

Abstract
Purpose
The focus of this study was to determine whether bone morphogenetic proteins (BMPs) trigger reactive gliosis in retinal astrocytes and/or Müller glial cells.
Methods
Retinal astrocytes and the Müller glial cell line MIO-M1 were treated with vehicle, BMP7, or BMP4. Samples from the treated cells were analyzed for changes in gliosis markers using reverse transcriptase – quantitative PCR (RT-qPCR) and western blotting. To determine potential similarities and differences in gliosis states, control and BMP-treated cells were compared to cells treated with sodium peroxynitrite (a strong oxidizing agent that will bring about some aspects of gliosis). Last, mature mice were microinjected intravitreally with BMP7 and analyzed for changes in gliosis markers using RT-qPCR, western blotting, and immunohistochemistry.
Results
Treatment of retinal astrocyte cells and Müller glial cells with BMP7 regulated various reactive gliosis markers. When compared to the response of cells treated with sodium peroxynitrite, the profiles of gliosis markers regulated due to exposure to BMP7 were similar. However, as expected, the profiles including the oxidative agent and growth factor were not identical. Treatment of cells with BMP4, however, showed an attenuated response in comparison to peroxynitrite and BMP7 treatment. Injection of BMP7 into the mouse retina also triggered a reactive gliosis response 7 days after injection.
Conclusions
BMP7 induced changes in levels of mRNA and protein markers typically associated with reactive gliosis in retinal astrocytes and Müller glial cells, including glial fibrillary acidic protein (GFAP), glutamine synthetase (GS), a subset of chondroitin sulfate proteoglycans (CSPGs), matrix metalloproteinases (MMPs), and other molecules.
Introduction
The mature mammalian retina contains several types of macroglial cells, including Müller glia, retinal astrocytes, and in some cases oligodendrocytes and nonastrocytic retinal glial cells (NIRG) [1-4]. The Müller glia arise from neural retinal progenitor cells late in retinal development. The Müller cells span nearly the entire width of the retina from the outer limiting membrane, where Müller processes form connections with photoreceptors, to the inner limiting membrane, where Müller and retinal astrocyte processes form the boundary between the retina and the vitreous [5]. The retinal astrocytes migrate into the retina from the developing optic nerve; their cell bodies populate the nerve fiber layer (NFL) and send processes into the ganglion cell layer (GCL) [6]. While the origins of NIRG cells are unknown, they have been hypothesized to migrate from the developing optic nerve [2]. Both the Müller glia and retinal astrocytes have been shown to play very important roles in supporting and protecting the retinal neurons. For instance, both are critical to the formation of the blood-retinal barrier, neurotransmitter recycling, removal of toxins, and growth factor supplementation of ganglion cells [7-10].
An important property of glial cells is their response to any damage/injury to nearby neurons; this response is known as reactive gliosis. The Müller glia and retinal and optic nerve astrocytes become reactive in various disease states such as glaucoma, retinal ischemia, and diabetes [11,12]. One particularly interesting aspect of gliosis is the molecular diversity in the reaction of the astrocytes to various disease states and injuries [13-15]. For instance, insult-dependent increases or decreases in the expression of glial fibrillary acidic protein (GFAP), vimentin, glutamine synthetase (GS), and extracellular matrix (ECM) molecules have been noted in Müller glial cells and astrocytes [11,16-18]. This variance has been hypothesized to be the result of the release of various factors that drive different aspects of reactive gliosis [14,19,20]. Further diversity in the response is introduced by virtue of the fact that reactive astrocytes fall on a continuum from mild to severe, in which cells display one or more of the following: hypertrophy, dedifferentiation, loss of function, proliferation, inflammation, and remodeling of the tissue and vasculature [21,22]. Furthermore, some of the cellular responses are dual in nature. An example of this is the hypertrophy of Müller glia and astrocytes that results from an increase in the intermediate filaments GFAP and vimentin. On one hand, the hypertrophied cellular processes may form a barrier around the injured region, inhibiting the spread of inflammatory molecules into healthy tissue; on the other, these processes can also block the regeneration of axons and synapses [22].
Severe reactive gliosis is also accompanied by tissue remodeling that includes excessive hypertrophy of the glial cell bodies and processes, as well as the turnover of the ECM to a regeneration-inhibitory matrix, together referred to as a glial scar. One of the major components of the ECM is chondroitin sulfate proteoglycans (CSPGs), a family of molecules that includes neurocan (Ncan), phosphacan (Pcan), versican, aggrecan, brevican, nerve/glial antigen 2 (NG2), and CD44 [23]. Although the mechanisms and triggers have not been completely characterized, a variety of growth factor signaling mechanisms have been found to be important in reactive gliosis, such as those of epidermal growth factor (EGF), fibroblast growth factor (FGF), tumor necrosis factor-α (TNF-α), ciliary neurotrophic factor (CNTF), insulin, and WNTs [20,22,24]. Data from several laboratories have indicated that each pathway may regulate specific characteristics associated with reactive gliosis. For instance, CNTF appears to be associated with the upregulation of GFAP, while EGF and FGF are associated with proliferation [17,25]. Studies using different central nervous system (CNS) injury models have shown that the bone morphogenetic protein (BMP) pathway is upregulated at the site of injury in the CNS and can trigger reactive gliosis [26-28]. Furthermore, BMP receptor 1A (BMPR1A) has been linked to hypertrophy, whereas BMPR1A has been associated with glial scars [29]. While the BMPs have been associated with penetrating wounds in the spinal cord, no information is available regarding the role of BMPs within the retina and in disease states of the CNS.
BMPs interact with two different types of receptors, namely type I and type II, both of which are serine-threonine kinases. Type II receptors phosphorylate and activate the type I receptors, and the type I receptors can then activate several intracellular pathways. The intracellular pathways are subdivided into different pathways, specifically the canonical pathway and several noncanonical pathways [30]. The canonical pathway consists of the activation of receptor SMADs (R-SMADs) 1, 5, and 8 by the type I BMP receptors. Once activated, the R-SMADs form heterodimers with the co-SMAD, SMAD4, whereupon they localize to the nucleus to interact with other transcription factors and regulate gene expression. BMPRIA and IB have also been shown to activate X-chromosome-linked apoptosis protein (XIAP). XIAP connects the BMP1A and IB to the kinase transforming growth factor-β (TGF-β) activated kinase 1 (TAK1), which activates downstream pathway members such as p38, nuclear factor κB (NFκB), and c-Jun N-terminal kinases (JNK) [31]. Another noncanonical pathway that has been identified is the FKBP12/rapamycin associated protein - signal transducer and activator of transcription (FRAP-STAT) pathway. Activation of this pathway occurs via phosphorylation of FK506 rapamycin binding protein (FKBP12) by BMPRIA or IB, eventually leading to the activation of STAT proteins, leading to changes in transcription [32].
In this study, we hypothesized that BMP7 plays a role in initiating reactive gliosis in glial cells of the retina. As a first step, we characterized the BMP receptors and intracellular signaling molecules present in the Müller glial cells of normal adult uninjured retinas. Using in vitro systems, we observed differential regulation of molecules typically associated with reactive gliosis in retinal astrocytes and human Müller glial cells following treatment with BMP7. Both retinal astrocytes and MIO-M1 Müller cells showed modest increased Gfap and paired-homeobox 2 (Pax2) levels following treatment with BMP7, but only retinal astrocytes appeared to show changes in matrix metalloproteinases (MMPs) and CSPGs. In contrast, the addition of BMP4 in vitro did not appear to lead to reactive gliosis in either retinal astrocytes or MIO-M1 Müller glial cells. Injection of BMP7 into the mature mouse eye led to more robust increases in molecules associated with reactive gliosis, such as Gfap, Pax2, Mmps, and CSPGs Ncan and Pcan. These results are consistent with the hypothesis that BMP7 acts as a trigger for reactive gliosis in disease states in the eye.
Methods
Tissue processing and fluorescent immunohistochemistry
Eyes from C57BL/6J mice (Jackson Laboratories, Bar Harbor, ME) were dissected from the heads of euthanized animals, washed in PBS (40 mM K2HPO4, 123.2 mM NaCl, 8.4 mM NaH2PO4; Fisher Scientific, Pittsburgh, PA), and fixed in 4% paraformaldehyde for immunohistochemistry. The eyes were then incubated in an ascending series of sucrose (5%, 10%, 15%, and 20%) made in 0.1M phosphate buffer, pH 7.4 overnight. The tissues were frozen in a 3:1 20% sucrose-in phosphate buffer and optimum cutting temperature (OCT) solution. Thick sections (10 μm) were cut using a Leica CM3050 S cryostat and placed on a Superfrost Plus slide (Fisher Scientific) treated with Vectabond (Vector Labs, Burlingame, CA), and were stored at −80 °C until used for immunohistochemistry. Antibodies used for immunohistochemistry are listed in Table 1. For immunohistochemistry, sections were fixed with 4% paraformaldehyde for 30 min. Sections were then washed in 1X PBS, permeabilized in methanol, and subjected to antigen retrieval by placing the sections in 1% sodium dodecyl sulfate (SDS, Fisher Scientific) in 0.01M PBS for 5 min. To aid in autofluorescence reduction, sections were treated with 1% sodium borohydride in PBS (Fisher Scientific) for 2 min at room temperature, then rinsed with PBS. Tissue was blocked by incubating with 10% serum in 1X PBS containing 0.25% Triton X-100 (Bio-Rad, Hercules, CA) at room temperature for 1 h and incubated with the primary antibody, diluted in 0.025% TritonX-100 PBS with 2% blocking serum, overnight at 4 °C. The following day, the slides were incubated in Dylight conjugated secondary antibody (Jackson Immunoresearch, West Grove, PA) at 1:800 diluted with 1X PBS, for 1 h at room temperature, then washed twice with 1X PBS for 5 min each rinse, and mounted with ProLong Gold with 4',6-diamidino-2-phenylindole (DAPI, Invitrogen, Grand Island, NY) or counterstained with Hoechst solution (diluted 1:500 in 1X PBS; Sigma-Aldrich, St. Louis, MO) and mounted with Aqua Poly/Mount (Vector Labs). For labeling of mouse tissue slides with GS, blocking and overnight incubation with primary antibody was performed as specified by the Vector mouse-on-mouse immunodetection kit (Vector Labs). For immunolabeling with NCAN, SMAD, BMPR1A, BMPR1B, and TAK1, biotin streptavidin amplification was performed. Following overnight incubation with the primary antibody, the sections were first incubated with biotinylated antibody (1:1,000, Vector Labs) for 1 h and then streptavidin-conjugated Dylight (1:33, Vector Labs) for 1 h at room temperature. For colabels of phospho-SMAD1/5/8 and phospho-TAK1 with sex determining region Y box 2 (SOX2), sections were subjected to heat antigen retrieval following methanol incubation. Briefly, the sections were incubated in sodium citrate buffer (10 mM sodium citrate, 0.05% Tween-20, pH 6.0) at 65 °C for 1 h, allowed to cool for 20 min, rinsed 3× in water and 1X in PBS, and blocked with 10% horse serum in 1X PBS containing 0.25% Triton X-100 for 1 h at room temperature. Following overnight incubation with the primary antibodies, the slides were incubated with biotinylated horse anti-rabbit (1:300, Vector Labs) for 1 h and then Alexa Fluor anti-goat (1:500, Invitrogen) and horseradish peroxidase (HRP, 1:500, Biolegend) for 1 h. The slides were then washed 2× in 0.1M Tris-HCl, 0.15M NaCl, 0.05% Tween-20 (TNT) buffer and incubated with fluorescein plus amplification reagent diluted 1:300 in 1X tyramide signal amplification (TSA) amplification diluent (Perkin Elmer, Waltham, MA) for 5 min. The slides were washed 2× in TNT buffer, 2X in PBS, counterstained with Hoechst solution, and mounted with Aqua polymount (Vector Labs). Slides were viewed under an Olympus Fluoview FV 1000 confocal microscope.
