91:2836–2845. doi: 10.1099/vir.0.024794-0. [DOI] [PubMed] [Google Scholar]
- 38.Poulos BT, Tang KFJ, Pantoja CR, Bonami JR, Lightner DV. Purification and characterization of infectious myonecrosis virus of penaeid shrimp. J Gen Virol. 2006;87:987–996. doi: 10.1099/vir.0.81127-0. [DOI] [PubMed] [Google Scholar]
- 39.Bruenn JA. A structural and primary sequence comparison of the viral RNA-dependent RNA polymerases. Nucleic Acids Res. 2003;31:1821–1829. doi: 10.1093/nar/gkg277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu H, Fu Y, Xie J, Cheng J, Ghabrial SA, Li G, Peng Y, Yi X, Jiang D. Evolutionary genomics of mycovirus-related dsRNA viruses reveals cross-family horizontal gene transfer and evolution of diverse viral lineages. BMC Evol Biol. 2012;12:91. doi: 10.1186/1471-2148-12-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ. The Most Recent Report of the ICTV: Virus Taxonomy: Classification and Nomenclature of Viruses: Ninth Report of the International Committee on Taxonomy of Viruses. San Diego: Elsevier Academic Press; 2012. [Google Scholar]
- 42.Won H, Rothman R, Ramachandran P, Hsieh YH, Kecojevic A, Carroll KC, Aird D, Gaydos C, Yang S. Rapid identification of bacterial pathogens in positive blood culture bottles by use of a broad-based PCR assay coupled with high-resolution melt analysis. J Clin Microbiol. 2010;48:3410–3413. doi: 10.1128/JCM.00718-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jeng K, Gaydos CA, Blyn LB, Yang S, Won H, Matthews H, Toleno D, Hsieh YH, Carroll KC, Hardick J, Masek B, Kecojevic A, Sampath R, Peterson S, Rothman RE. Comparative analysis of two broad-range PCR assays for pathogen detection in positive-blood-culture bottles: PCR-high-resolution melting analysis versus PCR-mass spectrometry. J Clin Microbiol. 2012;50:3287–3292. doi: 10.1128/JCM.00677-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li BS, Wang XY, Ma FL, Jiang B, Song XX, Xu AG. Is high resolution melting analysis (HRMA) accurate for detection of human disease-associated mutations? A meta analysis. PLoS One. 2011;6(12):e28078. doi: 10.1371/journal.pone.0028078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2008. [Google Scholar]
- 46.A collection of tools for computing, evaluating and manipulating multiple alignments of DNA, RNA, protein sequences and structures [http://www.tcoffee.org/]
- 47.Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ. Jalview Version 2 - a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Katoh K, Toh H. Parallelization of the MAFFT multiple sequence alignment program. Bioinformatics. 2010;26(15):1899–1900. doi: 10.1093/bioinformatics/btq224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Katoh K, Frith MC. Adding unaligned sequences into an existing alignment using MAFFT and LAST. Bioinformatics. 2012;28(23):3144–3146. doi: 10.1093/bioinformatics/bts578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Lu F, Marchler GH, Mullokandov M, Omelchenko MV, Robertson CL, Song JS, Thanki N, Yamashita RA, Zhang D, Zhang N, Zheng C, Bryant SH. CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 2011;v39(Database issue):D225–D229. doi: 10.1093/nar/gkq1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Edgar RRC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Abascal F, Zardoya R, Posada D. ProtTest: selection of best-fit models of protein evolution. Bioinformatics. 2005;21:2104–2105. doi: 10.1093/bioinformatics/bti263. [DOI] [PubMed] [Google Scholar]
- 53.Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- 54.Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61:539–542. doi: 10.1093/sysbio/sys029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29:1969–73. doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vienna RNA Package 2.0 [http://rna.tbi.univie.ac.at/cgi-bin/RNAz.cgi]
- 58.Washietl S, Hofacker IL, Stadler PF. Fast and reliable prediction of noncoding RNAs Proc. Natl Acad Sci. 2005;102:2454–2459. doi: 10.1073/pnas.0409169102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hofacker IL, Fekete M, Stadler PF. Secondary structure prediction for aligned RNA sequences. J Mol Biol. 2002;319:1059–1066. doi: 10.1016/S0022-2836(02)00308-X. [DOI] [PubMed] [Google Scholar]
- 60.Ising E. Beitrag zur Theorie des Ferromagnetismus. Z Phys. 1925;31:253–258. doi: 10.1007/BF02980577. [DOI] [Google Scholar]
- 61.Hill TL. Statistical Mechanics. New York: McGraw-Hill; 1956. [Google Scholar]
- 62.Wartell RM, Benight AS. Thermal denaturation of DNA molecules: a comparison of theory with experiment. Physics Rep. 1985;126:67–107. doi: 10.1016/0370-1573(85)90060-2. [DOI] [Google Scholar]
- 63.Tamura K, Peterson D, Peterson N. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Figure S1: Regions with conserved RNA secondary structures identified in GLV-like group and their respective melting curves. (A) Regions with secondary structures identified using RNAz software, from the alignment of ORF2 RNA sequences of GLV-like group members. (B) Secondary structure calculated using RNAfold, corresponding to each conserved region identified by RNAz. (C) Melting curves calculated from the conserved region, using the software RNAheat which considers ssRNA denaturation. (D) Melting curves calculated from the conserved region, using the software MELTSIM which considers dsDNA denaturation. (PPT 270 KB)
Additional file 2: Figure S2: Regions with conserved RNA secondary structures identified in GaRV-like group and their respective melting curves. (A) Regions with secondary structures identified using RNAz software, from the alignment of ORF2 RNA sequences of GaRV-like group members. (B) Secondary structure calculated using RNAfold, corresponding to each conserved region identified by RNAz. (C) Melting curves calculated from the conserved region, using the software RNAheat which considers ssRNA denaturation. (D) Melting curves calculated from the conserved region, using the software MELTSIM which considers dsDNA denaturation. (PPT 302 KB)
Additional file 3: Figure S3: Cluster analysis and dendogram of GLV-like group. The curves generated for each sequence were compared and clustered using a statistical inference. The proximity between individuals of groups indicated in the column (A) is due exclusively to the similarity between the melting curves generated in silico. Columns (B) and (C) shows the dendograms calculated from the curves generated by the programs RNAheat and MELSTSIM for the members of GLV group. (PPT 355 KB)