Table 1. Antibodies.
| Antigen | Antibody Cat No. | Supplier | IF concentration | WB concentration |
|---|---|---|---|---|
| ALK2 |
GTX88867 |
Genetex |
1:250 |
|
| b-tubulin |
T1098 |
Sigma |
1:1000 |
|
| BMPR IA |
AP2004D |
Abgent |
1:50 |
|
| BMPRIB |
SC25455 |
Santa Cruz Biotech |
1:100 |
|
| FKBP12 |
PA1–026A |
Thermo scientific |
1:100 |
|
| GFAP |
Z0334 |
DAKO |
1:250 |
1:600 |
| Glutamine synthetase |
MAB302 |
Millipore |
1:300 |
1:1000 |
| Neurocan |
AF5800 |
R&D |
1:100 |
|
| Pax2 |
P100859 |
Aviva |
1:500 |
|
| S100β |
AB52642 |
Abcam |
1:250 |
1:1000 |
| SMAD1 |
AB63356 |
Abcam |
1:100 |
|
| pSMAD 1/5/8 |
9511S |
Cell Signaling |
1:50 |
|
| Sox2 |
SC17320 |
Santa Cruz Biotech |
1:250 |
|
| TAK-1 |
AB79354 |
Abcam |
1:250 |
|
| pTAK-1 |
4531S |
Cell Signaling |
1:100 |
|
| TXNIP | SC-271238 | Santa Cruz Biotech | 1:250 | 1:250 |
Isolated retinas were processed and used for reverse transcriptase quantitative PCR (RT-qPCR) and western blot analysis. Briefly, animals were euthanized by CO2 exposure. Eyes were then dissected using sterile forceps and the retina isolated. The retinas were homogenized and processed for RNA using the RNeasy Mini Kit as per the manufacturer’s protocol (Qiagen, CA). Briefly, isolated retinal tissues or cells were homogenized in buffer RLT. Seventy percent ethanol was added and the solution centrifuged in an RNeasy spin column. The flow through was discarded, and 700 µl of buffer RW1 was added and centrifuged. Following this step, 500 µl of buffer RPE was added to the spin column and centrifuged. This step was repeated once, following which the RNA was eluted with 40 µl of RNase free water. RNA was quantified using Nanodrop 2000c (Thermoscientific, Rockford, IL). For protein isolation, retinas isolated were homogenized and incubated with radioimmunoprecipitation assay (RIPA) lysis buffer (0.1% SDS, 50 mM Tris-HCl pH 7.5, 1% Triton-X100) supplemented with 0.01 mM phenyl methyl sulfonyl fluoride (PMSF; Roche Diagnostics, Indianapolis, IN) and 1:25 protease inhibitor cocktail (RPI Corp., Mount Prospect, IL) for 2 h at 4 °C. The tube was then centrifuged at 13,680 × g for 20 min at 4 °C and the supernatant aspirated into a new tube.
Cell isolation, cytospinning, and immunocytochemistry
Retinal cells were isolated as previously described [33]. Briefly, mouse retinas were prepared in sterile 0.9% saline. Approximately 200 mg of retinal tissue was incubated with saline containing 0.4 mg/ml papain for 30 min at 37 °C. The tissues were triturated with a siliconized pipette and dissociated single cells were centrifuged at 500 × g for 10 min. The cells were fixed in 4% paraformaldehyde for 10 min, centrifuged, and washed with saline. The cells were then placed onto a Superfrost Plus slide (Fisher Scientific) treated with Vectabond (Vector Labs) using a Cytospin4 (Thermoscientific) and stored at −80 °C until used for immunocytochemistry. For immunocytochemistry, the cells were permeabilized in methanol, incubated with 1% SDS in 0.01M PBS, and washed in 1X PBS. The slides were then incubated with 1% sodium borohydride, blocked using the mouse on mouse kit (Vector Labs), and colabeled with GS and the BMP receptor antibodies (BMPR1A, BMPRIB, and activin-like kinase receptor 2 [ALK2]). Following overnight incubation with the primary antibodies, the slides were incubated with the appropriate secondary antibodies with biotin streptavidin amplification used for the receptor antibodies. The slides were then washed, counterstained with Hoechst solution, and mounted with Aqua polymount.
Cell culture
The mouse retinal astrocytes were isolated as previously stated [34,35]. Cells were cultured in DMEM (Catalogue # D1152, Sigma-Aldrich, St. Louis, MO) containing EC growth supplement (Sigma-Aldrich), 1% penicillin/streptomycin (Sigma-Aldrich), 1 mM sodium pyruvate (Life Technologies, Grand Island, NY), 20 mM HEPES (Sigma-Aldrich), 2 mM Glutamine (Life Technologies), 1X nonessential amino acids (Sigma-Aldrich), heparin (Sigma-Aldrich), 10% fetal bovine serum, and 44 U/ml of murine recombinant interferon γ (R&D Systems, Minneapolis, MN). The cells were grown on Cellbind™ dishes (Fisher Scientific) in an incubator with 5% CO2 at 33 °C and passaged every 3–4 days using trypsin-EDTA (Sigma-Aldrich). The human MIO-M1 Müller glial cell line was obtained from Dr. G.A. Limb at the Institute of Ophthalmology, University College London [36]. MIO-M1 cells were grown in DMEM containing 10% fetal bovine serum and 200 mM glutamine (Life Technologies). The cells were grown in sterilized tissue culture dishes (BD Falcon, Corning, NJ) in an incubator at 5% CO2 and 37 °C and passaged every 6–7 days using trypsin-EDTA and the medium was changed every 3–4 days.
Treatment of retinal astrocytes and MIO-M1 cultures in vitro with sodium peroxynitrite (Cayman Chemicals, Ann Arbor, MI) was performed as previously stated [37]. Briefly, confluent astrocyte cell cultures were washed 3× with PBS supplemented with 0.8 mM MgCl2, 1 mM CaCl2, and 5 mM glucose. They were then incubated in 1 ml of 1X PBS (50 mM Na2HPO4, 90 mM NaCl, 5 mM KCl, 0.8 mM MgCl2, 1 mM CaCl2, and 5 mM glucose, pH 7.4), followed by three additions of sodium peroxynitrite at a concentration of 0.15 mM. The first bolus of sodium peroxynitrite was added to one edge of the dish and the buffer was swirled to allow mixing of the peroxynitrite throughout the dish. This step was repeated twice while changing the edge at which the addition was made and then incubating for 5 min. The buffer was then removed, replaced with the respective growth medium, and placed in the appropriate incubator. The cells were then processed after 32 h for protein or RNA. Some cultures were treated with recombinant BMP7 and BMP 4 (R&D Systems) reconstituted in 0.4% HCl-PBS. Dishes were treated with either vehicle or 100 ng/ml of BMP7 or BMP4 for 24 or 36 h. Cells were then processed for either RNA or protein for analysis via RT-qPCR or western blotting, respectively.
Bone morphogenetic protein 7 injections in vivo
The mice were injected intravitreally with BMP7 (1 μl of 20 ng/μl). Intravitreal injections were performed with a pump microinjection apparatus (Harvard Apparatus) and pulled glass micropipettes. Each micropipette was calibrated to deliver 1 μl of vehicle or vehicle containing BMP7. The mice were anesthetized under a dissecting microscope; the sharpened tip of the micropipette was passed through the sclera, just behind the limbus, into the vitreous cavity. Once the micropipette was in place, the vehicle or BMP7 was injected using a PL-1000 picospritzer (Harvard Apparatus, Holliston, MA). D3 or D7 post-injection, the animals were sacrificed and retinas were dissected for RNA and protein preparations. Each experiment was performed in triplicate.
Western blot analysis
Antibodies used for western blot analyses are listed in Table 1. The total protein was quantified using a Pierce BCA Protein Assay Kit (Thermoscientific) following the manufacturer’s protocol. Briefly, 5 μl of sample was mixed with 495 μl of working reagent (prepared by mixing Pierce BCA Reagent A and B in 50: 1 ratio) and 500 μl of water. This solution was incubated at 65 °C for 30 min, followed by colorimetric analysis using a NanoDrop 2000c spectrophotometer (Thermoscientific). Fifty μg of total protein mixed with the loading dye at a 3:1 ratio was then loaded and run on a 4%–20% SDS polyacrylamide gel (Expedeon, San Diego, CA) at 125 V for 1 h. Proteins were transferred to a polyvinylidene fluoride (PVDF) membrane (Bio-Rad) via a wet transfer and subjected to immunoblotting. Prior to incubation with the antibody, the membrane was blocked using a 5% milk solution in Tris-buffered saline–Tween (TBST; composition – 20 mM Tris base, 137 mM sodium chloride, 1 M HCl, 0.1% Tween-20, at pH 7.6) for 1 h. The blots were then incubated with the primary antibody diluted in TBST at 4 °C overnight. The blots were washed twice with TBST and then incubated with a peroxidase-conjugated secondary antibody (Thermoscientific) diluted to 1:5,000 in TBST for 1 h in the dark at room temperature. The blots were incubated with either Pierce ECL Western Blotting Substrate (Thermoscientific) or SuperSignal West Femto Chemiluminescent Substrate (Thermoscientific), and the bands visualized on X-ray films (Thermoscientific). β-TUBULIN was used as a loading control and densitometry of the blots was performed using Image J software.
Real-time quantitative PCR
Prior to cDNA synthesis, RNA samples were run on a 1% agarose gel to confirm the overall quality of the total RNA. The cDNA was synthesized from 1 µg of total RNA with an iScript cDNA Synthesis Kit (Bio-Rad) according to the manufacturer’s protocol and diluted to 1:20 before adding to qPCR wells. Real-time quantitative PCR (RT-qPCR) was performed using the 7300 RT detection system (Applied Biosystems, Carlsbad, CA) with the Power SYBR green PCR master mix (Invitrogen). The primer pairs used have been listed in Table 2 (mouse) and Table 3 (human). The total volume for each reaction was 20 µl using the diluted cDNA, corresponding to 5 ng of initial total RNA and 0.4 mM of each primer. The cycler conditions used were as follows: initial denaturation at 95 °C for 10 min, 45 cycles of denaturation at 95 °C for 10 s, annealing at 60 °C for 20 s and extension at 72 °C for 20 s, followed by a final extension at 72 °C for 5 min. Efficiency of the primer sets was determined by the standard curve method, where efficiency E=((10(−1/Ct2 – Ct1)) – 1) × 100. A no template control and an internal control using primers specific for β-2 microglobulin (B2M) were used for each run. The amplified samples were run on a 2% agarose gel to confirm that the amplification was of the right size. The change in the gene expression levels was done using the 2 –ΔΔCt method, where “Ct” is the crossing threshold value.
Table 2. List of MOUSE primers used in qPCR.
| Gene | Accession number | Primer | Sequence | Product length | Efficiency (%) |
|---|---|---|---|---|---|
| Gfap2 |
NM_010277.3 |
Forward |
TAGCCCTGGACATCGAGATCGCC |
141 |
104.19 |
| [79] |
Reverse |
GGTGGCCTTCTGACACGGATTTGG |
|||
| Pax2 |
NM_011037.4 |
Forward |
ACCCTGGCAGGAATGGTGCCT |
70 |
90.39 |
| [80] |
Reverse |
AGGCGGTGTACTGGGGATGGC |
|||
| S100-Β |
NM_009115.3 |
Forward |
GACTGCGCCAAGCCCACACC |
142 |
92.25 |
| [81] |
Reverse |
TCCAGCTCGGACATCCCGGG |
|||
| Vim |
NM_011701.4 |
Forward |
AGGAAGCCGAAAGCACCCTGC |
78 |
92.6 |
| [79] |
Reverse |
TCCGTTCAAGGTCAAGACGTGCC |
|||
| Gs |
NM_008131.3 |
Forward |
GCGCTGCAAGACCCGTACCC |
145 |
105.87 |
| [82] |
Reverse |
GGGGTCTCGAAACATGGCAACAGG |
|||
| Egfr-1 |
NM_207655.2 |
Forward |
ACCTATGCCACGCCAACTGTACCT |
82 |
108.21 |
| [83] |
Reverse |
TGAACGTACCCAGATGGCCACACTT |
|||
| Tlr-4 |
NM_021297.2 |
Forward |
TGCCTGACACCAGGAAGCTTGA |
102 |
98.8 |
| [84] |
Reverse |
AGGAATGTCATCAGGGACTTTGCTG |
|||
| Pcan |
NM_001081306.1 |
Forward |
ATCCCTGAGTGGGGAAGGCACA |
96 |
90.01 |
| [85] |
Reverse |
AGCAGGGGATGCTGGGTGATGA |
|||
| Ncan |
NM_007789.3 |
Forward |
CCTGACAAGCGTCCATTCGCCA |
90 |
95.22 |
| [85] |
Reverse |
ACTGTCCGGTCATTCAGGCCGAT |
|||
| Timp2 |
NM_011594.3 |
Forward |
GCAACAGGCGTTTTGCAATG |
71 |
108.7 |
| [86] |
Reverse |
CGGAATCCACCTCCTTCTCG |
|||
| Mmp9 |
NM_013599.2 |
Forward |
TGTGCCCTGGAACTCACACGAC |
135 |
108.97 |
| [87] |
Reverse |
ACGTCGTCCACCTGGTTCACCT |
|||
| Mmp11 |
NM_008606.2 |
Forward |
ACTGACTGGCGAGGGGTACCTT |
128 |
106.7 |
| [87] |
Reverse |
GCAGATGGACCCCATGTTTGCTGT |
|||
| Mmp14 |
NM_008608.3 |
Forward |
TGGGCCCAAGGCAGCAACTT |
89 |
99.37 |
| [87] |
Reverse |
CGTTGTGTGTGGGTACGCAGGT |
|||
| Glast |
NM_148938.3 |
Forward |
GATTTGCCCTCCGACCGTAT |
185 |
97.64 |
| [88] |
Reverse |
ATAGACTACAGCGCGCATCC |
|||
| Ednrb |
NM_001136061.2 |
Forward |
TTGACCTCCCCATCAACGTG |
140 |
96.45 |
| [89] |
Reverse |
AGCACAGAGGTTCAAGACGG |
|||
| Lcn2 |
NM_008491.1 |
Forward |
AATGTCACCTCCATCCTGGTCA |
145 |
93.93 |
| [90] |
Reverse |
CCACTTGCACATTGTAGCTCT |
|||
| Asic1 |
NM_009597.1 |
Forward |
TACTGCAGGACAAAGCCAACTT |
176 |
102.39 |
| [91] |
Reverse |
GTAGCACTTTCCATAGCGTGTG |
|||
| Kir2.1 |
NM_008425.4 |
Forward |
GACCCTCCTCGGACCTTACG |
151 |
98.23 |
| [92] |
Reverse |
ACTGGCCGTTCTTCTTGACA |
|||
| Spp1 |
NM_001204201.1 |
Forward |
TCCTTGCTTGGGTTTGCAGT |
188 |
102.02 |
| [93] |
Reverse |
GTCACTTTCACCGGGAGGG |
|||
| Txnip |
NM_001009935.2 |
Forward |
CCTACAGCAGGTGAGAACGA |
103 |
101.78 |
| [94] |
Reverse |
TAAAGGATGTTCCCAGGGGC |
|||
| Gal3 |
NM_001145953.1 |
Forward |
GGCGGGTGGAGCACTAATC |
74 |
109.13 |
| [95] |
Reverse |
TAAGCGAAAAGCTGTCTGCC |
|||
| β2-Microglobulin |
NM_009735.3 |
Forward |
TCGCGGTCGCTTCAGTCGTC |
135 |
98.36 |
| Reverse | CATTCTCCGGTGGGTGGCGTG |
Table 3. List of HUMAN primers used in qPCR.
| Gene | Accession number | Primer | Sequence | Product length | Efficiency (%) |
|---|---|---|---|---|---|
| Gfap2 |
NM_002055.4 |
Forward |
GCACGCAGTATGAGGCAATG |
139 |
97.35 |
| Reverse |
TAGTCGTTGGCTTCGTGCTT |
||||
| Pax2 |
NM_003990.3 |
Forward |
CCAGTTGTGACTGGTCGTGA |
206 |
107.14 |
| Reverse |
GGCATTAGTAAGGCGGGGTT |
||||
| S100-Β |
NM_006272.2 |
Forward |
GACCAGGAAGGGGTGAGACA |
141 |
105.74 |
| Reverse |
TGATGAGCTCCTTCAGTTCGG |
||||
| Vim |
NM_003380.3 |
Forward |
GCAGGATTTCTCTGCCTCTTC |
198 |
103.251 |
| Reverse |
CTGCACTGAGTGTGTGCAATTT |
||||
| Gs |
NM_002065.5 |
Forward |
CTTAACCCACCAACCTGCCTG |
221 |
105.48 |
| Reverse |
AGGTGGTCATGGTGGAAGGT |
||||
| Egfr |
NM_005228.3 |
Forward |
AAACAACACCCTGGTCTGGA |
126 |
95.85 |
| Reverse |
GGGATCTTAGGCCCATTCGT |
||||
| Tlr-4 |
NM_138554.4 |
Forward |
GTGAGACCAGAAAGCTGGGA |
151 |
105.57 |
| Reverse |
TGCCTAAATGCCTCAGGGGA |
||||
| Pcan |
NM_002851.2 |
Forward |
TCCACAGATTTCAGTTTTGCAG |
198 |
98.81 |
| Reverse |
CCCCTCAGCTAGACCAATACG |
||||
| Ncan |
NM_004386.2 |
Forward |
GAGGTGCACTCAGATCCCTG |
196 |
96.51 |
| Reverse |
GGCAGACAAAGCCATTGACC |
||||
| Timp2 |
NM_003255.4 |
Forward |
GGCGTTTTGCAATGCAGATG |
72 |
105.47 |
| Reverse |
TCGTTTCCAGAGTCCACTTCC |
||||
| Mmp9 |
NM_004994.2 |
Forward |
CGACGTCTTCCAGTACCGA |
81 |
97.12 |
| Reverse |
TTCAACTCACTCCGGGAACTC |
||||
| Mmp11 |
NM_005940.3 |
Forward |
CTGGGAGAAGACGGACCTCA |
242 |
105.26 |
| Reverse |
TCTTGGGGAAGAAGGCATGG |
||||
| Mmp14 |
NM_004995.3 |
Forward |
GTGGTCTCGGACCATGTCTC |
143 |
92.26 |
| Reverse |
AGCCATATTGCTGTAGCCAGG |
||||
| Glast |
NM_004172.4 |
Forward |
GGCTAGCCTGCCTGCTTAC |
165 |
105.29 |
| Reverse |
TGTCTGGGAATCACCCACAG |
||||
| Ednrb |
NM_001201397.1 |
Forward |
TGCTTGCTTCATCCCGTTCA |
197 |
105.82 |
| Reverse |
TCCCGTCTCTGCTTTAGGTG |
||||
| Lcn2 |
NM_005564.3 |
Forward |
GACCCGCAAAAGATGTATGCC |
197 |
99.22 |
| Reverse |
CTCACCACTCGGACGAGGTA |
||||
| Asic1 |
NM_020039.3 |
Forward |
CGCAGCTTCAAACCCAAACC |
169 |
105.85 |
| Reverse |
GGCCCGAGTTGAACGTGTA |
||||
| Kir2.1 |
NM_000891.2 |
Forward |
ACTCTCGTCGGACCCTCC |
183 |
94.54 |
| Reverse |
GCGACTTGTTGCTCAGGTTG |
||||
| Spp1 |
NM_001251830.1 |
Forward |
AGCCAAACAACAAATGGGCA |
199 |
98.46 |
| Reverse |
AGATGGGTCAGGGTTTAGCC |
||||
| Txnip |
NM_006472.4 |
Forward |
TCAAGATGCCGAACCCTGTG |
130 |
96.45 |
| Reverse |
AGTGAGGGGCCAATATCCCT |
||||
| Gal3 |
NM_002306.3 |
Forward |
GCCTTCCACTTTAACCCACG |
173 |
105.55 |
| Reverse |
ACTGCAACCTTGAAGTGGTCA |
||||
| β2-Microglobulin |
NM_004048.2 |
Forward |
AGATGAGTATGCCTGCCGTG |
120 |
102.37 |
| Reverse | TCATCCAATCCAAATGCGGC |
Statistical analysis
Statistical analysis of RT-qPCR data was performed by unpaired t test between the control and treated groups. Statistical analysis of densitometry results was performed using the Student t test. All analyses were performed using SPSS software (IBM) and Excel 2010 (Microsoft).
Results
Müller glia express bone morphogenetic protein type IA, IB, and activin like kinase receptor 2 receptors
BMPs bind preferentially to three type I receptors; receptor IA, IB, or ALK2. Double-label immunofluorescence was used to determine whether any of the type I BMP receptors was expressed by Müller cells or retinal astrocytes. Sections through adult murine retina were colabeled with antibodies specific for GS, which labels Müller glial cells and retinal astrocytes, and BMPR1A, BMPR1B, or ALK2 (Figure 1). All three receptors showed similar labeling patterns in the retina with Müller glial cell processes labeled in the outer nuclear layer (ONL), outer plexiform layer (OPL), and GCL (Figure 1A-C). Photoreceptor outer segments (OS) and ganglion cell bodies were also clearly labeled for all three receptors. To confirm the label of Müller glial cells and their processes by antibodies to type I receptors, enzymatically dissociated cells were colabeled with antibodies to GS and BMPRIA, BMPR1B, or ALK2 (Figure 1D). Immunolabeled Müller glia were readily apparent in dissociated samples, as they had retained their shape throughout the processing. All three receptors appeared to colabel GS (+) cells, indicating that the cells are responsive to BMPs in vivo.
Figure 1.

Bone morphogenetic protein (BMP) type I receptors in the mature mouse retina. A: Retinal sections of 4 week-old retina double-labeled using antibodies that recognize Müller glial and retinal astrocyte marker glutamine synthetase (GS) and BMP receptor 1A (BMPR1A). Thin plane confocal microscopy with y,z (strips to right of panels), and x,z planes (strips at bottom of panels) are shown in the last panels in the row. BMPR1A was localized to the Müller glial processes in the outer nuclear layer (ONL) and in Müller glial cell or retinal astrocyte processes in the ganglion cell layer (GCL, arrows A-C). BMPR1A was also detectable in the photoreceptor outer segments, the inner plexiform layer (IPL), cell bodies within the ganglion cell layer, and the nerve fiber layer (NFL). B: Retinal sections of 4 week-old retina double-labeled using antibodies that recognize GS and BMPR1B. BMPR1B appeared to be localized to the outer segments (OS), outer plexiform layer (OPL) and IPL and, Müller glial cell processes as well as Müller glial/retinal astrocyte processes in the GCL and NFL (arrows). C: Retinal sections of 4 week-old retina double-labeled using antibodies that recognize GS and activin receptor like kinase 2 (ALK2). The distribution of ALK2 receptors was similar to that of BMPR1A, with the majority of signal being localized to the end feet within the GCL and NFL. D: Müller glia isolated from P30 retinas were co-labeled for GS and BMPR1A, 1B and ALK2. The isolated cells showed the distribution of receptors to be primarily in the processes and the end feet of the Müller glia, as seen in the P30 tissues. n=3 different eyes for each label. Scale bar A=50 μm applies to A, B, C.
Bone morphogenetic protein 7 signaling components in the retinal glia
The BMP signaling pathway members present in the Müller glial and retinal astrocytes were also investigated to determine which pathways might be activated in the presence of BMPs. To investigate the canonical BMP pathway, sections were colabeled with antibodies against Sox2, which labels Müller glial cells, retinal astrocytes, and cholinergic amacrine cells in the mature murine retina, and SMAD1. SMAD1 was localized throughout cells of the inner nuclear and GCLs, and was colocalized with SOX2 (+) cells (Figure 2A). To determine whether the BMP-kinase pathway could be activated in response to BMPs, sections through the adult retina were colabeled with antibodies to GS and TAK1 (Figure 2B). While TAK1 was prominently expressed in the retina, most notably in a subpopulation of inner nuclear layer (INL) cells and the GCL, little to no co-expression could be detected in GS (+) cells (Figure 2B). Last, potential involvement of the FRAP-STAT pathway in Müller glial BMP signaling was investigated by colabeling sections with GS and FKBP12 (Figure 2C). FKBP12 label was noted in the inner nuclear, ganglion cell, and inner plexiform layers (IPLs). No apparent colabel was detected of FKBP12 and GS (Figure 2C).
Figure 2.

Bone morphogenetic protein (BMP) signaling components in the Müller glia of adult mouse retina. A: Sections of 4 week-old retina were subjected to double-label immunofluorescence with antibodies specific for sex determining region Y box 2 (SOX2), which labels Müller glia, retinal astrocytes, and cholinergic amacrine cells, and intracellular members of the BMP pathway SMAD1 . SMAD1 (+) cells were localized to the inner nuclear layer (INL) and ganglion cell layer (GCL) of P30 retina. A subpopulation of SOX2 (+) Müller glia (arrows) and retinal astrocyte cells (arrowheads) were also positive for SMAD1. B: Sections of 4 week-old retina were subjected to double-label immunofluorescence with antibodies for SOX2, and TGF-β activated kinase 1 (TAK1). Cells positive for the protein TAK1 were found to be localized in the INL and GCL. C: Sections of 4 week-old retina were subjected to double-label immunofluorescence with antibodies for SOX2, and FK506 rapamycin binding protein (FKBP12). FKBP12, also part of the non-canonical BMP pathway, was found to be localized in the INL along with sparse localization in the GCL. Very little to no co-label was seen with glutamine synthetase (GS; +) and TAK1 or FKBP12 (+) cells (B, C). n=3 different eyes for each label. Scale Bar A=50 μm applies to all panels.
Bone morphogenetic protein 7 can trigger changes in retinal astrocytes and MIO-M1 Müller cells resembling mild reactive gliosis
The role of BMP7 in reactive gliosis was first tested by the addition of the factor to an in vitro system using isolated retinal astrocytes or a Müller glial cell line, MIO-M1 [34-36]. Previously characterized retinal astrocytes have been shown to express paired box 2 (PAX2), NG2, and GFAP [34,35]. To use these cells, their ability to exhibit changes associated with reactive gliosis first had to be determined. The cells were briefly treated with vehicle or sodium peroxynitrite, a strong oxidizing agent that is released by injured neurons and has been shown to trigger reactive gliosis [38]. Following treatment, levels of mRNAs known to be regulated during reactive gliosis were examined 16 (not shown) and 32 h after treatment using RT-qPCR (Figure 3). In the analysis of the RT-qPCR levels throughout this study, two thresholds had to be met in order for the change to be considered convincing: 1) The comparative change with controls had to be statistically significant, and 2) an average increase in levels had to reach 1.5-fold or above that of controls, or similarly, a decrease had to reach 0.5-fold or below. No statistically significant changes in expression were noted between vehicle and peroxynitrite 16 h following treatment (not shown). In contrast, there were statistically significant increases that reached the 1.5-fold threshold in the level of many mRNAs within the reactive gliosis panel in peroxynitrite-treated cells in comparison to vehicle-treated ones, including Pax2, lipocalin 2 (Lcn2), Mmp9, Pcan, S100β, toll-like receptor 4 (Tlr4), tissue inhibitor of matrix metalloproteinase 2 (Timp2), and Gfap (Figure 3A). There was also a statistically significant decrease in the glutamate aspartate transporter (Glast), which also reached the level of 0.5 and below threshold. Levels of protein were investigated by western blotting for three proteins, namely GFAP, thioredoxin-interacting protein (TXNIP), and PAX2 (Figure 3B). While there was a modest increase in GFAP and PAX2 protein in comparison to control vehicle-treated cells, there was no increase in levels of TXNIP 36 h following treatment (Figure 3B).
Figure 3.
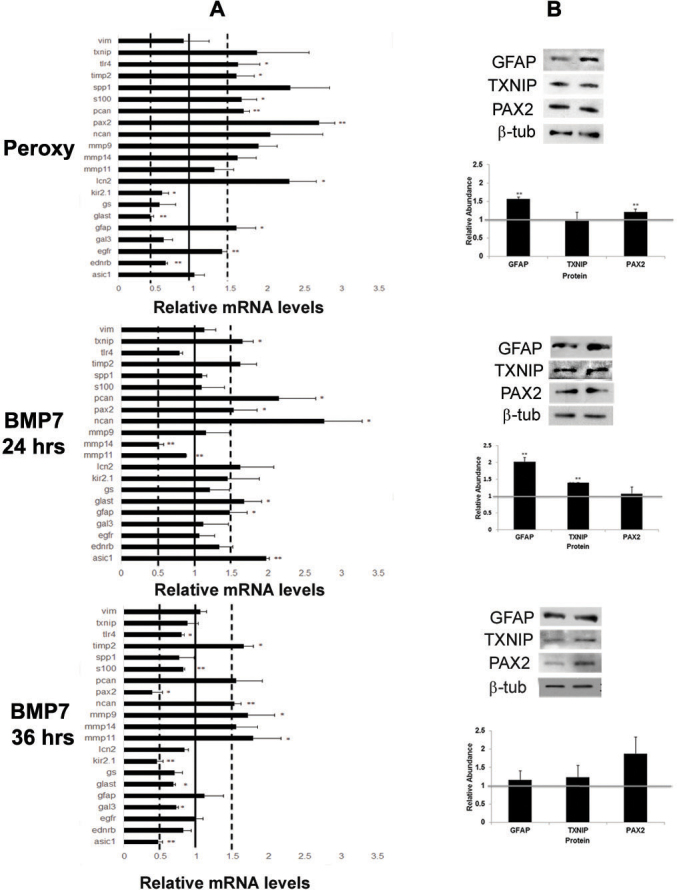
Bone morphogenetic protein 7 (BMP7) treatment of retinal astrocyte cells increases markers of glial scar formation. A: Expression patterns for a panel of markers associated with reactive gliosis were compared in murine retinal astrocytes treated with sodium peroxynitrite or BMP7 for 24 or 36 h. For each experimental treatment, cells were treated with vehicle, sodium peroxynitrite, or BMP7 and real-time quantitative PCR (RT-qPCR) was undertaken. Levels of mRNA were normalized to internal control β 2 Microglobulin, and values were plotted relative to control levels. Each sample was run in triplicate and experiment repeated 3 times for each gene. Values represented are means ±SEM. Unpaired t test was performed between the control and treated groups with * denoting a p value<0.05 and ** denoting a p value<0.005. Any change above or below 1.0 indicates a change relative to control values. Peroxynitrite-treated cells showed statistically significant increases above 1.5-fold in mRNA levels of paired-homeobox 2 (Pax2), lipocalin2 (Lcn2), matrix metalloproteinase 9 (Mmp9), phosphacan (Pcan), S100β, toll like receptor 4 (Tlr4), tissue inhibitor of matix metalloproteinases 2 (Timp2), and glial fibrillary acidic protein (Gfap). Decreases in mRNA levels below 0.5-fold were noted for glutamate-aspartate transporter (Glast) in peroxynitrite-treated cells. In comparison, 24 h of BMP treatment led to a modest increase above the 1.5-fold level in neurocan (Ncan), Pcan, acid sensing ion channel 1 (Asic1), Glast, thioredoxin-interacting protein (Txnip), Pax2, and Gfap mRNA levels. By 36 h post BMP7-addition, Gfap levels had returned to baseline and Asic1, potassium inwardly rectifying channel 2.1 (Kir2.1), and Pax2 mRNA levels were reduced in comparison to vehicle-treated cells. Levels of Mmp11, Timp2, and Ncan were still increased in comparison to vehicle-treated cells. B: western blot analysis was performed for GFAP, TXNIP, and PAX2, with β-TUBULIN used as a loading control. Densitometric data shown are means+/−SEM of 3 trials. Unpaired t test was performed between the control and treated groups with * denoting a p value<0.05 and ** denoting a p value<0.005. Densitometric analysis of the blots showed a statistically significant increase in protein levels of GFAP in the peroxynitrite and 24 h BMP7 treatments. Significant increases in levels of PAX2 and TXNIP was observed in the peroxynitrite and 24 h BMP7 treatments, respectively. No statistically significant change was observed in the 36 h BMP7 treatment.
To test whether BMP7 treatment could also trigger signs of reactive gliosis in vitro, retinal astrocytes were treated with either vehicle or BMP7 for 24 or 36 h. Following 24 h of vehicle or BMP7 treatment, retinal astrocytes treated with BMP7 showed a statistically significant increase in transcript levels above the 1.5-fold threshold for Ncan, Pcan, acid-sensing ion channel 1 (Asci1), Glast, Txnip, Pax2, and Gfap (Figure 3A). There was also a decrease in levels of Mmp14 in BMP-treated versus vehicle-treated retinal astrocytes that met the 0.5 threshold and was a statistically significant change in comparison to vehicle-treated cells (Figure 3A). Western blotting was used to assess the levels of a subset of proteins in vehicle- and BMP-treated cells. A statistically significant increase in protein levels of GFAP and TXNIP protein was noted for BMP7-treated cells compared to vehicle-treated cells at 24 h after treatment (Figure 3B). However, there was no detectable differences in the levels of TXNIP or PAX2 at 24 h (Figure 3B). Cells treated with BMP7 for 36 h showed changes in levels of a slightly different cohort of mRNAs; Mmp11, Timp2, Ncan, and Mmp9 were modestly increased, while Asic1, inwardly rectifying potassium channel 2.1 (Kir2.1), and Pax2 were decreased slightly (Figure 3A). By western blotting, small but statistically insignificant increases were observed in GFAP, TXNIP, and PAX2 (Figure 3B).
Similar to the retinal astrocyte cultures, treatment of the Müller glial cell line MIO-M1 with peroxynitrite also led to increases in some of the markers in the reactive gliosis panel (Figure 4A). Western blots showed a modest increase in protein levels of GFAP and S100β; however, no change was noted in PAX2 expression (Figure 4B). Treatment of MIO-M1 cells with BMP7 yielded similar, but slightly different changes in mRNA levels following treatment for 24 or 36 h. At 24 h, Gfap and Pax2 mRNA levels were increased in comparison to control vehicle-treated cells above the 1.5-fold threshold, while there was a decrease in Txnip, Gal3, Lcn2, and Mmp9 levels below the 0.5-fold threshold (Figure 4A). Western blotting showed statistically significant changes in GFAP, S100β, and TXNIP protein levels in comparison to controls; however, increases in PAX2 were not statistically significant (Figure 4B). Treatment of cells with BMP7 for 36 h yielded increases in Gfap and Tlr4 mRNA levels in comparison to control cultures and a decrease in secreted phosphoprotein 1 (Spp1; Figure 4A). By western blot, densitometric analysis showed a statistically significant decrease in PAX2 levels and no significant changes in GFAP, S100β, or TXNIP levels in comparison to vehicle-treated cells (Figure 4B).
Figure 4.
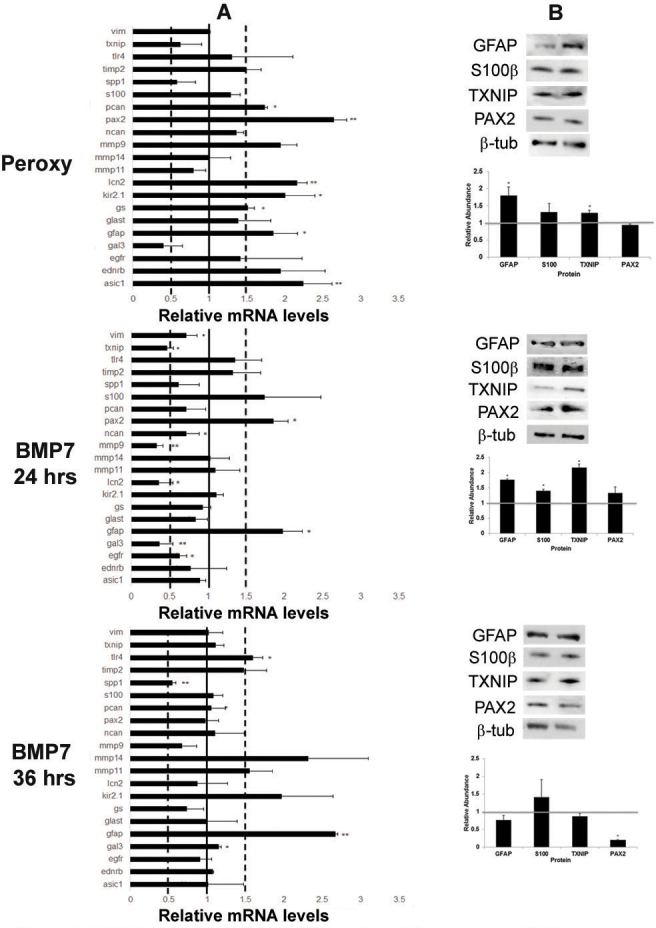
Bone morphogenetic protein 7 (BMP7) treatment of MIO-M1 Müller glial cell line increases glial fibrillary acidic protein (GFAP) expression. A: Expression patterns for a panel of markers associated with reactive gliosis were compared in human MIO-M1 Müller glial cells treated with sodium peroxynitrite, or BMP7 for 24 or 36 h. For each experimental treatment, cells were treated with vehicle, sodium peroxynitrite, or BMP7 and real-time quantitative PCR (RT-qPCR) was undertaken. Levels of mRNA were normalized to internal control β2 Microglobulin, and values were plotted relative to control levels. Each sample was run in triplicate and experiment repeated 3 times for each gene. Values represented are means±SEM. Unpaired t test was performed between the control and treated groups with * denoting a p value<0.05 and ** denoting a p value<0.005. Any change above or below 1.0 indicates a change relative to control values. Peroxynitrite-treated MIO-M1 cells showed a statistically significant increase above the 1.5-fold level in paired-homeobox 2 (Pax2), acid sensing ion channel 1 (Asic1), lipocalin2 (Lcn2), potassium inwardly rectifying channel 2.1 (Kir2.1), Gfap, and phosphacan (Pcan) in comparison to vehicle-treated cells. Twenty-four hours following BMP7 addition, the MIO-M1 cells only showed a statistically significant increase above the 1.5-fold level in Gfap and Pax2, and a decrease in Txnip, galectin 3 (Gal3), Lcn2, and matrix metalloproteinase 9 (Mmp9). At the 36 h time point Gfap, and toll-like receptor 4 (Tlr4) levels of mRNA were increased above the 1.5-fold level in comparison to vehicle-treated cells and Spp1 was decreased. B: western blot analysis was performed for GFAP, S100β, TXNIP, and PAX2, with β-TUBULIN used as a loading control. Densitometric data shown are means+/−SEM of 3 trials. Unpaired t test was performed between the control and treated groups with * denoting a p value<0.05 and ** denoting a p value<0.005. Densitometric analysis of the blots showed a statistically significant increase in protein levels of GFAP and TXNIP in the peroxynitrite treatment. A significant increase in levels of GFAP, S100β, and TXNIP was observed in the 24 h BMP7 treatment, while a significant decrease was observed in PAX2 levels in the 36 h bmp7 treatment.
To determine whether treatment of cells with another BMP family member, BMP4, led to similar changes in reactive gliosis markers, retinal astrocytes and MIO-M1 cells were treated with BMP4 for 24 or 36 h, and changes in mRNA levels were assessed by RT-qPCR. Overall, there were fewer statistically significant changes in BMP4-treated cells at or over the level of a 1.5-fold increase or a 0.5-fold decrease in comparison to vehicle-treated cells in both retinal astrocytes and MIO-M1 cells (Figure 5). In retinal astrocytes, Txnip and GS were increased more than 1.5-fold over control and the difference was statistically significant. Only Mmp9 showed a decrease in comparison to control at 24 h following treatment. In comparison, at the 36 h time point, only the decrease in Mmp9 was statistically significant and reached the 0.5-fold level. In the MIO-M1 cells, only Asic1 and Spp1 were upregulated at least 1.5-fold over levels of control mRNA at 24 h following addition. Further, at the 36 h time point, a statistically significant increase at the 1.5-fold level in endothelin receptor type B (Ednrb) and Asic1 were the only increases noted at 36 h following addition of BMP4 (Figure 5).
Figure 5.
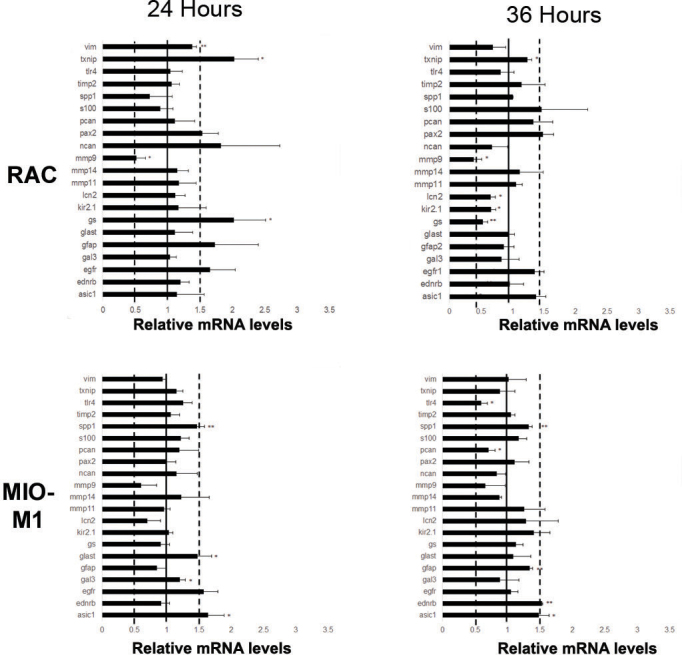
Retinal astrocytes and MIO-M1 cells show an attenuated response to bone morphogenetic protein 4 (BMP4). Retinal astrocytes (RAC) and MIO-M1 cells were treated with BMP4 and mRNA isolates from samples treated for 24 h or 36 h were subjected to real-time quantitative PCR (RT-qPCR) using the reactive gliosis panel established in previous experiments. Each sample was run in triplicate and experiment repeated 3 times for each gene. Values represented are means±SEM. Unpaired t test was performed between the control and treated groups with * denoting a p value<0.05 and ** denoting a p value<0.005. At 24 h, retinal astrocytes treated with BMP4 showed statistically significant increase above the 1.5-fold level in thioredoxin-interacting protein (Txnip) and glutamine synthetase (GS) in comparison to controls, as well as a decrease in Mmp9. By 36 h, statistically significant decreases in GS and matrix metalloproteinase 9 (Mmp9) in comparison to vehicle were noted. Similarly, MIO-M1 cells treated showed small changes in acid sensing ion channel 1 (Asic1), glutamate-aspartate transporter (Glast) and secreted phosphoprotein 1 (Spp1), following 24 h of BMP4 treatment. At 36 h, increases above 1.5-fold in endothelin receptor type B (Ednrb) and Asic1 in comparison to controls were noted.
Intravitreal injection of bone morphogenetic protein 7 induces reactive gliosis
To further explore the role of BMP7 in reactive gliosis, wild-type adult mouse eyes underwent intravitreal injections of vehicle or BMP7. To determine the BMP signaling cascade in BMP7-mediated gliosis, the vehicle- and BMP7-injected retinas were colabeled with SOX2 and phospho-SMAD 1/5/8 (Figure 6) or phospho-TAK1 (Figure 7). While the vehicle-injected retina showed little or no phospho-SMAD and SOX2 colabeling, the BMP7-injected retinas showed an increased colabel for phospho-SMAD 1/5/8 and the Müller glial marker SOX2 (Figure 6). Colabeling was also performed for SOX2 and phospho-TAK1 to determine the activation of the BMP-MAPK noncanonical signaling cascade. The vehicle-injected retina showed phospho-TAK1 label primarily in the GCL (Figure 7), while the BMP7-injected retina showed an increase in the phospho-TAK1 label in the INL and an increase in colabeling with SOX2 (Figure 7). Further, retinas at 3 and 7 days after injection were examined using RT-qPCR, immunofluorescence, and western blot analysis. Three days following injection, the only statistically significant change that reached the threshold was a decrease in Pax2 and Gal3 mRNA levels (Figure 8A). At the 7 day time point, however, statistically significant increases in mRNA levels above the threshold were observed for Txnip, Glast, Mmp9, Lcn2, GS, Kir2.1, and Mmp14. There were also several decreases noted at 7 days after injection, specifically in Tlr4, Pax2, and Egfr (Figure 8A). Immunohistochemistry on sections through the injected retinas at 3 days following injection revealed no readily apparent changes in GFAP, S100β, or NEUROCAN in BMP7-injected retinas as compared to vehicle-injected ones. In contrast, there was a marked increase in GFAP, S100β, and NEUROCAN in BMP7-injected retinas when compared with their control counterparts (Figure 8B). In addition, Nomarski optics of the injected retina showed some thickened Müller glial processes in the IPL, reaffirming the claim that BMP7 induces hypertrophy of at least a subpopulation of Müller cells at 7 days after injection (Figure 9). By western blot, small but significant increases were observed in S100β and TXNIP at 3 days following injection in BMP7-injected retinas, and while there were increases in GFAP, S100β, and TXNIP at 7 days after injection in BMP-injected retinas, none of the changes were statistically significant (Figure 8C).
Figure 6.

Canonical bone morphogenetic protein (BMP) signaling is activated in the Müller glia in BMP7 injected murine eyes. Sections of retina from adult mouse eyes injected with vehicle or BMP7 and harvested 3 or 7 days after injections were double-labeled with antibodies against phospho-SMAD1/5/8 (green) and the nuclear marker sex determining region Y box 2 (SOX2; red) which labels Müller glia, retinal astrocytes and cholinergic amacrine cells . While the vehicle-injected control showed phospho-SMAD 1/5/8 labeled cells primarily localized in the ganglion cell layer (GCL), with little or no co-label with the SOX2 (+) cells, in the D3 and D7 BMP7-injected retinas, phospho-SMAD1/5/8 label was also detectable in the inner nuclear layer (arrows INL). A sub population of the cells in the INL which were positive for phospho-SMAD1/5/8 were also SOX2 positive. n=3 different retinas for each immunolabel. Scale Bar A=50 μm applies to all panels.
Figure 7.
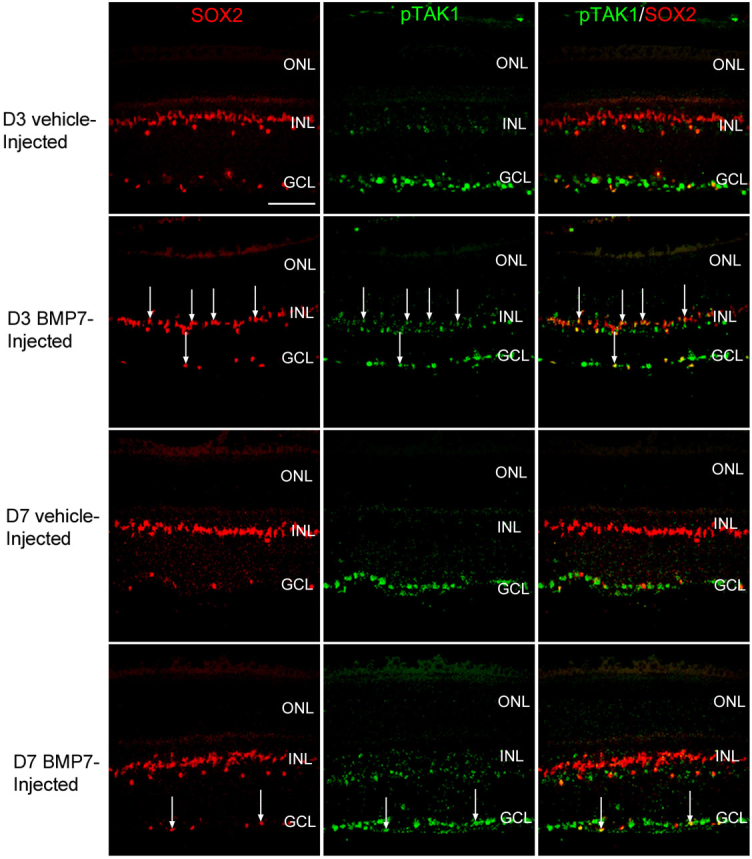
Non-canonical bone morphogenetic protein (BMP) signaling mediated via Transforming Growth Factor-β activated kinase (TAK) is upregulated in the Müller glia in BMP7 injected murine eyes. Retinal sections from mouse eyes processed 3 and 7 days following injection with either vehicle or BMP7 were co-labeled with antibodies against phospho-TAK1 (green) and Müller glial marker sex determining region Y box 2 (SOX2; red) . The vehicle injected control retinas for 3 and 7 days (D3 and D7 respectively) showed phospho-TAK1 label primarily cells in the ganglion cell layer (GCL), with no phospho-TAK1 labeling in the iner nuclear layer (INL) or outer nuclear layer (ONL). SOX2 (+) reinal astrocytes were observed in the GCL, however very little co-expression was observed with phospho-TAK1 in the vehicle injected retinas. The BMP7-injected retinas did show an increase in phospho-TAK1 expression in the INL, showing a comparatively higher expression in the BMP7-injected retina at 3 days than at 7 days. A subpopulation of the SOX2 (+) in the GCL and INL were co-labeled with phospho-TAK1 in the BMP7 injected retinas (arrows). n=3 different retinas for each immunolabel. Scale Bar A=50 μm applies to all panels.
Figure 8.
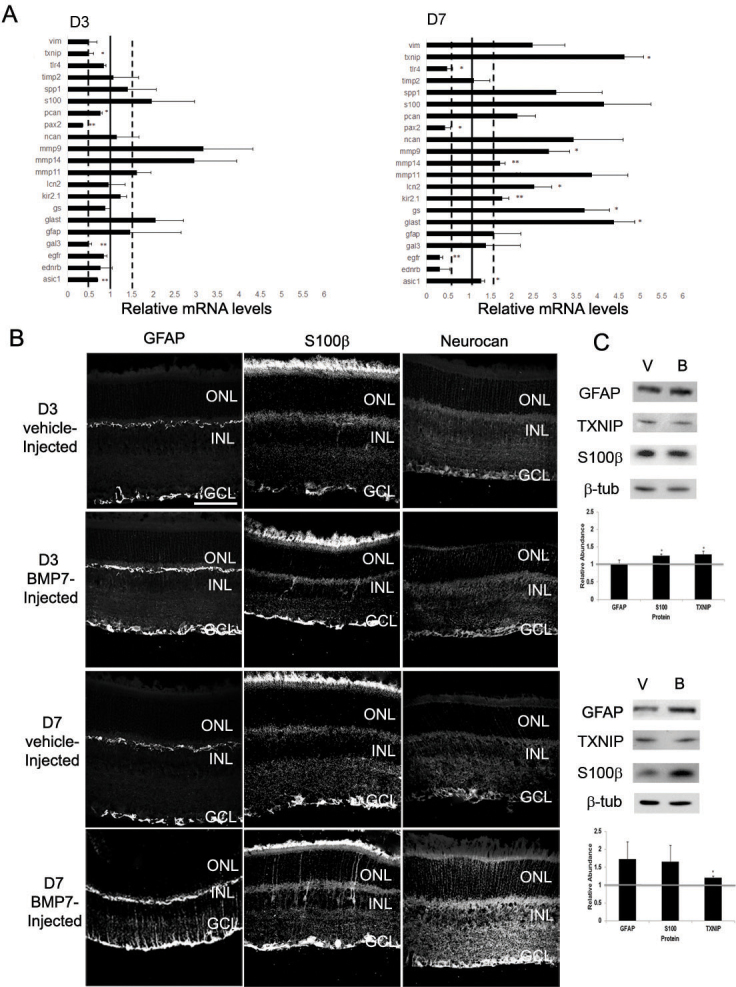
Intravitreal injection of bone morphogenetic protein 7 (BMP7) into murine eyes leads to reactive gliosis. A: RNA from adult eyes injected with vehicle or BMP7 was isolated 3 and 7 days following injections and real time quantitative PCR (RT-qPCR) was performed using the reactive gliosis panel described in previous figures. Graphs have been normalized to β2 Microglobulin and all values are relative to vehicle-treated eyes from the same post-injection day. Each sample was run in triplicate and experiment repeated 3 times for each gene. Values represented are means ±SEM. Unpaired t test was performed between the control and treated groups with * denoting a p value<0.05 and ** denoting a p value<0.005. Any value above 1.0 represents an increase in mRNA levels, while a level below 1.0 represents a decrease. At 3 days following injection, there was a significant decrease in paired-homeobox 2 (Pax2) and galectin 3 (Gal3). In contrast, by 7 days following injection there were significant increases in thioredoxin-interacting protein (Txnip), glutamate-aspartate transporter (Glast), glutamine synthetase (GS), matrix metalloproteinase 9 (Mmp9), lipocalin 2 (Lcn2), potassium inwardly rectifying channel 2.1 (Kir2.1), and matrix metalloproteinase 14 (Mmp14) and decreases in toll-like receptor 4 (Tlr4), Pax2, and epidermal growth factor receptor (Egfr). B: Immunolabel of retinas from 3 day and 7 day vehicle and BMP7 injected eyes for a subset of the gliosis markers. To confirm some of the changes seen at the mRNA level, eyes injected with vehicle or BMP7 were fixed 3 or 7 days after injection and immunolabeled for GFAP, S100β, or NEUROCAN (NCAN). At 3 days following injection, GFAP, S100β and NCAN immunolabel looks similar in vehicle- and BMP7-injected eyes. In contrast, by 7 days, sections immunolabeled for GFAP, S100β, and NCAN showed an increase in labeling in BMP7-injected eyes in comparison to vehicle-injected. n=3 different retinas for each label. Scale bar A=50 μm applies to all panels. C: western blot analysis was performed for GFAP, TXNIP, S100β, and PAX2, with β-TUBULIN used as a loading control. Densitometric data shown are means±SEM of 3 trials. Unpaired t test was performed between the control and treated groups with * denoting a p value<0.05 and ** denoting a p value<0.005. Densitometric analysis of the blots showed a statistical increase in protein levels of S100β at the 3 day stage. While there did appear to be an increase in GFAP, S100β, and TXNIP at 7 days following injection in the BMP7-treated retinas as compared to vehicle-injected, none of the changes in protein levels was statistically significant.
Figure 9.

Nomarski images of 3 day and 7 day vehicle and bone morphogenetic protein 7 (BMP7) injected retinal sections. Retinal sections of vehicle and BMP7 injected eyes were imaged by Nomarski microscopy on day 3 and day 7. The vehicle injected retinal sections showed no thickened Müller glial trunks at the 3 day and 7 day stage. The BMP7 injected retinas for the 3 day and 7 day stage showed very few thickened Müller glial trunks arrows), suggesting that BMP7 triggers mild gliosis in the retina. n=3 for each day and condition. Scale bar A=50 μm applies to all panels.
Discussion
Our findings indicate that the BMP7 pathway plays a role in triggering or maintaining reactive gliosis in the retina. The glial cells of the retina express the BMP receptors (BMPR1A, BMPR1B, and ALK2). While the downstream components for the canonical and noncanonical signaling pathways are present in the retina, only the SMADs appeared to be present in the retinal glia before BMP7 injection. Following injection, both activated SMADs and TAK1 were present, indicating that TAK1 had been upregulated following injection. Treatment of retinal astrocyte cells and Müller glial cells in vitro with BMP7 regulated various reactive gliosis markers. The profiles of gliosis markers regulated due to exposure to BMP7 were similar, although not identical, to that observed after exposure to peroxynitrite. Injection of BMP7 into the mouse retina also triggered a reactive gliosis response. Incubation of cells with BMP4, however, showed no significant increase or decrease in the levels of gliosis markers.
Regulation of glutamine synthetase during reactive gliosis
Reactive gliosis is a common and a complex response observed following neural injury [39]. One response typical of Müller cells undergoing gliosis is a change in levels of glutamine synthesis, the enzyme critical for glutamate-glutamine cycling in the nervous system. Under normal circumstances within the retina, GS prevents the excitotoxic effects of glutamate and is necessary for ammonium detoxification [40]. In the retina, GS is widely expressed in the astrocytes and Müller glial cells [41]. Levels of GS are exquisitely regulated by injury in an injury- or disease-state manner. For instance, expression of GS is downregulated in disease states in which photoreceptors (the major source of glutamate in the retina) degenerate [42,43]. However, in disease states where the presence of GS is necessary to detoxify the retina when high levels of ammonia, glutamate, or glutamate agonists are present (such as hepatic retinopathy or following kainic acid injection), expression levels of the enzyme increase [44,45]. Finally, levels of GS can remain stable in some injury and disease states, including diabetic retinopathy and following nerve crush [46,47]. In the studies presented here, GS levels increased following addition of BMP7 in vitro and in vivo, suggesting that the BMP7 addition mimics situations where there is an increase in glutamate or NH4+ concentrations in the retina. Further studies will be necessary to determine whether differential regulation of BMP7 occurs in a disease-dependent manner.
Bone morphogenetic protein 7 triggers reactive gliosis via the SMAD and the transforming growth factor-β activated kinase pathway
Reactive gliosis encompasses a wide range of responses, ranging from hypertrophy to regulation of transporter channels and enzymes, as well as cell migration, proliferation, and dedifferentiation [20]. The triggers for these different molecular and functional responses vary with the injury or disease state. Many growth factors have been implicated in regulating one or more of the changes associated with gliosis. For instance, epidermal growth factor has been associated with Müller glial proliferation, FGF2 with the scarring response, and CNTF with GFAP upregulation [17,23]. Further, inflammation is also a commonly observed response during gliosis. Factors such as TNF-α and CNTF have been known to play a role in inflammatory response of reactive gliosis, while TNF-α and some of the interleukins are also known to induce proliferative reactive gliosis [48-50].
BMPs have also been associated with various changes that occur during reactive gliosis; however, many of the studies to date have focused on the role of the BMPs following injury to the spinal cord [28,29,51]. Initial studies indicated that BMP signaling increased at the site of spinal cord lesions, and treatment of astrocytes in vitro with BMPs 4 or 7 increased expression of CSPGs typically associated with glial scars [52]. Further, inhibition of BMPs following spinal cord injury leads to a decrease in CSPGs and an increase in the ability of axons to regenerate over time [28,51]. Similarly, knockout studies for two of the type 1 BMP receptors have shown that BMPR1A is critical in the hypertrophy response that occurs early in reactive gliosis and BMPR1B is critical for some aspects of the scarring response. Little is known about the role of BMPs in injury or disease within the retina or optic nerve. Detailed descriptions of the expression patterns of the BMPs, BMP receptors, and several BMP-binding proteins have been described in the developing and adult optic nerve, the optic nerve head, and the trabecular meshwork [26,53,54]. An increase in BMP signaling within the trabecular meshwork has been implicated in blocking TGF-β-driven changes in gene expression [55-57]. Finally, BMP stimulation downstream of EGF signaling has recently been implicated in Müller glial cell proliferation [58].
In the studies detailed here, we showed that the Müller glial cells express BMPR1A, BMPR1B, and ALK2 receptors. While it is probable that astrocytes are also expressing the receptors, due to the intensity of the label in the ganglion cell and inner limiting membrane area, we were unable to determine this for certain. BMP7 preferentially binds the ALK2 receptor and transduces the signal via SMAD 1/5/8 [59]. To examine BMP signaling in the retinas, we first analyzed the expression and glial localization of the canonical and noncanonical BMP signaling components, namely SMAD, TAK and FRAP. While colabeling showed that SMAD and TAK were localized with Müller glia, with SMAD to a greater extent than TAK, FRAP showed no localization (Figure 2). Analysis of the activated forms of SMAD (phospho-SMAD) and TAK (phospho-TAK) revealed that there is activation of both in BMP7-injected retinas (Figure 6 and Figure 7). Apart from the glial cells, several other cells also showed positive labels for both phospho-SMAD and phospho-TAK. This upregulated BMP signaling in both macroglial and other cells of the retina suggests that BMP7 can regulate gliosis either by directly acting on the retinal glia or indirectly by regulating other retinal cells.
In comparison to BMP7, treatments with BMP4 did not appear to induce gliosis in retinal astrocytes or Müller cells in vitro. While BMP4 has been shown to have a neuroprotective effect when injected before a toxic insult, BMP7 was not shown to have the same protective effect [60]. The differential response of retinal neurons and macroglia to BMP4 and 7 is indicative of the involvement of different signaling pathways, but further study is necessary to elucidate the mechanism of this differential response.
Differences in response patterns
Apart from growth factors, various molecular factors such as adenosine tri-phosphate (ATP), nitric oxide (NO), and so on can also trigger reactive gliosis [20]. Although roles of specific factors have been implicated in certain aspects of gliosis such as GFAP, vimentin, and neurofilament expression [14,60], there does not seem to be a clear correlation between the various gliosis markers and its regulators. GFAP is used as the standard indicator of gliosis. However, it and other markers such as GS, MMPs, and others have been shown to be regulated differentially depending on the stress to which they are subjected [42,47,61,62]. In this study, RT-qPCR analysis of RNA from BMP7-injected retinal tissue compared to the RNA prepared from treated retinal astrocytes and Müller glial cells showed a greater deviation from their respective control treatments (Figure 3, Figure 4, and Figure 8). This could be attributed to the fact that the cells in vitro represented a pure population of cells, while the retinal tissue represented a mixed population of macroglia, microglia, neurons, and components of the vasculature. We also observed differences in the profile of regulated genes between the sodium peroxynitrite and BMP7-treated cells. This observation is consistent with the idea that various factors are responsible for driving specific aspects of the gliosis response [13,14,21].
In addition to differential expression between peroxynitrite and BMP7 treatments in vitro, as well as in vivo versus in vitro responses, we also noted that there were substantial differences between the regulation of various markers at the mRNA and protein level. This asymmetry has also been noted by other investigators who have compared mRNA and protein levels, indicating that there may be multiple levels of regulation involved in gliosis [63-67]. Although mRNA levels showed increased regulation both in vivo and in vitro, a similar change was not detected at the protein level. mRNA translation and protein levels are regulated in the cell by several systems, namely regulation of mRNA localization and translational repression mediated by RNA binding proteins, regulation via miRNAs, the ubiquitin proteasome system, and changes in protein stability [68,69]. Here, we did not observe a linear correlation between the mRNA and protein levels in the gliosis models analyzed. We thus hypothesize that protein is regulated at the mRNA translation level, mediated either by RNA binding proteins or by miRNAs. Furthermore, protein stability may also be regulated in BMP7-mediated gliosis, leading to differences in mRNA and protein levels. mRNA binding protein such as cytoplasmic polyadenylation element binding protein (CPEB) has been previously shown to upregulated in gliosis [70]. One of the downstream targets of the TAK-mediated BMP cascade is p38-MAPK [59,71]. p38-MAPK can regulate the phosphorylation, and thereby, the activation of CPEB [70]. miRNAs are small, 22–25 nucleotide long, noncoding RNAs that play an important role in cell fate and development. miRNAs have been found that are specific to the nervous system, as well as to the retina, and BMPs have also been shown to regulate miRNA levels [72-75]. Further, regulation of miRNAs has also been observed in astrocytes following injury to the spinal cord [76]. The miRNAs bind to their target RNA and can either direct it to degradation or repress translation. ceRNA interaction mediated by miRNA, which can target more than mRNA, has been previously reported in diabetic retinopathy in RPE cells [77]. Similar interactions may also be prevalent in the glial cells leading to translational regulation.
In this study, we observed a more severe response in gliosis in vivo as compared to in vitro. Immunohistochemistry for BMP receptors in the mouse retina showed a large population of neuronal cells other than Müller glia and retinal astrocytes to express them. Thus, binding of BMP7 to receptors in these cells and to Müller glia could amplify the gliosis signal, leading to a comparatively severe response in the mouse retina.
Extracellular matrix and reactive gliosis
One of the responses seen during reactive gliosis is ECM modifications. These changes in the matrix are primarily due to the regulation of MMPs, which remove ECM molecules, and the regeneration-inhibiting chondroitin sulfate, ultimately leading to the formation of a glial scar [20,78]. The formation of the scar serves to protect the injured site while also preventing axonal regrowth [25].We observe here an increase in RNA levels of ECM molecules such as Ncan, Pcan, and Mmps, both in vivo and in vitro (Figure 3, Figure 4, and Figure 8). Furthermore, immunohistochemistry of the BMP7-injected mouse retina also showed an increase in localization of Ncan in the INLs of the retina (Figure 8). Other members of the TGF-β family have been previously shown to increase proteoglycan production in reactive glial cells [16]. Here, we observed an increase in Ncan at the 7 day stage when compared to the earlier 3 day time point, a result similar to a previous study [16]. Further study is necessary to determine whether the effects of BMPs on ECM production are direct or indirect.
Acknowledgments
The authors would like to thank Dr. Gloria A Limb (Ocular Biology and Therapeutics, UCL Institute of Ophthalmology, London) for MIO-M1 cell line. The authors also gratefully acknowledge funding support for this project; TBA is supported by RO1EY019525 and 1R15EY020816, CMS is supported R21 EY023024, NS is supported by RC4 EY021357, P30 EY016665 from the National Institutes of Health, an unrestricted departmental award from Research to Prevent Blindness, Research Award from American Diabetes Association 1–10-BS-160 and Retina Research Foundation.
References
- 1.Ffrench-Constant C, Miller RH, Burne JF, Raff MC. Evidence That Migratory Oligodendrocyte-Type-2 Astrocyte (O-2a) Progenitor Cells Are Kept out of the Rat Retina by a Barrier at the Eye-End of the Optic-Nerve. J Neurocytol. 1988;17:13–25. doi: 10.1007/BF01735374. [DOI] [PubMed] [Google Scholar]
- 2.Fischer AJ, Zelinka C, Scott MA. Heterogeneity of glia in the retina and optic nerve of birds and mammals. PLoS ONE. 2010;5:e10774. doi: 10.1371/journal.pone.0010774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rompani SB, Cepko CL. A common progenitor for retinal astrocytes and oligodendrocytes. J Neurosci. 2010;30:4970–80. doi: 10.1523/JNEUROSCI.3456-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perry VH, Lund RD. Evidence That the Lamina-Cribrosa Prevents Intraretinal Myelination of Retinal Ganglion-Cell Axons. J Neurocytol. 1990;19:265–72. doi: 10.1007/BF01217304. [DOI] [PubMed] [Google Scholar]
- 5.Newman E, Reichenbach A. The Muller cell: a functional element of the retina. Trends Neurosci. 1996;19:307–12. doi: 10.1016/0166-2236(96)10040-0. [DOI] [PubMed] [Google Scholar]
- 6.Huxlin KR, Sefton AJ, Furby JH. The Origin and Development of Retinal Astrocytes in the Mouse. J Neurocytol. 1992;21:530–44. doi: 10.1007/BF01186955. [DOI] [PubMed] [Google Scholar]
- 7.Dubois-Dauphin M, Poitry-Yamate C, De Bilbao F, Julliard AK, Jourdan F, Donati G. Early postnatal Muller cell death leads to retinal but not optic nerve degeneration in NSE-HU-BCL-2 transgenic mice. Neuroscience. 2000;95:9–21. doi: 10.1016/s0306-4522(99)00313-9. [DOI] [PubMed] [Google Scholar]
- 8.Kuchler-Bopp S, Delaunoy JP, Artault JC, Zaepfel M, Dietrich JB. Astrocytes induce several blood-brain barrier properties in non-neural endothelial cells. Neuroreport. 1999;10:1347–53. doi: 10.1097/00001756-199904260-00035. [DOI] [PubMed] [Google Scholar]
- 9.Bringmann A, Iandiev I, Pannicke T, Wurm A, Hollborn M, Wiedemann P, Osborne NN, Reichenbach A. Cellular signaling and factors involved in Muller cell gliosis: Neuroprotective and detrimental effects. Prog Retin Eye Res. 2009;28:423–51. doi: 10.1016/j.preteyeres.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 10.Jadhav AP, Roesch K, Cepko CL. Development and neurogenic potential of Muller glial cells in the vertebrate retina. Prog Retin Eye Res. 2009;28:249–62. doi: 10.1016/j.preteyeres.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bringmann A, Pannicke T, Grosche J, Francke M, Wiedemann P, Skatchkov SN, Osborne NN, Reichenbach A. Muller cells in the healthy and diseased retina. Prog Retin Eye Res. 2006;25:397–424. doi: 10.1016/j.preteyeres.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 12.Hernandez MR, Miao H, Lukas T. Astrocytes in glaucomatous optic neuropathy. Prog Brain Res. 2008;173:353–73. doi: 10.1016/S0079-6123(08)01125-4. [DOI] [PubMed] [Google Scholar]
- 13.Bringmann A, Wiedemann P. Muller glial cells in retinal disease. Ophthalmologica Journal Augenheilkunde. 2012;227:1–19. doi: 10.1159/000328979. [DOI] [PubMed] [Google Scholar]
- 14.Fischer AJ, Omar G, Eubanks J, McGuire CR, Dierks BD, Reh TA. Different aspects of gliosis in retinal Muller glia can be induced by CNTF, insulin, and FGF2 in the absence of damage. Mol Vis. 2004;10:973–86. [PubMed] [Google Scholar]
- 15.Ridet JL, Malhotra SK, Privat A, Gage FH. Reactive astrocytes: cellular and molecular cues to biological function. Trends Neurosci. 1997;20:570–7. doi: 10.1016/s0166-2236(97)01139-9. [DOI] [PubMed] [Google Scholar]
- 16.Asher RA, Morgenstern DA, Fidler PS, Adcock KH, Oohira A, Braistead JE, Levine JM, Margolis RU, Rogers JH, Fawcett JW. Neurocan is upregulated in injured brain and in cytokine-treated astrocytes. J Neurosci. 2000;20:2427–38. doi: 10.1523/JNEUROSCI.20-07-02427.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nickerson PE, McLeod MC, Myers T, Clarke DB. Effects of epidermal growth factor and erythropoietin on Muller glial activation and phenotypic plasticity in the adult mammalian retina. J Neurosci Res. 2011;89:1018–30. doi: 10.1002/jnr.22629. [DOI] [PubMed] [Google Scholar]
- 18.Ekmark-Lewen S, Lewen A, Israelsson C, Li GL, Farooque M, Olsson Y, Ebendal T, Hillered L. Vimentin and GFAP responses in astrocytes after contusion trauma to the murine brain. Restor Neurol Neurosci. 2010;28:311–21. doi: 10.3233/RNN-2010-0529. [DOI] [PubMed] [Google Scholar]
- 19.Norton WT, Aquino DA, Hozumi I, Chiu FC, Brosnan CF. Quantitative aspects of reactive gliosis: a review. Neurochem Res. 1992;17:877–85. doi: 10.1007/BF00993263. [DOI] [PubMed] [Google Scholar]
- 20.Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009;32:638–47. doi: 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Belecky-Adams TL, Chernoff EC, Wilson JM, Dharmarajan S. Reactive Muller Glia as Potential Retinal Progenitors. 2013;04:24. [Google Scholar]
- 22.Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119:7–35. doi: 10.1007/s00401-009-0619-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee MJ, Chen CJ, Huang WC, Huang MC, Chang WC, Kuo HS, Tsai MJ, Lin YL, Cheng H. Regulation of chondroitin sulphate proteoglycan and reactive gliosis after spinal cord transection: effects of peripheral nerve graft and fibroblast growth factor 1. Neuropathol Appl Neurobiol. 2011;37:585–99. doi: 10.1111/j.1365-2990.2011.01182.x. [DOI] [PubMed] [Google Scholar]
- 24.Nakazawa T, Matsubara A, Noda K, Hisatomi T, She H, Skondra D, Miyahara S, Sobrin L, Thomas KL, Chen DF, Grosskreutz CL, Hafezi-Moghadam A, Miller JW. Characterization of cytokine responses to retinal detachment in rats. Mol Vis. 2006;12:867–78. [PubMed] [Google Scholar]
- 25.Silver J, Miller JH. Regeneration beyond the glial scar. Nat Rev Neurosci. 2004;5:146–56. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- 26.Setoguchi T, Yone K, Matsuoka E, Takenouchi H, Nakashima K, Sakou T, Komiya S, Izumo S. Traumatic injury-induced BMP7 expression in the adult rat spinal cord. Brain Res. 2001;921:219–25. doi: 10.1016/s0006-8993(01)03123-7. [DOI] [PubMed] [Google Scholar]
- 27.Hampton DW, Asher RA, Kondo T, Steeves JD, Ramer MS, Fawcett JW. A potential role for bone morphogenetic protein signalling in glial cell fate determination following adult central nervous system injury in vivo. Eur J Neurosci. 2007;26:3024–35. doi: 10.1111/j.1460-9568.2007.05940.x. [DOI] [PubMed] [Google Scholar]
- 28.Fuller ML, DeChant AK, Rothstein B, Caprariello A, Wang R, Hall AK, Miller RH. Bone morphogenetic proteins promote gliosis in demyelinating spinal cord lesions. Ann Neurol. 2007;62:288–300. doi: 10.1002/ana.21179. [DOI] [PubMed] [Google Scholar]
- 29.Sahni V, Mukhopadhyay A, Tysseling V, Hebert A, Birch D, McGuire TL, Stupp SI, Kessler JA. BMPR1a and BMPR1b signaling exert opposing effects on gliosis after spinal cord injury. J Neurosci. 2010;30:1839–55. doi: 10.1523/JNEUROSCI.4459-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bragdon B, Moseychuk O, Saldanha S, King D, Julian J, Nohe A. Bone morphogenetic proteins: a critical review. Cell Signal. 2011;23:609–20. doi: 10.1016/j.cellsig.2010.10.003. [DOI] [PubMed] [Google Scholar]
- 31.Dai L, Thu CA, Liu XY, Xi JJ, Cheung PCF. TAK1, more than just innate immunity. IUBMB Life. 2012;64:825–34. doi: 10.1002/iub.1078. [DOI] [PubMed] [Google Scholar]
- 32.Rajan P, Panchision DM, Newell LE, McKay RDG. BMPs signal alternately through a SMAD or FRAP-STAT pathway to regulate fate choice in CNS stem cells. J Cell Biol. 2003;161:911–21. doi: 10.1083/jcb.200211021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Iandiev I, Wurm A, Pannicke T, Wiedemann P, Reichenbach A, Robson SC, Zimmermann H, Bringmann A. Ectonucleotidases in Muller glial cells of the rodent retina: Involvement in inhibition of osmotic cell swelling. Purinergic Signal. 2007;3:423–33. doi: 10.1007/s11302-007-9061-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sehgal R, Sheibani N, Rhodes SJ, Belecky Adams TL. BMP7 and SHH regulate Pax2 in mouse retinal astrocytes by relieving TLX repression. Dev Biol. 2009;332:429–43. doi: 10.1016/j.ydbio.2009.05.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scheef E, Wang S, Sorenson CM, Sheibani N. Isolation and characterization of murine retinal astrocytes. Mol Vis. 2005;11:613–24. [PubMed] [Google Scholar]
- 36.Limb GA, Salt TE, Munro PM, Moss SE, Khaw PT. In vitro characterization of a spontaneously immortalized human Muller cell line (MIO-M1). Invest Ophthalmol Vis Sci. 2002;43:864–9. [PubMed] [Google Scholar]
- 37.Cassina P, Peluffo H, Pehar M, Martinez-Palma L, Ressia A, Beckman JS, Estevez AG, Barbeito L. Peroxynitrite triggers a phenotypic transformation in spinal cord astrocytes that induces motor neuron apoptosis. J Neurosci Res. 2002;67:21–9. doi: 10.1002/jnr.10107. [DOI] [PubMed] [Google Scholar]
- 38.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fitch MT, Silver J. CNS injury, glial scars, and inflammation: Inhibitory extracellular matrices and regeneration failure. Exp Neurol. 2008;209:294–301. doi: 10.1016/j.expneurol.2007.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bringmann A, Pannicke T, Biedermann B, Francke M, Iandiev I, Grosche J, Wiedemann P, Albrecht J, Reichenbach A. Role of retinal glial cells in neurotransmitter uptake and metabolism. Neurochem Int. 2009;54:143–60. doi: 10.1016/j.neuint.2008.10.014. [DOI] [PubMed] [Google Scholar]
- 41.Derouiche A, Rauen T. Coincidence of L-glutamate/L-aspartate transporter (GLAST) and glutamine synthetase (GS) immunoreactions in retinal glia: evidence for coupling of GLAST and GS in transmitter clearance. J Neurosci Res. 1995;42:131–43. doi: 10.1002/jnr.490420115. [DOI] [PubMed] [Google Scholar]
- 42.Grosche J, Hartig W, Reichenbach A. Expression of glial fibrillary acidic protein (GFAP), glutamine synthetase (GS), and Bcl-2 protooncogene protein by Muller (glial) cells in retinal light damage of rats. Neurosci Lett. 1995;185:119–22. doi: 10.1016/0304-3940(94)11239-f. [DOI] [PubMed] [Google Scholar]
- 43.Lewis GP, Erickson PA, Guerin CJ, Anderson DH, Fisher SK. Changes in the expression of specific Muller cell proteins during long-term retinal detachment. Exp Eye Res. 1989;49:93–111. doi: 10.1016/0014-4835(89)90079-1. [DOI] [PubMed] [Google Scholar]
- 44.Reichenbach A, Stolzenburg JU, Wolburg H, Hartig W, el-Hifnawi E, Martin H. Effects of enhanced extracellular ammonia concentration on cultured mammalian retinal glial (Muller) cells. Glia. 1995;13:195–208. doi: 10.1002/glia.440130306. [DOI] [PubMed] [Google Scholar]
- 45.Chang ML, Wu CH, Jiang-Shieh YF, Shieh JY, Wen CY. Reactive changes of retinal astrocytes and Muller glial cells in kainate-induced neuroexcitotoxicity. J Anat. 2007;210:54–65. doi: 10.1111/j.1469-7580.2006.00671.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mizutani M, Gerhardinger C, Lorenzi M. Muller cell changes in human diabetic retinopathy. Diabetes. 1998;47:445–9. doi: 10.2337/diabetes.47.3.445. [DOI] [PubMed] [Google Scholar]
- 47.Chen H, Weber AJ. Expression of glial fibrillary acidic protein and glutamine synthetase by Muller cells after optic nerve damage and intravitreal application of brain-derived neurotrophic factor. Glia. 2002;38:115–25. doi: 10.1002/glia.10061. [DOI] [PubMed] [Google Scholar]
- 48.Cui M, Huang Y, Tian C, Zhao Y, Zheng J. FOXO3a inhibits TNF-alpha- and IL-1beta-induced astrocyte proliferation:Implication for reactive astrogliosis. Glia. 2011;59:641–54. doi: 10.1002/glia.21134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fontaine V, Mohand-Said S, Hanoteau N, Fuchs C, Pfizenmaier K, Eisel U. Neurodegenerative and neuroprotective effects of tumor Necrosis factor (TNF) in retinal ischemia: opposite roles of TNF receptor 1 and TNF receptor 2. J Neurosci. 2002;22:RC216. doi: 10.1523/JNEUROSCI.22-07-j0001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xue W, Cojocaru RI, Dudley VJ, Brooks M, Swaroop A, Sarthy VP. Ciliary neurotrophic factor induces genes associated with inflammation and gliosis in the retina: a gene profiling study of flow-sorted, Muller cells. PLoS ONE. 2011;6:e20326. doi: 10.1371/journal.pone.0020326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Matsuura I, Taniguchi J, Hata K, Saeki N, Yamashita T. BMP inhibition enhances axonal growth and functional recovery after spinal cord injury. J Neurochem. 2008;105:1471–9. doi: 10.1111/j.1471-4159.2008.05251.x. [DOI] [PubMed] [Google Scholar]
- 52.Fuller ML, DeChant AK, Rothstein B, Caprariello A, Wang R, Hall AK, Miller RH. Bone morphogenetic proteins promote gliosis in demyelinating spinal cord lesions. Ann Neurol. 2007;62:288–300. doi: 10.1002/ana.21179. [DOI] [PubMed] [Google Scholar]
- 53.Masumoto A, Hirooka Y, Shimokawa H, Hironaga K, Setoguchi S, Takeshita A. Possible involvement of Rho-kinase in the pathogenesis of hypertension in humans. Hypertension. 2001;38:1307–10. doi: 10.1161/hy1201.096541. [DOI] [PubMed] [Google Scholar]
- 54.Masumoto A, Hirooka Y, Hironaga K, Eshima K, Setoguchi S, Egashira K, Takeshita A. Effect of pravastatin on endothelial function in patients with coronary artery disease (cholesterol-independent effect of pravastatin). Am J Cardiol. 2001;88:1291–4. doi: 10.1016/s0002-9149(01)02090-2. [DOI] [PubMed] [Google Scholar]
- 55.Fuchshofer R, Yu AH, Welge-Lussen U, Tamm ER. Bone morphogenetic protein-7 is an antagonist of transforming growth factor-beta2 in human trabecular meshwork cells. Invest Ophthalmol Vis Sci. 2007;48:715–26. doi: 10.1167/iovs.06-0226. [DOI] [PubMed] [Google Scholar]
- 56.Wordinger RJ, Fleenor DL, Hellberg PE, Pang IH, Tovar TO, Zode GS, Fuller JA, Clark AF. Effects of TGF-beta2, BMP-4, and gremlin in the trabecular meshwork: implications for glaucoma. Invest Ophthalmol Vis Sci. 2007;48:1191–200. doi: 10.1167/iovs.06-0296. [DOI] [PubMed] [Google Scholar]
- 57.Zode GS, Clark AF, Wordinger RJ. Activation of the BMP canonical signaling pathway in human optic nerve head tissue and isolated optic nerve head astrocytes and lamina cribrosa cells. Invest Ophthalmol Vis Sci. 2007;48:5058–67. doi: 10.1167/iovs.07-0127. [DOI] [PubMed] [Google Scholar]
- 58.Ueki Y, Reh TA. EGF stimulates Muller glial proliferation via a BMP-dependent mechanism. Glia. 2013;61:778–89. doi: 10.1002/glia.22472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Miyazono K, Kamiya Y, Morikawa M. Bone morphogenetic protein receptors and signal transduction. J Biochem. 2010;147:35–51. doi: 10.1093/jb/mvp148. [DOI] [PubMed] [Google Scholar]
- 60.Fischer AJ, Schmidt M, Omar G, Reh TA. BMP4 and CNTF are neuroprotective and suppress damage-induced proliferation of Muller glia in the retina. Mol Cell Neurosci. 2004;27:531–42. doi: 10.1016/j.mcn.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 61.Tucker B, Klassen H, Yang L, Chen DF, Young MJ. Elevated MMP Expression in the MRL Mouse Retina Creates a Permissive Environment for Retinal Regeneration. Invest Ophthalmol Vis Sci. 2008;49:1686–95. doi: 10.1167/iovs.07-1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ganesh BS, Chintala SK. Inhibition of reactive gliosis attenuates excitotoxicity-mediated death of retinal ganglion cells. PLoS ONE. 2011;6:e18305. doi: 10.1371/journal.pone.0018305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Steward O, Kelley MS, Torre ER. The process of reinnervation in the dentate gyrus of adult rats: temporal relationship between changes in the levels of glial fibrillary acidic protein (GFAP) and GFAP mRNA in reactive astrocytes. Exp Neurol. 1993;124:167–83. doi: 10.1006/exnr.1993.1187. [DOI] [PubMed] [Google Scholar]
- 64.Inman DM, Horner PJ. Reactive nonproliferative gliosis predominates in a chronic mouse model of glaucoma. Glia. 2007;55:942–53. doi: 10.1002/glia.20516. [DOI] [PubMed] [Google Scholar]
- 65.Liu C, Li Y, Lein PJ, Ford BD. Spatiotemporal patterns of GFAP upregulation in rat brain following acute intoxication with diisopropylfluorophosphate (DFP). Current Neurobiology. 2012;3:90–7. [PMC free article] [PubMed] [Google Scholar]
- 66.Wu X, Hsuchou H, Kastin AJ, Mishra PK, Pan W. Upregulation of astrocytic leptin receptor in mice with experimental autoimmune encephalomyelitis. J Mol Neurosci. 2013;49:446–56. doi: 10.1007/s12031-012-9825-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wong RW, Hagen T. Mechanistic target of rapamycin (mTOR) dependent regulation of thioredoxin interacting protein (TXNIP) transcription in hypoxia. Biochem Biophys Res Commun. 2013;433:40–6. doi: 10.1016/j.bbrc.2013.02.070. [DOI] [PubMed] [Google Scholar]
- 68.Baek D, Villen J, Shin C, Camargo FD, Gygi SP, Bartel DP. The impact of microRNAs on protein output. Nature. 2008;455:64–71. doi: 10.1038/nature07242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Di Liegro CM, Schiera G, Di Liegro I. Regulation of mRNA transport, localization and translation in the nervous system of mammals. Int J Mol Med. 2014;33:747–62. doi: 10.3892/ijmm.2014.1629. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kim KC, Hyun Joo S, Shin CY. CPEB1 modulates lipopolysaccharide-mediated iNOS induction in rat primary astrocytes. Biochem Biophys Res Commun. 2011;409:687–92. doi: 10.1016/j.bbrc.2011.05.065. [DOI] [PubMed] [Google Scholar]
- 71.Cargnello M, Roux PP. Activation and Function of the MAPKs and Their Substrates, the MAPK-Activated Protein Kinases (vol 75, pg 50, 2011). Microbiol Mol Biol Rev. 2012;76:496. doi: 10.1128/MMBR.00031-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hackler L, Wan J, Swaroop A, Qian J, Zack DJ. MicroRNA Profile of the Developing Mouse Retina. Invest Ophthalmol Vis Sci. 2010;51:1823–31. doi: 10.1167/iovs.09-4657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.de Sousa É, Walter LT, Higa GSV, Augusto O, Casado N, Kihara AH. Developmental and Functional Expression of miRNA-Stability Related Genes in the Nervous System. PLoS ONE. 2013;8 doi: 10.1371/journal.pone.0056908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Davis BN, Hilyard AC, Lagna G, Hata A. SMAD proteins control DROSHA-mediated microRNA maturation. Nature. 2008;454:56–61. doi: 10.1038/nature07086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ning G, Liu X, Dai M, Meng A, Wang Q. MicroRNA-92a upholds Bmp signaling by targeting noggin3 during pharyngeal cartilage formation. Dev Cell. 2013;24:283–95. doi: 10.1016/j.devcel.2012.12.016. [DOI] [PubMed] [Google Scholar]
- 76.Bhalala OG, Srikanth M, Kessler JA. The emerging roles of microRNAs in CNS injuries. Nat Rev Neurol. 2013;9:328–39. doi: 10.1038/nrneurol.2013.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ling S, Birnbaum Y, Nanhwan MK, Thomas B, Bajaj M, Ye YM. MicroRNA-dependent cross-talk between VEGF and HIF1 alpha in the diabetic retina. Cell Signal. 2013;25:2840–7. doi: 10.1016/j.cellsig.2013.08.039. [DOI] [PubMed] [Google Scholar]
- 78.Pizzi MA, Crowe MJ. Matrix metalloproteinases and proteoglycans in axonal regeneration. Exp Neurol. 2007;204:496–511. doi: 10.1016/j.expneurol.2006.12.012. [DOI] [PubMed] [Google Scholar]
- 79.Lewis GP, Fisher SK. Up-regulation of glial fibrillary acidic protein in response to retinal injury: its potential role in glial remodeling and a comparison to vimentin expression. Int Rev Cytol. 2003;230:263–90. doi: 10.1016/s0074-7696(03)30005-1. [DOI] [PubMed] [Google Scholar]
- 80.Ziman MR, Rodger J, Chen P, Papadimitriou JM, Dunlop SA, Beazley LD. Pax genes in development and maturation of the vertebrate visual system: Implications for optic nerve regeneration. Histol Histopathol. 2001;16:239–49. doi: 10.14670/HH-16.239. [DOI] [PubMed] [Google Scholar]
- 81.Kim D, Kim MJ, Lee JH, Im JO, Won YJ, Yoon SY, Hong HN. Concomitant distribution shift of glial GABA transporter and S100 calcium-binding proteins in the rat retina after kainate-induced excitotoxic injury. Neurosci Lett. 2003;353:17–20. doi: 10.1016/j.neulet.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 82.Zou J, Wang YX, Mu HJ, Xiang J, Wu W, Zhang B, Xie P. Down-regulation of glutamine synthetase enhances migration of rat astrocytes after in vitro injury. Neurochem Int. 2011;58:404–13. doi: 10.1016/j.neuint.2010.12.018. [DOI] [PubMed] [Google Scholar]
- 83.Liu B, Neufeld AH. Activation of epidermal growth factor receptors in astrocytes: From development to neural injury. J Neurosci Res. 2007;85:3523–9. doi: 10.1002/jnr.21364. [DOI] [PubMed] [Google Scholar]
- 84.Kigerl KA, Lai WM, Rivest S, Hart RP, Satoskar AR, Popovich PG. Toll-like receptor (TLR)-2 and TLR-4 regulate inflammation, gliosis, and myelin sparing after spinal cord injury. J Neurochem. 2007;102:37–50. doi: 10.1111/j.1471-4159.2007.04524.x. [DOI] [PubMed] [Google Scholar]
- 85.Tang X, Davies JE, Davies SJA. Changes in distribution, cell associations, and protein expression levels of NG2, neurocan, phosphacan, brevican, versican V2, and tenascin-C during acute to chronic maturation of spinal cord scar tissue. J Neurosci Res. 2003;71:427–44. doi: 10.1002/jnr.10523. [DOI] [PubMed] [Google Scholar]
- 86.Pizzi MA, Crowe MJ. Matrix metalloproteinases and proteoglycans in axonal regeneration. Exp Neurol. 2007;204:496–511. doi: 10.1016/j.expneurol.2006.12.012. [DOI] [PubMed] [Google Scholar]
- 87.Tucker B, Klassen H, Yang L, Chen DF, Young MJ. Elevated MMP expression in the MRL mouse retina creates a permissive environment for retinal regeneration. Invest Ophthalmol Vis Sci. 2008;49:1686–95. doi: 10.1167/iovs.07-1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Woldemussie E, Wijono M, Ruiz G. Muller cell response to laser-induced increase in intraocular pressure in rats. Glia. 2004;47:109–19. doi: 10.1002/glia.20000. [DOI] [PubMed] [Google Scholar]
- 89.Tonari M, Kurimoto T, Horie T, Sugiyama T, Ikeda T, Oku H. Blocking Endothelin-B Receptors Rescues Retinal Ganglion Cells from Optic Nerve Injury through Suppression of Neuroinflammation. Invest Ophthalmol Vis Sci. 2012;53:3490–500. doi: 10.1167/iovs.11-9415. [DOI] [PubMed] [Google Scholar]
- 90.Bi F, Huang C, Tong JB, Qiu G, Huang B, Wu QX, Li F, Xu ZS, Bowser R, Xia XG, Zhou HX. Reactive astrocytes secrete lcn2 to promote neuron death. Proc Natl Acad Sci USA. 2013;110:4069–74. doi: 10.1073/pnas.1218497110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Friese MA, Craner MJ, Etzensperger R, Vergo S. AWemmie J, Welsh MJ, Vincent A, Fugger L. Acid-sensing ion channel-1 contributes to axonal degeneration in autoimmune inflammation of the central nervous system. Nat Med. 2007;13:1483–9. doi: 10.1038/nm1668. [DOI] [PubMed] [Google Scholar]
- 92.Eberhardt C, Amann B, Feuchtinger A, Hauck SM, Deeg CA. Differential expression of inwardly rectifying K+ channels and aquaporins 4 and 5 in autoimmune uveitis indicates misbalance in Muller glial cell-dependent ion and water homeostasis. Glia. 2011;59:697–707. doi: 10.1002/glia.21139. [DOI] [PubMed] [Google Scholar]
- 93.Küry P, Zickler P, Stoll G, Hartung HP, Jander S. Osteopontin, a macrophage-derived matricellular glycoprotein, inhibits axon outgrowth. FASEB J. 2005;19:398. doi: 10.1096/fj.04-1777fje. [DOI] [PubMed] [Google Scholar]
- 94.Devi TS, Lee I, Huttemann M, Kumar A, Nantwi KD, Singh LP. TXNIP Links Innate Host Defense Mechanisms to Oxidative Stress and Inflammation in Retinal Muller Glia under Chronic Hyperglycemia: Implications for Diabetic Retinopathy. Exp Diabetes Res. 2012;2012:438238. doi: 10.1155/2012/438238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yan YP, Lang BT, Vemuganti R, Dempsey RJ. Galectin-3 mediates post-ischemic tissue remodeling. Brain Res. 2009;1288:116–24. doi: 10.1016/j.brainres.2009.06.073. [DOI] [PubMed] [Google Scholar]